

Abstract
The aggressiveness of pancreatic cancer urgently requires more efficient treatment options. Because the sigma-2 (σ2) receptor was recently proposed as a promising target for pancreatic cancer therapy, we explored our previously developed multifunctional thiosemicarbazones, designed to synergistically impair cell energy levels, by targeting σ2 and P-gp proteins and chelating Iron. A deconstruction approach was herein applied by removing one function at a time from the potent multifunctional thiosemicarbazones 1 and 2, to investigate the contribution to cytotoxicity of each target involved. The results from in vitro (panel of pancreatic tumor cells) and in vivo experiments (C57BL/6 bearing KP02 tumor), suggest that while the multifunctional activity was not required for the antitumor activity of these thiosemicarbazones, σ2-targeting appeared to allow alternative tumor cell death mechanisms, leading to potent and less toxic off-targets toxicities compared to other thiosemicarbazones devoid of σ2-targeting.
TOC image
1. Introduction
According to the latest Cancer Statistics’ report [1], cancer death rates have been decreasing by 23% since 1991. Despite that, cancer overtook cardiovascular diseases becoming the first cause of death in 21 states within US, while death rates are increasing for cancers of the liver and pancreas. Pancreatic cancer is one of the most aggressive diseases characterized by a rapid progression, high probability of local recurrence and occurence of early liver metastases. [2] It represents the fourth cause of death for cancer related mortalities with a very poor prognosis [3], and less than 8% five years survival rate. Currently, for patients that are diagnosed early enough, surgery represents the preferred treatment option, further supported by radiation- and gemcitabine-based chemotherapy, but all of these treatment regimens generally result in heterogeneous response rates with a high probability of developing recurrent disease [4–6]. Pancreatic cancer ultimately develops resistance to gemcitabine and thus requires an urgent need of identifying more efficient treatment options as well as better understanding the deranged molecular pathways characteristic for this debilitating malignancy.
In recent years, many attempts to harness the σ2 receptor as a tumor-specific molecular target for pancreatic cancer therapy have emerged. The σ2 receptor belongs to the σ receptors family, divided in σ1 and σ2 proteins. After being proposed as a histone [7,8], the σ2 receptor was later idientified as the progesterone receptor membrane component 1 (PGRMC1) protein complex [9]. However, mounting evidence suggested that the σ2 receptor might actually not be related to PGRMC1 [10–12] and has thus been recently proposed to be the endoplasmic reticulum ER-resident membrane protein TMEM97 [13]. Despite such a controversial identification, the σ2 receptor has been increasingly studied due to its overexpression and activity in a number of human tumors. σ2 Ligands are able to selectively induce tumor cells death through mechanisms that may involve caspase-dependent and -independent apoptosis, lysosomal membrane permeabilization, generation of reactive oxygen species (ROS), and autophagy [14–20].
Recently, the mitochondrial superoxide pathway has been recognized as σ2 receptor-activated process in pancreatic cancer cells [21]. Importantly, certain σ2 receptor agonists have shown efficacy in preclinical tumor models of pancreatic cancer [22–25] highlighting the notion that this receptor is a promising target for cancer therapy. In an effort to develop new drugs that target multiple biologic functions with a single molecule (i.e. multi-target approach), we recently developed a novel class of σ2 receptor ligands, capable of chelating metal ions through their thiosemicarbazone moiety [20]. These molecules were also able to interact with the P-glycoprotein (P-gp) efflux pump, i.e. the most well-described ABC ‘polyspecific’ transporter, whose overexpression is associated with poor prognosis and poor quality of life in cancer patients. Our choice to focus on these particular cellular targets was made based on recent evidence that cancer cells are exquisitely vulnerable to changes in energy levels due to their increased metabolism to sustain rapid cell proliferation. Accordingly, a simultaneous action on the three targets would synergistically compromise cells energy levels. Accordingly, engaging these three drug targets simultaneously was projected to compromise the cellular energy levels in a synergistic manner, because i) σ2 receptor ligand cytotoxicity is correlated with lysosomal membrane permeabilization and an increase in ROS production [18,25]; ii) metal chelators that complex endogenous ironII/III are known as redox cycling agents that also result in increased production of ROS; and iii) P-gp modulation with activation of the futile ATP-cycle would lead to increases in oxidative phosphorylation, subsequently increasing the level of ROS production [26]. Therefore, our multifunctional thiosemicarbazones were anticipated to combine the cytotoxic properties of σ2 agonists with the ironII/III chelating and potentially P-gp modulating capacity within a single drug conjugate, thereby creating a highly lethal and cancer-directed therapy option.
