

Abstract
Ipomoeassin F, a plant-derived macrolide, exhibited single-digit nanomolar growth inhibition activity against many cancer cell lines. In this report, a series of 5-oxa/aza analogues was prepared and screened for cytotoxicity. Replacement of 5-CH2 with O/NH simplified the synthesis and led to only a small activity loss. N-methylation almost completely restored the potency. Further studies with additional 5-oxa analogues suggested, for the first time, that size and flexibility of the ring also significantly influence the bioactivity of ipomoeassin F.
Keywords: Resin glycosides, Ipomoeassin F, Bioisosteric ester/amide analogues, Macrolide, Cytotoxicity
TOC image
1. Introduction
Structurally and functionally unique natural products constitute a precious resource for developing novel drugs and conducting fundamental biomedical research. Notably, resin glycosides are underexplored plant-derived macrocyclic glycolipids. Their molecular skeleton with carbohydrates as part of the ring has no structural overlap with any clinical drugs and almost all other natural products. Despite a broad range of therapeutic applications in folk medicine, no systematic biological investigation has yet been reported for any single family of resin glycosides, presumably due to their low material availability.
To date, more than 250 resin glycosides have been isolated.[1] Structural diversity of those natural products largely comes from their carbohydrate components and their peripheral ester decorations. On the other hand, fatty acid aglycones share a very high degree of similarity. Jalapinolic acid (11S-hydroxyhexadecanoic acid) and convolvulinolic acid (11S-hydroxytetradecanoic acid) are most common in virtually all members of resin glycosides. Although these structural features imply that carbohydrates and their surrounding ester functionalities are determinants for a broad range of bioactivities of this series, it remains unclear about whether or how modifications in the evolutionally conserved fatty acid aglycone could confer resin glycosides improved or even new biological functions.
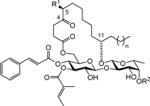
In this regard, the ipomoeassins are a distinctive family of resin glycosides that was isolated from the leaves of Ipomoea squamosa in the Suriname rainforest by Kingston’s group in 2005 (ipomoeassin A–E)[2] and 2007 (ipomoeassin F)[3]. The presence of oxygenation at C-4 for all the members and at C-5 for C–E (Table 1) is rare compared to other families of resin glycosides. Although we showed that the ketone group at C-4 had little impact on the cytotoxicity of ipomoeassin F (up to 2–3 fold activity loss),[4] deoxygenation or acetylation of the free hydroxyl group at C-5 leads to varied conclusions. For example, switching R1 from H to OAc (A vs. D) significantly enhanced the potency by up to 14-fold against the A2780 human ovarian cancer cell line (Table 1), whereas the opposite result was observed for B vs. E against the same cell line, that is, 8-fold activity loss. Moreover, extension of the lipid tail by two methylene units (F vs. A) consistently increased the activity for the six tested cancer cell lines by up to 14-fold (Table 1). Taken together, it is of great interest to know how modifications of the fatty acid moiety would affect biological activities of the ipomoeassins. Unfortunately, to date, only one synthetic analogue with an unnatural aglycone (the 11R epimer of ipomoeassin F from our group[5]) has been reported. This research gap also holds true for all other resin glycosides.
Table 1.
Structures and IC50 Values of Ipomoeassins A-F4
| structure
|
IC50 (nM)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ipomoeassin | R1 | R2 | n | Hela | L-929 | A2780 | U937 | HT-29 | MDA-MB-435 | H522-T1 | |
|
|
|||||||||||
|
A | H | Ac | 1 | 64 | 77.8 | 500 | 20.2 | 46.1 | 42.6 | 108.9 |
| B | H | H | 1 | 2500 | N/A= | 400 | 134 | 396 | 2700 | 1070 | |
| C | OH | Ac | 1 | 1500 | >1000 | 2900 | N/A | N/A | N/A | N/A | |
| D | OAc | Ac | 1 | 32 | 135 | 35 | 7.9 | 11.8 | 19.9 | 23.2 | |
| E | OAc | H | 1 | 4300 | >1000 | 3300 | 163 | 393 | 1633 | 967 | |
| F | H | Ac | 3 | N/A | 7.4 | 36 | 2.6 | 4.2 | 9.4 | 12.9 | |
N/A: not available
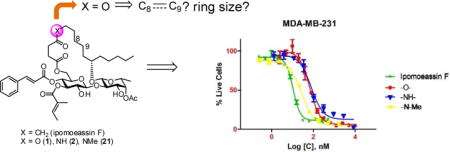
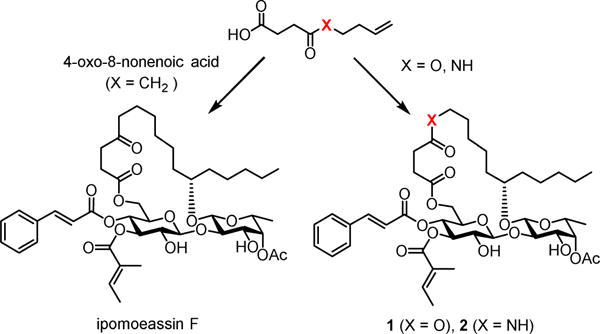
Attracted by single-digit nanomolar IC50 values against several cancer cell lines (Table 1), four total syntheses of ipomoeassin F have been accomplished by three independent research teams including ourselves.[5–9] In recent NCI-60 cell line screens, we further confirmed both high potency (the average GI50 of ~30 nM) and potential novel functional mechanism (the correlation index < 0.5 in the compare analyses) of ipomoeassin F,[6] which suggests that the ipomoeassins are unique chemical space for studying biology. To accelerate future ipomoeassin research in chemical biology and even drug discovery, however, analogues that can be synthesized more easily but have biological activity similar or superior to ipomoeassin F are still highly desirable. Therefore, in conjunction with our interest in modifying the conserved aglycone, we focused our attention on 4-oxo-8-nonenoic acid (Figure 1). In this report, we describe the evolution of the design and synthesis of several new 5-oxa/aza analogues of ipomoeassin F, e.g. 1 and 2 in Figure 1, and also some critical new SAR (structure–activity relationship) information derived from biological tests of their ability to inhibit cancer cell growth.
