

Abstract
Targeting host functions essential for viral replication has been considered as a broad spectrum and resistance-refractory antiviral approach. However, only a few host functions have, thus far, been validated as broad-spectrum antiviral targets in vivo. ER α-glucosidases I and II have been demonstrated to be essential for the morphogenesis of many enveloped viruses, including members from four families of viruses causing hemorrhagic fever. In vivo antiviral efficacy of various iminosugar-based ER α-glucosidase inhibitors has been reported in animals infected with Dengue, Japanese encephalitis, Ebola, Marburg and influenza viruses. Herein, we established Huh7.5-derived cell lines with ER-α-glucosidase I or II knockout using CRISPR/Cas9 and demonstrated that the replication of Dengue, Yellow fever and Zika viruses was reduced by only 1–2 logs in the knockout cell lines. The results clearly indicate that only a partial suppression of viral replication can possibly be achieved with a complete inhibition of ER-α-glucosidases I or II by their inhibitors. We therefore explore to improve the antiviral efficacy of a lead iminosugar IHVR-19029 through combination with another broad-spectrum antiviral agent, favipiravir (T-705). Indeed, combination of IHVR-19029 and T-705 synergistically inhibited the replication of Yellow fever and Ebola viruses in cultured cells. Moreover, in a mouse model of Ebola virus infection, combination of sub-optimal doses of IHVR-19029 and T-705 significantly increased the survival rate of infected animals. We have thus proved the concept of combinational therapeutic strategy for the treatment of viral hemorrhagic fevers with broad spectrum host- and viral- targeting antiviral agents.
1. Introduction
Viral hemorrhagic fevers can be caused by four distinct viral families, including Arenaviridae (Junin, Lassa, Machupo and Pichinde viruses), Bunyaviridae (Rift Valley fever, Crimean-Congo hemorrhagic fever, Hanta viruses), Filoviridae (Ebola and Marburg viruses), and Flaviviridae (Yellow fever, Dengue and Zika viruses). Hemorrhagic fever diseases caused by viruses from different families usually present with similar clinical manifestations. Moreover, in the tropical areas, in practice, febrile and hemorrhagic syndromes are often treated based on clinical and epidemiologic information without precise etiological diagnosis. While cost is one of the issues, time-consumption is another important factor since early treatment is essential for favorable clinical outcomes (Chang et al., 2013a; Watanabe et al., 2016). Although these viruses are geographically restricted to the areas where their host/vector species live, hemorrhagic fever caused by different viruses can be co-epidemic in the same geographic areas, due in part, to the vectors commonly used by different viruses. For example, Dengue, Yellow fever and Zika viruses are all transmitted by Aedes aegypti mosquitoes. Accordingly, development of broad-spectrum antiviral agents that are active against multiple hemorrhagic fever viruses is an optimal solution. In recent years, infections with emerging hemorrhagic fever viruses, such as dengue, Ebola and the most recently re-emerging Zika virus, against which no vaccines (or limited use) or antiviral therapies are approved, have become an increasing global risk. In fact, more than 10 hemorrhagic fever viruses have been listed as agents causing emerging, and re-emerging infectious diseases (http://www.niaid.nih.gov/about/whoWeAre/profile/fy2004/Documents/research_emerging_reemerging.pdf). Therefore, a broad-spectrum strategy is also desirable to prepare for the next hemorrhagic fever epidemic, which could also potentially emerge from as-yet unidentified or existing neglected viruses.
Although the hemorrhagic fever viruses all have RNA genomes, their genome sequences and structures are significantly distinct from each other. Effective broad spectrum antivirals directly targeting the viral proteins are rare, except for a few examples of nucleotide/nucleoside analogs targeting the viral polymerases of more than one viral species (Boldescu et al., 2017; De Clercq and Li, 2016). Interestingly, one common feature of all these hemorrhagic fever viruses is that they are all enveloped by viral encoded N-linked glycoproteins that appear to require processing by host ER α-glucosidases. By sequentially cleaving glucose residues from N-linked glycans attached to the immature glycoproteins in the endoplasmic reticulum, the host cellular carbohydrate processing enzymes, ER α-glucosidases I and II, are proved to be critical for viral envelope proteins to properly fold, oligomerize and ultimately form the envelope of the virion particles (Alonzi et al., 2017; Chang et al., 2013a; Chang et al., 2013b; Warfield et al., 2016b). Although these enzymes are also used in the processing of host glycoproteins, there is an apparent greater dependency upon certain viruses that is greater than of host functions, resulting in selectivity for inhibition of viral morphogenesis. Perhaps the assembly of infectious virion particles relies on highly coordinated interaction of multiple copies of envelope glycoproteins, and thus misfolding of a small fraction of viral glycoproteins may lead to a dramatic defect in virion assembly and infectivity (Alonzi et al., 2017; Chang et al., 2015; Dwek et al., 2002).
Consistent with the critical role of ER α-glucosidases in the life cycle of enveloped viruses, Sadat et al reported that cells from patients genetically deficient in the gene encoding ER α-glucosidase I are naturally resistant to productive infection of multiple enveloped viruses (Sadat et al., 2014). It has also been well documented that two families of small molecule ER α-glucosidase inhibitors, indolizidine and 1-deoxynojirimycin (DNJ)-derived iminosugar compounds, suppressed many enveloped viruses in vitro, including one or multiple members from each of the four families of viruses causing hemorrhagic fever (Chang et al., 2013a; Chang et al., 2013c; Perry et al., 2013; Schul et al., 2007; Tyrrell et al., 2017; Warfield et al., 2017), and in vivo in animal models of flavivirus and filovirus infections such as dengue, Ebola and Marburg viruses (Chang et al., 2013a; Chang et al., 2013b; Chang et al., 2011a; Chang et al., 2011b; Schul et al., 2007; Warfield et al., 2016b; Whitby et al., 2005). In addition to hemorrhagic fever viruses, the in vivo efficacy of small molecule ER glucosidase inhibitors has also been demonstrated in animal models of Influenza virus and Japanese encephalitis virus infection (Warfield et al., 2016a; Wu et al., 2002).
In this study, to evaluate the maximum extent of antiviral potency by targeting ER glucosidase I and II, we examined Dengue virus (DENV), Yellow fever virus (YFV) and Zika virus (ZIKV) infections in human hepatoma derived cell lines with either ER α-glucosidases I or II knockout, and demonstrated that only partial inhibition of these viruses were achieved in the absence of ER glucosidase I or II. IHVR-19029 is a lead N-alkyl analog of DNJ with demonstrated broad-spectrum antiviral efficacies against at least one member from each of the four families of hemorrhagic fever viruses in vitro, and partial protection against Ebola virus (EBOV) and Marburg virus (MARV) in lethal infection mouse models (Chang et al., 2013c; Du et al., 2013a; Du et al., 2013b; Ma et al., 2017). In an effort to further improve the therapeutic efficacy, we demonstrated that combination of IHVR-19029 with another broad spectrum antiviral agent favipiravir (T-705) synergistically inhibited YFV and EBOV in vitro and increased the survival rates of lethal EBOV infected mice.
2. Materials and methods
2.1. Cells and chemicals
HEK293 cells (ATCC), Vero cells (ATCC) and Huh7.5 cells (a gift of Dr. Charles M. Rice at Rockefeller University) were maintained in Dulbecco’s modified minimal essential medium (DMEM, Corning) supplemented with 10% fetal bovine serum (FBS). HeLa (CCL-2) (ATCC) was cultured in DMEM supplemented with 10% FBS, 1% L-glutamine, 10 mM HEPES and 1% non-essential amino acids. TLR3-expressing HEK293 (293TLR3HA, Invivogen)-derived stable reporter cell line expressing firefly luciferase under the control of a human IFN-β promoter (293TLR3/IFNβLuc) were established and cultured as described previously (Guo et al., 2012). IHVR-19029 was synthesized in house with 95% purity (Ma et al., 2017). favipiravir (T-705, 6-fluoro-3-hydroxy-2-pyrazinecarboxamide) was either purchased from AdooQ Bioscience, or obtained from Medivector, Inc. (for EBOV studies).
2.2. Generation of glucosidase I and II knockout cell lines
The gene mannosyl-oligosaccharide glucosidase (MOGS) encodes ER α-glucosidase I, the first enzyme in the N-linked oligosaccharide processing pathway. The gene glucosidase II alpha subunit (GANAB) encodes the alpha subunit of ER α-glucosidase II. To generate glucosidase I and II knockout cell lines, Huh7.5 cells were first transduced with Cas9 expressing plasmid, hEF1a-Blast-Cas9 (GE Heathcare Dharmacon). Two days after transduction, the cells were re-seeded at a density of 104 cells per 10-cm dish and cultured in medium containing 5 μg/ml of blasticidin. Single cell clones expressing Cas9 were expanded into cell lines (Huh7.5 Cas9). The expression of Cas9 was determined by Western blot assay. sgRNA CRISPR lentiviral vectors were purchased from Applied Biological Materials. The target sequence for MOGS is GGTCCTCTAGTAGGTCAAGG, and for GANAB, GAGTCGGCAGGCAACACAGG. Lentiviral particles were packaged in Lenti-X 293T cells using Lenti-X Packaging Single Shots according to the procedure provided by the manufacturer (Clontech). Huh7.5 Cas9 cells generated above were seeded for 50% confluence and transduced twice by using 1:1 ratio of cell culture media and lentivirus-containing supernatants. The cells were allowed to grow to confluence and then selected with the corresponding selective agent (5μg/ml blasticidin or 400μg/ml G418 sulfate, respectively) to generate single clone-derived cell lines.
2.3. Western blot assay
Cell lysates were resolved by electrophoresis in 4–12% Bis-Tris gel and transferred onto PVDF membrane (Invitrogen). The membranes were blocked and probed with antibodies against Cas9 (Cell Signaling 14697) at a concentration of 1:1000, ER α-glucosidase I (Sigma HPA011969) at 1:250, ER α-glucosidase II (Abcam ab68814) at 1:1000 or β-actin (Cell Signaling 3700) at 1:1000, and followed by incubation with IRDye secondary antibodies and imaging with LI-COR Odyssey system (LI-COR Biotechnology).
