

Abstract
The copper hydride (CuH)-catalyzed enantioselective reduction of α,β-unsaturated carboxylic acids to saturated aldehydes is reported. This protocol provides a new method to access a variety of β-chiral aldehydes in good yields, with high levels of enantioselectivity and broad functional group tolerance. A reaction pathway involving a ketene intermediate is proposed based on preliminary mechanistic studies and density functional theory calculations.
Graphical Abstract

Aldehydes featuring β-stereogenic centers represent a class of important building blocks for the synthesis of various natural products and pharmaceuticals.1 Due to their utility, numerous strategies for their preparation have been devised, including the asymmetric transfer-hydrogenation of unsaturated aldehydes,2 the conjugate addition of nucleophilic reagents to enals,3, 4 and the enantioselective isomerization of allylic alcohols (Figure 1a).5 As an alternative, the direct conversion of α,β-unsaturated carboxylic acids to β-chiral aldehydes would be valuable due to the ready accessibility and stability of the starting acids. An elegant catalytic reduction process of unsaturated acids to racemic saturated aldehydes has been reported by Breit,6 which proceeds through the consecutive Rh-catalyzed hydroformylation and decarboxylation reactions (Figure 1b). Herein we report a CuH-catalyzed asymmetric reduction protocol of α,β-unsaturated carboxylic acids as an approach to access enantiomerically enriched aldehydes (Figure 1c).
Figure 1.

(a) General Access to β-Chiral Aldehydes (b) Racemic and (c) Asymmetric Transformation from α,β-Unsaturated Carboxylic Acids to Saturated Aldehydes (This work)
Over the past two decades, the CuH-catalyzed asymmetric 1,4-reduction of α,β-unsaturated carbonyl compounds has been extensively developed as a method to deliver optically active ketones and esters.7 A hallmark of this chemistry is the 1,4-chemoselectivity provided by bisphosphine-ligated CuH catalysts, which preserves the oxidation state at the carbonyl carbon.8
We recently disclosed an asymmetric olefin hydroacylation reaction involving the direct coupling of α,β-unsaturated acids with aryl alkenes to form α-chiral ketones.9 During this study, when we attempted to couple a trisubstituted alkene with crotonic acid, no desired ketone product was generated. Instead, the silyl enol ether of n-butanal was observed by 1H NMR (Scheme 1a). When the alkene was excluded, further experiments demonstrated that this process is general for a wide range of unsaturated acids. Based on this and our prior results, we reasoned that for substrates with two different β-substituents, we could exploit this reduction process to access β-chiral aldehydes with high levels of enantiopurity.
Scheme 1. (a) Discovery and (b) Model Reaction of Unsaturated Acid Reduction Protocola, 10, 11.
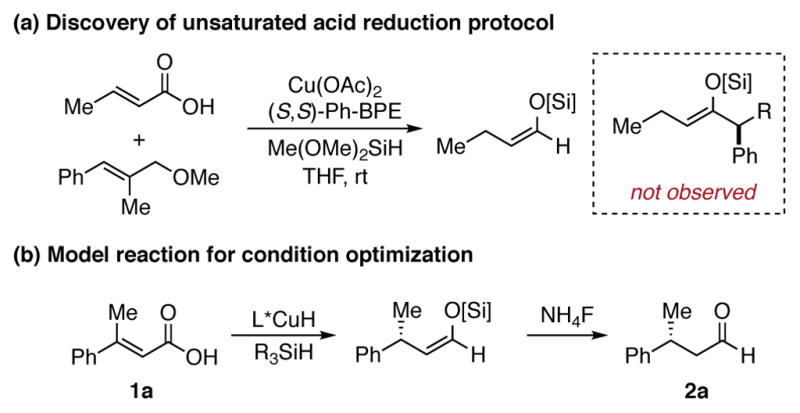
a (S,S)-Ph-BPE = 1,2-Bis((2S,5S)-2,5-diphenylphospholano)ethane.
We initiated our study of the asymmetric acid reduction reaction by subjecting (E)-3-phenylbut-2-enoic acid (1a) to conditions similar to those used for the previously reported hydroacylation reaction (4 mol% catalyst). The silyl enol ether shown was formed in 73% yield (1H NMR). After work-up with NH4F, the corresponding chiral aldehyde 2a was obtained in 86% ee (Scheme 1b). Optimization of the reaction temperature and solvent revealed that increased yield and enantiomeric excess could be achieved when the reaction was run at 40 °C in toluene. We also found that for reactions conducted on a 1 mmol scale, 1 mol% catalyst was sufficient to obtain the desired β-chiral aldehyde without diminished yield or enantioselectivity.