Figure 1.
Structures of ipomoeassin F and its bioisosteric 5-oxa/aza analogues
2. Results and discussion
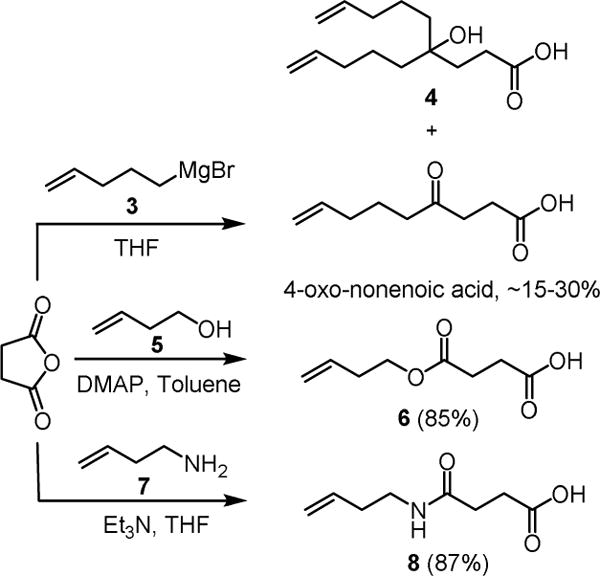
4-Oxo-nonenoic acid is an essential late-stage building block involved in all the reported total syntheses of ipomoeassin F. Although its preparation seemed to be straightforward by reacting succinic anhydride with the in situ generated Grignard reagent 3 (Scheme 1),[10] in fact, this single-step transformation suffered from low efficiency. A significant amount of dialkylated alcohol product 4 was obtained due to the inherently higher reactivity of the ketone functional group in 4-oxo-nonenoic acid than succinic anhydride. With very careful control of reaction conditions, such as temperature and addition speed of 1, 4-oxo-nonenoic acid could be obtained in a yield of up to ~30% in our hands; however, in many cases, the yields were only ~20% or even lower.
Scheme 1.
Synthesis of 4-Oxo-nonenoic Acid and Its Bioisosteric Analogues 4 and 6
To overcome this limitation, we thought that 4-oxo-nonenoic acid might be replaced with its ester/amide counterparts 6 and 8 (Scheme 1). An oxygen atom or an NH group has been considered a bioisostere of methylene according to the Grimm’s Hydride Displacement Law.[11 ] Bioisosteric replacement is a strategy commonly utilized in medicinal chemistry for optimization of bioactive molecules to enhance activity, selectivity, and/or stability while reducing metabolism.[12] We speculated that such small changes from a ketone to an ester or amide would not affect the activity much.
Before embarking on the syntheses, the molecular conformations of the various bioisosteric molecules were first investigated with density functional theory (DFT) to ensure the proposed modifications would indeed not affect the overall molecular shape significantly. One particular concern is the hydrogen of the secondary amide in analogue 2, which may potentially form an intramolecular hydrogen bond with one of the oxygen atoms around the disaccharide core, thereby altering the 3-dimensional shape of the molecule. The DFT modeling was accomplished with the B3LYP exchange correlation functional[13, 14] with the D2 empirical dispersion correction from Grimme and coworkers.[15] The 6-311G(d,p) basis set was used in an all electron calculation.[16] All the structures were optimized in vacuo since the protein binding sites of small molecules likely have relatively low dielectric constants. The lowest energy states for ipomoeassin F and the 5-oxa/aza analogues 1 and 2 are presented in Figure 2. It can be seen that all the three analogues shared virtually identical spatial orientation. The carbon, oxygen, and nitrogen atoms in ipomoeassin F, 1 and 2 (indicated by the yellow text and arrow in Figure 2) almost completely overlapped. To our surprise, both the glucose and the fucose moieties adopted the less favorable twisted-boat conformation, probably due to the favorable hydrophobic attraction between the lipid tail and the cinnamate moiety. Such hydrophobic interaction has been reported before in a crystal structure.[17] The influence of the rigid carbohydrate rings on the overall conformation of the molecule may explain why fucose-truncated analogues are inactive.[18]
Figure 2.
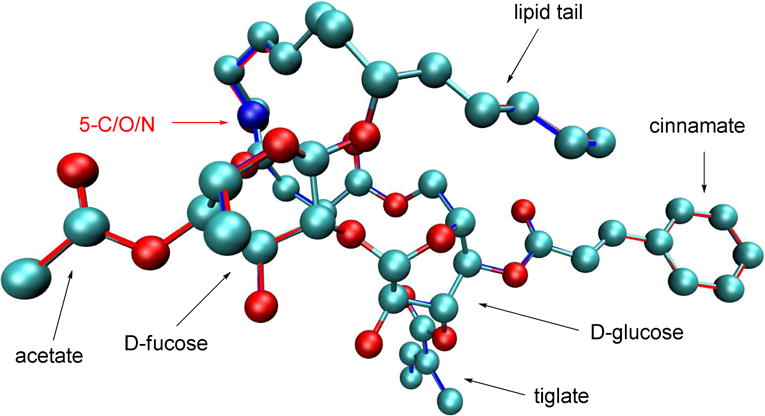
B3LYP optimized structures for ipomoeassin F, the 5-oxa analogue 1 and the 5-aza analogue 2. Ipmoeassin F is shown as ball-and-stick with the CPK coloring scheme. Analogues 1 and 2 are shown only as red and blue outlines, respectively. All the three structures overlap with each other with very minor differences. (Hydrogen atoms are not shown for clarity.)