2.4. Virus infection and analyses
DENV, YFV and ZIKV infection
YFV 17D and DENV serotype 2 (New Guinea C) virus stocks were produced as reported previously (Guo et al., 2016; Qu et al., 2011) by electroporation of Huh7.5 cells with in vitro transcribed RNA from pACNR/FLYF-17Dx (a gift of Dr. Charles M. Rice (Bredenbeek et al., 2003; Rice et al., 1985)) and pACYC177-NGC-DENV-2 (a gift of Dr. Pei-Yong Shi (Xie et al., 2013)), respectively. ZIKV (PRVABC59) was purchased from ATCC. For YFV, DENV or ZIKV infection, cells seeded in 96-well plates at a density of 4×104/well in 200 μl of medium/well were infected with the virus at 0.1 MOI for one hour and then washed gently with phosphate buffer saline (Genesee Scientific) twice, after which medium (with or without compound) was added and cultured for 48 hrs. To detect intracellular viral RNA, total cellular RNAs were extracted using NucleoSpin 96 RNA kit (Macherey-Nagel). Intracellular viral RNA was detected by one step qRT-PCR with SuperScript II Platinum SYBR Green Kit (Invitrogen) on LightCycler 480II (Roche) using viral specific primers as reported previously (Adcock et al., 2016; Guo et al., 2016; Ma et al., 2017). β-actin mRNA was also determined in parallel to serve as an internal control. Viral titers in the cell culture media were determined in Vero cells by plaque assay. Briefly, Vero cells were seeded at a density of 2×104 cells/well (1 ml medium/well) in a 24-well plate overnight. The culture media containing viral particles were diluted in a 10-fold scale from 10−1 to 10−6 in 200 μl/well Opti-Mem medium (Gibco). Vero cells were infected for 1 hr followed by incubation in DMEM medium with 0.75% methylcellulose for 5–7 days until the plaque became visible. To report the virus replication using IFN-β promoter driven Luciferase reporter assay, 293TLR3/IFNβLuc cells were seeded in 96-well plate and infected with YFV at 0.1 MOI followed by treatment of indicated concentrations of compounds for 48 hrs. The firefly luciferase activities were measured using Steady-Glo (Promega), followed by luminometry in a TopCounter (Perkin Elmer) (Guo et al., 2014).
EBOV infection
Hela cells were plated in 40μl of culture media at 2,000 cells/well into 384 well plates (Aurora 384 IQ-EB/NB) and incubated for 20–24 hrs before compound treatment and infection. Cells were treated with indicated concentrations of compounds 24 hrs prior to infection and then transferred to the BSL-4 suite and infected with EBOV for 48 hrs (Makona isolate at a MOI of 1.5 in antiviral assay shown in Fig. 2, or EBOV Zaire at MOI of 2.5 in synergistic study shown in Fig. 3). Immunostaining was performed to visualize infected cells using antigen specific monoclonal antibody 6D8 anti-GP (Wilson et al., 2000) and DyLight 488 anti-mouse-IgG (Thermo). Draq5 (Biostatus) was used to stain the cell nuclei. Images are acquired on the Opera imaging instrument (Perkin Elmer) using 10x Air objective and four images/well were typically acquired. Signals of virus staining were detected at 488nm emission wavelength and nuclei at 640nm. Image analysis was performed using PE Acapella algorithms.
Figure 2. Antiviral activity of IHVR-19029 against DENV, YFV, ZIKV and EBOV in vitro.
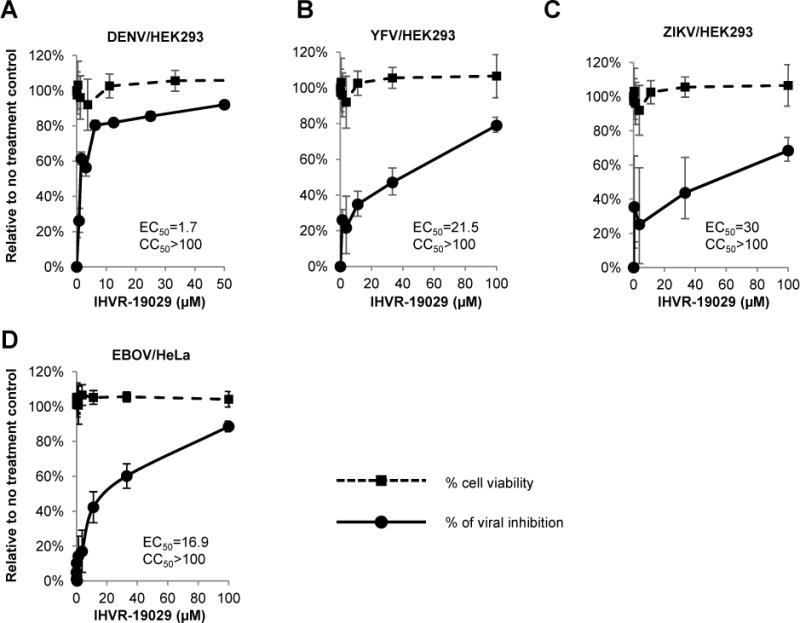
HEK293 cells were infected with viruses (DENV, YFV or ZIKV) at MOI of 0.1 for 1 hr and treated with concentrations of IHVR-19029 for additional 48 hrs. The levels of DENV RNA (A), YFV RNA (B) and ZIKV RNA (C) were determined by qRT-PCR using β-actin as internal control. Values were the percentages of inhibition on viral RNA relative to no treatment controls (mean± standard deviation, n=4). Cell viability was determined by MTT assay and expressed as percentage of no treatment controls (mean± standard deviation, n=3). (D) Hela cells were treated with indicated concentrations of IHVR-19029 2 hrs prior to infection with EBOV at MOI of 1.5. Immunostaining assay was performed to quantify EBOV GP protein. Values were the percentages of inhibition on florescence intensities relative to no treatment control (mean± standard deviation, n=4). Cell viability was determined by immunostaining of cell nuclei and expressed as percentage of no treatment control (mean± standard deviation, n=4).
Figure 3. Combination of ER α-glucosidase inhibitor IHVR-19029 and T-705 synergistically inhibited YFV and EBOV infection in vitro.
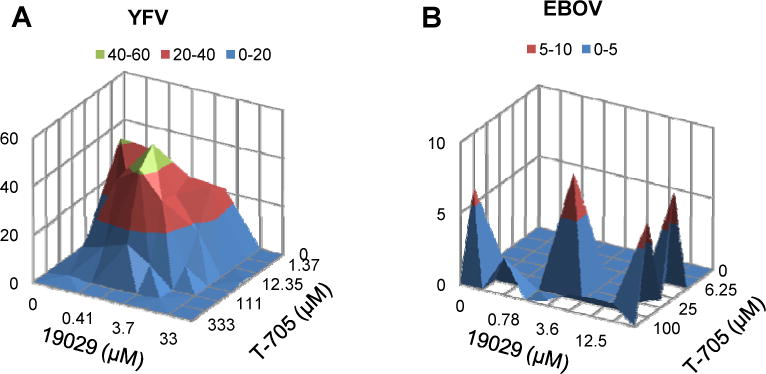
Antiviral activities of indicated doses of 19029 and T-705, either alone or in combination in a checkerboard Matrix format, were determined. (A) 293TLR3IFNβ Luc cells were seeded in 96-well plate for overnight and infected with either YFV at MOI of 0.1 for 1 hr. The cells were then treated with indicated concentrations of compounds or their combinations for additional 48 hrs followed by quantification of luciferase activity (n=6). (B) Hela cells were seeded in 384-well plate and treated with indicated concentrations of compounds or their combination for 2 hr. The cells were then infected with EBOV (Zaire) at MOI of 2.5 for 48 hrs. Immunostaining assay was performed to quantify EBOV GP protein (n=4). Synergy analysis was performed using MacSyngergy II. The peak above the surface represents the degree of synergy with 95% confidence interval.
2.5. Cytotoxicity Assay
To determine the cell viability, a MTT assay (Sigma) was performed. HEK293 cells were set up and incubated with various concentrations of compounds under condition that was identical to that used for antiviral assays of DENV, YFV and ZIKV, except that cells were not infected (Chang et al., 2009). In EBOV infected Hela cells, cytotoxicity was measured by number of cells in each treated well and expressed as percentage of viability relative to untreated control well using GeneData Scanner.
2.6 Pharmacokinetics and tolerability studies in mice
Pharmacokinetics and tolerability studies in mice were performed by QPS, LLC. Male Balb/c mice weighed 21.6 g to 25.9g were used for the studies. IHVR-19029 was formulated in a vehicle of phosphate buffered saline (PBS, pH = 7.4) and administrated by intraperitoneal injection (IP). In a single dosing study, each mouse was administrated with IHVR-19029 at 75 mg/kg. Blood as well as tissue samples (heart, kidney, liver, lung and spleen) were collected at pre-dose, 10 min, 30 min, 90 min, and 2, 4, 6, 8 hr post-dose. Three mice were included in each time point. In a repeated dosing study, each mouse was administered three times per day (TID, every 8 hours) of IHVR19029 via IP for 5 days at 50, 75, 100, 150 or 200 mg/kg/dose. At 4 hr post final dosing on day 5, blood as well as tissue samples (heart, kidney, liver, lung and spleen) were collected and weighted. Organs were snap-frozen by placing on aluminum foil in the petri dish over the vapor phase liquid for 5–10 min. Terminal plasma and frozen tissue samples were analyzed by LC/MS/MS to determine the concentration of IHVR19029. Three mice were included in each dosing group. Observations of tolerability were recorded for all animals during the acclimation period, at least once pre-dose and once post-dose. Cageside observations included morbidity, mortality, injury, and the availability of food and water. Clinical Observations included, but were not limited to, evaluation of the skin, fur, eyes, ears, nose, oral cavity, thorax, abdomen, external genitalia, limbs and feet, respiratory and circulatory effects, autonomic effects such as salivation, and nervous system effects including tremors, convulsions, reactivity to handling, and bizarre behavior. Food consumption was measured and recorded once daily during the dosing phases.