Having identified suitable reduction conditions, we next examined the scope of this asymmetric reduction protocol. As shown in Table 1, a variety of β-alkyl, β-aryl disubstituted unsaturated carboxylic acids were efficiently reduced to the aldehydes in good yields, and the observed enantioselectivity is uniformly high regardless of the electronic properties of the substituents. A diverse set of functional groups, including aryl chlorides (2b), heteroaryl bromides (2g), and thioethers (2c), were tolerated. Substrates containing heterocycles, such as pyridine (2f), pyrazole (2g), and benzofuran (2h), are all readily converted under the current conditions. In addition to substrates bearing a methyl β-substituent, those with more sterically-demanding ones such as ethyl (2i), cyclopropyl (2j), functionalized propyl (2l, 2m), N-pyrrolyl (2n) groups, and an exocyclic double bond (2k), were also successfully converted to the aldehydes with similarly high levels of enantioselectivity.
Table 1.

|
All yields represent average isolated yields of two runs, performed with 1 mmol of acid.
4 mol% Cu(OAc)2 and 4.4 mol% (S, S)-Ph-BPE were used.
The reactivity of β,β-diaryl acrylic acids was also investigated (Table 2). When (E)-3-(4-methoxyphenyl)-3-phenylacrylic acid (1o) was subjected to the above-described reduction conditions, the desired aldehyde was formed in high yield, but with a moderate level of enantioselectivity (70% ee). We reasoned that this might be due to poor differentiation of the two similar aryl substituents by the ligand Ph-BPE. We thus re-examined a range of common chiral ligands. Of these, the Josiphos-type ligand 3 provided the best results when the reaction was conducted at room temperature.14 Under the new reaction conditions, acids containing halides (2p, 2r), a heterocycle (2q), and an ortho-substituted arene (2r), were all efficiently converted to 3,3-diarylpropanals in high yields and with excellent enantioselectivity.
Table 2.
Examples of the Asymmetric Reduction of β,β-Diaryl Unsaturated Carboxylic Acidsa

|
Yields represent average isolated yields of two runs, performed with 1 mmol of acid.
We next set out to elucidate the mechanism of this new reduction process. One possibility is proposed in Scheme 2a. First, the carboxylic acid is silylated in the presence of CuH catalyst and silane. Subsequent 1,2-reduction of the silyl ester would give an α,β-unsaturated aldehyde, which could be further reduced to silyl enol ether by the L*CuH species. In order to test this hypothesis, the corresponding unsaturated aldehyde of 1a was prepared and subjected to the standard reaction conditions (Scheme 2b). In contrast to the 18:1 Z/E ratio of silyl enol ether produced directly from the acid, the reduction of the aldehyde favored the formation of E isomer in <1:20 Z/E ratio. This result rules out the involvement of this unsaturated aldehyde as an intermediate.
Scheme 2.

Proposed Mechanisms and Preliminary Mechanistic Studies
Based on the unusual Z-preference observed in this reduction reaction, as well as previous studies of CuH chemistry,7a,15 we postulated an alternative mechanism, shown in Scheme 2c. Instead of 1,2-reduction of the silyl ester, 1,4-reduction would result in the formation of a Cu enolate,16 from which a β-alkoxide elimination could generate a ketene intermediate. Rapid interception of this ketene by L*CuH followed by σ-bond metathesis with a hydrosilane would afford the silyl enol ether and regenerate the CuH catalyst. A simple stereochemical model for ketene reduction indeed predicts a preference for the formation of the Z silyl enol ether isomer in order to minimize steric interactions in the transition state.
Density functional theory (DFT) calculations indicated that the ketene pathway is energetically possible, with comparable barriers for the initial hydrocupration of silyl ester (18.2 kcal/mol ) and the collapse of the Cu enolate to release the ketene (16.9 kcal/mol, see the Supporting Information for details). Furthermore, the reduction reaction was performed on a series of unsaturated carboxylic acids, and the Z/E ratio of silyl enol ethers was measured by 1H NMR spectroscopy (Scheme 2d). These values closely match the computationally predicted ratios for ketene reduction by L*CuH, providing further evidence for the presence of this ketene intermediate in the reaction mechanism17, 18 (see the Supporting Information for details). Thus far, we have not been able to directly observe a ketene intermediate, presumably due to its expected high reactivity with L*CuH.