With the confidence from the computational analyses, we then initiated our experimental work. The syntheses of 6 and 8 were highly efficient by reacting succinic anhydride with 3-butenol (5) or 3-butenamine (7, see the Supporting Information for its synthesis), respectively (Scheme 1).
The diol compound 9 (Scheme 2) is a key intermediate in our first total synthesis of ipomoeassin F.[5] With 6 and 8 in hand, they were selectively coupled with the primary hydroxyl group of 9 using DCC-mediated Steglich esterification to give the corresponding diene 10 and 11. Subsequently, the ring-closing metathesis reaction was performed with Hoveyda-Grubbs catalyst (II)[19, 20] (20 mol%) to afford the macrolide 12 and 13 as a mixture of E and Z stereoisomers. To achieve better chemoselectivity between di- and tri-substituted alkenes, Wilkinson’s catalyst was then employed for hydrogenation of 12 and 13 to furnish the saturated macrocycle 14 and 15 in good yield. The penultimate step of introducing the cinnamate moiety to produce 16 and 17 was realized by Steglich esterification of 14 and 15 with cinnamic acid. For fast removal of the two TBS groups, we first tried to treat 16 and 17 with TBAF at −10 °C for three hours. The 5-oxa analogue 1 was obtained smoothly; however, compound 18 was isolated instead of the desired 5-aza analogue 2 (Scheme 2). The process for the formation of 18 could be proposed as follows. The basic fluoride anion first removed the amide proton to form Z-5-iminodihydro-2-furanone and simultaneously release the C-6 alkoxide anion, which subsequently caused the migration of the cinnamate moiety. The Z-geometry was confirmed by a NOESY (Nuclear Overhauser Effect Spectroscopy) experiment because of missing correlation between 3-CH2 and 6-CH2 (see the Supporting Information). To prevent this side reaction, acetic acid was added to decrease the basicity of the reaction mixture. By performing the desilylation reaction at room temperature, the 5-aza analogue 2 was obtained in good yield.
Scheme 2.
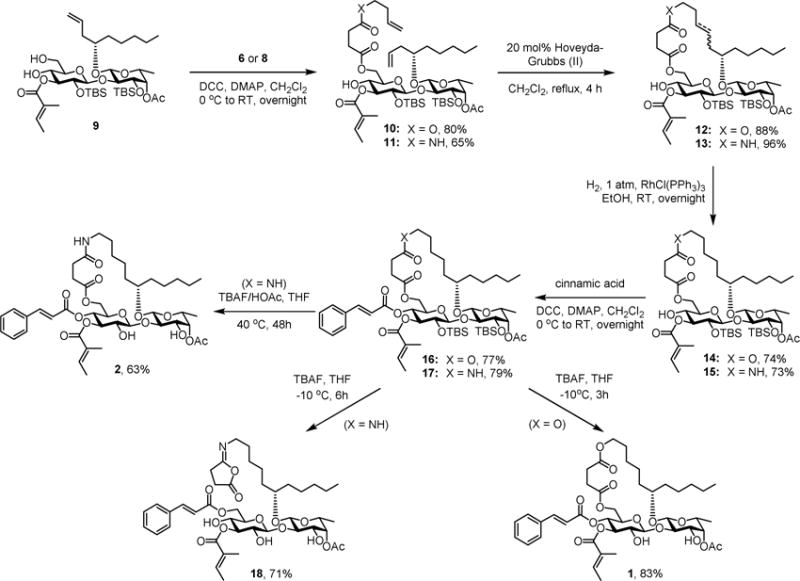
Synthesis of Bioisosteric 5-Oxa/Aza Analogues (1 and 2) of Ipomoeassin F
Using the fluorescent Alamar Blue or colorimetric MTT assay, the cytotoxicity of 1 and 2 was evaluated against two breast cancer cell lines (MDA-MB-231 and MCF7) using ipomoeassin F as the positive control and vehicle-treated cells as the negative control. The results are presented in Table 2. As we expected, the ester analogue 1 was still potent, although about 4- and 2-fold less so than ipomoeassin F. Compared to 1, the amide analogue 2 was slightly less active against MDA-MB-231cells (IC50 = 48.7 nM) but significantly less active against MCF7 cells (IC50 = 351.3 nM). Given the large activity differences (up to 80-fold) between ipomoeassin C and ipomoeassin A/D (Table 1), the relatively low activity of 2 is not that surprising, presumably due to the increased local polarity or hydrophilicity of the amide group. The ring-opened side product 18 was essentially inactive.
Table 2.
Cytotoxicity (IC50 nM)a, b of Ipomoeassin F and Its Analogues 1, 2, 18 and 21
| MDA-MB-231c | MCF7d | MCF-10Ac | |
|---|---|---|---|
| Ipomoeassin F | 7.7 | 36.4 | 5.1 |
| 1 | 31.5 | 81.2 | 26.9 |
| 2 | 48.7 | 351.3 | 75.2 |
| 18 | >25000 | >25000 | –c |
| 21 | 14.4 | 69.2 | 17.5 |
| 22 | 68.3 | 306.9 | 27.2 |
| 23 | 808.5 | 3280.7 | –c |
| 24 | 259.9 | 816.5 | –c |
| 25 | 290.3 | 710.5 | –c |
3-day treatment;
The data were obtained from at least two independent experiments, and the standard errors are within 20%;
Alamar Blue;
MTT;
“–” = not tested.