2.7 Glycan profiling in tissue samples isolated from mice treated with IHVR-19029
Isolation of Free Oligosaccharides (FOS)
25mg of the frozen mouse spleen was mixed with 1 ml of distilled water and frozen and thawed using a dry ice/methanol bath before homogenization. Samples were centrifuged for 5 minutes at maximum g in an Eppendorf centrifuge at 4°C. The supernatant was run through a porous graphitized carbon (PGC) column (Thermo Scientific), sequentially pre-equilibrated with methanol, water and acetonitrile (ACN) with 0.1% trifluoroacetic acid (TFA). Oligosaccharides were eluted with 2 ml of 50% ACN, 0.1% TFA and then dried in stages (300ul, 50ul, 10ul) in a speed-vac (Thermo Scientific). The free oligosaccharides were labeled with 10μl of aminobenzoic acid (2-AB), which was prepared with GlycoProfile 2-AB Labeling Kit (Sigma Aldrich), and then vortexed and incubated at 65°C for 2–3 hours. Labeled FOS were then spotted onto Whatman 3 MM paper which was then placed in a beaker containing acetonitrile and chromatographed until the solvent front reached the top of the paper. The paper was dried, and UV light was used to check for efficient removal of free 2-AB. The 2-AB-labeled glycans were then eluted and dried by speed-vac over various stages to ensure the labeled FOS were concentrated at the bottom of the tube, and then resuspended in 30μl of distilled water.
FOS analysis
Purified 2-AB-labelled oligosaccharides were separated by Normal Phase High-performance liquid chromatography (NP-HPLC) (Waters) using a 4.6×250mm TSKgel Amide-80 column (Fisher Scientific). Glycan compositions were determined following comparison with a 2-AB-labeled glucose oligomer ladder external standard. Peaks were analyzed using Waters Empower software.
2.8 In vivo efficacy study in mice infected with EBOV
8–12 week old C57Bl/6 mice were infected with 1000 pfu mouse-adapted EBOV-Zaire via IP injection. Treatment was initiated immediately after infection and typically completed within 120 mins for all the infected animals. In the T-705 dose-finding experiment, mice were randomly assigned into 5 groups with either mock treated or treated with one of the dose groups: 40mg/kg, 8mg/kg, 1.6 mg/kg or 0.325 mg/kg. T-705 was formulated in individual suspensions in sterile with 0.4% carboxymethylcellulose and administrated PO in 100 μl volume, once daily for 10 days. Each group included 10 mice. In the IHVR-19029 and T-705 combination experiment, mice were randomly assigned into 9 groups: mock treated, or treated with T-705 (at 0.325 or 1.6 mg/kg), IHVR-19029 (at 50 or 75 mg/kg) or the combinations of the two compounds at each dose for 10 days. IHVR-19029 was administrated via IP injection in 100 μl volume, twice daily. 0.4% carboxymethylcellulose and/or PBS were used as vehicle(s) in groups of animals mock treated or treated with one compound. Each group included 9 mice. Health and weight were monitored for 14 days. The mouse efficacy studies were conducted according to research protocols approved by the USAMRIID Institutional Animal Care and Use Committee (IACUC). This was in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council. The BSL-4 facility where this research was conducted is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
2.9 Statistics
P values were calculated using 2-tailed student’s t-test. Synergy effect was evaluated using MacSyngergy II. Log-rank (Mantel-cox) test was performed for animal survival analysis (GraphPad Prism 7).
3 Results
3.1 Replication of hemorrhagic fever viruses in cells with ER α-glucosidase I or II knockout
Previously we performed target validation studies using siRNAs of ER glucosidase I and II, which only resulted in a partial knockdown of ER α-glucosidase I and II expression and a less than 1 log reduction of hepatitis C virus (Qu et al., 2011) and DENV (data not shown). In this study, we used CRISPR/Cas9 technology to establish Huh7.5-derived cell lines with knockout of either MOGS or GANAB gene encoding ER α-glucosidase I or II, respectively (Huh7.5/Gluc I KO or Huh7.5 Gluc II KO). Fig. 1A and B shows the successful knockout of ER α-glucosidase I or II expression in the two cell lines, as determined by western blot assay. The ER α-glucosidase I and II knockout cells were infected with DENV, YFV, or ZIKV. As shown in Fig. 1C to H, the replication levels of all three viruses were significantly reduced in α-glucosidase I and/or II knockout cells, as judged by the reduction of intracellular viral RNA and virus titers in the culture media. Thus, using α-glucosidase I and II knockout cells, we firmly validated α-glucosidase I and II as antiviral targets for these three hemorrhagic fever viruses. Nevertheless, even in absence of either ER α-glucosidase I or II, the replication of DENV, YFV, or ZIKV could only be partially blocked, with the maximum reduction of virus titers ranging from one to two logs (Fig. 1D, F and H), with exception of a three logs reduction in virus titer in Gluc I KO cells infected with YFV, and no significant reduction in Gluc II KO cells infected with ZIKV. These results suggest that there is an upper limit to what can be expected in inhibiting these viruses by pharmacologically targeting ER glucosidases I and II. In addition, we also attempted to establish a cell line with both α-glucosidase I and II knockout but without success (data not shown). The failure to obtain α-glucosidase I and II double knockout cells indicates that complete inhibition of both enzymes may be lethal to the cells. Accordingly, a combinational therapeutic strategy may be a reasonable approach, in order to improve the antiviral efficacy of ER glucosidase inhibitors.
Figure 1. DENV, YFV and ZIKV replication in ER α-glucosidase I and II knockout cells.
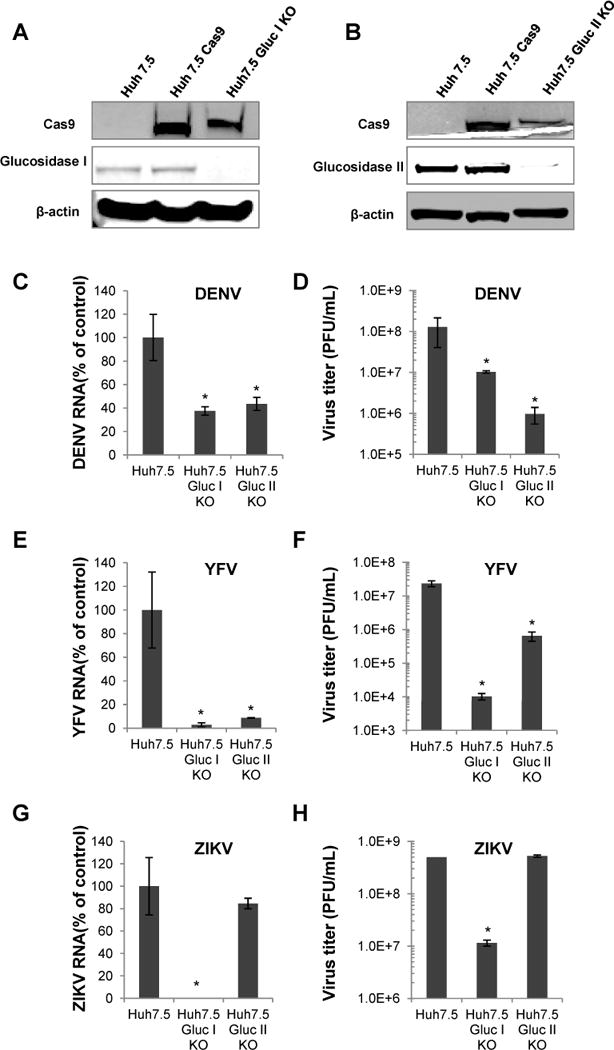
The expression of ER α-glucosidase I (A) or II (B) in parental Huh7.5 cells, Huh7.5 cells expressing Cas9, and cells with ER glucosidase I or II knockout (Huh7.5 Gluc I KO or Huh7.5 Gluc II KO) were examined by a Western blot assay. β-actin served as a loading control. (C to H) Parental Huh7.5 cells and derived ER α-glucosidase I and II knockout cell lines were infected with DENV (C and D), YFV (E and F) or ZIKV (G and H) at MOI of 0.1 for 1 hr and cultured for additional 48 hrs. The levels of DENV RNA (C), YFV RNA (E) or ZIKV RNA (G) were determined by qRT-PCR using β-actin as internal control and expressed as the percentage relative to that in Huh7.5 cells. The viral titers of DENV (D), YFV (F) or ZIKV (H) in culture supernatant were determined by plaque assay. Data were presented as mean from four independent replicates (mean ± standard deviation). * p<0.05 compared to that in parental cells.
3.2 ER α-glucosidase inhibitor IHVR-19029 can be potentiated by T-705 against yellow fever virus and Ebola virus in tissue culture
Previously, we reported that IHVR-19029 can inhibit the replication of several hemorrhagic fever viruses, such as DENV and Rift valley fever virus in vitro (Chang et al., 2013c). In this study, we further tested its antiviral activity against YFV, ZIKV and EBOV. As shown in Fig. 2A to C, IHVR-19029 most potently inhibited DENV, but to a lesser extent, YFV and ZIKV in HEK293 cells infected with the viruses at MOI of 0.1. While there is no previous report of ER glucosidase inhibitor against ZIKV, the lower sensitivity of YFV to IHVR-19029, is consistent with a previous report using another family of ER glucosidase inhibitor, Castanospermine (Whitby et al., 2005). The discrepancy in the antiviral potency of IHVR-19029 against these three flaviviruses could be due to the number and/or location of glycosylation site(s) within their envelope proteins. For example, while DENV serotype 2 virus has two conserved glycosylation sites in the envelope protein located at amino acid residues 67 and 153, only the N153 site is maintained in the envelope protein of PRVABC59 strain of ZIKV (Dai et al., 2016). Interestingly, while none of these two conserved glycosylation sites in DENV envelope exist in the envelope protein of 17D YFV, there is a potential N-linked glycosylation site located at amino acid residue 308, which has not been experimentally confirmed (Hahn et al., 1987). Nevertheless, it was previously reported that N-linked glycosylation in YFV NS1 protein is also important for virus replication and may also confer sensitivity of the virus to ER α-glucosidase inhibition (Muylaert et al., 1996).