Finally, the synthetic utility of this transformation is demonstrated via a one-pot reductive amination reaction starting from carboxylic acids (Scheme 3). Several highly enantioenriched amines with γ-stereocenters were prepared in good yield, further highlighting the application of our method to access interesting compounds.
Scheme 3. One-pot Transformation from α, β-Unsaturated Carboxylic Acids to Chiral Aminesa.
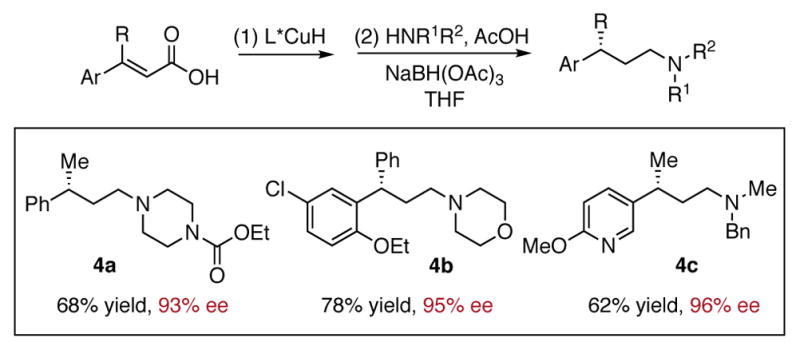
a Reactions were conducted on 0.50 mmol scale. See the Supporting Information for detailed conditions.
In conclusion, we have developed a new asymmetric reduction process that transforms α,β-unsaturated carboxylic acids to saturated aldehydes. β-chiral aldehydes with a variety of substitution patterns and functional groups can be accessed from readily available starting materials in an efficient process (1 mol% catalyst). Detailed mechanistic studies and the development of other CuH-catalyzed reactions involving carboxylic acids are ongoing.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health (GM46059, GM122483). We also thank the NIH for a supplemental grant for the purchase of supercritical fluid chromatography (SFC) equipment (GM058160-17S1) and for a postdoctoral fellowship for J. S. B. (GM112197). We acknowledge Michael Gribble for helpful discussion on reaction mechanisms. We thank Dr. Nicholas White for advice on this manuscript.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website. Experimental procedures and characterization data for all compounds (PDF).
References
- 1.(a) Brossi A, Grethe G, Teltel S, Wildman WC, Bailey DT. J Org Chem. 1970;35:1100. [Google Scholar]; (b) Welch WM, Kraska AR, Sarges R, Koe BK. J Med Chem. 1984;27:1508. doi: 10.1021/jm00377a021. [DOI] [PubMed] [Google Scholar]; (c) Hyttel J, Larsen JJ. J Neurochem. 1985;44:1615. doi: 10.1111/j.1471-4159.1985.tb08803.x. [DOI] [PubMed] [Google Scholar]; (d) Nilvebrant L, Andersson KE, Gillberg PG, Stahl M, Sparf B. Eur J Pharmacol. 1997;327:195. doi: 10.1016/s0014-2999(97)89661-6. [DOI] [PubMed] [Google Scholar]; (e) Andersson PG, Schink HE, Österlund K. J Org Chem. 1998;63:8067. [Google Scholar]; (f) Selenski C, Pettus TRR. J Org Chem. 2004;69:9196. doi: 10.1021/jo048703c. [DOI] [PubMed] [Google Scholar]
- 2.For reviews and selected examples, see: Zheng C, You SL. Chem Soc Rev. 2012;41:2498.Phillips AMF, Pombeiro AJL. Org Biomol Chem. 2017;15:2307. doi: 10.1039/c7ob00113d.Ouellet SG, Tuttle JB, MacMillan DWC. J Am Chem Soc. 2005;127:32. doi: 10.1021/ja043834g.Yang JW, Hechavarria Fonseca MT, List B. Angew Chem, Int Ed. 2004;43:6660. doi: 10.1002/anie.200461816.Mayer S, List B. Angew Chem, Int Ed. 2006;45:4193. doi: 10.1002/anie.200600512.