N-methylation is a viable strategy to improve bioavailability of peptides as drug candidates.[21] We envisioned that N-methylation of the analogue 2 could also decrease polarity and enhance hydrophobicity of the secondary amide, therefore improving the potency. To test this idea, we next designed the N-methylated analogue of ipomoeassin F (21, Scheme 3).
Scheme 3.
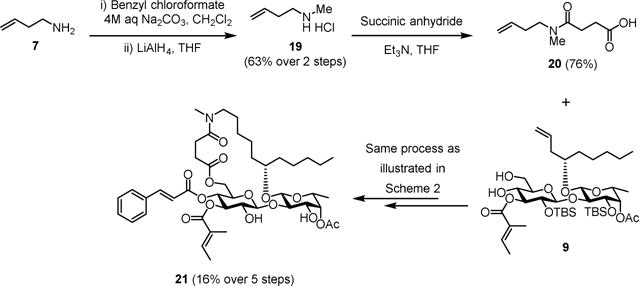
Synthesis of Methylated 5-Aza Analogue (21) of Ipomoeassin F
Given the well-established 5-step process for transformation of 9 to a final product (Scheme 2), the carboxylic acid building block 19 was key to the synthesis of 21. In brief, 3-butenamine 7 was first reacted with benzyl chloroformate. Without further purification, the resulting crude benzyl carbamate compound was fully reduced by LiAlH4 to give N-methyl-3-butenamine 19 in good yield over two steps. Subsequently, the treatment of 19 with succinic anhydride under basic conditions smoothly produced the desired carboxylic acid 20. Although the overall yield was relatively low, the conversion of 20 and 9 to make 21 did not encounter any major problems (see the Supporting Information for synthetic details). Instead, interpretation of the NMR spectra for 20 and several of its succeeding derivatives was challenging due to the coexistence of cis-trans amide bond rotamers. Nevertheless, the final product 21 was fully characterized and confirmed the ratio of the two rotamers was almost 1:1. To our delight, after screening against the two breast cancer cell lines, analogue 21 regained biological activity within 2-fold of the original natural product (Table 2).
With a better synthetic profile for the 5-oxa modification, we decided to carry out two more studies that were very challenging for carbon-chain algycone analogues of ipomoeassin F. An alkene is a common functionality in many macrocylic natural products. Because a double bond was created by RCM during the total synthesis of ipomoeassin F, we had tried hard to isolate stereochemically pure components along the way to the end. Due to the extremely close polarity of all the intermediates, however, the final product 22 (Figure 3) could be obtained only as a mixture of E/Z isomers. Fortunately, the E/Z isomers of 5-oxa intermediate 12 (Scheme 2) could be separated by column chromatography. Therefore, we were able to obtain the pure 5-oxa trans- and cis-mimics of ipomoeassin F (23 and 24, Figure 3) (see the Supporting Information for synthetic details). Due to the signal overlap, it was not possible to confirm the correlation between 7-CH2 and 10-CH2 in 24 from the NOESY spectrum (available in the Supporting Information); however, the trans-isomer 23 was assigned because the desired correlation was clearly absent. The slightly larger coupling constant between 8-CH= and 9-CH= in 23 (J = 15.4 Hz) than in 24 (J = 10.5 Hz) also supports our assignment. Next, biological assays of analogues 22–24 were conducted (Table 2). Although the Z-analogue 24 was ~3-fold more potent than the E-counterpart 23, all three compounds showed lower activity (8–20 fold) compared to ipomoeassin F and 1. Therefore, for the first time, we demonstrated that increasing rigidity around C-8/C-9 was disfavored.
Figure 3.
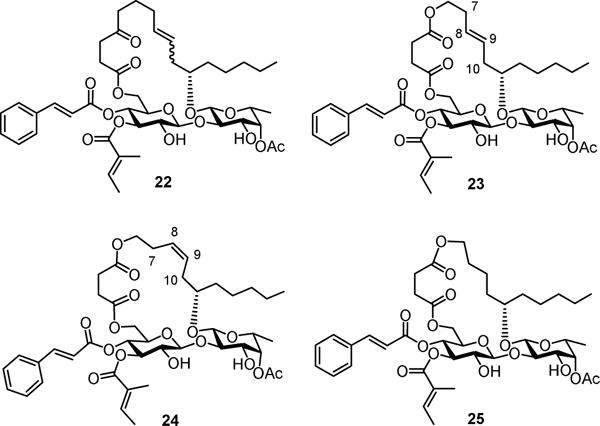
Structures of aglycone-modified ipomoeassin F analogues 22–25.
Despite the poor activity of 18, our earlier studies showed that unlike many other macrolides, the cyclic structure of ipomoeassin F is not absolutely crucial because one ring-opened analogue showed only a 2~3-fold cytotoxicity loss against seven tested cell lines.[4] Hence, we surmised that we might be able to change the ring size from the 20-membered ring for all the ipomoeassins to improve existing or even search for new biological activity. However, this idea was quite hard to test because of the low efficiency in making pure carbon chain analogues of 4-oxo-nonenoic acid with various chain lengths. Since the 5-oxa analogue 1 was found to be active, it would be possible for us to explore the effect of ring size more efficiently by using 1 as the surrogate of ipomoeassin F. Therefore, as a preliminary experiment, we synthesized the 19-membered 5-oxa analogue 25 (Figure 3) by replacing 3-butenol with allyl alcohol (see the Supporting Information for synthetic details). Somewhat to our surprise, compared to the ring-opened analogue, removing only one methylene unit inside the ring caused a significantly greater activity loss (9-fold for both MBA-BD-231 and MCF7 when compared to 1, Table 2). Although further studies are needed to elucidate the role of the ring size, this indicates for the first time that the ring size is also an important parameter for the biological activity of the ipomoeassins.
