

Abstract
SB269652 is a negative allosteric modulator of the dopamine D2 receptor (D2R) yet possesses structural similarity to ligands with a competitive mode of interaction. In this study, we aimed to understand the ligand-receptor interactions that confer its allosteric action. We combined site-directed mutagenesis with molecular dynamics simulations using both SB269652 and derivatives from our previous structure activity studies. We identify residues within the conserved orthosteric binding site (OBS) and a secondary binding pocket (SBP) that determine affinity and cooperativity. Our results indicate that interaction with the SBP is a requirement for allosteric pharmacology, but that both competitive and allosteric derivatives of SB269652 can display sensitivity to the mutation of a glutamate residue (E952.65) within the SBP. Our findings provide the molecular basis for the differences in affinity between SB269652 derivatives, and reveal how changes to interactions made by the primary pharmacophore of SB269652 in the orthosteric pocket can confer changes in the interactions made by the secondary pharmacophore in the SBP. Our insights provide a structure-activity framework towards rational optimization of bitopic ligands for D2R with tailored competitive versus allosteric properties.
Keywords: G protein-coupled receptor, dopamine receptor, allosteric modulation, bitopic ligands, molecular dynamics simulations, mutagenesis
Graphical Abstract
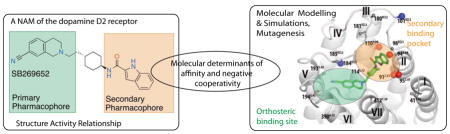
1. Introduction
The dopamine D2 receptor (D2R), a prototypical class A G protein-coupled receptor (GPCR), is a therapeutic target for a variety of central nervous system disorders [1]. To date, drug discovery at the D2R has focused on targeting the conserved orthosteric binding site (OBS), either with agonists for the treatment of the motor symptoms of Parkinson’s disease, or antagonists for the treatment of the positive symptoms of schizophrenia [2]. However, this approach can be associated with significant adverse side effects. In particular, the orthosteric blockade of the D2R by typical antipsychotics, while effective in treating the positive symptoms of schizophrenia, is associated with extrapyramidal side effects [2].
It is now apparent that many class A GPCRs possess topographically distinct, allosteric binding sites that have the potential to be targeted by small molecules [3–5]. Allosteric targeting may offer several advantages over purely orthosteric ligands, including increased subtype selectivity afforded by the lower degree of sequence conservation at these sites, and maintenance of the spatiotemporal patterns associated with endogenous neurohumoral signalling [6]. These potential advantages make allosteric targeting of the D2R an attractive approach for the treatment of schizophrenia, for which partial blockade by a negative allosteric modulator (NAM) with limited negative cooperativity could be safer and associated with fewer side effects. However, despite the potential of allosteric modulators, this approach has yet to be exploited therapeutically. This is due, in part, to the paucity of allosteric scaffolds that act at the D2R. In addition, there is only limited information about the location and druggability of allosteric binding site(s) within this receptor [7].
Recently, we demonstrated that the compound SB269652 is a NAM of the D2R, and presented a novel allosteric mechanism whereby SB269652 adopts a bitopic pose at one protomer of a D2R dimer to modulate the binding of dopamine at the adjacent protomer [8]. To this end, molecular modelling and site-directed mutagenesis experiments have revealed a secondary binding pocket (SBP) at the extracellular ends of transmembrane segments (TMs) 2 and 7 that accommodates the indole-2-carboxamide secondary pharmacophore (SP) of SB269652 [8,9]. Our molecular modelling studies predict that E952.65 (Ballesteros-Weinstein nomenclature [10]), which sits at the water-accessible surface at the extracellular end of TM2, forms a hydrogen-bond interaction with the indolic NH of SB269652. This prediction was validated by the mutation E952.65 to alanine, which results in a significant loss in SB269652 affinity and negative cooperativity with dopamine [8]. The observation that methylation of the indole NH, thus abrogating the ability to hydrogen bond with E952.65, results in a derivative that displays pharmacology best fitted by a competitive model provided further evidence that this interaction plays a critical role in determining the allosteric pharmacology of SB269652 [8].
The antagonist-bound crystal structure of the highly homologous D3R revealed a SBP comprising residues from the extracellular ends of TMs 1, 2, 3, and 7 and extracellular loops (ECLs) 1 and 2 [11]. Subsequent studies have revealed that the subtype-selectivity of extended ligands such as R-22 is conferred by the interaction of the SP of such ligands with this SBP [12]. It should be noted that although R-22 displays competitive pharmacology, the 1H-indole-2-carboxamide SP of R-22 is identical to that of SB269652. Thus, the allosteric action of SB269652 is not simply conferred by its extension into this SBP but instead must be conferred by differences in the receptor-ligand interactions made by these two structurally related-ligands [12]. In agreement with this observation, our structure-activity studies of SB269652 revealed that relatively subtle changes to the orthosteric tetrahydroisoquinoline (THIQ) primary pharmacophore, the linker region, or the SP all had profound affects upon cooperativity and/or affinity, and could even lead to a change from allosteric to apparently competitive pharmacology [9]. Therefore, to exploit this novel class of allosteric modulator at the D2R, it is important to understand the ligand-receptor interactions that confer this allosteric pharmacology and the differences in the binding modes of structurally similar competitive antagonists. In this study, we investigated the structural basis for the allosteric mechanism of SB269652 by combining site-directed mutagenesis with molecular modelling and simulations of D2R in complex with either SB269652 or its selected derivatives.
2. Methods and Materials
2.1 Materials
SB269652 and all derivatives were synthesised as previously described and were shown to be >95% pure [8,9]. Dopamine was purchased from Sigma-Aldrich (Castle Hill, NSW, Australia) and was >98% pure as indicated by the supplier. Dulbecco’s modified Eagle’s medium (DMEM), hygromycin B, and FlpIn CHO cells were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from ThermoTrace (Melbourne, VIC, Australia). [3H]Spiperone, AlphaScreen reagents, Ultima gold scintillation cocktail, and 384-well proxiplates were purchased from PerkinElmer (Boston, MA). All the other reagents were purchased from Sigma-Aldrich (Castle Hill, NSW, Australia).
2.2 Synthesis of compounds
All compounds were synthesized as described in Shonberg et. al. 2015 with the exception of MIPS1868 which was synthesised as follows.
N-((trans)-4-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl)cyclohexyl)-7-fluoro-1H-indole-2-carboxamide (MIPS1868).
To a stirred solution of 2-(2-((trans)-4-aminocyclohexyl)ethyl)-1,2,3,4-tetrahydroisoquinoline-7-carbonitrile (126 mg, 444 μmol) and 7-fluoro-1H-indole-2-carboxylic acid (95.5 mg, 533 μmol) in a minimal volume of anhydrous DMF (3–4 mL) was added the coupling reagent, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU, 253 mg, 666 μmol) and an excess of DIPEA. The reaction was stirred at room temperature overnight after which time, LCMS revealed complete consumption of starting material. The reaction mixture was then diluted with 30 mL of a 1:1 mixture of a saturated solution of NaHCO3 and water and left to stir for a further 30 min. The resulting precipitate was filtered, washed with water, dried and then recrystallised via hot filtration in a mixture of MeOH and water to yield the title compound as a beige crystalline solid (112 mg, 57%). 1H NMR (d6-DMSO) δ 11.95 (s, 1H), 8.22 (d, J = 7.9 Hz, 1H), 7.59 – 7.54 (m, 2H), 7.45 – 7.40 (m, 1H), 7.31 (d, J = 8.4 Hz, 1H), 7.19 (d, J = 2.5 Hz, 1H), 7.04 – 6.96 (m, 2H), 3.75 (m, 1H), 3.57 (s, 2H), 2.88 (t, J = 5.6 Hz, 2H), 2.66 (t, J = 5.8 Hz, 2H), 2.55 – 2.44 (m, 2H), 1.86 (dd, J = 32.7, 10.8 Hz, 4H), 1.45 (dd, J = 14.5, 6.8 Hz, 2H), 1.40 – 1.23 (m, 3H), 1.06 (dd, J = 24.4, 10.5 Hz, 2H). 13C NMR (d6-DMSO) δ 160.0 (C), 149.7 (d, 1JCF = 245.1 Hz, C), 141.2 (C), 137.2 (C), 134.0 (C), 131.3 (d, 4JCF = 5.7 Hz, C), 130.8 (CH), 130.1 (CH), 129.9 (CH), 125.0 (d, 3JCF = 13.4 Hz, C), 120.4 (d, 4JCF = 6.0 Hz, CH), 119.6 (C), 118.0 (d, 4JCF = 3.4 Hz, CH), 108.6 (C), 108.3 (d, 3JCF = 16.2 Hz, CH), 104.6 (CH), 55.8 (CH2), 55.3 (CH2), 50.3 (CH2), 48.8 (CH), 35.2 (CH), 34.0 (CH2), 32.7 (CH2), 32.1 (CH2), 29.4 (CH2).HPLC: tR 6.36 min, >95% purity. HRMS (m/z): C27H30FN4O requires [M+H]+ 445.2404; found 445.2405.
2.3 Molecular biology
cDNA in pcDNA3.1+ encoding the long isoform of the wild-type human dopamine D2 receptor (D2LR)was obtained from Missouri University of Science and Technology (http://www.cdna.org/). An N-terminal c-Myc epitope tag (EQKLISEEDL) was inserted into the sequence of the D2LR via overlap extension PCR. The receptor construct in pcDNA3.1+ was transferred into the hygromycin resistance gene containing pEF5/frt/V5/dest vector using the LR clonase enzyme mix (Invitrogen). Oligonucleotides were purchased from GeneWorks (Hindmarsh, Australia). Desired mutations were introduced using the Quikchange site-directed mutagenesis kit (Agilent, Santa Clara, CA). The manufacturer’s instructions were followed with the exception of the number of oligonucleotides used, their concentration and the concentration of the template DNA. Either two primers (200 nM of each) were used with 50 ng of c-Myc D2LR template, or alternatively one primer (1 μM) with 200 ng of c-Myc D2LR template was used. All mutations were confirmed by DNA sequencing (Australian Genome Research Facility, Melbourne, Australia).