- 3.For selected examples of transition-metal-catalyzed asymmetric conjugated addition reactions, see: Itooka R, Iguchi Y, Miyaura N. J Org Chem. 2003;68:6000. doi: 10.1021/jo0207067.Paquin JF, Defieber C, Stephenson CRJ, Carreira EM. J Am Chem Soc. 2005;127:10850. doi: 10.1021/ja053270w.Hayashi T, Tokunaga N, Okamoto K, Shintani R. Chem Lett. 2005;34:1480.Bräse S, Höfener S. Angew Chem, Int Ed. 2005;44:7879. doi: 10.1002/anie.200501732.Nishimura T, Sawano T, Hayashi T. Angew Chem, Int Ed. 2009;48:8057. doi: 10.1002/anie.200904486.Ibrahem I, Ma G, Afewerki S, Córdova A. Angew Chem, Int Ed. 2013;52:878. doi: 10.1002/anie.201208634.
- 4.For selected examples of organocatalysis catalyzed asymmetric conjugated addition, see: Paras NA, MacMillan DWC. J Am Chem Soc. 2002;124:7894. doi: 10.1021/ja025981p.Ooi T, Doda K, Maruoka K. J Am Chem Soc. 2003;125:9022. doi: 10.1021/ja0352810.Wang W, Li H, Wang J. Org Lett. 2005;7:1637. doi: 10.1021/ol0503337.Wu F, Li H, Hong R, Khan J, Liu X, Deng L. Angew Chem, Int Ed. 2006;45:4301. doi: 10.1002/anie.200600867.Brandau S, Landa A, Franzén J, Marigo M, Jørgensen KA. Angew Chem, Int Ed. 2006;45:4305. doi: 10.1002/anie.200601025.Chen YK, Yoshida M, MacMillan DWC. J Am Chem Soc. 2006;128:9328. doi: 10.1021/ja063267s.Lee S, MacMillan DWC. J Am Chem Soc. 2007;129:15438. doi: 10.1021/ja0767480.Akagawa K, Kudo K. Angew Chem, Int Ed. 2012;51:12786. doi: 10.1002/anie.201206916.
- 5.For reviews and selected examples, see: Cahard D, Gaillard S, Renaud JL. Tetrahedron Lett. 2015;56:6159.Li H, Mazet C. Acc Chem Res. 2016;49:1232. doi: 10.1021/acs.accounts.6b00144.Tanaka K, Qiao S, Tobisu M, Lo MMC, Fu GC. J Am Chem Soc. 2000;122:9870.Doppiu A, Salzer A. Eur J Inorg Chem. 2004:2244.Mantilli L, Gérard D, Torche S, Besnard C, Mazet C. Angew Chem, Int Ed. 2009;48:5413. doi: 10.1002/anie.200901863.Quintard A, Alexakis A, Mazet C. Angew Chem, Int Ed. 2011;50:2354. doi: 10.1002/anie.201007001.Wu R, Beauchamps MG, Laquidara JM, Sowa JR. Angew Chem, Int Ed. 2012;51:2106. doi: 10.1002/anie.201107910.Arai N, Sato K, Azuma K, Ohkuma T. Angew Chem, Int Ed. 2012;51:12786.Larionov E, Lin L, Guénée L, Mazet C. J Am Chem Soc. 2014;136:16882. doi: 10.1021/ja508736u.
- 6.(a) Šmejkal T, Breit B. Angew Chem, Int Ed. 2008;47:3946. doi: 10.1002/anie.200800956. [DOI] [PubMed] [Google Scholar]; (b) Diab L, Gellrich U, Breit B. Chem Commun. 2013;49:9737. doi: 10.1039/c3cc45547e. [DOI] [PubMed] [Google Scholar]
- 7.For reviews and selected examples, see: Deutsch C, Krause N, Lipshutz BH. Chem Rev. 2008;108:2916. doi: 10.1021/cr0684321.Lipshutz BH. Synlett. 2009;4:0509.Appella DH, Moritani Y, Shintani R, Ferreira EM, Buchwald SL. J Am Chem Soc. 1999;121:9473.Moritani Y, Appella DH, Jurkauskas V, Buchwald SL. J Am Chem Soc. 2000;122:6797.Lipshutz BH, Servesko JM. Angew Chem, Int Ed. 2003;42:4789. doi: 10.1002/anie.200352313.Hughes G, Kimura M, Buchwald SL. J Am Chem Soc. 2003;125:11253. doi: 10.1021/ja0351692.Lipshutz BH, Servesko JM, Taft BR. J Am Chem Soc. 2004;126:8352. doi: 10.1021/ja049135l.Lipshutz BH, Servesko JM, Petersen TB, Papa PP, Lover AA. Org Lett. 2004;6:1273. doi: 10.1021/ol0400185.