We investigated the relationship between the overall lipophilicity of different analogues based on their HPLC retention times and their bioactivity (see Charts S1 and S2 in the Supporting Information). No correlations were observed from the charts. However, local lipophilicity/hydrophilicity and/or H-bonding network around the ketone moiety at C-4 influence the biological activity of the natural product.
As a final note, all the active compounds with an IC50 value lower than 200 nM against MDA-MB-231 and 500nM against MCF7 were also assayed against one immortalized normal human mammary epithelial cell line (MCF-10A) to check selectivity between tumor and non-tumor cells. Unfortunately, no significant selectivity between the selected cell lines was observed for most of the cytotoxic analogues reported here (Table 2). Only the amide analogue 2 showed slight selectivity towards MDA-MB-231 cells.
3. Conclusions
Using the previously established synthetic route, a panel of 5-oxa/aza analogues of ipomoeassin F was efficiently synthesized over 4/5 steps from the common diol precursor 9. They represent the first series of synthetic analogues with modifications in the carbon backbone of the aglycone, not only for the ipomoeassin class specifically but also for all resin glycosides in general. The exchange of a methylene unit by O/NH streamlined the synthesis while retaining low nanomolar bioactivity, which may accelerate ipomoeassin research in chemical biology and drug discovery. The findings here convey an important message that the fatty acid backbone is modifiable, which will open new avenues for engineering the structure of ipomoeassin F in the future.
4. Experimental section
General Methods
Reactions were carried out in oven-dried glassware. All reagents were purchased from commercial sources and were used without further purification unless noted. Unless stated otherwise, all reactions were carried out under a nitrogen atmosphere and monitored by TLC using silica gel GF254 plates with detection by charring with 5% (v/v) H2SO4 in EtOH or by visualizing in UV light (254 nm). Column chromatography was performed on silica gel (230–450 mesh). The ratio between silica gel and crude product ranged from 100 to 50:1 (w/w). 1H NMR spectra were obtained in deuteriochloroform (CDCl3) with chloroform (CHCl3, δ = 7.27 for 1H) as an internal reference. 13C NMR spectra were proton decoupled and were in CDCl3 with CHCl3 (δ = 77.0 for 13C) as an internal reference. Chemical shifts are reported in ppm (δ). Data are presented in the form: chemical shift (multiplicity, coupling constants, and integration). 1H data are reported as though they were first order. The errors between the coupling constants for two coupled protons were less than 0.5 Hz, and the average number was reported. Proton assignments, when made, were done so with the aid of COSY NMR spectra. For some compounds, HSQC and HMBC NMR were also applied to assign the proton signals. Optical rotations were measured at 25 ± 1 °C for solutions in a 1.0 dm cell. High resolution mass spectra (HRMS) were performed with an ion cyclotron resonance analyzer by electrospray ionization. The purity of the obtained analogues was determined by HPLC analysis using a C18 column (150 mm × 4.6 mm, 5 μm) and a UV detector at the wavelength of 280 nm. The mobile phase was a MeCN/H2O mixture. The tested compounds were at least 95% pure.
General procedure for Steglich esterification
DCC (1.5 equiv) was added in one portion to a 0°C CH2Cl2 (3 mL) solution of alcohol (1 equiv), cinnamic acid (1.5 eq) and 4-dimethylaminopyridine (0.5 equiv). The reaction was allowed to warm to ambient temperature and stirred overnight. At this point, TLC (silica, EtOAc–hexanes) showed the reaction was complete. The reaction mixture was diluted with ether (5 mL) and hexanes (2 mL), stirred for 20 minutes then filtered thru a pad of Celite using ether (5 mL) as the eluent, and the filtrate was concentrated in vacuo. The residue was purified by flash column chromatography (silica, EtOAc–hexanes) to give the cinnamate derivatives as a colorless syrup.
General procedure for Mukaiyama esterification
2-Chloro-1-methylpyridinium iodide (CMPI, 2 equiv), N,N-dimethylaminopyridine (DMAP, 0.5 equiv) and Et3N (10 eq) were added to a solution of alcohol (1 equiv) and cinnamic acid (2 equiv) in dry CH2Cl2 (2 mL) at 0 °C. The reaction was allowed to warm to ambient temperature and stirred for 24 h. At this point, TLC (silica, EtOAc–hexanes) showed the reaction was complete. Evaporation of the solvent followed by purification of the residue by column chromatography (silica, EtOAc–hexanes) gave the cinnamate derivatives as a colorless syrup.
General procedure for TBS removal using TBAF
To a solution of TBS protected compounds (1 equiv) in THF (2 mL) was added TBAF (1M solution in THF, 6 equiv) at –10 °C. The reaction mixture was stirred at the same temperature for 3–5 h at which point TLC (silica, EtOAc–hexanes) showed it was complete. The reaction mixture was diluted with Et2O (20 mL), washed with 1M HCl (10 mL), saturated NaHCO3 (10 mL), brine (10 mL). The aqueous layer was extracted with Et2O (20 mL). The combined organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc–hexanes) gave analogues.