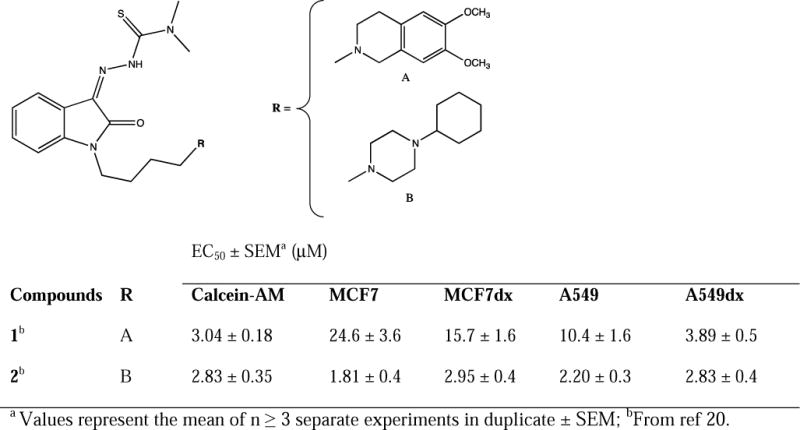
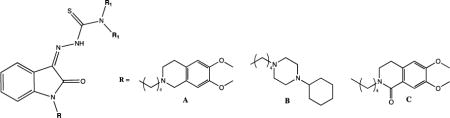
Among all the novel σ2 thiosemicarbazone ligands, two compounds, i.e. (Z)-2-(1-(4-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-2-oxoindolin-3-ylidene)-N,N-dimethylhydrazinecarbothioamide 1 and (Z)-2-[1-[4-(4-Cyclohexylpiperazin-1-yl)butyl]-2-oxoindolin-3-ylidene]-N,N-dimethylhydrazinecarbothioamide 2 (Figure 1), stood out due to their promising cytotoxic activity in two cell line pairs: MCF7 breast cancer cells, A549 lung cancer cells and their corresponding doxorubicin-resistant cell lines, namely MCF7dx and A549dx, both overexpressing P-gp (EC50s, Figure 1) [20]. With the aim of contributing to the proposal of alternative strategies for treating pancreatic cancer, herein we studied the effects of 1 and 2 in diverse pancreatic cancer cells. Because of the multifunctional nature of these molecules, we aimed to study the impact of each target involved to the overall effect, as well as on the respective mechanism leading to cell death. In order to do that, we applied a structural deconstruction approach on thiosemicarbazones 1 and 2. Structural modifications were carried out so that the metal chelating moiety or the interaction with σ2 receptor and/or with P-gp were alternatively removed. Besides their affinity to the σ2 receptor, all ‘deconstructed’ analogues were evaluated for their interaction capacity with P-gp, primarily because it has been recently shown that P-gp could play a critical role in the acquired resistance of pancreatic cancer to gemcitabine [27]. For all of the compounds, cytotoxic activity and pathways activated (e.g. caspase-3 activity, ROS and mitochondrial superoxide generation) were studied in a panel of pancreatic cancer cells, with the attempt to shed new light on the contribution of the targets involved in the mechanisms of cells death which also depend on the different sensitivities of diverse cells. The most promising compounds were tested in a pre-clinical tumor model of syngeneic KP02 pancreatic cancer cells, which are known for closely mimicking the human disease with regard to a highly fibrotic tumor stroma, providing valuable hints about the beneficial effects of a multifunctional, σ2 ligand-based cancer therapeutics.
Figure 1.
Novel σ2 thiosemicarbazone ligands and their antiproliferative activity
2. Results and Discussion
2.1. Chemistry
The synthetic scheme for the synthesis of isatin-β-thiosemicarbazones 7, 8 and 9 is depicted in Scheme 1. 3,4-Dihydroisoquinolin-1(2H)-one 3 [28] was alkylated with 1-bromo-4-chlorobutane using NaH as a base to afford 2-(3-chlorobutyl)-3,4-dihydroisoquinolin-1(2H)-one 4 which was used to alkylate isatin 5 providing intermediate 6. Isatins 5 or 6, were dissolved in hot ethanol and treated with thiosemicarbazide or N,N-dimethyl-3-thiosemicarbazide to provide the final thiosemicarbazones 7, 8 and 9 respectively. In Scheme 2 the synthesis of the final compounds 10, 11, 13 and 14 is illustrated. The already known compounds 13 and 14 [29] were obtained through a slightly modified procedure. Treatment of indole with 1-bromo-4-chlorobutane in the presence of KOH gave the intermediate 12. Final compounds 10-14 were obtained upon alkylation of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline or 1-cyclohexylpiperazine with 1-bromo-butane or with intermediate 12 in the presence of K2CO3.
Scheme 1. Synthesis of Thiosemicarbazones 7–9a.