Ebola virus has been predicted to be sensitive to ER glucosidase inhibition since its glycosylated envelope proteins GP1 and GP2 have more than 10 predicted glycosylation sites (Lennemann et al., 2014; Lennemann et al., 2015). Previously we reported that IHVR-19029 inhibited the assembly and infectivity of EBOV glycosylated envelope protein pseudotyped-lentiviral particles (Chang et al., 2013c). Here we further demonstrated that in an immunofluorescent-based quantitative assay, IHVR-19029 inhibited EBOV in Hela cells with an EC50 of 16.9 μM (Fig. 2D). The antiviral potency could be underestimated since a high MOI of infection (MOI of 1.5) was needed in order to achieve optimal florescent signals in this assay.
To overcome the limited antiviral potency associated with glucosidase inhibition, we tested whether the antiviral activity of IHVR-19029 can be potentiated by combination with other broad-spectrum antivirals. Favipiravir (T-705) is a broad-spectrum viral polymerase inhibitor that is active against YFV and Ebola in vitro and in vivo (Baz et al., 2017; Best et al., 2017b; Julander et al., 2009). As shown in Fig. 3A and B, combination of IHVR-19029 and T-705 in a checkerboard matrix format in cells infected with YFV or EBOV achieved a significant synergistic antiviral effect as demonstrated by the peaks above the surface analyzed by MacSynergy software (Prichard and Shipman, 1996).
3.3 Pharmacokinetics (PK), pharmacodynamics (PD) and tolerability of IHVR-19029 in mice
Previously we reported the PK profile of IHVR-19029 using IP and oral administration, which showed that the compound has very low oral bioavailability and short half-life via IP route (Chang et al., 2013c). Nevertheless, IP injection of IHVR-19029 at 75 mg/kg twice daily for 10 days partially protected the lethal infection in mice infected with EBOV or Marburg virus. In order to further optimize the dose and dosing frequency of IHVR-19029 in efficacy studies via IP injection route, we set out to analyze the PK profile with specific focus on tissue distribution of the compound, which should be more relevant to the antiviral efficacy. The result from a single dose pilot study using IP injection of IHVR19029 at 75 mg/kg in male Balb/c mice was used for the design of the repeat dose PK and tolerability study. In the single dose experiment, a tendency of higher level accumulation of IHVR-19029 in tissues compared to plasma was observed. For example, the compound concentration in the liver was 15-fold higher at 8 hr post injection (224ng/g, equivalent to 0.5μM) compared to that in plasma (Fig. 4A). Accordingly, three times daily (TID) treatment at 8 hr apart was selected in the following repeated dosing experiment.
Figure 4. Pharmacokinetics and tissue distribution of IHVR19029 in mice.
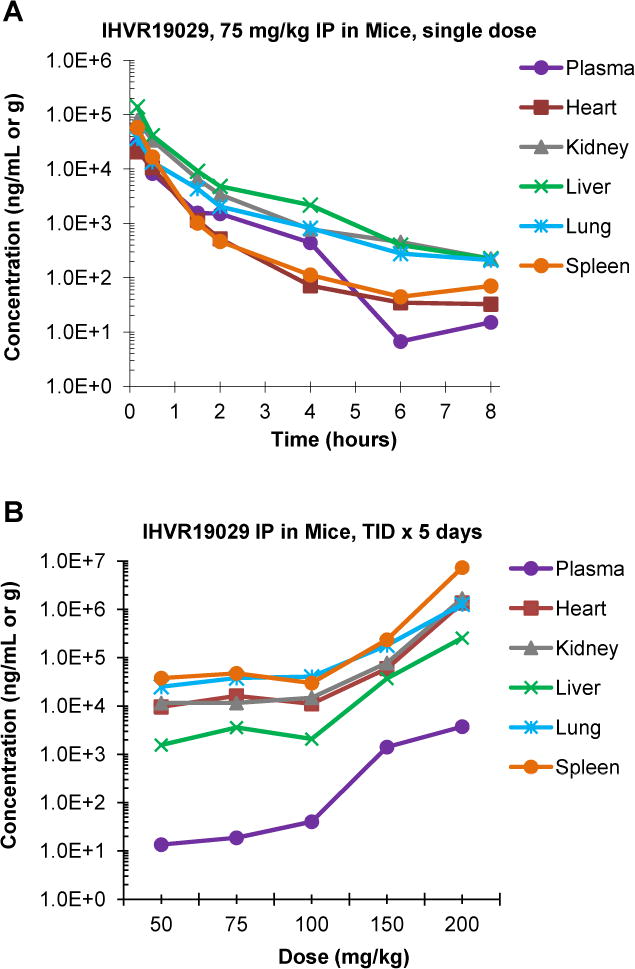
(A) Male Balb/c mice were treated with single dose of IHVR-19029 via IP injection at 75 mg/kg. At indicated time points post injection, Plasma samples together with heart, kidney, liver, lung and spleen samples were collected. IHVR-19029 concentrations in plasma and tissues were determined and expressed as average from 3 animals (ng/ml in plasma or ng/g in tissue). (B) Male Balb/c mice were randomly assigned into six groups, three animals per group: one control group and five treated groups at 50, 75, 100, 150 or 200 mg/kg. The animals were either mock treated or treated with IHVR-19029 via IP injection, three times daily at 8 hr interval for five days. Plasma samples together with heart, kidney, liver, lung and spleen samples were collected at 4 hr post the last dosing at day 5 except for samples from animals treated with 200mg/kg which were collected at day 4 before the animals were euthanized due to toxicity. IHVR-19029 concentrations in plasma and tissues were determined and expressed as average from 5 animals (ng/ml in plasma or ng/g in tissues).
A five-day repeated dosing experiment was designed to evaluate the PK, PD, and tolerability. Animals were randomly assigned into 5 dose groups of 50, 75, 100, 150 and 200 mg/kg/dose, TID via IP injection. Doses of 50–100 mg/kg TID for 5 days appeared to be very well tolerated by all the animals, but mice that received ≥ 150 mg/kg TID for 5 days developed observable signs of toxicity. At day 3, all 5 mice in the 200mg/kg group appeared to be lethargic, had swollen abdomens, and lower skin turgor, a characteristic that is consistent with dehydration. All the mice were euthanized for humane reasons at day 4. At the time of necropsy, the average weight of heart, kidney and spleen was 15% to 50% lower in the group of animals treated with 200 mg/kg (data not shown), which is another indication of dehydration. Mice in the 150 mg/kg group had peri-anal staining and soft feces after day 3, but eventually survived the 5-day dosing. This observed adverse effect at 150mg/kg group may be due to potential off-target inhibition of carbohydrate-metabolizing glucosidases located in the gastrointestinal lumen (Dwek et al., 2002).
An examination of the tissue to plasma ratios for each dosing group showed that IHVR19029 partitioned highly into the spleen, lung, kidney, heart, and liver. As shown in Fig. 4B, 4 hr post final injection, in the 50, 75 and 100 mg/kg dose groups, the mean drug concentrations were up to 3 logs higher in the spleen and 2 logs higher in the liver, compared to that in plasma. These results suggested there was accumulation of IHVR19029 in tissues after multiple IP doses of IHVR19029, especially in the heart, lung, and spleen. Interestingly, treating mice with nontoxic doses of 50 to 100 mg/kg TID for 5 days resulted in steady-state tissue concentrations of IHVR-19029, ranging from 2076 ng/g in the liver to 47401 ng/g in the spleen (approximately equal to 5 to100μM), which are close to or above the targeted therapeutic concentrations for various virus infections. The concentrations of IHVR19029 in tissues and plasma appeared to increase with higher doses, which could be related to the dehydration status of the animals due to toxicity.
Given the high level accumulation of IHVR-19029 in tissues, especially in the spleen, a relevant organ with multiple hemorrhagic fever infection (Geisbert et al., 2003), we set out to determine whether IHVR-19029 changed the protein glycan profile in the spleen. Consistent with the known function of ER glucosidases I and II, which sequentially trim the three terminal tri-glucose moieties on the N-linked glycans attached to nascent glycoproteins, treatment of the mice with IHVR-19029 resulted in retention of the terminal glucose structure of N-linked glycans in the free oligosaccharides isolated from the spleen. Fig. 5 shows representative chromatograms of total cellular FOS from the spleen of untreated and treated mice. In contrast to untreated control (Fig. 5A), there were multiple florescent peaks consistent with mono- (G1M4N and G1M5N), di- (G2M4N) or tri-(G3M5N and G3M6N) glucose retention in FOS isolated from the spleen of IHVR-19029 treated mouse (Fig. 4B). This result indicates that IHVR-19029 is not only accumulated in tissues of treated animals, but also functional against its target enzymes ER glucosidase I and II.
Figure 5. Changing protein glycan profiles in mice treated with IHVR-19029.
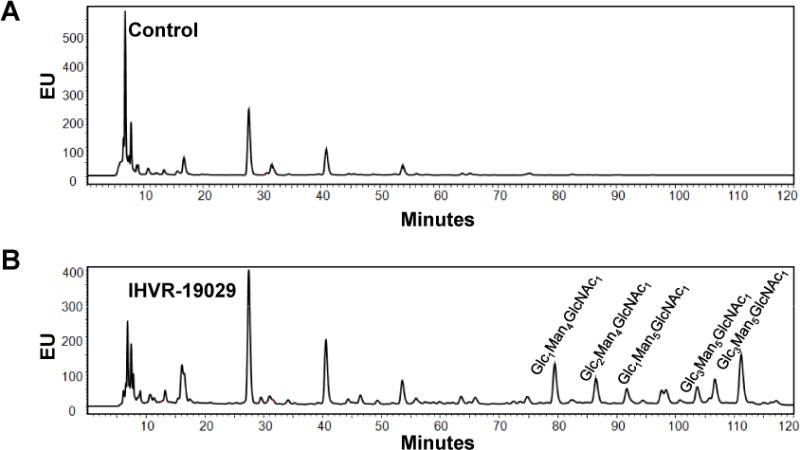
Male Balb/c mice were either mock treated or treated with IHVR-19029 via IP injection at 100 mg/kg, three times daily at 8 hr interval for five days. Free Oligosaccharides (FOS) in the spleen of untreated and treated animals were purified and labeled. Different FOS were separated using NP-HPLC and expressed as fluorescent value (EU) as a function of time. The compositions of corresponding glycans in the FOS peaks were labeled at the top. Glc, glucose; Man, mannose; GlcNAc, N-Acetylglucosamine.