General procedure for TBS removal using TBAF/AcOH
To a solution of TBS protected compounds (1 equiv) in THF (2 mL) was added AcOH (100 equiv) and TBAF (1M solution in THF, 50 equiv) at room temperature. The reaction was then stirred at 25–40 °C for 12–48 h. At this point, TLC (silica, EtOAc–hexanes) showed the reaction was complete. The reaction mixture was diluted with Et2O (20 mL), washed with 1M HCl (10 mL), saturated aqueous NaHCO3 (10 mL), brine (10 mL). The aqueous layer was extracted with Et2O (20 mL). The combined organic layer was dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (silica, EtOAc–hexanes) to give analogues.
Analogue 1
Steglich esterification of 14 (27.6 mg, 0.030 mmol) with cinnamic acid gave ester 16 (24.1 mg, 77%) as a colorless syrup. Removal of TBS in 16 (22.4 mg, 0.0221 mmol) using TBAF gave analogue 1 (14.6 mg, 83%) as a white foam. [α]D25 −39.5° (c 0.5 CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 16.0 Hz, 1H, Ph-CH=C-), 7.54–7.45 (m, 2H, 2 × ArH), 7.44–7.35 (m, 3H, 3 × ArH), 6.95–6.85 (m, 1H, Me-CH-C(Me)-C=O), 6.32 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 5.49 (t, J = 10.0 Hz, 1H, H-4-Glup), 5.20–5.05 (m, 2H, H-3-Glup, H-4-Fucp), 4.86–4.52 (m, 2H, H-1-Glup, OH), 4.39 (d, J = 7.6 Hz, 1H, H-1-Fucp), 4.30 (dd, J = 12.8, 2.4 Hz, 1H, H-6-Glup), 4.15 (dd, J = 12.4, 2.0 Hz, 1H, H-6-Glup), 4.15–4.00 (m, 3H, -C(O)O-CH2-, OH), 3.91 (dd, J = 9.6, 3.6 Hz, 1H, H-3-Fucp), 3.83–3.55 (m, 5H, H-2-Glup, H-5-Glup, H-2-Fucp, H-5-Fucp, -CH2-CH-CH2-), 2.84–2.69 (m, 2H, aglycone-CH2), 2.68–2.53 (m, 2H, aglycone-CH2), 2.18 (s, 3H, CH3-C=O), 1.82–1.70 (m, 6H, CH3-CH-C(CH3)-C=O), 1.68–1.47 (m, 8H, aglycone-CH2), 1.44–1.22 (m, 8H, aglycone-CH2), 1.18 (d, J = 6.4 Hz, 3H, H-6-Fucp), 0.90 (t, J = 6.8 Hz, 3H, aglycone-CH3). 13C NMR (100 MHz, CDCl3) δ 172.3, 171.8, 171.3, 169.0, 165.2, 146.3, 140.0, 133.9, 130.7, 128.9 (×2), 128.2 (×2), 127.5, 116.5, 106.7, 101.8, 84.3, 81.4, 76.0, 74.3, 72.7, 72.6, 72.1, 68.7, 67.2, 64.5, 62.3, 34.9, 34.7, 31.9, 29.8, 29.4, 28.4, 26.5, 25.1, 24.4, 22.6, 20.9, 16.4, 14.6, 14.1, 12.0. HRMS (ESI) m/z calcd for C43H60NaO16 [M+Na]+ 855.3774, found: 855.3770. Purity: 97.9%, tR = 15.04 min.
Analogue 2
Removal of TBS in 17 (25.9 mg, 0.0244 mmol) using TBAF/AcOH gave analogue 2 (12.8 mg, 63%) as a white film. [α]D25 −41.5° (c 0.2, CHCl3). 1H NMR (400 MHz, CDCl3). 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 7.56–7.47 (m, 2H, 2 × ArH), 7.45–7.34 (m, 3H, 3 × ArH), 6.96–6.85 (m, 1H, Me-CH-C(Me)-C=O), 6.34 (d, J = 16.0 Hz, 1H, Ph-CH=CH-), 6.01–5.90 (m, 1H, NH), 5.45 (t, J = 9.6 Hz, 1H, H-4-Glup), 5.18 (t, J = 9.6 Hz, 1H, H-3-Glup), 5.15–5.07 (m, 1H, H-4-Fucp), 4.69–4.57 (m, 2H, H-1-Glup, OH), 4.40 (d, J = 7.6 Hz, 1H, H-1-Fucp), 4.31 (dd, J = 12.4, 2.0 Hz, 1H, H-6-Glup), 4.19 (dd, J = 12.2, 1.8 Hz, 1H, H-6-Glup), 3.96–3.88 (m, 1H, H-3-Fucp), 3.84–3.75 (m, 2H, H-5-Glup, OH), 3.74–3.58 (m, 4H, H-2-Glup, H-2-Fucp, H-5-Fucp, -CH2-CH-CH2-), 3.52–3.40 (m, 1H, NHCH2), 3.13–3.00 (m, 1H, NHCH2), 2.88–2.77 (m, 1H, aglycone-CH2), 2.75–2.65 (m, 1H, aglycone-CH2), 2.61–2.50 (m, 1H, aglycone-CH2), 2.48–2.38 (m, 1H, aglycone-CH2), 2.18 (s, 3H, CH3-C=O), 1.82–1.71 (m, 6H, CH3-CH-C(CH3)-C=O), 1.58–1.15 (m, 19H, H-6-Fucp, aglycone-CH2), 0.90 (t, J = 6.8 Hz, 3H, aglycone-CH3). 13C NMR (100 MHz, CDCl3) δ 173.1, 172.0, 171.9, 168.8, 165.3, 146.4, 139.8, 133.9, 130.8, 129.0 (×2), 128.3 (×2), 127.5, 116.5, 106.5, 101.3, 83.9, 80.6, 75.6, 74.4, 72.8, 72.5, 72.0, 68.7, 67.5, 62.7, 38.6, 34.4, 34.2, 32.0, 31.9, 31.1, 29.2, 26.6, 25.1, 24.0, 22.7, 20.9, 16.4, 14.6, 14.1, 12.0. HRMS (ESI) m/z calcd for C43H61NaO [M+Na]+ 854.3933, found: 854.3930. Purity: 98.9%, tR = 6.47 min.