- 8.For examples of chemoselective 1, 2-reduction of α,β-unsaturated carbonyl compounds, see: Moser R, Bošković ŽV, Crowe CS, Lipshutz BH. J Am Chem Soc. 2010;132:7852. doi: 10.1021/ja102689e.Voigtritter KR, Isley NA, Moser R, Aue DH, Lipshutz BH. Tetrahedron. 2012;68:3410.Shi SL, Wong ZL, Buchwald SL. Nature. 2016;532:353. doi: 10.1038/nature17191.Malkov AV. Angew Chem, Int Ed. 2010;49:9814. doi: 10.1002/anie.201004701.Gallagher BD, Taft BR, Lipshutz BH. Org Lett. 2009;11:5374. doi: 10.1021/ol9020404.Waidmann CR, Silks LA, Wu R, Gordon JC. Catal Sci Technol. 2013;3:1240.
- 9.Zhou Y, Bandar JS, Buchwald SL. J Am Chem Soc. 2017;139:8126. doi: 10.1021/jacs.7b04937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For detailed information about the safe handling of di-methoxy(methyl)silane (DMMS), see the Supporting Information.
- 11.For applications of BPE ligands, see: Burk MJ. Acc Chem Res. 2000;33:363. doi: 10.1021/ar990085c.Lennon IC, Pilkington CJ. Synthesis. 2003;11:1639.. Also see reference 15 and references therein.
- 12.β,β-Dialkyl unsaturated carboxylic acids provided less satisfactory results under the current conditions. When geranic acid was used, the corresponding aldehyde was isolated in 58% yield, with 70% ee.
- 13.The reactivity of α-substituted unsaturated acids was also examined. For instance, when the reduction reaction was conducted with (E)-2-methyl-3-phenylacrylic acid, the corresponding silyl enol ether was obtained in 30% NMR yield, with 1.4:1 Z/E ratio.
- 14.For applications of Josiphos ligands, see: Blaser HU, Brieden W, Pugin B, Spindler F, Studer M, Togni A. Top Catal. 2002;19:3.Blaser H-U, Pugin B, Spindler F, Mejia E, Togni A. Josiphos Ligands: From Discovery to Technical Application. In: Zhou Q-L, editor. Privileged Chiral Ligands and Catalyst. Wiley-VCH; Weinheim, Germany: 2011. pp. 93–136. and references therein
- 15.For recent reviews and mechanistic studies, see: Pirnot MT, Wang YM, Buchwald SL. Angew Chem, Int Ed. 2016;55:48. doi: 10.1002/anie.201507594.Mohr J, Oestreich M. Angew Chem, Int Ed. 2016;55:12148. doi: 10.1002/anie.201606701.Jordan AJ, Lalic G, Sadighi JP. Chem Rev. 2016;116:8318. doi: 10.1021/acs.chemrev.6b00366.Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ. Science. 2016;354:300. doi: 10.1126/science.aah5133.Bandar JS, Pirnot MT, Buchwald SL. J Am Chem Soc. 2015;137:14812. doi: 10.1021/jacs.5b10219.Issenhuth JT, Notter FP, Dagorne S, Dedieu A, Bellemin-Laponnaz S. Eur J Inorg Chem. 2010;2010:529.Tobisch S. Chem Eur J. 2016;22:8290. doi: 10.1002/chem.201600230.Dang L, Zhao H, Lin Z, Marder TB. Organometallics. 2007;26:2824.Xi Y, Hartwig JF. J Am Chem Soc. 2017;139:12758. doi: 10.1021/jacs.7b07124.Lee J, Radomkit S, Torker S, del Pozo J, Hoveyda AH. Nat Chem. 2018;10:99. doi: 10.1038/nchem.2861.
- 16.DFT calculations indicate that 1,2-hydrocupration to form the C-enolate is more facile than 1,4-hydrocupration to form the O-enolate. However, DFT also predicts that the C- and O-enolate species are similar in energy and can interconvert quickly.
- 17.A disilyl ketene acetal species may also be a reactive intermediate. However, the barrier to hydrocupration of this acetal is predicted to be significantly higher than for alternative pathways. Calculations also indicate that subsequent elimination will preferentially afford the E silyl enol ether product. See the Supporting Information for details.
- 18.Methylphenyl ketene was subjected to the standard reduction conditions. The corresponding silyl enol ether was observed by 1H NMR (10% yield). See the Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.