2.4 Cell lines and transfections
Flp-In-CHO cells were grown in DMEM supplemented with 10% fetal bovine serum and maintained at 37°C in a humidified incubator containing 5% CO2. The Flp-In-CHO cells were transfected with the pOG44 vector encoding Flp recombinase and the pDEST vector encoding the wild-type or mutant c-Myc-D2LR at a ratio of 9:1 using polyethylenimine as transfection reagent. 24 hours after transfection the cells were subcultured and the medium was supplemented with 700 μg/ml hygromycin B as selection agent to obtain cells stably expressing the c-Myc-D2LR.
2.5 ERK1/2 Phosphorylation assay
In our Flp-In-CHO cell line stably expressing the human D2LR, the pERK1/2 response is abrogated by pertussis toxin treatment (data not shown). Thus, this provides a convenient downstream measurement of Gαi/o protein activation. ERK1/2 phosphorylation was measured using the AlphascreenTM SureFire ERK kit (PerkinElmer, Waltham, USA). Cells were seeded into 96-well plates at a density of 50,000 cells/well. After 4–6 h, cells were washed with PBS and incubated in serum-free DMEM (pH 7.4) overnight before assaying. Interaction experiments were performed for each ligand at 37 °C in the presence of 0.1 % ascorbic acid. SB269652 or derivative of interest was pre-equilibrated with cells for 30 min prior to agonist stimulation. Stimulation of the cells was terminated after 5 minutes of agonist exposure by removing the media and the addition of 100 μl of SureFire lysis buffer to each well. The plate was shaken for 5 minutes at room temperature before transferring 5 μl of the lysates to a white 384-well Proxiplate (PerkinElmer, Waltham, USA). Then 8 μl of a 240:1440:7:7 mixture of Surefire activation buffer: Surefire reaction buffer: Alphascreen acceptor beads: Alphascreen donor beads was added to the samples and incubated in the dark at 37 °C for 1.5 h. Plates were read using a Fusion-TM plate reader (PerkinElmer, Waltham, USA).
2.6 Membrane preparation
Flp-In-CHO cells stably expressing the wild-type or mutant c-Myc-D2LR were grown to 90% confluency in 175 cm2 cell culture flasks or 500 cm2 cell culture dishes (Corning, Corning, NY). The cells were harvested in PBS containing 2mM EDTA and centrifuged at 300 g for 3 min. The resulting pellet was resuspended in assay buffer (20 mM HEPES, 100 mM NaCl, 6 mM MgCl2, 1 mM EGTA and 1 mM EDTA, pH 7.4, 4 °C) and the centrifugation step was repeated. The intact cell pellet was then resuspended in assay buffer and homogenized using a Polytron homogenizer. After centrifugation (350 g, 5 min) the pellet was discarded, and the supernatant was recentrifuged at 30,000 g for 1 h at 4 °C using a Sorvall Evolution RC ultracentrifuge (Thermo Scientific, Waltham, MA). The resulting pellet was resuspended in assay buffer and stored in 500 μl aliquots at −80 °C. Membrane protein concentration was determined using the method of Bradford.
2.7 [3H]spiperone binding assays
All radioligand binding experiments were conducted in a 1 ml reaction volume in assay buffer (20 mM HEPES, 100 mM NaCl, 6 mM MgCl2, 1 mM EGTA and 1 mM EDTA, pH 7.4) containing 100 μM GppNHp and 0.1% ascorbic acid. In all cases non-specific binding was determined in the presence of 10 μM haloperidol. To obtain affinity estimates of unlabelled agonists, competition-binding experiments were performed at equilibrium. The ability of increasing concentrations of the agonists to compete with 0.1 nM [3H]spiperone for binding to the wild-type or mutant c-Myc-D2LR was tested. The membranes (5 μg) were incubated with the drugs for 3 h at 37 °C. After this incubation period, bound and free radioligand were separated by fast-flow filtration through GF/B filters using a Brandel harvester followed by three washes with ice-cold 0.9% NaCl. Filter bound radioactivity was measured by scintillation spectrometry after the addition of 3.5 ml of Ultima Gold (PerkinElmer) using a Tri-Carb 2900TR liquid scintillation counter (PerkinElmer).
2.8 Molecular modelling and simulations
The binding mode of SB269652 and its derivatives in the D2R were investigated based on our previous study [8]. Briefly, assuming similar binding modes of the THIQ moiety in the near-identical OBSs of D3R and D2R, the pose from the induced-fit docking (IFD) [13] trial with our previous equilibrated D2R model [11,12] that is closest to the reference pose in our D3R model was selected. The full-length SB269652 was then docked into the D2R model by a core-restrained IFD protocol [12][13] with restraints on the heavy atoms (heavy-atom RMSD deviation <2.0 Å) of the selected THIQ pose. Representative poses for the SB269652 derivative compounds, were acquired similarly by using the core-restrained IFD protocol, where restraints were applied on the THIQ moiety of the full-length SB269652, assuming that this moiety adopts a similar pose in SB269652 derivatives as well.
Molecular dynamics (MD) simulations of the D2R–ligand complexes were performed in the explicit water and 1-palmitoyl-2-oleoylphosphatidylcholine lipid bilayer solvent environment using the Desmond MD system (version 4.5–4.8; D. E. Shaw Research, New York, NY) with the CHARMM36 protein force field [14–16], CHARMM36 lipid force field [17], and TIP3P water model. The ligand parameters were obtained from the GAAMP server [18], with the initial force field based on CGenFF assigned by ParamChem [19]. The system charges were neutralized, and a solvent concentration of 150 mM NaCl was added. The average system size is ~111,000 atoms. Each system was first minimized and then equilibrated with restraints on the ligand heavy atoms and protein backbone atoms, followed by an isothermal–isobaric simulation at 310 K with all atoms unrestrained, as described previously [20,21]. We collected multiple trajectories for each complex, and the total simulation lengths for each complex were summarized in Table 1.
Table 1.
MD simulations of D2R bound to SB269652 derivatives.
| Ligand | Number of trajectories | Total Simulation Length (μs) |
|---|---|---|
| MIPS1588 | 3 | 4.2 |
| MIPS1576 | 2 | 3.9 |
| MIPS1529 | 3 | 4.8 |
| MIPS1546 | 3 | 4.8 |
| MIPS1278 | 2 | 3.6 |
| MIPS1556 | 2 | 4.8 |
| Total | 15 | 26.1 |
2.9 Data analysis
Radioligand binding data
For radioligand saturation binding data, the following equations were globally fitted to total binding (1) and nonspecific binding (2) data respectively:
| (1) |
| (2) |
Where Y is radioligand binding, Bmax is the total receptor density, [A] is the free radioligand concentration, KA is the equilibrium dissociation constant of the radioligand, and NS is the fraction of nonspecific radioligand binding.
Competition-binding curves between [3H]spiperone and dopamine in the absence or presence of SB269652 were fitted to a one-site binding equation [22]:
| (3) |
Where Y is percentage (vehicle control) binding, [A], [B], and [I] are the concentrations of [3H]spiperone, SB269652, and dopamine, respectively, KA and KB are the equilibrium dissociation constants of [3H]spiperone and SB269652, respectively, KI is the equilibrium dissociation constant of dopamine and α′ and α represent the cooperativity between SB269652 and [3H]spiperone or dopamine, respectively. Values of α (or α′) >1 denote positive cooperativity; values <1 (but >0) denote negative cooperativity, and values = 1 denote neutral cooperativity.
The concentration of ligand that inhibited half of the [3H]spiperone binding (IC50) was determined using the following equation [23]:
| (4) |
where Y denotes the percentage-specific binding, Top and Bottom denote the maximal and minimal asymptotes, respectively, IC50 denotes the X-value when the response is midway between Bottom and Top, and nH denotes the Hill slope factor. IC50 values obtained from the inhibition curves were converted to Ki values using the Cheng and Prusoff equation.
Competition-binding curves between [3H]spiperone and SB269652 could be fit to the allosteric ternary complex model using the following equation [24]:
| (5) |
Where Y is percentage (vehicle control) binding; [A] and [B] are the concentrations of [3H]spiperone and SB269652, respectively; KA and KB are the equilibrium dissociation constants of [3H]spiperone and SB269652, respectively; α is the cooperativity between SB269652 and [3H]spiperone Values of α >1 denote positive cooperativity; values <1 (but >0) denote negative cooperativity, and values = 1 denote neutral cooperativity.
Functional data
A logistic equation of competitive agonist-antagonist interaction was globally fitted to data from functional experiments measuring the interaction between dopamine and all analogues of SB269652 [25]:
| (6) |
Where s represents the Schild slope for the antagonist, and pA2 represents the negative logarithm of the molar concentration of antagonist that makes it necessary to double the concentration of agonist needed to elicit the original submaximal response obtained in the absence of antagonist.