Analogue 21
Steglich esterification of alcohol S2 (see SI, 60.0 mg, 0.0635 mmol) with cinnamic acid gave ester S3 (see SI, 41.0 mg, 60%) as a colorless syrup. Removal of TBS in S3 (39.0 mg, 0.036 mmol) using TBAF gave analogue 21 (20.2 mg, 66%) as a colorless syrup. 1H NMR (400 MHz, CDCl3). 1H NMR (400 MHz, CDCl3) δ 7.63, 7.62 (2d, J = 16.0 Hz, 1H, Ph-CH=CH-), 7.55–7.46 (m, 2H, 2 × ArH), 7.44–7.34 (m, 3H, 3 × ArH), 6.96–6.84 (m, 1H, Me-CH-C(Me)-C=O), 6.34, 6.33 (2d, J = 16.0 Hz, 1H, Ph-CH=CH-), 5.38, 5.29 (2t, J = 9.8 Hz, 1H, H-4-Glup), 5.20–5.00 (m, 2H, H-3-Glup, H-4-Fucp), 4.79–4.08 (m, 5H, H-1-Glup, H-1-Fucp, H-6-Glup, OH), 4.04–3.85 (m, 2H, H-3-Fucp, OH), 3.80–3.20 (m, 7H, H-2-Glup, H-5-Glup, H-2-Fucp, H-5-Fucp, -CH2-CH-CH2-, N(CH3)CH2), 3.05, 2.92 (2s, 3H, NCH3), 2.86–2.52 (m, 4H, aglycone-CH2), 2.18, 2.17 (2s, 3H, CH3-C=O), 1.80–1.69 (m, 6H, CH3-CH-C(CH3)-C=O), 1.66–1.14 (m, 19H, H-6-Fucp, aglycone-CH2), 0.94–0.85 (m, 3H, aglycone-CH3). 13C NMR (100 MHz, CDCl3) δ 172.4, 171.9, 171.8, 171.6, 171.6, 171.1, 169.4, 168.8, 165.4, 165.3, 146.2, 146.1, 140.4, 139.8, 134.0, 133.9, 130.7, 130.6, 128.9, 128.8, 128.3, 128.2, 127.6, 127.4, 116.7, 106.2, 106.0, 101.3, 100.8, 83.8, 83.7, 81.1, 80.4, 76.9, 75.8, 74.3, 72.9, 72.7(4), 72.6(8), 72.6, 72.4, 72.3, 68.9, 68.7, 67.6, 66.7, 62.1, 61.3, 49.3, 46.7, 35.2, 34.7, 34.6, 34.3, 33.9, 33.5, 31.9, 31.8, 30.6, 30.4, 28.2, 27.7, 27.3, 27.2, 26.5, 26.4, 25.5, 24.7, 24.0(9), 24.0(6), 22.7, 22.6, 20.9, 16.4, 16.3, 14.7, 14.6, 14.1(1), 14.1(0), 12.0, 11.9. HRMS (ESI) m/z calcd for C44H63NNaO15 [M+Na]+ 868.4090, found 868.4095. Purity: 95.9%, tR = 8.63 min.
Biological evaluation
The cytotoxicity of the analogues was evaluated using the fluorescent Alamar Blue or colorimetric MTT assay. The procedure for the Alamar Blue assay is shown below. The procedures for cell culture and the colorimetric MTT assay are described in the Supplementary data.
Alamar Blue Cytotoxicity Assay
Counting of viable cells was performed before each experiment. Experiments were done in triplicate. First, 100 μL of cell suspension at the density of 50,000 cells/mL was seeded in a 96-well plate (5,000 cells/well), which was incubated at 37 °C in 5% CO2 for 24 hours. The compounds were dissolved in DMSO (dimethyl sulfoxide) to make drug stocks (10 mM). The stock solutions were diluted with the complete DMEM/HIGH medium to make a series of gradient fresh working solutions right before each test. The highest amount of DMSO was controlled to be lower than 0.5%. Subsequently, the cells were treated with 100 μL of the freshly made gradient working solution in the total volume of 200 μL/well for 72 hours. After that, the media were discarded and 200 μL of the fresh complete medium containing 10% of AlamarBlue (resazurin) stock solution (3 mg/27.15mL) (5 mg/mL) was added to each well. The plate was then incubated at 37 °C in 5% CO2 atmosphere for another 3 hours. Next, 180 μL of the medium was discarded from each well. The formed formazan crystals were dissolved with 180 μL of DMSO. Absorbance of formazan was detected by a microplate reader (BioTek Synergy H1) at excitation 580 nm with emission 620 nm as the reference wavelength. The percentage of viability compared to the negative control (DMSO-treated cells) was determined.
Activities of synthesized compounds against breast cancer cell line MDA-MB-231 and human breast epithelial cell line MCF-10A were tested by the alamarBlue cytotoxicity assay. GraphPad Prism 6 software was used to make a plot of % viability versus sample concentration and to calculate the concentration at which a compound exhibited 50% cytotoxicity (IC50).