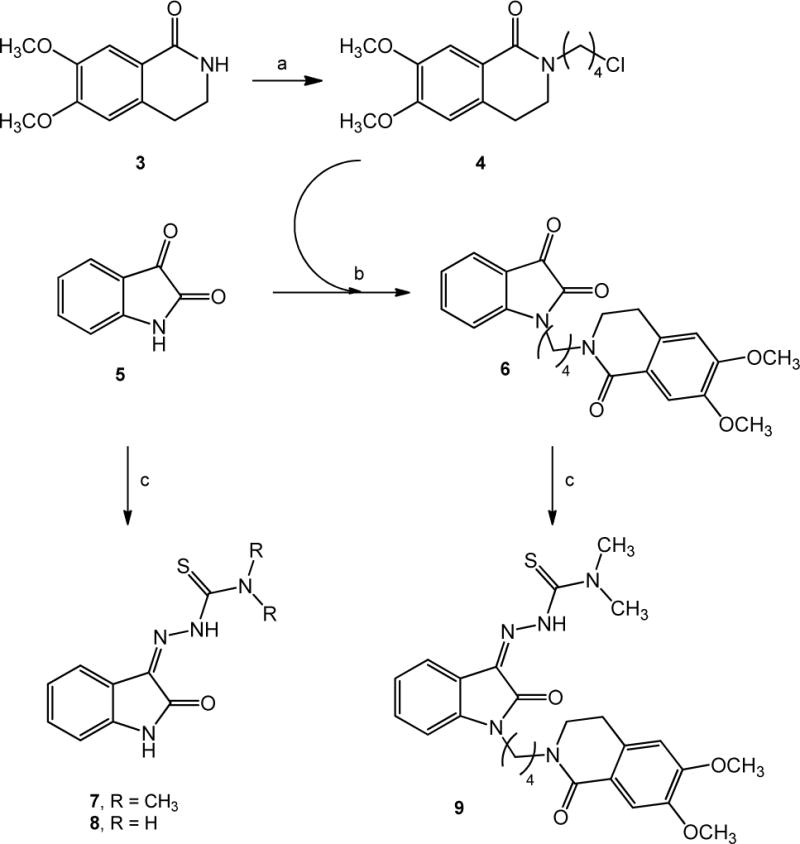
aReagents and Conditions: (a) 1-Bromo-4-chlorobutane, NaH, DMF; (b) K2CO3, CH3CN; (c) appropriate thiosemicarbazide, EtOH.
Scheme 2. Synthesis of 11–14a.
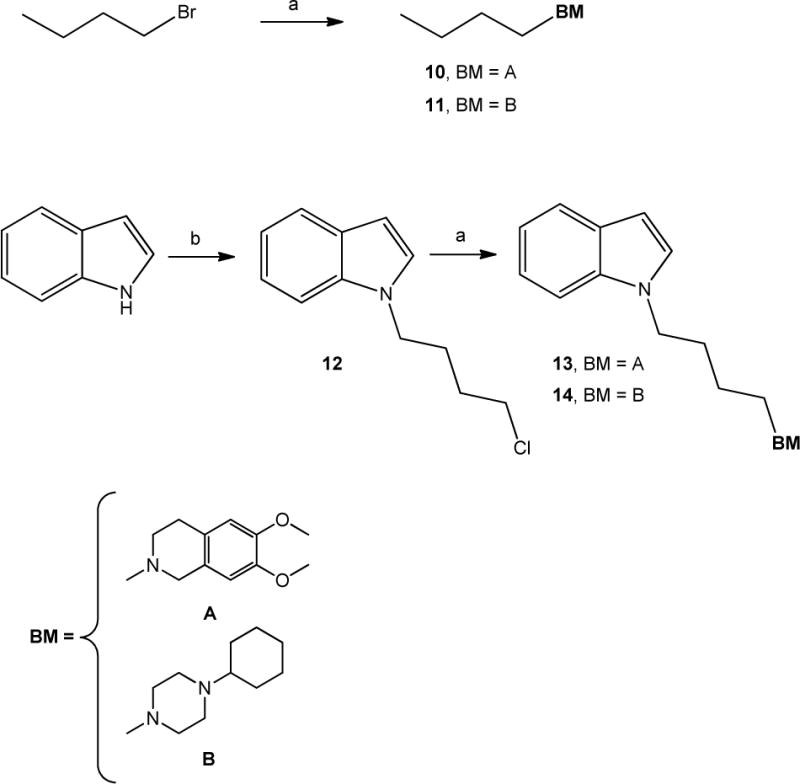
aReagents and Conditions: (a) 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline or 1-cyclohexylpiperazine, K2CO3, CH3CN; (b) 1-Bromo-4-chlorobutane, KOH, TBAB, DMF.