Functional data describing the interaction between all SB269652 analogues and dopamine analysed according to the allosteric ternary complex model [3]:
| (7) |
Where Em is the maximum possible cellular response, [A] and [B] are the concentrations of orthosteric and allosteric ligands, respectively, and KB are the equilibrium dissociation constant of the orthosteric and allosteric ligands, αβ is a composite cooperativity parameter between the orthosteric and allosteric ligand that includes effects upon orthosteric ligand affinity and efficacy and nH is the Hill slope of the orthosteric agonist concentration-response curve. Values of α and/or β greater than 1 denote allosteric potentiation, whereas values less than 1 (but greater than 0) denote allosteric inhibition.
For each of the compounds the two equations (models) were then compared for their fit using an extra-sum-of-squares F test, whereby the simpler model was selected unless the P value was less than 0.05.
3. Results
3.1 Effect of mutations upon the actions of orthosteric ligands
In order to characterize the effects of the various OBS and SBP mutations on orthosteric ligands, we performed radioligand binding assays using the antagonist [3H]spiperone, and measured phosphorylation of ERK1/2 (pERK1/2) as a downstream readout of D2R activation. The mutation D1143.32A abolished both [3H]spiperone binding and the functional effect of dopamine, consistent with previous studies [30]. Saturation-binding experiments revealed that the remaining 31 mutants were expressed to similar levels to WT (Table 2). The affinity of [3H]spiperone was not significantly different from WT for most of the mutant receptors (Table 2). However, both I184ECL2A and S1935.42A significantly increased the affinity of [3H]spiperone (3-fold and 4-fold, respectively, Table 2), whilst D1143.32E and F3906.52A significantly reduced this affinity (6-fold and 5-fold, respectively, Table 2). In addition, 11 of the D2R mutants significantly decreased dopamine potency (Table 2), several of which (F1103.28, S1935.42, S1945.43, F3906.52 and H3936.55) have been previously shown to be important for agonist binding and efficacy [30–32]. The conserved TM5 serine residues (5.42, 5.43 and 5.46) interact with the meta-OH and para-OH moieties of catecholamine agonists [33,34]. No dopamine response was observed at the D1143.32E, S1935.42A, or S1975.46A mutants (Table 2). It is interesting to note that mutation of Leu411.39 and Thr4127.39 resulted in significantly reduced dopamine potency, as did three mutations in ECL2 (L174ECL2A, I184ECL2A, and N186ECL2A, Table 2). While these effects require further investigation, the binding affinity of dopamine at the T4127.39A mutant was significantly higher that for the WT whereas it is unchanged for I184ECL2A (Table 2). This suggests that the decrease in potency observed at the two mutants is not caused by a decrease in agonist affinity but rather may reflect a role for these residues in signal transduction.
Table 2. The effect of mutations on the pharmacology of the orthosteric antagonist [3H]spiperone and agonist dopamine.
Binding parameters were determined in [3H]spiperone saturation and competition (dopamine) binding assays. Functional parameters for dopamine were measured in an assay of ERK1/2 phosphorylation. Values are expressed as mean ± S.D. from three separate experiments.
| Radioligand binding ([3H]spiperone) | pERK1/2 (dopamine) | |||
|---|---|---|---|---|
| pKD ([3H]Spiperone) | Bmax (fmol.mg−1) | pKi (dopamine) | pEC50 | |
| WT | 10.41 ± 0.11 | 257 ± 16 | 5.10 ± 0.05 | 8.30 ± 0.09 |
| Y371.35A | 10.37 ± 0.16 | 226 ± 23 | 5.21 ± 0.07 | 8.39 ± 0.07 |
| L411.39A | 10.23 ± 0.03 | 384 ± 46 | - | 6.94 ± 0.06* |
| V912.61A | ND | ND | - | 8.60 ± 0.14 |
| L942.64A | 9.93 ± 0.04 | 285 ± 86 | - | 7.90 ± 0.05 |
| E952.65A | 10.13 ± 0.16 | 216 ± 16 | 5.66 ± 0.05* | 8.18 ± 0.07 |
| G98ECL1A | 10.07 ± 0.12 | 234 ± 60 | - | 7.85 ± 0.06 |
| K101ECL1A | 10.28 ± 0.07 | 174 ± 56 | - | 7.90 ± 0.05 |
| F1103.28A | 10.48 ± 0.06 | 171 ± 23 | - | 6.64 ± 0.08* |
| D1143.32E | 9.62 ± 0.07* | 390 ± 81 | - | ND |
| F1644.54A | 10.35 ± 0.15 | 250 ± 43 | - | 7.98 ± 0.06 |
| C1684.58A | 10.39 ± 0.16 | 276 ± 52 | - | 8.10 ± 0.07 |
| L174ECL2A | 10.64 ± 0.14 | 241 ± 49 | - | 7.01 ± 0.09* |
| N180ECL2A | 10.33 ± 0.04 | 266 ± 72 | - | 8.30 ± 0.08 |
| E181ECL2A | 10.36 ± 0.05 | 328 ± 47 | 5.31 ± 0.05 | 8.67 ± 0.10 |
| I183ECL2A | 10.31 ± 0.13 | 346 ± 23 | - | 7.73 ± 0.07 |
| I184ECL2A | 10.85 ± 0.04* | 363 ± 79 | 5.24 ± 0.04 | 7.02 ± 0.05* |
| A185ECL2S | 10.45 ± 0.09 | 199 ± 17 | - | 8.08 ± 0.05 |
| N186ECL2A | 10.19 ± 0.05 | 116 ± 24 | - | 7.18 ± 0.05* |
| S1935.42A | 11.01 ± 0.08* | 227 ± 86 | 4.21 ± 0.04* | 5.13 ± 0.03* |
| S1945.43A | 10.58 ± 0.09 | 209 ± 22 | 4.36 ± 0.04* | 6.05 ± 0.06* |
| S1975.46A | 10.43 ± 0.10 | 453 ± 109 | - | ND |
| F3906.52A | 9.75 ± 0.12* | 152 ± 14 | - | 7.04 ± 0.07* |
| H3936.55A | 10.40 ± 0.01 | 461 ± 106 | - | 5.90 ± 0.12* |
| H3936.55F | 10.50 ± 0.13 | 237 ± 24 | 4.41 ± 0.07* | 6.47 ± 0.12* |
| Y4087.35A | ND | ND | - | ND |
| S4097.36A | 10.14 ± 0.11 | 262 ± 43 | 5.49 ± 0.04* | 8.16 ± 0.06 |
| F4117.38A | 10.21 ± 0.13 | 283 ± 36 | - | 8.00 ± 0.11 |
| T4127.39A | 10.28 ± 0.11 | 255 ± 39 | 5.48 ± 0.05* | 7.15 ± 0.05* |
| Y4167.43A | ND | ND | - | ND |
P<0.05, significantly different from the wild-type receptor determined by a one-way ANOVA, Dunnett post-hoc test
ND, either no binding of [3H]spiperone was detected up to a concentration of 10 nM or no dopamine effect was detected
3.2 The interaction of SB269652 with the SBP is important for both affinity and cooperativity
The 1H-indole-2-carboxamide SP of SB269652 extends into a SBP between the extracellular ends of TMs 1, 2 and 7, and ECL1 and ECL2 [8]. To identify the receptor-ligand contacts within the SBP that are important for the binding and cooperativity of SB269652, SBP residues were mutated to alanine. The mutation Y371.35A did not significantly alter the affinity or cooperativity of SB269652 whereas the mutation L411.39A, a turn down from Y371.35, caused a significant 11-fold increase in functional affinity of SB269652 (Figure 1, Table 3). T4127.39A, at the extracellular end of TM7, also significantly increased both binding and functional affinity of SB269652 (14- and 9-fold, respectively, Table 3). Interestingly, in binding experiments, there was a significant enhancement in the negative cooperativity between SB269652 and [3H]spiperone at both L411.39A and T4127.39A (3-fold, Table 3). In contrast, the affinity and negative cooperativity of SB269652 was not altered at S4097.36A or F4117.38A (Table 3). In agreement with our previous study, we observed a significant loss of binding affinity, functional affinity and negative cooperativity upon mutation of E952.65A at Table 3). In addition, the mutation L942.64A causes a 3-fold decrease in negative cooperativity with dopamine. F1103.28 and V912.61 line the hydrophobic interface between the OBS and the SBP. Mutation of these residues causes a significant loss in negative cooperativity with dopamine but no significant change in affinity (Table 3).
Figure 1.

A homology model of the D2R with SB269652 docked. Residues are highlighted for which mutation to alanine significantly alters the affinity of SB269652 (pKB, A) or cooperativity with [3H]spiperone (Logα, B) in a radioligand binding assay (A & B), or the affinity (pKB, C) of SB269652 and/or its cooperativity with dopamine (Logαβ, D) as determined in an assay measuring phosphorylation of ERK1/2 (C & D). Residues are highlighted for which there was significant increase (blue) or decrease (red) between parameter at wild type versus mutant D2R as determine by a one-way ANOVA with Dunnetts post-hoc test, P < 0.05 (Tables 3 & 4).
Table 3. The effect of mutations within the SBP and extracellular loops of the D2R upon the pharmacology of SB269652.