Supplementary Material
Highlights.
A panel of 5-oxa/aza analogues of ipomoeassin F were synthesized.
The 5-oxa/aza analogues showed low nanomolar cytotoxicity.
The evolutionarily-conserved fatty acid backbone is modifiable.
Ring size and aglycone flexibility have significant influence on the bioactivity.
Acknowledgments
This work was primarily supported by the startup funds from the University of Arkansas and also in part by Grant Number R15GM116032 and P30 GM103450 from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) and by seed money from the Arkansas Biosciences Institute (ABI). We wish to thank Dr. Jianjun Zhang at CAU (China Agricultural University) for providing per-acetylated D-fucopyranose (NKT R&D Program of China, 2015BAK45B01). RB was the recipient of a Student Undergraduate Research Fellowship (SURF) from the Arkansas Department of Higher Education.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Notes: The authors report no conflicts of interest.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at. See DOI:.
References
- 1.Pereda-Miranda R, Rosas-Ramirez D, Castaneda-Gomez J. Resin glycosides from the morning glory family. Prog Chem Org Nat Prod. 2010;92:77–153. doi: 10.1007/978-3-211-99661-4_2. [DOI] [PubMed] [Google Scholar]
- 2.Cao S, Guza RC, Wisse JH, Miller JS, Evans R, Kingston DGI. Ipomoeassins A-E, cytotoxic macrocyclic glycoresins from the leaves of Ipomoea squamosa from the Suriname rainforest. J Nat Prod. 2005;68:487–492. doi: 10.1021/np049629w. [DOI] [PubMed] [Google Scholar]
- 3.Cao S, Norris A, Wisse Jan H, Miller James S, Evans R, Kingston David GI. Ipomoeassin F, a new cytotoxic macrocyclic glycoresin from the leaves of Ipomoea squamosa from the Suriname rainforest. Nat Prod Res. 2007;21:872–876. doi: 10.1080/14786410600929576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zong G, Aljewari H, Hu Z, Shi WQ. Revealing the Pharmacophore of Ipomoeassin F through Molecular Editing. Org Lett. 2016;18:1674–1677. doi: 10.1021/acs.orglett.6b00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zong G, Barber E, Aljewari H, Zhou J, Hu Z, Du Y, Shi WQ. Total Synthesis and Biological Evaluation of Ipomoeassin F and Its Unnatural 11R-Epimer. J Org Chem. 2015;80:9279–9291. doi: 10.1021/acs.joc.5b01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zong G, Whisenhunt L, Hu Z, Shi WQ. Synergistic Contribution of Tiglate and Cinnamate to Cytotoxicity of Ipomoeassin F. J Org Chem. 2017;82:4977–4985. doi: 10.1021/acs.joc.7b00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Postema MHD, TenDyke K, Cutter J, Kuznetsov G, Xu Q. Total Synthesis of Ipomoeassin F. Org Lett. 2009;11:1417–1420. doi: 10.1021/ol900086b. [DOI] [PubMed] [Google Scholar]
- 8.Nagano T, Pospisil J, Chollet G, Schulthoff S, Hickmann V, Moulin E, Herrmann J, Mueller R, Fuerstner A. Total Synthesis and Biological Evaluation of the Cytotoxic Resin Glycosides Ipomoeassin A-F and Analogues. Chem - Eur J. 2009;15:9697–9706. doi: 10.1002/chem.200901449. [DOI] [PubMed] [Google Scholar]
- 9.Zong G, Shi WQ. Total Synthesis of Ipomoeassin F and Its Analogs for Biomedical Research. In: Harmata M, editor. Strategies Tactics Org Synth. Academic Press; 2017. pp. 81–110. [Google Scholar]
- 10.Lhommet G, Freville S, Thuy V, Petit H, Celerier JP. A general and versatile synthesis of 4- and 5-oxo acids. Synth Commun. 1996;26:3897–3901. [Google Scholar]
- 11.Grimm HG. Structure and size of the non-metallic hydrides. Z Elektrochem Angew Phys Chem. 1925;31:474–480. [Google Scholar]
- 12.Patani GA, LaVoie EJ. Bioisosterism: A rational approach in drug design. Chem Rev (Washington, D C) 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- 13.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 14.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 15.Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J Comput Chem. 2006;27:1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 16.Krishnan R, Binkley JS, Seeger R, Pople JA. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys. 1980;72:650–654. [Google Scholar]
- 17.Sakurai K, Fukumoto T, Noguchi K, Sato N, Asaka H, Moriyama N, Yohda M. Three-Dimensional Structures of OSW-1 and Its Congener. Org Lett. 2010;12:5732–5735. doi: 10.1021/ol1025519. [DOI] [PubMed] [Google Scholar]
- 18.Zong G, Hirsch M, Mondrik C, Hu Z, Shi WQ. Design, synthesis and biological evaluation of fucose-truncated monosaccharide analogues of ipomoeassin F. Bioorg Med Chem Lett. 2017;27:2752–2756. doi: 10.1016/j.bmcl.2017.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gessler S, Randl S, Blechert S. Synthesis and metathesis reactions of a phosphine-free dihydroimidazole carbene ruthenium complex. Tetrahedron Lett. 2000;41:9973–9976. [Google Scholar]
- 20.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
- 21.Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelnick R, Gilon C, Hoffman A, Kessler H. Improving oral bioavailability of peptides by multiple N-methylation: somatostatin analogues. Angew Chem, Int Ed. 2008;47:2595–2599. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.