3.4 T-705 potentiates IHVR-19029 in vivo in a mouse model of EBOV infection
Encouraged by the synergistic antiviral effects between T-705 and IHVR-19029 against yellow fever and EBOV in tissue culture, as well as the optimal PK and PD profile in the tissues of animals treated with IHVR-19029 via IP route, we tested the in vivo combination effect of T-705 and IHVR-19029 in a mouse model of EBOV infection. First of all, we determined the suboptimal doses of T-705 being 0.325mg/kg and 1.6mg/kg (Fig. 6A). Next, we demonstrated, when two suboptimal doses of each compound were combined, there was always an increase in the survival rates (Fig.6B–E). Statistical analysis using Log-rank (Mantel-cox) test indicated that there are statistically significant differences in 5 out of the 8 comparisons between combination treatment and mono-treatment. Specifically, low and high dose of T-705 both potentiated low dose of IHVR-19029 (Fig. 6 B and D). Low and high dose of IHVR-19029 both potentiated low dose of T-705 (Fig. 6 B and C). In addition, high dose of IHVR-19029 also potentiated high dose of T-705 (Fig. 6 E). This result are consistent with the data obtained from in vitro study, suggesting T-705 can also potentiate the antiviral activity of IHVR-19029 in vivo in a mouse model of EBOV infection.
Figure 6. Combination of IHVR-19029 and T-705 against EBOV in a mouse model.
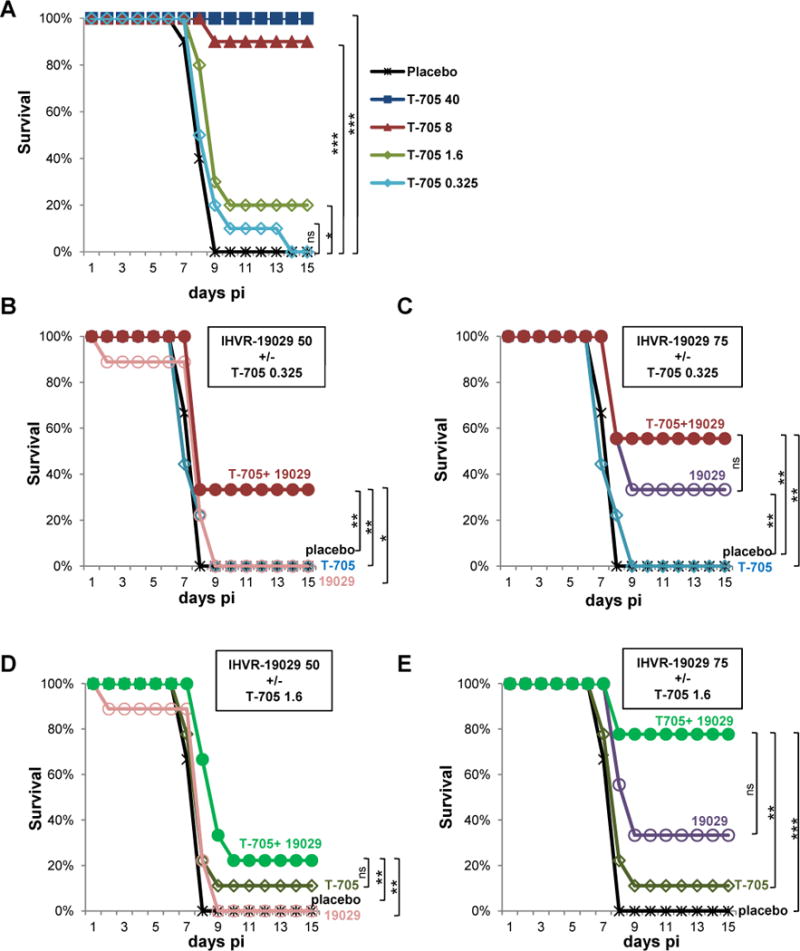
(A) Survival rate of EBOV infected mice treated with doses of T-705 ranging from 0.325 mg/kg to 40 mg/kg or 0.4% carboxymethylcellulose for placebo control once daily via oral route for 10 days immediately after infection (n=10 per group). (B and C) Survival rate of EBOV infected mice treated with 0.325 mg/kg of T-705 once daily orally, 50 or 75 mg/kg of IHVR-19029 twice daily via IP injection, either alone or in combination for 10 days immediately after infection. (D and E) Survival rate of EBOV infected mice treated with 1.6 mg/kg of T-705, 50 or 75 mg/kg of IHVR-19029, either alone or in combination for 10 days immediately after infection. Animals treated with vehicles served as placebo controls (n=9 per group). P values were calculated with Log-rank (Mantel-cox) test. *P<0.05; **P<0.01; ***P<0.001; ns, no significance.
4. Discussion
It has been thirty years since the first report of ER α-glucosidase inhibitor as an anti-retroviral agent (Sunkara et al., 1987). Numerous studies have since then demonstrated that ER glucosidase inhibitors are broad-spectrum antivirals with in vitro and in vivo activity against many members of enveloped viruses, including several hemorrhagic fever viruses (reviewed in (Chang et al., 2013a)), and have a confirmed high genetic barrier against escape mutations in vivo (Plummer et al., 2015). However, the clinical development of ER glucosidase inhibitors has been limited by low potency when treating patients with chronic infections. In clinical trials of treating patients with chronic infections of hepatitis C and human immunodeficiency viruses, long term treatment only resulted in modest reduction in viremia, and thus lacks definitive clinical benefits (Chang et al., 2013a; Durantel, 2009; Fischl et al., 1994). Unlike chronic viral infections where long-term treatment and potent suppression of viral replication is required for achieving a beneficial clinical efficacy, clinical data suggested that suppression of viremia by merely 1 to 2 logs in the early phase of an acute viral infection, such as dengue hemorrhagic fever, can be life-saving (Vaughn et al., 2000). Nevertheless, a prodrug of indolizidine, Celgosivir (6-O-butanoyl castanospermine), which is highly protective when given twice daily to AG129 mice infected with dengue virus, did not significantly reduce serum viral loads in patients in a Phase Ib proof-of-concept clinical trial when given in a similar regimen (Low et al., 2014; Watanabe et al., 2012).
Great effort has been exerted to improve the therapeutic efficacy of glucosidase inhibitors. In particular, for the indolizidine family compound celgosivir, optimizing treatment protocol was performed in mouse models of dengue virus infection to search for a window of potential therapeutic efficacy including increasing dose, dosing frequency, optimized time of initiation of treatment, which should provide guidance for the planning of another clinical trial (Watanabe et al., 2016). Other approaches are mainly focusing on the chemical modification of DNJ-derived glucosidase inhibitors to improve potency and/or tolerability (Chang et al., 2013a; Chang et al., 2013b; Chang et al., 2009; Chang et al., 2013c; Du et al., 2013a; Du et al., 2013b; Ma et al., 2017; Yu et al., 2012). Recently, the structures of mammalian ER α-glucosidase II were reported, which capture the binding modes of iminosugar, and thus provide a template for rational design of more potent inhibitors (Caputo et al., 2016). UV-4B, an N-alkyl-derivative of DNJ, with in vivo activity against influenza and dengue viruses (Stavale et al., 2015; Warfield et al., 2016a; Warfield et al., 2016b), is currently in Phase I clinical trial treating dengue disease (NCT02061358). Unfortunately, the pre-clinical study attempting to expand the antiviral spectrum of UV-4B to Ebola virus infection failed in mice, guinea pigs and nonhuman primates (Warfield et al., 2017). These studies highlighted the complexity and challenges that need to be addressed in the advanced preclinical and clinical development of ER glucosidase inhibitors as broad-spectrum antiviral candidates.
Given the long standing issue of low efficacy associated with many of the ER glucosidase inhibitors, in this paper, we first set out to determine the maximum extent of antiviral potency in human hepatoma derived cell lines with either ER α-glucosidases I or II knockout. On average, around 1 to 2 logs reduction of viral titers were observed in DENV, YFV, and ZIKV infection in the absence of either glucosidase I or II (Fig. 1C to H). These results are consistent with a previous report using cells derived from two patients with genetic defects in ER α-glucosidase I that only demonstrated reduced ability to support productive infection of multiple enveloped viruses (Sadat et al., 2014). Together, these results provide independent genetic validation for the critical role of ER glucosidases I and II in the morphogenesis and infectivity of a broad-spectrum of enveloped viruses. But more importantly, the results also suggest that viruses can manage to replicate in the absence of ER glucosidase I or II, which explains, at least in part, the observed low efficacy of glucosidase inhibitors in in vivo or clinical studies.
IHVR-19029 is another lead molecule of N-alkyl-DNJ derivative, which showed partial protection via IP injection against Ebola and Marburg in mouse models. The preclinical development of IHVR-19029 has been limited by its low oral bioavailability and dose-limiting gastrointestinal side effects via oral route (Chang et al., 2013c). In our effort to improve the pharmacokinetic properties of IHVR-19029, prodrugs of IHVR-19029 were recently developed which improved its oral availability as well as side effects associated with oral administration (Ma et al., 2017). These features, as a whole, have been predicted to improve the in vivo antiviral efficacy via oral route. Although IHVR-19029 treatment provided partial protection in Ebola and Marburg virus infected mice via injection route, it has a short plasma half-life (Chang et al., 2013c). In order to further modify the treatment protocol and improve the in vivo efficacy, in this study we examined its tissue distribution and tolerability after 5 days of repeated dosing. Interestingly, we showed that at the end of the 5 days repeated dosing, the drug concentrations were 2 to 3 logs higher in the major tissues as compared to that in the plasma. In addition, treatment with IHVR-19029 at 50mg/kg to 100mg/kg resulted in a steady state level of IHVR-19029 at or above the predicted antiviral therapeutic values in target organs such as spleen, kidney and liver. The accumulation of IHVR-19029 in tissues after repeated IP dosing could well explain the in vivo efficacy of IHVR-19029 at a dose of 75mg/kg. However, this partial efficacious dose is very close to its toxic dose at 150mg/kg, which means improving antiviral efficacy cannot be achieved simply by increasing the dose. Moreover, it is also impractical to have a dosing frequency of more than three times a day in a BSL-4 environment. In this regards, as an alternative approach to further explore the therapeutic potential of IHVR-19029 via injection, in the current study, we explored the possibility of combining IHVR-19029 with another broad antiviral agent.