Binding affinity and cooperativity values were obtained in competition or interaction binding experiments using the radioligand [3H]spiperone. Functional parameters for the interaction of SB269652 at WT and mutant D2R were determined in an assay measuring phosphorylation of ERK1/2. Values are expressed as mean ± S.D. from four separate experiments.
| Binding ([3H]spiperone) | Function (pERK1/2) | ||||
|---|---|---|---|---|---|
| pKB a (KB, nM) | Logαb (α) | Logα′c (α′) | pKB d (KB, nM) | Logαβe (αβ) | |
| WT | 6.11 ± 0.06 (776) | −0.54 ± 0.03 (0.28) | −0.54 ± 0.11 (0.28) | 6.26 ± 0.09 (550) | −1.23 ± 0.14 (0.06) |
| Y371.35A | 6.68 ± 0.18 (209) | −0.41 ± 0.04 (0.39) | −0.38 ± 0.12 (0.42) | 6.18 ± 0.14 (661) | −0.90 ± 0.02 (0.13) |
| L411.39A | 6.91 ± 0.17* (123) | −0.95 ± 0.08* (0.11) | - | 7.30 ± 0.19* (50) | −1.05 ± 0.06 (0.09) |
| V912.61A | - | - | - | 6.08 ± 0.08 (830) | −0.48 ± 0.16* (0.33) |
| E952.65A | 5.34 ± 0.22* (4570) | −0.21 ± 0.05* (0.62) | −0.33 ± 0.07 (0.47) | 5.14 ± 0.28* (7240) | −0.32 ± 0.14* (0.48) |
| L942.64A | 6.49 ± 0.04 (324) | −0.28 ± 0.06 (0.52) | - | 6.26 ± 0.20 (550) | −0.70 ± 0.08* (0.20) |
| G98ECL1A | 6.34 ± 0.08 (457) | −0.19 ± 0.11* (0.64) | - | 7.43 ± 0.14* (37) | −1.15 ± 0.07 (0.07) |
| K101ECL1A | 6.52 ± 0.04 (302) | −0.29 ± 0.07 (0.51) | - | 7.22 ± 0.10* (60) | −1.78 ± 0.06* (0.02) |
| F1103.28A | 6.74 ± 0.07 (182) | −0.30 ± 0.05 (0.50) | - | 6.31 ± 0.28 (490) | −0.38 ± 0.07* (0.42) |
| L174ECL2A | 6.45 ± 0.12 (354) | −0.40 ± 0.08 (0.40) | - | 6.57 ± 0.19 (269) | −1.04 ± 0.11 (0.09) |
| N180ECL2A | 5.99 ± 0.04 (1023) | −0.54 ± 0.10 (0.29) | - | 6.98 ± 0.14* (105) | −1.34 ± 0.09 (0.05) |
| E181ECL2A | 6.34 ± 0.24 (457) | −0.47 ± 0.10 (0.34) | −0.37 ± 0.03 (0.42) | 7.29 ± 0.13* (51) | −1.53 ± 0.12 (0.03) |
| I183ECL2A | 6.47 ± 0.24 (339) | −0.45 ± 0.05 (0.35) | - | 6.43 ± 0.18 (370) | −0.88 ± 0.08 (0.13) |
| I184ECL2A | 7.06 ± 0.14* (87) | −1.53 ± 0.03* (0.03) | −1.15 ± 0.13* (0.07) | 7.23 ± 0.08* (59) | −1.64 ± 0.08* (0.02) |
| A185ECL2S | 6.85 ± 0.24* (141) | −0.68 ± 0.08 (0.21) | - | 6.20 ± 0.12 (631) | −1.27 ± 0.08 (0.05) |
| N186ECL2A | 6.23 ± 0.13 (590) | −0.38 ± 0.06 (0.42) | - | 6.03 ± 0.17 (933) | −0.85 ± 0.09 (0.14) |
| S4097.36A | 6.03 ± 0.17 (933) | −0.77 ± 0.03 (0.17) | −0.87 ± 0.22 (0.13) | 6.03 ± 0.17 (933) | −1.08 ± 0.11 (0.08) |
| F4117.38A | 6.40 ± 0.10 (398) | −0.53 ± 0.06 (0.30) | - | 6.59 ± 0.10 (257) | −1.29 ± 0.07 (0.05) |
| T4127.39A | 7.25 ± 0.09* (56) | −1.04 ± 0.11* (0.09) | −0.93 ± 0.23 (0.12) | 7.20 ± 0.11* (63) | −1.55 ± 0.09 (0.02) |
Estimate of the negative logarithm of the equilibrium dissociation constant of SB269652
Logarithm of the affinity cooperativity factor between SB269652 and [3H]spiperone
Logarithm of the affinity cooperativity factor between SB269652 and dopamine
Estimate of the negative logarithm of the equilibrium dissociation constant of SB269652 eLogarithm of the net cooperativity factor between SB269652 and dopamine
P<0.05, significantly different from the wild-type receptor parameter determined by a one-way ANOVA, Dunnett post-hoc test
There is evidence that interaction with ECLs1 and 2 determines the subtype selectivity of SB269652 [26]. In addition ECL1 and 2 residues form part of an allosteric pocket at muscarinic acetylcholine receptors, contribute directly to orthosteric ligand binding at the D2R, and line the pathway of ligand entry and egress at aminergic receptors [27–29]. To understand the importance of these loops for the binding and action of SB269652, we extended our mutagenesis study to ECL 1 and 2 residues. The mutations G98A or K101A in ECL1 significantly increased the functional affinity (15- and 9-fold, respectively) of SB269652, and K101A significantly increased negative cooperativity with dopamine (3-fold, Table 3). In contrast, these mutations had no effect on the binding affinity of SB269652, but G98A caused a 2-fold decrease in cooperativity with [3H]spiperone. The mutations N180A and E181A in ECL2 significantly improved SB269652’s functional affinity (5- and 11-fold, respectively) but not binding affinity (Table 3). Together these data are consistent with the occupation of the SBP by SB269652 as predicted by our modelling studies and that this interaction is an important determinant of affinity and cooperativity (Figure 1).
3.3 SB269652 engages residues within the OBS of the D2R
We next turned our attention to identifying OBS residues that interact with SB269652 or that are important for its negative cooperativity with dopamine. Our molecular modelling predicts that the protonated aliphatic amine in the THIQ moiety of SB269652 forms a salt bridge with the conserved D1143.32, an interaction made by most orthosteric dopaminergic ligands. [8]. We observed no significant loss in binding affinity of SB269652 at the D1143.32E mutant but a significant 2-fold decrease in negative cooperativity with [3H]spiperone (Figure 1, Table 4). The mutation D1143.32E extends this residue by one carbon and retains the ability to form a salt bridge with the tertiary amine of dopaminergic ligands. The lack of an effect by the mutation D1143.32E upon the binding affinity of SB269652 suggests that the small THIQ core can accommodate the introduction of this slightly larger residue into the OBS. Indeed, while the D1143.32A mutation abrogated [3H]spiperone binding, the D114E mutation caused only a modest 6-fold decrease in [3H]spiperone binding affinity (Table 1). The significant change in negative cooperativity of SB269652 at the D1143.32E mutant compared to WT, however, suggests that the orientation of SB269652 within the OBS pocket is a determinant of cooperativity (Figure 1). Mutation of H3936.55 (H3936.55A or H3936.55F), which lies at the periphery of the OBS, causes no significant change in SB269652 affinity or cooperativity (Table 4). F3906.52 is positioned slightly deeper within the core of the OBS. F3906.52A resulted in a significant 6-fold increase in the functional affinity of SB269652, but no significant change in negative cooperativity with dopamine but a significant 2-fold increase in negative cooperativity with [3H]spiperone (Table 4).
Table 4. The effect of mutations within the OBS of the D2R upon the pharmacology of SB269652.
Functional parameters for the interaction of SB269652 at WT and mutant D2R were measured in an ERK1/2 phosphorylation assay. Binding affinity and cooperativity values were obtained in competition or interaction binding experiments using the radioligand [3H]spiperone. Values are expressed as mean ± S.D. from three separate experiments.
| Binding ([3H]spiperone) | Function (pERK1/2) | ||||
|---|---|---|---|---|---|
| pKB a (KB, nM) | Logαb (α) | Logα′c (α′) | pKB d (KB nM) | Logαβe (αβ) | |
| WT | 6.11 ± 0.06 (776) | −0.54 ± 0.03 (0.28) | −0.54 ± 0.11 (0.28) | 6.26 ± 0.09 (550) | −1.23 ± 0.14 (0.06) |
| D1143.32E | 6.39 ± 0.06 (407) | −0.22 ± 0.09* (0.60) | - | - | - |
| S1935.42A | 7.60 ± 0.15* (25) | −1.88 ± 0.04* (0.01) | −1.47 ± 0.15* (0.03) | - | - |
| S1945.43A | 6.79 ± 0.22 (162) | −0.14 ± 0.03* (0.72) | −0.18 ± 0.31 (0.66) | 6.58 ± 0.15 (263) | −0.52 ± 0.09* (0.30) |
| S1975.46A | 6.73 ± 0.27 (186) | −0.45 ± 0.05 (0.35) | - | - | - |
| F3906.52A | 6.82 ± 0.03* (151) | −0.92 ± 0.11* (0.12) | - | 7.01 ± 0.18* (98) | −0.95 ± 0.09 (0.11) |
| H3936.55A | 6.80 ± 0.16 (158) | −0.81 ± 0.07 (0.15) | - | 6.35 ± 0.09 (447) | −1.23 ± 0.05 (0.06) |
| H3936.55F | 6.63 ± 0.37 (234) | −0.62 ± 0.03 (0.24) | −0.54 ± 0.18 (0.30) | 5.67 ± 0.20 (2137) | −1.01 ± 0.24 (0.09) |
Estimate of the negative logarithm of the equilibrium dissociation constant of SB269652
Logarithm of the affinity cooperativity factor between SB269652 and [3H]spiperone
Logarithm of the affinity cooperativity factor between SB269652 and dopamine
Estimate of the negative logarithm of the equilibrium dissociation constant of SB269652
Logarithm of the net cooperativity factor between SB269652 and dopamine
P<0.05, significantly different from the wild-type receptor parameter determined by a one-way ANOVA, Dunnett post-hoc test
We next explored the influence of the three conserved serine residues in TM5. The mutation S1975.46A had no effect, whereas S1945.43A caused a significant 5-fold decrease in negative cooperativity with dopamine in the functional assay and significant 3-fold decrease in negative cooperativity with [3H]spiperone but no change in affinity (Figure 1, Table 4). Of note, we observed a significant increase in SB269652 binding affinity (31-fold) and negative cooperativity with both dopamine (9-fold) and [3H]spiperone (28-fold, Table 4) at the S1935.42A mutant. The results of our MD simulations indicate that the cyano group of SB269652 points towards S1935.42 but does not form a polar interaction. Thus, we speculate that the S1935.42A may increase SB269652 affinity by better accommodating the cyano group. This is supported by the corresponding changes from the ligand side where a derivative of SB269652 that lacks the cyano group displays increased affinity at the WT D2R (see below and Discussion).