2.2. σ2 Receptor Binding
For all the compounds, σ2 receptor affinity was evaluated by radioligand binding assays and the results are reported as inhibition constants (Ki values) in Table 1. Removal of the N-butyl linked basic moieties (7 and 8), as well as elimination of the basic properties of the N-atom (9), dramatically reduced the σ2 receptor affinity (7, 8 and 9, Ki >10000). This outcome was anticipated because of the lack of structural molecule domains that are essential for an interaction with σ2 receptor (i.e. basic moieties). On the other hand, butyl-6,7-dimethoxytetrahydroisoquinoline and butyl-cyclohexylpiperazine showed appreciable σ2 affinity values (10 and 11, Ki = 15.0 nM and 65.2 nM respectively) despite the lack of an important hydrophobic portion required for the optimal interaction with the σ2 receptors, according to the σ2 receptors pharmacophoric models [30–32]. The presence of the indole hydrophobic portion in the 6,7-dimethoxytetrahydroisoquinoline 13 and cyclohexylpiperazine 14 derivatives, that mimicks the isatin ring in 1 and 2, led a 3- to 30-fold increase in the σ2 affinity (Ki = 3.66 nM and 1.90 nM respectively) compared to 10 and 11, in agreement with the σ2 receptor’s pharmacophore.
Table 1.
σ2 Receptor affinity and P-gp activity.
2.3. Calcein-AM assay
For all the novel deconstructed analogues of thiosemicarbazones 1 and 2, interaction with P-gp was investigated employing the Calcein-AM assay. Results as EC50 values are reported in Table 1. Lead compounds 1 and 2 moderately inhibited P-gp (EC50 = 3.04 μM and 2.83 μM, respectively) [20], whereas the absence of the N-butyl-linked basic moiety at the isatin nucleus (7 and 8) completely abolished the interaction with the P-gp, demonstrating how the sole isatin-thiosemicarbazone portion is unable to interact with the efflux pump (Table 1). In accordance with previous SAfiR analyses [33], removal of the basic character of the N-atom in the tetrahydroisoquinoline ring through the replacement of 6,7-dimethoxytetrahydroisoquinoline with the corresponding amide 3,4-dihydrisoquinolin-(2H)-1-one (9), kept a moderate interaction with the efflux pump (EC50 = 11.0 μM) [33]. On the other hand, 2-butyl-6,7-dimethoxytetrahydroisoquinoline (10) and 1-butyl-cyclohexylpiperazine (11) respectively showed modest (EC50 = 76.4 μM) or a complete loss of interaction with P-gp. Conversly, the corresponding indole derivatives 13 and 14 modulated the efflux pump in the low micromolar or submicromolar range (EC50 = 6.09 μM and 0.46 μM, respectively), indicative of a key importance of the the hydrophobic portion within these derivatives for the interaction with P-gp.
2.4. Cytotoxic activity
The cytotoxic activities of 1 and 2 and their deconstructed analogues were in vitro measured in a panel of human (MIAPaCa-2, BxPC3, AsPC1 and Panc-1) and mouse (Panc02, KP02 and KCKO) pancreatic cancer cell lines, which were anticipated to display differential sensitivities to structurally diverse σ2 receptor ligands, as already shown [21]. Cytotoxic activities of the ligands are expressed as EC50 values in Table 2. Thiosemicarbazones 1 and 2 generated potent cytotoxic effects in all the pancreatic cells studied, in line with the potent efficacy showed in the MCF7/MCF7dx and A549/A549dx cell lines pairs (Figure 1) [20]. These compounds displayed a potent activity even in the human pancreatic cancer cell line PANC-1 for which other σ2 ligands previously developed, exhibited no or only minimal cytotoxic activity [21]. Therefore, combination of the σ2/P-gp targeting moieties with ironII/III chelator properties of the thiosemicarbazone group in a single structure (1 and 2) resulted in cytotoxic agents with potent activity in pancreatic tumors (EC50 values ranging from 1.17 μM to 14.66 μM). In order to evaluate the contribution of the metal chelator properties alone on the overall activity profile of 1 and 2, the σ2/P-gp targeting moiety (i.e. N-butyl-linked portions) was removed from the isatin-N-atom, and the thiosemicarbazone structure was kept, in the N,N-dimethylated (7), and non-dimethylated forms (8), both inactive at σ2 receptor and at P-gp. Compound 7 determined a strong cytotoxic activity in most of the cell lines studied (EC50 values ranging from 1.21 μM to 3.14 μM) except for MIAPaCa-2 (EC50 > 18.3 μM) and Panc-1, where curiously no cytotoxic activity was detected (EC50 > 100 μM). In contrast, the not-dimethylated compound 8 did not show cytotoxic activity in the cell lines tested (EC50 > 100 μM). This result is consistent with our previous data, in which N-terminal-dimethylation in thiosemicarbazones resulted in enhanced antitumor activity in comparison to the un-substituted and mono-substituted variants, similarly to what has been reported for other thiosemicarbazones [34–36]. Therefore, independently from the presence of the σ2 targeting moiety, the absence of the dimethyl substitution at the thiosemicarbazone moiety abolished the cytotoxic activity also in pancreatic cancer cells. Removal of the sole σ2-mediated activity from the overall action of 1, was obtained in compound 9, in which the metal chelator and P-gp targeting moieties were retained. Thiosemicarbazone 9 displayed good efficacy in all the pancreatic cancer cell lines tested (EC50 values ranging from 2.18 μM to 10.30 μM). MIAPaCa-2 and Panc-1 responded to this compound the least, which was not unexpected given the lack of sensitivity to compounds 1 and 2 and suggests that these cell lines are less sensitive to thiosemicarbazones in general. The sole contribution of the σ2 targeting moieties with respect to the overall activity profile of 1 and 2 was studied through the N-butyl-6,7-dimethoxytetrahydroisoquinoline and 1-butyl-4-cyclohexylpiperazine (10 and 11), and more reliably through their indole-bearing analogues (13 and 14), which are characterized by higher σ2 receptor affinities. While 13 and 14 displayed only a weak activity profile in KP02, KCKO and MIAPaCa-2 cells (EC50 values ranging from 40 μM to 79 μM), and even further reduced activity in the other pancreatic cancer cell lines studied (EC50 > 100 μM), 10 and 11 were inactive in all the cell lines tested in our current study (EC50 > 100 μM). Since a number of other σ2 ligands devoid of metal chelator moieties show cytotoxic activity in pancreatic cancer cells [21,23], the lack of cytotoxic effects in these σ2 ligands may suggest an antagonist activity at the σ2 receptor (generally referred as lack of activity).