Favipiravir, or T-705, is a pyrazinecarboxamide derivative with activity against many RNA viruses, including influenza viruses, as well as members of flaviviruses, arenaviruses, bunyaviruses, filoviruses, and alphaviruses (Furuta et al., 2013; Furuta et al., 2009; Gowen et al., 2014). In 2014, favipiravir was approved in Japan for stockpiling against influenza pandemics [9]. For viruses causing hemorrhagic fever, in vivo efficacy of favipiravir has been demonstrated against yellow fever virus in a hamster model (Julander et al., 2009), Junin virus in a guinea pig model, Pichinde virus in a hamster model (Westover et al., 2016), Crimean-Congo hemorrhagic fever virus in a mouse model (Oestereich et al., 2014b), and Ebola virus in two mouse models (Oestereich et al., 2014a; Smither et al., 2014). On the other hand, favipiravir showed limited or no efficacy against Zika virus in vitro, and a mathematic model predicted that a very high therapeutic efficacious dose is required in vivo (Baz et al., 2017; Best et al., 2017a; Lanko et al., 2017). Clinically, a historically controlled, single-arm proof-of-concept trial in Guinea (NCT02329054) treating patients with Ebola virus infection showed that favipiravir monotherapy was only efficacious in patients with low viremia, but not in those with high viremia (Sissoko et al., 2016). A follow-up study of the trial indicated that the favipiravir concentrations in the plasma of treated individuals were not sufficient to strongly inhibit the viral replication (Nguyen et al., 2017).
Given the overlapping antiviral spectrum between IHVR-19029 and T-705 and the common feature of low or insufficient potency in vivo and/or in clinical trials, it is reasonable to consider the combination of these two drugs. Indeed, in this study we demonstrated that combination of IHVR-19029 and T-705 synergistically inhibited YFV and EBOV in vitro. Moreover, combination of sub-optimal doses of IHVR-19029 and T-705 significantly improved the survival rates in EBOV infected mice. In the future, detailed analysis of in vivo toxicity should be performed to assess the potential additive toxicity by combining IHVR-19029 and T-705, which may allow for applying different dose combinations of the two compounds to further improve the therapeutic benefits. While clinical trials with iminosugar therapy are still ongoing, and it is not yet known whether or not monotherapy is adequate, this study certainly sets the foundation for combinational strategy as potential alternative in future pre-clinical and clinical studies against EBOV and other hemorrhagic fever viruses such as DENV, YFV and ZIKV. Furthermore, such combinational approach may be generalized for other iminosugar-based ER glucosidase inhibitors such as UV-4 and celgosivir, both of which are in more advanced pre-clinical development stage, as well as other broad-spectrum viral RNA polymerase inhibitors such as sofosbuvir, GS-5734, or BCX 4430 (Bekerman and Einav, 2015; Bullard-Feibelman et al., 2017; Warren et al., 2016; Warren et al., 2014; Xu et al., 2017). Other approaches to explore combinational therapeutic of EBOV infection have been reported, such as combination of three FDA-approved drugs that synergistically block EBOV entry (Sun et al., 2017; Sun et al., 2016).
Highlights.
Targeting ER α-glucosidase I/II is a broad spectrum and resistance-refractory antiviral approach
Gene knockout study suggests that there is a potency limitation by pharmacological targeting ER α-glucosidases
Combination of ER-α-glucosidase inhibitor IHVR-19029 with Favipiravir improved the antiviral efficacy in vitro and in vivo
We proved the concept of combination strategy for viral hemorrhagic fevers with two broad spectrum antiviral agents
Acknowledgments
We thank Dr. Richard R. Drake at Medical University of South Carolina for providing technical support. A cDNA clone of serotype 2 dengue virus, pACYC177-NGC-DENV-2, was a gift from Dr. Pei-Yong Shi at University of Texas Medical Branch. A cDNA clone of Yellow fever virus 17D strain, pACNR/FLYF-17Dx, was a gift from Dr. Charles M. Rice at Rockefeller University. This work was supported by grant from the National Institutes of Health, USA (AI104636), Unither Virology, LLC, and the Commonwealth of Pennsylvania through the Hepatitis B Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adcock RS, Chu YK, Golden JE, Chung DH. Evaluation of anti-Zika virus activities of broad-spectrum antivirals and NIH clinical collection compounds using a cell-based, high-throughput screen assay. Antiviral Res. 2016;138:47–56. doi: 10.1016/j.antiviral.2016.11.018. [DOI] [PubMed] [Google Scholar]
- Alonzi DS, Scott KA, Dwek RA, Zitzmann N. Iminosugar antivirals: the therapeutic sweet spot. Biochem Soc Trans. 2017;45:571–582. doi: 10.1042/BST20160182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baz M, Goyette N, Griffin BD, Kobinger GP, Boivin G. In vitro susceptibility of geographically and temporally distinct zika viruses to favipiravir and ribavirin. Antivir Ther. 2017 doi: 10.3851/IMP3180. [DOI] [PubMed] [Google Scholar]
- Bekerman E, Einav S. Infectious disease. Combating emerging viral threats. Science. 2015;348:282–283. doi: 10.1126/science.aaa3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best K, Guedj J, Madelain V, de Lamballerie X, Lim SY, Osuna CE, Whitney JB, Perelson AS. Zika plasma viral dynamics in nonhuman primates provides insights into early infection and antiviral strategies. Proc Natl Acad Sci U S A. 2017a;114:8847–8852. doi: 10.1073/pnas.1704011114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best K, Guedj J, Madelain V, de Lamballerie X, Lim SY, Osuna CE, Whitney JB, Perelson AS. Zika plasma viral dynamics in nonhuman primates provides insights into early infection and antiviral strategies. Proc Natl Acad Sci U S A. 2017b doi: 10.1073/pnas.1704011114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldescu V, Behnam MAM, Vasilakis N, Klein CD. Broad-spectrum agents for flaviviral infections: dengue, Zika and beyond. Nat Rev Drug Discov. 2017 doi: 10.1038/nrd.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredenbeek PJ, Kooi EA, Lindenbach B, Huijkman N, Rice CM, Spaan WJ. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J Gen Virol. 2003;84:1261–1268. doi: 10.1099/vir.0.18860-0. [DOI] [PubMed] [Google Scholar]
- Bullard-Feibelman KM, Govero J, Zhu Z, Salazar V, Veselinovic M, Diamond MS, Geiss BJ. The FDA-approved drug sofosbuvir inhibits Zika virus infection. Antiviral Res. 2017;137:134–140. doi: 10.1016/j.antiviral.2016.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo AT, Alonzi DS, Marti L, Reca IB, Kiappes JL, Struwe WB, Cross A, Basu S, Lowe ED, Darlot B, Santino A, Roversi P, Zitzmann N. Structures of mammalian ER alpha-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals. Proc Natl Acad Sci U S A. 2016;113:E4630–4638. doi: 10.1073/pnas.1604463113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Block TM, Guo JT. Antiviral therapies targeting host ER alpha-glucosidases: current status and future directions. Antiviral Res. 2013a;99:251–260. doi: 10.1016/j.antiviral.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Block TM, Guo JT. Viral resistance of MOGS-CDG patients implies a broad-spectrum strategy against acute virus infections. Antivir Ther. 2015;20:257–259. doi: 10.3851/IMP2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Guo JT, Du Y, Block T. Imino sugar glucosidase inhibitors as broadly active anti-filovirus agents. Emerg Microbes Infect. 2013b;2:e77. doi: 10.1038/emi.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Schul W, Butters TD, Yip A, Liu B, Goh A, Lakshminarayana SB, Alonzi D, Reinkensmeier G, Pan X, Qu X, Weidner JM, Wang L, Yu W, Borune N, Kinch MA, Rayahin JE, Moriarty R, Xu X, Shi PY, Guo JT, Block TM. Combination of alpha-glucosidase inhibitor and ribavirin for the treatment of dengue virus infection in vitro and in vivo. Antiviral Res. 2011a;89:26–34. doi: 10.1016/j.antiviral.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Schul W, Yip A, Xu X, Guo JT, Block TM. Competitive inhibitor of cellular alpha-glucosidases protects mice from lethal dengue virus infection. Antiviral Res. 2011b;92:369–371. doi: 10.1016/j.antiviral.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Wang L, Ma D, Qu X, Guo H, Xu X, Mason PM, Bourne N, Moriarty R, Gu B, Guo JT, Block TM. Novel imino sugar derivatives demonstrate potent antiviral activity against flaviviruses. Antimicrob Agents Chemother. 2009;53:1501–1508. doi: 10.1128/AAC.01457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Warren TK, Zhao X, Gill T, Guo F, Wang L, Comunale MA, Du Y, Alonzi DS, Yu W, Ye H, Liu F, Guo JT, Mehta A, Cuconati A, Butters TD, Bavari S, Xu X, Block TM. Small molecule inhibitors of ER alpha-glucosidases are active against multiple hemorrhagic fever viruses. Antiviral Res. 2013c;98:432–440. doi: 10.1016/j.antiviral.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Song J, Lu X, Deng YQ, Musyoki AM, Cheng H, Zhang Y, Yuan Y, Song H, Haywood J, Xiao H, Yan J, Shi Y, Qin CF, Qi J, Gao GF. Structures of the Zika Virus Envelope Protein and Its Complex with a Flavivirus Broadly Protective Antibody. Cell Host Microbe. 2016;19:696–704. doi: 10.1016/j.chom.2016.04.013. [DOI] [PubMed] [Google Scholar]