Substituted-cysteine accessibility studies at the D2R and the recent crystal structure of the D3R showed that ECL2 residues, and in particular I184, forms part of the OBS [11,27]. In this study, while the mutation of the nearby residue, I183ECL2A has no effect, SB269652 gains significant affinity (9-fold) and negative cooperativity with dopamine (3-fold) at the I184ECL2A mutant, an effect observed in both binding and functional assays (Table 3). In addition, this mutation increases negative cooperativity with [3H]spiperone 9-fold. Note that both S1935.42A and I184ECL2A increase the affinity of [3H]spiperone and cause a decrease in dopamine potency that may result from the stabilisation of a receptor conformation that favours antagonist binding (Table 2, Figure 1). Stabilisation of such a conformation may also contribute to the increase affinity of SB269652 observed at these mutants.
Our mutagenesis results reveal residues within the OBS and the SBP that determine the affinity of SB269652 and its cooperativity with orthosteric ligands, consistent with the extended bitopic binding pose predicted for SB269652. To understand how such a pose might explain the results of our previous SAR study, we next investigated the impact of mutations to a key residue within the OBS (D1143.32) and a key residue within the SBP (E952.65) upon the action of a selected series of SB269652 derivatives and fragments.
3.4 Fragments and derivatives of SB269652 that contain the THIQ moiety interact with D1143.32 within the OBS
It was interesting to note that the significant loss of cooperativity with [3H]spiperone at the D1143.32E mutant was not accompanied by a change in the affinity of SB269652. To gain further insight into the interaction of the THIQ primary pharmacophore with the OBS we extended our study to two fragments of SB269652: 2-propyl-1,2,3,4-tetrahydroisoquinoline-7-carbonitrile (MIPS1071), and N-((trans)-4-(2-(7-cyano-3,4-dihydroisoquinolin-2(1H)yl)ethyl)cyclohexyl)acetamide (MIPS1059, Table 6). Both fragments contain the THIQ core, act as competitive antagonists of dopamine, and displayed a significant loss of affinity at the D1143.32E mutant (230-fold and 100-fold, respectively), highlighting the importance of this residue for the binding of the THIQ scaffold within the OBS (Table 5) [8].
Table 6. The effect of E952.65A on the pharmacology of SB269652 derivatives.
Functional parameters for the interaction of SB269652 at WT and E952.65A D2R were measured in an assay of dopamine-mediated phosphorylation of ERK1/2. Values are expressed as mean ± S.D. from four separate experiments.
| WT | E952.65A | |||||
|---|---|---|---|---|---|---|
| pKB a (KB, nM) | Log αβb (αβ) | Schild slope | pKB a (KB, nM) | Log αβb (αβ) | Schild slope | |
| SB269652 | 6.26 ± 0.09 (550) | −1.23 ± 0.14 (0.06) | - | 5.14 ± 0.28* (7240) | −0.32 ± 0.14* (0.49) | - |
| MIPS1529 | 7.15 ± 0.07 (71) | −1.34 ± 0.06 (0.05) | - | 6.22 ± 0.24* (602) | −0.35 ± 0.06* (0.44) | - |
| MIPS1546 | 6.82 ± 0.08 (151) | - | 1.15 ± 0.08 | 6.75 ± 0.11 (177) | - | 0.97 ± 0.05 |
| MIPS1564 | 6.80 ± 0.05 (158) | −1.39 ± 0.09 (0.04) | - | 5.81 ± 0.46* (1550) | −0.55 ± 0.18* (0.28) | - |
| MIPS1565 | 7.48 ± 0.12 (33) | −1.63 ± 0.18 (0.02) | - | 6.36 ± 0.24* (437) | −1.50 ± 0.23 (0.03) | - |
| MIPS1531 | 6.92 ± 0.25 (83) | −0.45 ± 0.13 (0.35) | - | 5.52 ± 0.26* (3020) | −0.15 ± 0.16 (0.71) | - |
| MIPS1588 | 7.32 ± 0.10 (47) | −1.42 ± 0.09 (0.03) | - | 5.90 ± 0.20 (1260)* | −1.12 ± 0.16 (0.08) | - |
| MIPS1576 | 8.43 ± 0.20 (4) | - | 1.02 ± 0.08 | 7.02 ± 0.15 (95)* | −1.24 ± 0.10 (0.06) | - |
| MIPS1278 | 7.06 ± 0.07 (87) | −1.04 ± 0.14 (0.09) | - | 4.21 ± 0.15* (61660) | −0.30 ± 0.20* (0.50) | - |
| MIPS1556 | 6.14 ± 0.09 (71) | - | 1.31 ± 0.07 | 5.96 ± 0.12 (110) | - | 1.08 ± 0.09 |
| MIPS1528 | 7.68 ± 0.13 (21) | - | 0.79 ± 0.10 | 5.92 ± 0.07* (1202) | - | 1.68 ± 0.10 |
| MIPS1587 | 7.59 ± 0.14 (26) | - | 0.95 ± 0.08 | 6.41 ± 0.12* (389) | 0.77 ± 0.05 | |
| MIPS1590 | 7.25 ± 0.17 (56) | - | 0.98 ± 0.06 | 5.94 ± 0.20* (1148) | - | 0.97 ± 0.15 |
Estimate of the negative logarithm of the equilibrium dissociation constant of SB269652,
Logarithm of the net cooperativity factor between SB269652 and dopamine,
P<0.05, significantly different from the corresponding WT parameter, Student’s t-test
Table 5. The effect of D1143.32E on selected SB269652 derivatives.
Binding affinity and cooperativity values were obtained in competition or interaction binding experiments using the radioligand [3H]spiperone. Values are expressed as mean ± S.D. from four separate experiments.
| WT | D1143.32E | |||
|---|---|---|---|---|
| pKB a (KB, μM) | Logαb (α) | pKB (KB, μM) a | Logαb (α) | |
SB269652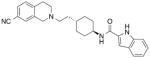
|
6.11 ± 0.06 (0.78) | −0.54 ± 0.11 (0.28) | 6.39 ± 0.06 (0.41) | −0.22 ± 0.09* (0.60) |
MIPS1059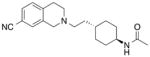
|
6.56 ± 0.06 (0.28) | - | 4.19 ± 0.11* (64) | - |
MIPS1071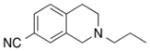
|
5.90 ± 0.07 (1.26) | - | 3.89 ± 0.14* (128) | - |
MIPS1868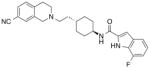
|
6.79 ± 0.09 (0.11) | −1.85 ± 0.32 (0.01) | 6.16 ± 0.09 * (0.69) | −1.39 ± 0.41 (0.04) |
Estimate of the negative logarithm of the equilibrium dissociation constant of SB269652,
Logarithm of the affinity cooperativity factor between SB269652 and [3H]spiperone,
P<0.05, significantly different from the corresponding WT receptor parameter, Student’s t-test
The cooperativity of SB269652 with [3H]spiperone is relatively weak, resulting in a small window to observe the effect of mutations, which may mask subtle effects. Therefore, we extended our study to MIPS1868, a higher-affinity SB269652 derivative that displays greater negative cooperativity with [3H]spiperone (KB = 160 nM, α = 0.01, Table 5). Furthermore, MIPS1868 possesses the structural characteristics of SB269652 that we have found to be necessary for allosteric pharmacology (THIQ core, trans-cyclohexylene linker, hydrogen-bond donating heteroatom, and aromatic bicyclic tail) and differs from SB269652 simply through the substitution of a fluorine at the 7-position of the indole-2-carboxamide moiety [9]. MIPS1868 displayed a significant 4-fold loss in binding affinity at the D1143.32E mutant (Table 5), consistent with a salt bridge interaction formed between this residue and the protonated aliphatic amine within the THIQ core of MIPS1868.
3.5 Linker length is critical for the correct engagement of the indole tail with E952.65
Derivatives of SB269652 in which the cyclohexylene spacer moiety was replaced with flexible polymethylene linkers were shown to display improved affinity as compared to SB269652. However, the length of the carbon spacer bridging the PP and SP was shown to determine allosteric pharmacology. A linker length of 3 or 4 carbon atoms conferred allosteric pharmacology but a 5-carbon linker length resulted in pharmacology best-fit by a competitive model [9]. We hypothesized that this difference in pharmacology could be conferred by a distinct orientation of the SP of the 5-carbon linker derivative, such that it does not interact with the SBP and in particular with E952.65.