Table 2.
Viability screening on pancreatic cancer cell lines following 48 hr treatment.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Activity, EC50 (μM)a
|
|||||||||
| Cmpd | R | R1 | Panc02 | KP02 | KCKO | MIAPaCa-2 | BxPC3 | AsPC1 | Panc-1 |
| 1 | A | CH3 | 1.33±0.2 | 6.92±0.9 | 6.49±1.0 | 14.6±2.2 | 2.34±0.8 | 2.01±0.6 | 14.6±2.1 |
| 2 | B | CH3 | 1.17±0.4 | 7.32±0.8 | 6.18±0.3 | 10.8±1.3 | 6.15±0.6 | 3.86±0.2 | 8.73±0.8 |
| 7 | H | CH3 | 1.21 ±0.1 | 2.83±0.6 | 3.14±0.4 | 18.3±2.1 | 2.52±0.5 | 2.17±0.3 | >100 |
| 8 | H | H | >100 | >100 | >100 | >100 | >100 | >100 | n.d.b |
| 9 | C | CH3 | 2.18±0.3 | 5.62±1.1 | 4.51±0.8 | 10.3±1.9 | 5.93±0.7 | 5.74±0.5 | n.d.b |
|
| |||||||||
| Activity, EC50 (μM)a
|
|||||||||
| Cmpd | X | BM | Panc02 | KP02 | KCKO | MIAPaCa-2 | BxPC3 | AsPC1 | Panc-1 |
| 10 | H | A | >100 | >100 | >100 | >100 | >100 | >100 | n.d.b |
| 11 | H | B | >100 | >100 | >100 | >100 | >100 | >100 | n.d.b |
| 13 | C | A | >100 | 49±2.5 | 77±5.2 | 40±3.5 | >100 | >100 | n.d.b |
| 14 | C | B | >100 | 70±4.6 | 67±4.5 | 79±6.1 | >100 | >100 | n.d.b |
Values represent the mean of n ≥ 3 separate experiments in duplicate ± SEM
n.d. = not determined.
In summary, the results obtained in pancreatic cancer with our lead thiosemicarbazones 1 and 2 and their deconstructed analogues show that while different pancreatic cell lines respond with differential sensitivity to structurally diverse σ2 ligands, their σ2 receptor affinity and/or their capacity to modulate P-gp does not seem to be required for their cytotoxic activity profiles. In these molecules, the N,N-dimethyl-substituted-thiosemicarbazone appears to play a dominant role for their augmented cytotoxic action, likely because of a good compromise in metal chelation and cell permeation. In any case, it appears that a multitarget approach represents a beneficial constellation, as the σ2 receptor is overexpressed in a number of diverse cancers and, in addition to the direct cytotoxic effects of its ligands toward the cancer cells, the σ2 receptor could be harnessed for the targeted delivery of drugs to tumor foci. In this regard, the combination of metal chelation and σ2-targeting may be a promising strategy for the treatment of human malignancies.
2.5. Studies on cell mechanisms involved in the activity
We next investigated the contribution of the diverse targets hit by the thiosemicarbazones 1 and 2 to the pathways activated that eventually lead to tumor cell death. Therefore, thiosemicarbazones 1 and 2 and their analogues displaying relevant cytotoxic activity (7 and 9) were evaluated using mouse KP02 pancreatic cancer cells, characterized by forming a stroma-rich tumor microenvironment in vivo and thus more closely mimic the human disease.
2.5.1. P-gp expression in KP02 tumor cells
In this cell line, where σ2 ligands previously displayed promising in vitro and in vivo efficacy [37], we assessed the expression level of P-gp as a mean to determine what role, if any, this efflux pump plays in the context of the cytotoxicity of our drugs. Western blot analysis clearly showed no expression of the efflux pump in KP02 cells, so that activity at the P-gp of these compounds did not seem to be involved in their action in this cell line (Figure S1, Supporting Information). Nevertheless, knowledge about the interaction of these compounds with P-gp may provide useful information in case of their administration in P-gp overexpressing tumor cells.