- De Clercq E, Li G. Approved Antiviral Drugs over the Past 50 Years. Clin Microbiol Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Ye H, Gill T, Wang L, Guo F, Cuconati A, Guo JT, Block TM, Chang J, Xu X. N-Alkyldeoxynojirimycin derivatives with novel terminal tertiary amide substitution for treatment of bovine viral diarrhea virus (BVDV), Dengue, and Tacaribe virus infections. Bioorg Med Chem Lett. 2013a;23:2172–2176. doi: 10.1016/j.bmcl.2013.01.108. [DOI] [PubMed] [Google Scholar]
- Du Y, Ye H, Guo F, Wang L, Gill T, Khan N, Cuconati A, Guo JT, Block TM, Chang J, Xu X. Design and synthesis of N-alkyldeoxynojirimycin derivatives with improved metabolic stability as inhibitors of BVDV and Tacaribe virus. Bioorg Med Chem Lett. 2013b;23:4258–4262. doi: 10.1016/j.bmcl.2013.04.052. [DOI] [PubMed] [Google Scholar]
- Durantel D. Celgosivir, an alpha-glucosidase I inhibitor for the potential treatment of HCV infection. Curr Opin Investig Drugs. 2009;10:860–870. [PubMed] [Google Scholar]
- Dwek RA, Butters TD, Platt FM, Zitzmann N. Targeting glycosylation as a therapeutic approach. Nat Rev Drug Discov. 2002;1:65–75. doi: 10.1038/nrd708. [DOI] [PubMed] [Google Scholar]
- Fischl MA, Resnick L, Coombs R, Kremer AB, Pottage JC, Jr, Fass RJ, Fife KH, Powderly WG, Collier AC, Aspinall RL, et al. The safety and efficacy of combination N-butyl-deoxynojirimycin (SC-48334) and zidovudine in patients with HIV-1 infection and 200–500 CD4 cells/mm3. J Acquir Immune Defic Syndr. 1994;7:139–147. [PubMed] [Google Scholar]
- Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res. 2013;100:446–454. doi: 10.1016/j.antiviral.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 2009;82:95–102. doi: 10.1016/j.antiviral.2009.02.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Hensley LE, Larsen T, Young HA, Reed DS, Geisbert JB, Scott DP, Kagan E, Jahrling PB, Davis KJ. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am J Pathol. 2003;163:2347–2370. doi: 10.1016/S0002-9440(10)63591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowen BB, Juelich TL, Sefing EJ, Brasel T, Smith JK, Zhang L, Tigabu B, Hill TE, Yun T, Pietzsch C, Furuta Y, Freiberg AN. Favipiravir (T-705) inhibits Junin virus infection and reduces mortality in a guinea pig model of Argentine hemorrhagic fever. PLoS Negl Trop Dis. 2014;7:e2614. doi: 10.1371/journal.pntd.0002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Mead J, Aliya N, Wang L, Cuconati A, Wei L, Li K, Block TM, Guo JT, Chang J. RO 90-7501 Enhances TLR3 and RLR Agonist Induced Antiviral Response. PLoS One. 2012;7:e42583. doi: 10.1371/journal.pone.0042583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Wu S, Julander J, Ma J, Zhang X, Kulp J, Cuconati A, Block TM, Du Y, Guo JT, Chang J. A Novel Benzodiazepine Compound Inhibits Yellow Fever Virus Infection by Specifically Targeting NS4B Protein. J Virol. 2016 doi: 10.1128/JVI.01253-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Zhao X, Gill T, Zhou Y, Campagna M, Wang L, Liu F, Zhang P, DiPaolo L, Du Y, Xu X, Jiang D, Wei L, Cuconati A, Block TM, Guo JT, Chang J. An interferon-beta promoter reporter assay for high throughput identification of compounds against multiple RNA viruses. Antiviral Res. 2014;107:56–65. doi: 10.1016/j.antiviral.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn CS, Dalrymple JM, Strauss JH, Rice CM. Comparison of the virulent Asibi strain of yellow fever virus with the 17D vaccine strain derived from it. Proc Natl Acad Sci U S A. 1987;84:2019–2023. doi: 10.1073/pnas.84.7.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julander JG, Shafer K, Smee DF, Morrey JD, Furuta Y. Activity of T-705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T-1106. Antimicrob Agents Chemother. 2009;53:202–209. doi: 10.1128/AAC.01074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanko K, Eggermont K, Patel A, Kaptein S, Delang L, Verfaillie CM, Neyts J. Replication of the Zika virus in different iPSC-derived neuronal cells and implications to assess efficacy of antivirals. Antiviral Res. 2017;145:82–86. doi: 10.1016/j.antiviral.2017.07.010. [DOI] [PubMed] [Google Scholar]
- Lennemann NJ, Rhein BA, Ndungo E, Chandran K, Qiu X, Maury W. Comprehensive functional analysis of N-linked glycans on Ebola virus GP1. MBio. 2014;5:e00862–00813. doi: 10.1128/mBio.00862-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennemann NJ, Walkner M, Berkebile AR, Patel N, Maury W. The Role of Conserved N-Linked Glycans on Ebola Virus Glycoprotein 2. J Infect Dis. 2015;212(Suppl 2):S204–209. doi: 10.1093/infdis/jiv201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JG, Sung C, Wijaya L, Wei Y, Rathore AP, Watanabe S, Tan BH, Toh L, Chua LT, Hou Y, Chow A, Howe S, Chan WK, Tan KH, Chung JS, Cherng BP, Lye DC, Tambayah PA, Ng LC, Connolly J, Hibberd ML, Leo YS, Cheung YB, Ooi EE, Vasudevan SG. Efficacy and safety of celgosivir in patients with dengue fever (CELADEN): a phase 1b, randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet Infect Dis. 2014;14:706–715. doi: 10.1016/S1473-3099(14)70730-3. [DOI] [PubMed] [Google Scholar]
- Ma J, Wu S, Zhang X, Guo F, Yang K, Guo J, Su Q, Lu H, Lam P, Li Y, Yan Z, Kinney W, Guo JT, Block TM, Chang J, Du Y. Ester Prodrugs of IHVR-19029 with Enhanced Oral Exposure and Prevention of Gastrointestinal Glucosidase Interaction. ACS Med Chem Lett. 2017;8:157–162. doi: 10.1021/acsmedchemlett.6b00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muylaert IR, Chambers TJ, Galler R, Rice CM. Mutagenesis of the N-linked glycosylation sites of the yellow fever virus NS1 protein: effects on virus replication and mouse neurovirulence. Virology. 1996;222:159–168. doi: 10.1006/viro.1996.0406. [DOI] [PubMed] [Google Scholar]
- Nguyen TH, Guedj J, Anglaret X, Laouenan C, Madelain V, Taburet AM, Baize S, Sissoko D, Pastorino B, Rodallec A, Piorkowski G, Carazo S, Conde MN, Gala JL, Bore JA, Carbonnelle C, Jacquot F, Raoul H, Malvy D, de Lamballerie X, Mentre F. Favipiravir pharmacokinetics in Ebola-Infected patients of the JIKI trial reveals concentrations lower than targeted. PLoS Negl Trop Dis. 2017;11:e0005389. doi: 10.1371/journal.pntd.0005389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestereich L, Ludtke A, Wurr S, Rieger T, Munoz-Fontela C, Gunther S. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res. 2014a;105:17–21. doi: 10.1016/j.antiviral.2014.02.014. [DOI] [PubMed] [Google Scholar]
- Oestereich L, Rieger T, Neumann M, Bernreuther C, Lehmann M, Krasemann S, Wurr S, Emmerich P, de Lamballerie X, Olschlager S, Gunther S. Evaluation of antiviral efficacy of ribavirin, arbidol, and T-705 (favipiravir) in a mouse model for Crimean-Congo hemorrhagic fever. PLoS Negl Trop Dis. 2014b;8:e2804. doi: 10.1371/journal.pntd.0002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry ST, Buck MD, Plummer EM, Penmasta RA, Batra H, Stavale EJ, Warfield KL, Dwek RA, Butters TD, Alonzi DS, Lada SM, King K, Klose B, Ramstedt U, Shresta S. An iminosugar with potent inhibition of dengue virus infection in vivo. Antiviral Res. 2013;98:35–43. doi: 10.1016/j.antiviral.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Plummer E, Buck MD, Sanchez M, Greenbaum JA, Turner J, Grewal R, Klose B, Sampath A, Warfield KL, Peters B, Ramstedt U, Shresta S. Dengue Virus Evolution under a Host-Targeted Antiviral. J Virol. 2015;89:5592–5601. doi: 10.1128/JVI.00028-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN, Shipman C., Jr Analysis of combinations of antiviral drugs and design of effective multidrug therapies. Antivir Ther. 1996;1:9–20. [PubMed] [Google Scholar]
- Qu X, Pan X, Weidner J, Yu W, Alonzi D, Xu X, Butters T, Block T, Guo JT, Chang J. Inhibitors of Endoplasmic Reticulum {alpha}-Glucosidases Potently Suppress Hepatitis C Virus Virion Assembly and Release. Antimicrob Agents Chemother. 2011;55:1036–1044. doi: 10.1128/AAC.01319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CM, Lenches EM, Eddy SR, Shin SJ, Sheets RL, Strauss JH. Nucleotide sequence of yellow fever virus: implications for flavivirus gene expression and evolution. Science. 1985;229:726–733. doi: 10.1126/science.4023707. [DOI] [PubMed] [Google Scholar]