We carried out MD simulations of D2R models in complex with either 4-carbon or 5-carbon linker derivatives (MIPS1529 and MIPS1546, respectively). The simulation results show that the 4-carbon linker (MIPS1529) permits an interaction of the indole moiety with the SBP and in particular the hydrogen-bond interaction between the indolic NH and E952.65, while this interaction could not form in the simulations of the D2R bound with the 5-carbon linker derivative (MIPS1546, Figure 2). Instead the indole moiety of this longer derivative is predicted to extend towards the sub-pocket of the SBP between TMs 2 and 3. Supporting this prediction, the 4-carbon linker derivative (MIPS1529, Figure 2) displays a significant loss in both functional affinity (8-fold) and negative cooperativity (9-fold) at the E952.62A mutant (Table 6, Figure 2). Conversely, the 5-carbon linker derivative (MIPS1546, Figure 2) behaves as a competitive antagonist at the WT D2R (151 nM, Schild slope = 0.97 ± 0.05, Table 6) and its affinity is not significantly altered at the E952.65A mutant (KB = 177 nM, Table 6). Consistent with these data, analysis of our MD simulation results shows a greater distance between the indolic NH and E952.65 for the 5-carbon linker derivative (MIPS1546) as compared to the 4-carbon linker derivative (MIPS1529). This is associated with a change in the orientation of the amide NH relative to E952.65 and the THIQ core relative to the Cβ atom of Asp2.50 at the intracellular side the OBS (Figure 2E). Interestingly, the 3- and 6-carbon linker derivatives, MIPS1564 and MIPS1565, both displayed allosteric pharmacology and were sensitive to the mutation of E952.65A, exhibiting a significant loss in functional affinity (Table 6). These results suggest that a linker length of 3 is sufficient to enable this interaction and allow the indole ring to be accommodated by the sub-pocket of SBP between TMs 2 and 7; and that extension of the linker length from 5 to 6 atoms may again allow a similar interaction, perhaps through additional flexibility within the linker region.
Figure 2.

(A) Derivatives of SB269652 in which the trans cyclohexylene spacer was replaced by polymethylene linkers of differing lengths, between the primary pharmacophore (PP) and SP. (B) In an assay measuring ERK1/2 phosphorylation, the derivatives with linkers lengths of 3, 4 and 6 carbons displayed allosteric pharmacology, decreased affinity and lower cooperativity (3 and 4 carbon linker only) at the E952.65A mutant. In contrast, the 5-carbon linker derivative displayed competitive pharmacology and was insensitive to this mutation, where NA signifies that cooperativity could not be determined as data were best fit by a competitive model. Data represents the mean ± SD of four independent experiments. * P < 0.05 between parameter at wild type versus mutant D2R as determined by a one-way ANOVA with Dunnett’s post-hoc test. (C–E) Our molecular modelling and docking experiments revealed that our predicted poses allow the NH indole of MIPS1529 (C, 4-carbon linker) can participate in a hydrogen-bond interaction with E952.65, the indole moiety of MIPS1546 (D, 5-carbon linker does not make this interaction and is instead oriented towards TM3. (E) Distribution of the distance between the N1 and carboxyl oxygen atoms of D1143.32 (i) and the minimum distance between the ligand PP and Cβ atom of D802.50 (ii) show that the PP of MIPS1529 and MIPS1546 have subtly different orientations in the OBS. Distribution of the minimum distances between N3 (iii) or N4 (iv) atoms of ligands and the carboxyl oxygen atoms of E952.65 show that MIPS1529 forms stable hydrogen bonds with E952.65 with both of the N3 and N4 atoms, whereas MIPS1546 cannot.
3.6 The influence of the SBP residue E952.65 on the affinity and cooperativity of SB269652 derivatives
Relatively subtle modifications to the 1H-indole-2-carboxamide SP of SB269652 cause a change in pharmacology from allosteric to that best fitted by a competitive model [9]. In particular, an N-methyl amide derivative retains weak negative cooperativity suggesting that the presence of a hydrogen bond donating indolic NH but not the amide NH group was required for allosteric pharmacology [8,9]. In order to understand how such subtle modifications can confer allosteric versus competitive pharmacology, we wanted to determine how such SP modifications changed the interaction with SBP residues and in particular E952.65.
The N-methyl amide derivative of SB269652, MIPS1531 exhibits a significant decrease of functional affinity at the E952.65A mutant compared to WT, consistent with the loss of a hydrogen bond interaction between the indole NH of this derivative and E952.65. Replacement of the indole moiety with an 7-azaindole moiety (MIPS1588, Figure 3, Table 6) that retains the constituents of the parent scaffold (a hydrogen-bond donating heteroatom and aromaticity of a biaryl system) causes a 12-fold improvement in affinity as compared to SB269652 and similar weak negative cooperativity with dopamine [9]. Furthermore, the results of our MD simulations indicate a significantly stronger interaction between the NH of the 7-azaindole moiety and E952.65 that may contribute to the higher affinity of this derivative compared to SB269652. Consistent with this computational finding, the affinity of this derivative is 27-fold lower at the E952.65A mutant compared to the 13-fold loss observed for SB269652 (Table 6, Figure 3).
Figure 3.

(A) N-methylated amide (MIPS1531), 7-azaindole (MIPS1588), and pyrrole (MIPS1576) derivatives of SB269652. (B) In an assay measuring ERK1/2 phosphorylation MIPS1531 and MIPS1588 displayed allosteric pharmacology and a decreased affinity at the E952.65A mutant. The pyrrole derivative (MIPS1576) displayed pharmacology best fit by a competitive model at the wild type receptor but displayed a lower affinity at the E952.65A mutant and weak negative cooperativity with dopamine. NA signifies that cooperativity could not be determined as data were best fit by a competitive model. Data represents the mean ± SD of four independent experiments. * P < 0.05 between parameter at wild type versus E952.65A D2R as determined by unpaired two-tailed Student’s test. (C) The switch of MIPS1576 from apparently competitive (WT) to allosteric (E952.65A) pharmacology is graphically illustrated by a Schild plot. Data represents the mean ± SD of four independent experiments. Note that this Schild plot was not used to generate the values displayed in table 6. Molecular modelling and docking experiments revealed that the indolic NH and the pyrrolic NH of MIPS1588 (D) and MIPS1576 (E) were able to participate in a hydrogen bond with E952.65A.
Consistent with our previous SAR study [9], replacement of the indole-2-carboxamide SP with pyrrole-2-carboxamide (MIPS1576, Figure 3) results in pharmacology best fitted by a competitive rather than allosteric model (KB = 4 nM, Schild slope = 1.02 ± 0.08, Table 6). These ligands differ in the size of the SP whereby the pyrrole of MIPS1576 is smaller than the indole of SB269652. Despite this difference, our MD simulations show that the orientation of the aryl and amide NH groups of MIPS1588 and MIPS1576 relative to E952.65 are similar, although MIPS1576 made these interactions slightly more frequently during our simulations (Figure 3). Supporting this prediction, the pyrrole derivative displays a significant 24-fold loss in affinity at the E952.65A mutant. Interestingly, however, MIPS1576 showed pharmacology consistent with the action of a NAM at the E952.65A mutant (αβ = 0.06, Table 6).
3.7 Size of the orthosteric head group modulates the interaction with E952.65
We have previously demonstrated that the nature and size of the orthosteric head group is critical for the allosteric pharmacology of SB269652 [9]. Our results above indicate that the interaction of the SP with the SBP is a critical determinant of the negative cooperativity exerted by SB269652. We speculated that changes in the size of the orthosteric head group might confer a change in orientation of the SP within the SBP that would result in a change in sensitivity to the mutation E952.65A. Replacement of the cyano group at the 7-position of the THIQ moiety with a hydrogen (MIPS1278) improves affinity and retains the weak cooperativity displayed by SB269652 (KB = 87 nM, αβ = 0.09, Table 6) [9]. The increased affinity is complementary to the effect of S1935.42A mutation and supports our predicted SB269652 binding pose (see Discussion). Both the affinity and negative cooperativity of MIPS1278 (Table 6) are significantly attenuated at the E952.65A mutant compared to WT (709-fold and 6-fold, respectively). This loss is consistent with an interaction between E95 2.65 and the indole NH for this derivative (Figure 4). Furthermore, the loss of affinity for MIPS1278 at the E952.65A mutant is over 50-fold greater than that observed for SB269652, suggesting that without the cyano group the interaction between the indolic NH and E952.65 makes a greater relative contribution to affinity. Replacement of the cyano substituent with halogens of increasing size resulted in a progressive decrease in cooperativity [9]. However, the largest halogen introduced, a bromine substitution at the 7-position (MIPS1556), yielded pharmacology best fitted by a competitive model (KB = 724 nM, Schild slope = 1.31 ± 0.07, Table 6). We modelled and simulated MIPS1556 in a similar pose as SB269652 in the OBS, i.e., with the 7-Br substitution pointing towards S1935.42. Although an interaction between the indole group of MIPS1556 and E952.65 is persistently formed during our accumulated 4.8 μs MD simulations of the D2R/MIPS1556 complex (Figure 4), we found the larger 7-Br substitution could not be accommodated in the same way as 7-CN and distorted the TM5 conformation (Figure 4E). We conclude then that 7-Br-THIQ must adopt an alternative pose in the OBS, resulting in significantly different positions of the linker and thereby the SP, which does not favour the interaction between its SP and E952.65. Consistent with this conclusion, no significant change in pharmacological parameters is observed at the E952.65A mutant for MIPS1556 (Table 6).
Figure 4.

(A) Derivatives of SB269652 in which the 7-cyano substitution of the THIQ moiety was replaced with hydrogen (MIPS1278) or bromine (MIPS1576). (B) In an assay measuring ERK1/2 phosphorylation MIPS1278 displayed allosteric pharmacology and a decreased affinity and cooperativity at the E952.65A mutant. The 7-bromo substituted derivative (MIPS1576) displayed pharmacology best fit by a competitive model at the wild type receptor and was insensitive to the E952.65A mutation. Data represents the mean ± SD of four independent experiments. * P < 0.05 between parameter at wild type versus E952.65A D2R as determine by unpaired two-tailed Student’s test. Molecular modelling and docking experiments revealed that the indolic NH of both MIPS1278 (C) and MIPS1556 (D) were predicted to participate in a hydrogen bond with E952.65A. (E) Comparison of the D2R when either MIPS1278 and MIPS1556 were bound revealed that the larger 7-Br substitution of MIPS1556 caused a distortion in TM5.