2.5.2 ROS involvement in the antiproliferative activity
We next explored ROS involvement in KP02 tumor cells employing lipid antioxidant α-tocopherol and the hydrophilic precursor of glutathione N-acetyl-L-cysteine (NAC). The effect of antioxidants was evaluated 48 hours after treatment with 4 μM of 1, 2, 7 and 9 (Figure 2, panel A). Addition of 100 μM α-tocopherol 1 hour prior to drug treatment, rescued the pancreatic cancer cells, thus effectively reducing cell death caused by 1, 2 and 9. By contrast, no cells were rescued by α-tocopherol upon treatment with 7, with no differences between the α-tocopherol treated and untreated cells. The same results were also obtained in AsPC1 and Panc02 tumor cells upon treatment with the same compounds (Figure S2, Supporting Information). Interestingly, ROS production was reduced following treatment with 1, 2 and 9 in the presence of α-tocopherol but not in the presence of NAC. Cell death caused by the four compounds administered at a 4 μM concentration was not rescued by NAC in the KP02 cell line, and even higher levels of cell death were recorded when cells were treated with NAC and 7 or 9 (Figure 2, panel A). These results are in agreement with data obtained upon treatment of pancreatic cancer cells with σ2 ligands and NAC [21]. We used two different antioxidants on the basis of their different mechanisms: while NAC is a hydrophilic antioxidant, precursor of glutathione that provides SH-groups to prevent oxidation, α-tocopherol is a chain-breaking lipophilic antioxidant localized in cellular membranes responsible of their integrity through inhibition of lipid peroxidation. Taken together, these results support the hypothesis that generation of ROS is at least partially responsible for the mechanism of action of these compounds in all the cell lines studied, except for 7. Therefore, the strong cytotoxic activity of 7 can only be explained with a mechanism of action that does not seem to involve ROS production. These data also suggest that, despite of sharing the same thiosemicarbazone-β-isatin structure, ROS generating pathways do not depend on it, as the presence of the N-butyl-linked moieties in thiosemicarbazones 1, 2 and 9 triggers a pathway that is different from that of compound 7.
Figure 2.
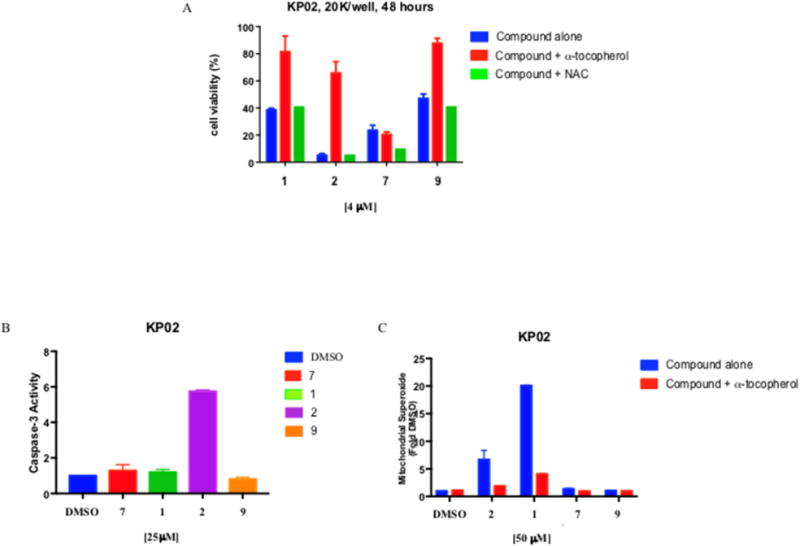
Caspase-3 activation, involvement of Reactive Oxygen Species (ROS) and Mitochondrial Superoxide detection in KP02 cell lines.
(A) Indirect measurement of ROS involvement following 48 hours treatment with 4 μM 1, 2, 7 or 9 in the presence of 100 μM lipophilic antioxidant α-tocopherol or hydrophilic antioxidant N-acetyl-L-cysteine (NAC) in KP02 cells p < 0.001; (B) Caspase-3 activation was measured by Caspase-GloR Assay in KP02 cells treated with 25 μM of different compounds for 5 hours and expressed relative to vehicle. Cells treated with 2 had significant increase in caspase-3 p < 0.0001. (C) Mitochondrial Superoxide detection in KP02 after 2 hours treatment with 50 μM of compounds alone or in combination with the lipid antioxidant α-tocopherol (1 mM), 1 generated high mitochondrial superoxide production p < 0.0001. Values are the means of n ≥ 3 independent experiments in triplicates with SEM.