- Sadat MA, Moir S, Chun TW, Lusso P, Kaplan G, Wolfe L, Memoli MJ, He M, Vega H, Kim LJY, Huang Y, Hussein N, Nievas E, Mitchell R, Garofalo M, Louie A, Ireland DC, Grunes C, Cimbro R, Patel V, Holzapfel G, Salahuddin D, Bristol T, Adams D, Marciano BE, Hegde M, Li Y, Calvo KR, Stoddard J, Justement JS, Jacques J, Priel DAL, Murray D, Sun P, Kuhns DB, Boerkoel CF, Chiorini JA, Di Pasquale G, Verthelyi D, Rosenzweig SD. Glycosylation, hypogammaglobulinemia, and resistance to viral infections. N Engl J Med. 2014;370:1615–1625. doi: 10.1056/NEJMoa1302846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schul W, Liu W, Xu HY, Flamand M, Vasudevan SG. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J Infect Dis. 2007;195:665–674. doi: 10.1086/511310. [DOI] [PubMed] [Google Scholar]
- Sissoko D, Laouenan C, Folkesson E, M’Lebing AB, Beavogui AH, Baize S, Camara AM, Maes P, Shepherd S, Danel C, Carazo S, Conde MN, Gala JL, Colin G, Savini H, Bore JA, Le Marcis F, Koundouno FR, Petitjean F, Lamah MC, Diederich S, Tounkara A, Poelart G, Berbain E, Dindart JM, Duraffour S, Lefevre A, Leno T, Peyrouset O, Irenge L, Bangoura N, Palich R, Hinzmann J, Kraus A, Barry TS, Berette S, Bongono A, Camara MS, Chanfreau Munoz V, Doumbouya L, Souley H, Kighoma PM, Rene L, Loua CM, Massala V, Moumouni K, Provost C, Samake N, Sekou C, Soumah A, Arnould I, Komano MS, Gustin L, Berutto C, Camara D, Camara FS, Colpaert J, Delamou L, Jansson L, Kourouma E, Loua M, Malme K, Manfrin E, Maomou A, Milinouno A, Ombelet S, Sidiboun AY, Verreckt I, Yombouno P, Bocquin A, Carbonnelle C, Carmoi T, Frange P, Mely S, Nguyen VK, Pannetier D, Taburet AM, Treluyer JM, Kolie J, Moh R, Gonzalez MC, Kuisma E, Liedigk B, Ngabo D, Rudolf M, Thom R, Kerber R, Gabriel M, Di Caro A, Wolfel R, Badir J, Bentahir M, Deccache Y, Dumont C, Durant JF, El Bakkouri K, Gasasira Uwamahoro M, Smits B, Toufik N, Van Cauwenberghe S, Ezzedine K, D’Ortenzio E, Pizarro L, Etienne A, Guedj J, Fizet A, Barte de Sainte Fare E, Murgue B, Tran-Minh T, Rapp C, Piguet P, Poncin M, Draguez B, Allaford Duverger T, Barbe S, Baret G, Defourny I, Carroll M, Raoul H, Augier A, Eholie SP, Yazdanpanah Y, Levy-Marchal C, Antierrens A, Van Herp M, Gunther S, de Lamballerie X, Keita S, Mentre F, Anglaret X, Malvy D. Experimental Treatment with Favipiravir for Ebola Virus Disease (the JIKI Trial): A Historically Controlled, Single-Arm Proof-of-Concept Trial in Guinea. PLoS Med. 2016;13:e1001967. doi: 10.1371/journal.pmed.1001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smither SJ, Eastaugh LS, Steward JA, Nelson M, Lenk RP, Lever MS. Post-exposure efficacy of oral T-705 (Favipiravir) against inhalational Ebola virus infection in a mouse model. Antiviral Res. 2014;104:153–155. doi: 10.1016/j.antiviral.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Stavale EJ, Vu H, Sampath A, Ramstedt U, Warfield KL. In vivo therapeutic protection against influenza A (H1N1) oseltamivir-sensitive and resistant viruses by the iminosugar UV-4. PLoS One. 2015;10:e0121662. doi: 10.1371/journal.pone.0121662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, He S, Martinez-Romero C, Kouznetsova J, Tawa G, Xu M, Shinn P, Fisher E, Long Y, Motabar O, Yang S, Sanderson PE, Williamson PR, Garcia-Sastre A, Qiu X, Zheng W. Synergistic drug combination effectively blocks Ebola virus infection. Antiviral Res. 2017;137:165–172. doi: 10.1016/j.antiviral.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Sanderson PE, Zheng W. Drug combination therapy increases successful drug repositioning. Drug Discov Today. 2016;21:1189–1195. doi: 10.1016/j.drudis.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkara PS, Bowlin TL, Liu PS, Sjoerdsma A. Antiretroviral activity of castanospermine and deoxynojirimycin, specific inhibitors of glycoprotein processing. Biochem Biophys Res Commun. 1987;148:206–210. doi: 10.1016/0006-291x(87)91096-5. [DOI] [PubMed] [Google Scholar]
- Tyrrell BE, Sayce AC, Warfield KL, Miller JL, Zitzmann N. Iminosugars: Promising therapeutics for influenza infection. Crit Rev Microbiol. 2017;43:521–545. doi: 10.1080/1040841X.2016.1242868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP, Raengsakulrach B, Rothman AL, Ennis FA, Nisalak A. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000;181:2–9. doi: 10.1086/315215. [DOI] [PubMed] [Google Scholar]
- Warfield KL, Barnard DL, Enterlein SG, Smee DF, Khaliq M, Sampath A, Callahan MV, Ramstedt U, Day CW. The Iminosugar UV-4 is a Broad Inhibitor of Influenza A and B Viruses ex Vivo and in Mice. Viruses. 2016a;8:71. doi: 10.3390/v8030071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield KL, Plummer EM, Sayce AC, Alonzi DS, Tang W, Tyrrell BE, Hill ML, Caputo AT, Killingbeck SS, Beatty PR, Harris E, Iwaki R, Kinami K, Ide D, Kiappes JL, Kato A, Buck MD, King K, Eddy W, Khaliq M, Sampath A, Treston AM, Dwek RA, Enterlein SG, Miller JL, Zitzmann N, Ramstedt U, Shresta S. Inhibition of endoplasmic reticulum glucosidases is required for in vitro and in vivo dengue antiviral activity by the iminosugar UV-4. Antiviral Res. 2016b;129:93–98. doi: 10.1016/j.antiviral.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield KL, Warren TK, Qiu X, Wells J, Mire CE, Geisbert JB, Stuthman KS, Garza NL, Van Tongeren SA, Shurtleff AC, Agans KN, Wong G, Callahan MV, Geisbert TW, Klose B, Ramstedt U, Treston AM. Assessment of the potential for host-targeted iminosugars UV-4 and UV-5 activity against filovirus infections in vitro and in vivo. Antiviral Res. 2017;138:22–31. doi: 10.1016/j.antiviral.2016.11.019. [DOI] [PubMed] [Google Scholar]
- Warren TK, Jordan R, Lo MK, Ray AS, Mackman RL, Soloveva V, Siegel D, Perron M, Bannister R, Hui HC, Larson N, Strickley R, Wells J, Stuthman KS, Van Tongeren SA, Garza NL, Donnelly G, Shurtleff AC, Retterer CJ, Gharaibeh D, Zamani R, Kenny T, Eaton BP, Grimes E, Welch LS, Gomba L, Wilhelmsen CL, Nichols DK, Nuss JE, Nagle ER, Kugelman JR, Palacios G, Doerffler E, Neville S, Carra E, Clarke MO, Zhang L, Lew W, Ross B, Wang Q, Chun K, Wolfe L, Babusis D, Park Y, Stray KM, Trancheva I, Feng JY, Barauskas O, Xu Y, Wong P, Braun MR, Flint M, McMullan LK, Chen SS, Fearns R, Swaminathan S, Mayers DL, Spiropoulou CF, Lee WA, Nichol ST, Cihlar T, Bavari S. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531:381–385. doi: 10.1038/nature17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren TK, Wells J, Panchal RG, Stuthman KS, Garza NL, Van Tongeren SA, Dong L, Retterer CJ, Eaton BP, Pegoraro G, Honnold S, Bantia S, Kotian P, Chen X, Taubenheim BR, Welch LS, Minning DM, Babu YS, Sheridan WP, Bavari S. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature. 2014;508:402–405. doi: 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Chan KW, Dow G, Ooi EE, Low JG, Vasudevan SG. Optimizing celgosivir therapy in mouse models of dengue virus infection of serotypes 1 and 2: The search for a window for potential therapeutic efficacy. Antiviral Res. 2016;127:10–19. doi: 10.1016/j.antiviral.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Rathore AP, Sung C, Lu F, Khoo YM, Connolly J, Low J, Ooi EE, Lee HS, Vasudevan SG. Dose- and schedule-dependent protective efficacy of celgosivir in a lethal mouse model for dengue virus infection informs dosing regimen for a proof of concept clinical trial. Antiviral Res. 2012;96:32–35. doi: 10.1016/j.antiviral.2012.07.008. [DOI] [PubMed] [Google Scholar]
- Westover JB, Sefing EJ, Bailey KW, Van Wettere AJ, Jung KH, Dagley A, Wandersee L, Downs B, Smee DF, Furuta Y, Bray M, Gowen BB. Low-dose ribavirin potentiates the antiviral activity of favipiravir against hemorrhagic fever viruses. Antiviral Res. 2016;126:62–68. doi: 10.1016/j.antiviral.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby K, Pierson TC, Geiss B, Lane K, Engle M, Zhou Y, Doms RW, Diamond MS. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J Virol. 2005;79:8698–8706. doi: 10.1128/JVI.79.14.8698-8706.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, Hart MK. Epitopes involved in antibody-mediated protection from Ebola virus. Science. 2000;287:1664–1666. doi: 10.1126/science.287.5458.1664. [DOI] [PubMed] [Google Scholar]
- Wu SF, Lee CJ, Liao CL, Dwek RA, Zitzmann N, Lin YL. Antiviral effects of an iminosugar derivative on flavivirus infections. J Virol. 2002;76:3596–3604. doi: 10.1128/JVI.76.8.3596-3604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Gayen S, Kang C, Yuan Z, Shi PY. Membrane topology and function of dengue virus NS2A protein. J Virol. 2013;87:4609–4622. doi: 10.1128/JVI.02424-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HT, Colby-Germinario SP, Hassounah SA, Fogarty C, Osman N, Palanisamy N, Han Y, Oliveira M, Quan Y, Wainberg MA. Evaluation of Sofosbuvir (beta-D-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine) as an inhibitor of Dengue virus replication() Sci Rep. 2017;7:6345. doi: 10.1038/s41598-017-06612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Gill T, Wang L, Du Y, Ye H, Qu X, Guo JT, Cuconati A, Zhao K, Block TM, Xu X, Chang J. Design, Synthesis, and Biological Evaluation of N-Alkylated Deoxynojirimycin (DNJ) Derivatives for the Treatment of Dengue Virus Infection. J Med Chem. 2012;55:6061–6075. doi: 10.1021/jm300171v. [DOI] [PMC free article] [PubMed] [Google Scholar]