3.8 Interaction with E952.65 does not determine allosteric versus competitive pharmacology
In our previous SAR study, removal of a hydrogen-bond donating heteroatom from the biaryl tail group of SB269652 appeared to confer competitive pharmacology [9]. While our initial study proposed an interaction between this hydrogen-bond donating heteroatom and E952.65, the mutation E952.65A does not cause a switch in the pharmacology of SB269652 from allosteric to competitive, but instead decreases both affinity and cooperativity. To investigate the importance of the interaction as a determinant of affinity and cooperativity of SB269652 and understand the effect of the E952.65A mutation, we extended our study to derivatives that retain the aromaticity and planarity of the indole moiety yet lack a hydrogen-bond donating hetero-atom (Figure 5, Table 6). Both the benzo[d]oxazole-2-carboxamide derivative (MIPS1528) and the indene analogue (MIPS1587) are unable to form a hydrogen bond between the SP and E952.65, due to incorporation of an oxygen into the indole ring, or isoteric replacement of the indole with CH2 respectively [9]. Both MIPS1528 and MIPS1587 display pharmacology best fitted by a competitive model at the WT D2R. While both ligands display a significant reduction in affinity at the E952.65A mutant they nonetheless retain an apparently competitive mode of action (Table 6).
Figure 5.

(A) Derivatives of SB269652 were generated in which the indole moiety of SB269652 was replaced with benzoxazole (MIPS1528), indene (MIPS1587) or D-proline derivatives (MIPS1590). (B) MIPS1528, MIPS1587 and MIPS1590 all displayed competitive pharmacology at both the wild type and E952.65A mutant D2R in an assay measuring ERK1/2 phosphorylation, yet displayed lower affinity at this mutant. Data represents the mean ± SEM of four independent experiments. * P <0.05 between parameter at wild type versus E952.65A D2R as determine by unpaired two-tailed Student’s test.
Replacement of the indole moiety with a D-proline (MIPS1590) retains a NH- group within the SP that can participate in a hydrogen bond with E952.65 [9]. Furthermore, this prolyl NH will be positively charged at pH 7.4 as it is a secondary aliphatic amine. Indeed, while MIPS1590 exhibited apparent competitive pharmacology at both WT and E952.65A, there was significant loss of affinity at E952.65A that may reflect the loss of a salt bridge formed between the prolyl NH2+ and this residue (Table 6). Therefore, the difference between the allosteric pharmacology of SB269652 and the competitive pharmacology of MIPS1590 is not solely related to an ability to interact with E952.65, but instead suggests that the lack of planarity, aromaticity or the additional positive charge of the proline moiety as compared to the indole moiety prevents additional interactions required for the weak negative cooperativity of SB269652.
4. Discussion
The structure of the NAM SB269652 is similar to many ligands that display competitive pharmacology at the D2R [11,12,36]. The aim of this study was to systematically characterize the receptor-ligand interactions that govern the allosteric pharmacology of SB269652. Our recent SAR study found that changes to the 1H-indole-2-carboxamide SP of SB269652 resulted in significant changes in both affinity and cooperativity [9]. This study reveals that mutation of residues within the SBP predicted to interact with the SP caused significant decreases in negative cooperativity (V912.61A, L942.64A, E952.65A, and F1103.28A) and affinity (E952.65A). In contrast, the mutations L411.39A and T4127.39A increased both the affinity and negative cooperativity of SB269652, perhaps by allowing the indole moiety of SB269652 to be better accommodated within the SBP. Together, these data provide further evidence that the interaction between the SP of SB269652 and the SBP is a critical determinant of affinity and negative cooperativity at the D2R. In addition, a derivative of SB269652 in which the cyclohexylene linker was replaced with a 5-carbon methylene linker displayed competitive pharmacology and was insensitive to the E952.65A mutation. Our MD simulations show that the SP of this derivative is oriented away from the SBP and instead occupies a subpocket of the SBP between TMs 2 and 3. Combined with our previous findings that the cis-isomer of SB269652 and truncated orthosteric derivatives of SB269652 that lack the SP all display competitive pharmacology, we conclude that interaction of the SP with the SBP between TMs 2 and 7 is a requirement for allosteric pharmacology [8].
A hydrogen-bond interaction between the indolic NH of SB269652 and E952.65 within the SBP was proposed to be a particularly important determinant of affinity and cooperativity [8,9]. In this study, all SB269652 derivatives that displayed allosteric pharmacology at the WT D2R exhibited a loss of affinity and negative cooperativity at the E952.65A mutant. However, some derivatives of SB269652 that display competitive pharmacology, including those that lack a hydrogen-bond donor within the SP, were also sensitive to the E952.65A mutation. This sensitivity may indicate that E952.65 can still interact with the amide NH of these derivatives. Alternatively, the E952.65A mutation may change the size or shape of the SBP and weaken the ability to accommodate such extended compounds. Indeed, results from MD simulations provide evidence that the hydrogen-bond interaction between the indole moiety and E952.65 is not a strong one. Instead, the E952.65A mutation significantly affects the overall shape and size of the SBP, resulting in new poses for SB269652 (Verma et al., manuscript in preparation) that likely contribute to changes in affinity and cooperativity at this mutant. Thus, while interaction with the SBP appears to be required for allosteric pharmacology, sensitivity to this mutation alone is not a predictor of allosteric versus competitive action.
Our SAR study, based on observations that proline and pyrrole derivatives display apparently competitive pharmacology at the WT D2R, provided evidence that the size, aromaticity and planarity of the 1H-indole-2-carboxamide tail of SB269652 are determinants of the allosteric pharmacology of SB269652 [8]. Both derivatives display lower affinity at the E952.65A D2R. The lack of aromaticity of the proline derivative and/or the positive charge of the prolyl NH confers apparently competitive behaviour but retains an interaction with E952.65. However, the pyrrole derivative displays allosteric pharmacology at this mutant. Our molecular modelling experiments predict that the positions of the amide and aryl NH groups of the pyrrole derivative (MIPS1576) relative to E952.65 within the SBP are largely similar to those of the 7-azaindole derivate (MIPS1588). It should be noted that, within this study, our definition of competitive versus allosteric pharmacology is operational rather than mechanistic. Derivatives of SB269652 were termed competitive if they caused a limitless dextral shift of the dopamine concentration response curve within the concentration range used, whereby a competitive model best fit these data. Accordingly, very high negative cooperativity cannot be distinguished from competitive antagonism if the limit of compound solubility did not allow the saturable effect of the modulator to be observed. The mutation E952.65A caused a significant decrease in the negative cooperativity between SB269652 and [3H]spiperone [8]. If, rather than being competitive with dopamine, MIPS1576 displays high negative cooperativity, the switch in pharmacology of MIPS1576 from apparently competitive (WT) to allosteric (E952.65A) may be consistent with the decrease in negative cooperativity observed for SB269652. If this mechanism is correct, this would suggest that, rather conferring competitive antagonism, the smaller size of the pyrrole-2-carboxamide (MIPS1576) SP leads to an increase in affinity and negative cooperativity.
Our previous modelling study predicted a salt bridge interaction between the highly conserved OBS residue D1143.32 and the protonated tertiary amine of the THIQ moiety of SB269652. Evidence for such an interaction was inferred from the competitive action of fragments of SB269652 (MIPS1059 and MIPS1071) that contain the THIQ moiety [8]. In agreement with this hypothesis, the mutation D1143.32E confers a 100-fold loss of affinity for these fragments. However, while we observe a close interaction between this protonated aliphatic amine and D1143.32 for all full-length SB269652 derivatives studied in our modelling experiments (Figures 1–4), SB269652 exhibited a loss of cooperativity but no loss of affinity at this mutant. It should be noted, however, that D1143.32E is a relatively conservative mutation and the loss of affinity for the orthosteric antagonist [3H]spiperone at this mutant was a modest 6-fold. Thus it is feasible that the protonated tertiary amine of the THIQ moiety of SB269652 may be able to maintain an interaction with this residue and/or that the loss of affinity conferred by this mutation may be compensated by the gain of other interactions. In agreement with this hypothesis, MIPS1868, a full-length derivative with preserved allosteric properties displayed a modest 4-fold loss of affinity at this moiety. This suggests that the THIQ moiety of this extended compound makes a salt bridge interaction with D1143.32, and that the addition of the SP confers additional contacts within the SBP and/or a reorientation of the PP within the OBS that reduces the impact of this mutation relative to the larger effect observed for the truncated orthosteric fragments. Therefore, the interaction of the SP of SB269652 with the SBP likely minimises the impact of the D1143.32E mutation upon affinity.