2.5.3. Caspase-3 activation
σ2 Ligands have been reported to activate mechanisms that are cell and ligand specific and can cause in some cases caspase-dependent and in others caspase-independent apoptosis [21,25,38–40]. Therefore, activation of caspase-3 was evaluated for thiosemicarbazones 1, 2, 7 and 9 in mouse KP02 tumor cells [41]. Pancreatic mouse adenocarcinoma cells were treated with the ligands (25 μM) for 5 hours and assayed for cleavage of the proluminescent caspase-3 substrate and subsequent generation of a glow-type luminescent signal. Compound 2 induced a good activation of caspase-3, increasing caspase-3 activity by 6-fold (p < 0.001) while compounds 1, 7, and 9 did not activate the caspase-3 at all (Figure 2, panel B). Despite targeting the same biological targets, thiosemicarbazones 1 and 2, which both increase ROS, did not share caspase-3 activation, in accordance with previously reported σ2 ligands, whose activation of caspase-3 appeared to be both cell dependent and molecule dependent. Similar results were obtained in AsPC1 and Panc02 tumor cells upon the treatment with the same compounds (Figure S3, Supporting Information).
2.5.4. Superoxide radical detection in the mitochondria of KP02
Since mitochondrial superoxide production was recently identified as a novel mechanism of σ2 receptor-mediated cell death [21], we wished to study superoxide radical production in mitochondria of KP02 pancreatic cancer cell lines. MitoSOX™ Red reagent is a novel fluorogenic dye specifically targeted to mitochondria in live cells, which is readily oxidized by superoxide but not by other ROS- or reactive nitrogen species (RNS)- generating systems (e.g. such as peroxides, hydroxyl radical, singlet oxygen, nitric oxide and peroxynitrite). KP02 cells were treated with 50 μM of 1, 2, 7 and 9 for 2 hours (Figure 2, panel C). While σ2 ligands 1 and 2 induced a strong mitochondrial ROS production with the mean fluorescence intensities increasing by 6-fold and 20-fold respectively, thiosemicarbazones 7 and 9 were not capable of superoxide radical production in the mitochondria of KP02 tumor cells. With the aim of obtaining a more complete picture, we assessed the mitochondrial superoxide production in the presence of the lipid antioxidant α-tocopherol, and found that it was strongly reduced when cells were treated with σ2 ligands in the presence of α-tocopherol, according to the rescue of cell viability displayed in the MTT assay (Figure 2, panel A). Using the same compounds, similar results were obtained in Panc02 and AsPC1 cells (Figure S4, Supporting Information). Taken together, these results demonstrate that the cytotoxic activity of thiosemicarbazones 7 and 9, devoid of σ2 affinity, does not rely on superoxide radical production in the mitochondria. On the other hand, we have confirmed that the mitochondrial superoxide production is as a common pathway shared by the two σ2-targeting thiosemicarbazones 1 and 2 in pancreatic cancer cells, in accordance with the results shown by other σ2 ligands in the same cells [21].
2.6. Thiosemicarbazones reduces tumor volume in preclinical model of pancreatic cancer
Based on the strong in vitro antitumor activity and diversity of death pathways activation patterns induced by the most promising thiosemicarbazones 1, 2 and 7, we next investigated these promising drug candidates for efficacy in a syngeneic, genetically-engineered murine stroma-dense cancer model, closely mimicking the human disease. Using this configuration, the in vivo efficacy of the two σ2/P-gp mixed ligands and iron chelators 1 and 2 (that share ROS and mithocondrial superoxide production capacity, but not caspase-3 activation), could be compared with that of the iron chelator 7, that does not act through ROS increase. For these three compounds, interaction with the σ1 receptor, which is also endowed with antitumor properties, was ruled out through a flow cytometry experiment previously set up (Figure S6, Supporting Information) [42].
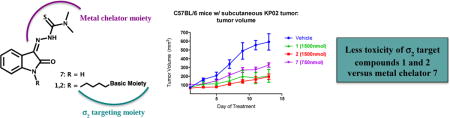
C57BL/6 female mice were inoculated subcutaneously with 2.5 × 105 KP02 cells and a week later, when tumors reached ~ 5-6 mm in diameter, mice were randomized into control and treatments groups of (n = 7-10). Vehicle consisting of 25% Cremophor in water, multifunctional compounds 1 and 2 (750 nmol/100μL vehicle), or 7 (750 nmol/100μL vehicle) were given by i.p. injection daily for two weeks. After conclusion of treatment, tumors were smaller for mice treated with iron chelator 7 (mean = 328 mm3), compared to vehicle (mean = 596 mm3) (Figure 3 panel A; p < 0.0001). Mice treated with compounds 1 and 2 had both tumor volumes that were statistically similar to vehicle, without experiencing any treatment-related deaths. Therefore, we repeated the experiment by employing higher concentrations of compounds 1 and 2 (1500 nmol/100μL vehicle). At this concentration, both multifunctional compounds were capable of reducing the mean tumor volume (mean = 196 mm3 for compound 2 and mean = 205 mm3 for compound 1) compared to vehicle treated group (mean = 592 mm3) (Figure 3, panel B; p > 0.001). Moreover, compounds 1 and 2 turned out to be more effective in reducing tumor burden than gemcitabine when compared to mice bearing a KP02 orthotopic tumor and treated twice weekly by i.p. injections with 20 mg/kg gemcitabine [37].