In the absence of a radiolabelled derivative of SB269652, or biophysical approaches to measure the binding of SB269652 directly, we must rely on the use of orthosteric ‘probe’ ligands to detect the effect of mutations upon the pharmacology of SB269652. This limits our choice of mutations to those that do not abrogate probe binding – indeed, with the exception of D1143.32E and F3906.52A, none of the selected mutations decreased the affinity of [3H]spiperone. It is not surprising, then, that with the exception of E952.65A (and D1143.32E on MIPS1868) none of the mutations decreased the affinity of SB269652. Instead, we observed changes in cooperativity and/or increases in affinity. Such mutations may change the orientation of the ligand within both the OBS and SBP such that loss of a contact within one pocket is compensated by gains of contacts within the other pocket leading to increases in affinity and changes in negative cooperativity. The mutation S1935.42A conferred an increase in the affinity of SB269652, as did the replacement of the 7-cyano substitution of SB269652 with hydrogen (MIPS1278). Our MD simulations indicate that the 7-cyano substituent is orientated towards S1935.42 and we speculate that both changes allow the THIQ moiety to adopt a more favourable position within the OBS. Consistent with the hypothesis that the change in orientation within the OBS will modulate interactions within the SBP, MIPS1278 displayed a 50-fold higher sensitivity to the SBP mutation E952.65A than SB269652. Indeed, the results of our MD simulations suggest the 7-cyano substitution may weaken the interaction between the indolic NH and E952.65 (Figure 4 and Draper-Joyce et al. manuscript in preparation). Thus, we speculate that stabilizing this interaction in the SBP by removing the 7-cyano substituent may, in part, contribute to the increased affinity of MIPS1278 as compared to SB269652.
In summary, by integrating the findings of our previous SAR results of SB269652 with this mutagenesis study we have systematically characterized a bitopic-binding mode that extends from the OBS into a SBP. We provide further evidence that interaction of the SP of SB269652 with the SBP is a requirement for allosteric pharmacology and show how changes to the THIQ moiety or the SP can lead to increases in affinity. We also reveal how changes to interactions within one pocket can modulate interactions in the other pocket. The orientation of SB269652 within both pockets likely determines allosteric pharmacology. A recent study of extended D2R partial agonists illustrated how the interactions made by a secondary pharmacophore within the SBP can alter the orientation of the orthosteric moiety within the OBS that, in turn, modulated intrinsic agonist efficacy [12]. Extended or bitopic D2R compounds have been shown to display distinct and desirable characteristics such as subtype-selectivity, biased agonism and allostery [8,12,37–39]. Understanding how such extended modes of interaction dictate these different pharmacological profiles is important for the design of novel selective ligands for this receptor.
Acknowledgments
This research was supported by Project Grant 1049564 of the National Health and Medical Research Council (NHMRC), Program Grant 1055134 (NHMRC), and the Intramural Reseach Program of the National Institutes of Health, National Institute on Drug Abuse (M.M., R.K.V., and L.S.). J.R.L. is a Larkin’s Fellow (Monash University, Australia). C.D.-J. acknowledges an Australian Postgraduate Awards.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beaulieu JM, Gainetdinov RR. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacological Reviews. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 2.Seeman P. Targeting the dopamine D2 receptor in schizophrenia. Expert Opin Ther Targets. 2006;10:515–531. doi: 10.1517/14728222.10.4.515. [DOI] [PubMed] [Google Scholar]
- 3.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Ann Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 4.Christopoulos A. Advances in G Protein-Coupled Receptor Allostery: From Function to Structure. Mol Pharmacol. 2014;86:463–478. doi: 10.1124/mol.114.094342. [DOI] [PubMed] [Google Scholar]
- 5.Jeffrey Conn P, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Reviews Drug Discovery. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nature Reviews Drug Discovery. 2013;12:630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- 7.Michino M, Beuming T, Donthamsetti P, Newman AH, Javitch JA, Shi L. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol Rev. 2015;67:198–213. doi: 10.1124/pr.114.009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lane JR, Donthamsetti P, Shonberg J, Draper-Joyce CJ, Dentry S, Michino M, et al. A new mechanism of allostery in a G protein--coupled receptor dimer. Nat Chem Biol. 2014;10:745–752. doi: 10.1038/nchembio.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shonberg J, Draper-Joyce C, Mistry SN, Christopoulos A, Scammells PJ, Lane JR, et al. Structure--Activity Study of N-((trans)-4-(2-(7-Cyano-3,4-dihydroisoquinolin-2(1 H)-yl)ethyl)cyclohexyl)-1 H-indole-2-carboxamide (SB269652), a Bitopic Ligand That Acts as a Negative Allosteric Modulator of the Dopamine D 2Receptor. J Med Chem. 2015;58:5287–5307. doi: 10.1021/acs.jmedchem.5b00581. [DOI] [PubMed] [Google Scholar]
- 10.Ballesteros JA, Weinstein H. Methods in Neurosciences. Elsevier; 1995. [DOI] [Google Scholar]
- 11.Chien EYT, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, et al. Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, et al. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J Med Chem. 2012;55:6689–6699. doi: 10.1021/jm300482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 14.MacKerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 15.MacKerell DA, Brooks B, Brooks CL, Nilsson L, Roux B, Won Y, et al. CHARMM: The Energy Function and Its Parameterization. John Wiley & Sons, Inc; n.d. [Google Scholar]
- 16.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang L, Roux B. AUTOMATED FORCE FIELD PARAMETERIZATION FOR NON-POLARIZABLE AND POLARIZABLE ATOMIC MODELS BASED ON AB INITIO TARGET DATA. J Chem Theory Comput. 2013;9 doi: 10.1021/ct4003477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michino M, Free RB, Doyle TB, Sibley DR, Shi L. Structural basis for Na(+)-sensitivity in dopamine D2 and D3 receptors. Chem Commun (Camb) 2015;51:8618–8621. doi: 10.1039/c5cc02204e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michino M, Boateng CA, Donthamsetti P, Yano H, Bakare OM, Bonifazi A, et al. Toward Understanding the Structural Basis of Partial Agonism at the Dopamine D3 Receptor. J Med Chem. 2017;60:580–593. doi: 10.1021/acs.jmedchem.6b01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nawaratne V, Leach K, Felder CC, Sexton PM, Christopoulos A. Structural determinants of allosteric agonism and modulation at the M4 muscarinic acetylcholine receptor: identification of ligand-specific and global activation mechanisms. Journal of Biological Chemistry. 2010;285:19012–19021. doi: 10.1074/jbc.M110.125096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 24.Kenakin T, Christopoulos A. Analytical pharmacology: the impact of numbers on pharmacology. Trends Pharmacol Sci. 2011;32:189–196. doi: 10.1016/j.tips.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Motulsky HJ, Christopoulos A. Fitting Models to Biological Data Using Linear and Nonlinear Regression: A Practical guide to Curve Fitting. GraphPad Software Inc; San Diego CA: 2003. http://www.graphpad.com. [Google Scholar]
- 26.Silvano E, Millan MJ, Mannoury LA, Cour C, Han Y, Duan L, Griffin SA, et al. The tetrahydroisoquinoline derivative SB269,652 is an allosteric antagonist at dopamine D3 and D2 receptors. Mol Pharmacol. 2010;78:925–934. doi: 10.1124/mol.110.065755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci USA. 2004;101:440–445. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, et al. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci USA. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108/-/DCSupplemental/pnas.1104614108_SI.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Javitch JA, Fu D, Chen J, Karlin A. Mapping the binding-site crevice of the dopamine D2 receptor by the substituted-cysteine accessibility method. Neuron. 1995;14:825–831. doi: 10.1016/0896-6273(95)90226-0. [DOI] [PubMed] [Google Scholar]
- 31.Javitch J, Li X, Kaback J, Karlin A. A cysteine residue in the third membrane-spanning segment of the human D2 dopamine receptor is exposed in the binding-site crevice. Proc Natl Acad Sci U S A. 1994;91:10355–10359. doi: 10.1073/pnas.91.22.10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tschammer N, Bollinger S, Kenakin T, Gmeiner P. Histidine 6.55 Is a Major Determinant of Ligand-Biased Signaling in Dopamine D2L Receptor. Mol Pharmacol. 2011;79:575–585. doi: 10.1124/mol.110.068106. [DOI] [PubMed] [Google Scholar]
- 33.Cox BA, Henningsen RA, Spanoyannis A, Neve RL, Neve KA. Contributions of conserved serine residues to the interactions of ligands with dopamine D2 receptors. Journal of Neurochemistry. 1992;59:627–635. doi: 10.1111/j.1471-4159.1992.tb09416.x. [DOI] [PubMed] [Google Scholar]
- 34.Javitch J, Fu D, Chen J. Residues in the fifth membrane-spanning segment of the dopamine D2 receptor exposed in the binding-site crevice. Biochemistry. 1995;34:16433–16439. doi: 10.1021/bi00050a026. [DOI] [PubMed] [Google Scholar]
- 35.Guo W, Urizar E, Kralikova M, Mobarec JC, Shi L, Filizola M, et al. Dopamine D2 receptors form higher order oligomers at physiological expression levels. The EMBO Journal. 2008;27:2293–2304. doi: 10.1038/emboj.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ehrlich K, Götz A, Bollinger S, Tschammer N, Bettinetti L, Härterich S, et al. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J Med Chem. 2009;52:4923–4935. doi: 10.1021/jm900690y. [DOI] [PubMed] [Google Scholar]
- 37.Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, et al. Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci USA. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weichert D, Banerjee A, Hiller C, Kling RC, Hübner H, Gmeiner P. Molecular determinants of biased agonism at the dopamine D2 receptor. J Med Chem. 2015;58:2703–2717. doi: 10.1021/jm501889t. [DOI] [PubMed] [Google Scholar]
- 39.Bonifazi A, Yano H, Ellenberger MP, Muller L, Kumar V, Zou MF, et al. Novel Bivalent Ligands Based on the Sumanirole Pharmacophore Reveal Dopamine D2 Receptor (D2R) Biased Agonism. J Med Chem. 2017;60:2890–2907. doi: 10.1021/acs.jmedchem.6b01875. [DOI] [PMC free article] [PubMed] [Google Scholar]