Figure 3.
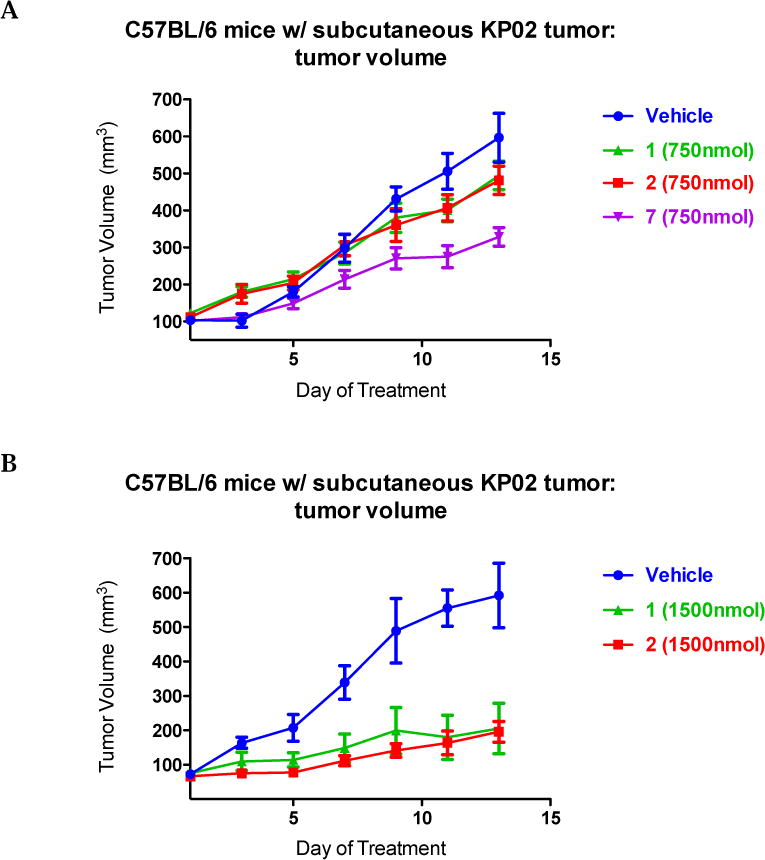
Thiosemicarbazones efficacy in C57BL/6 bearing KP02 tumors.
2.5 × 105 KP02 cells were inoculated subcutaneously into female, 8 weeks old C57BL/6 mice and when tumors had reached a mean diameter of 5-6 mm, daily thiosemicarbazones treatment (750nmol/100μL 1, 2 and 7 (A); 1500 nmol/100μL 1 and 2 (B)) began by i.p. injection. 7 treatment (A) decreased tumor volume p < 0.001, while at the same dosage 1 and 2 treatment (A) was not effective p > 0.05. Treatment with a higher concentration of 1 and 2 (B) decreased significantly tumor volume p = 0.004 and p = 0.012 respectively. Data represent Means ± SEM, n = 7-10 per group.
Importantly, we did not observe treatment-related deaths or gross abnormalities in mouse behavior. In order to assess for more subtle toxicities, serum chemistries (AST, ALT, BUN, total protein, glucose, and Cr) and complete blood counts were analyzed with no significant differences noted when compared to the control group (Table 3). Necropsy revealed no difference in mouse weights (vehicle: 19.5± 0.5 grams; 7: 19.5 ± 0.5 grams; 1: 17.5 ± 0.5 grams; 2: 19 ± 1 grams; Figure 4; p > 0.05). There were no significant lesions in the brain, heart, alimentary tract, kidneys, liver, or pancreas. Only mild peritonitis was identified at the site of repeated drug injections. However, the compound 7 treated group revealed foci of pulmonary metastases of the implanted tumor and a mild chronic progressive nephropathy that were not noticed in mice treated with compounds 1 and 2.
Table 3.
Thiosemicarbazones do not induce changes in blood cytology (CBC) following treatment of KP02 tumor-bearing C57BL/6 mice.
| A.
| ||||
|---|---|---|---|---|
| Control | 7 | 1 | 2 | |
| WBC (10 3 /μL) | 7.8 ± 0.55 | 7.93 ± 1.35 | 5.75 ± 0.45 | 8.87 ± 0.63 |
| RBC (10 6 /μL) | 9.35 ± 0.11 | 8.63 ± 0.71 | 9.19 ± 0.15 | 9.94 ± 0.92 |
| HGB (g/dL) | 13.8 ± 0.1 | 12.9 ± 1.1 | 13.45 ± 0.05 | 14.9 ± 0.9 |
| PCV (%) | 47.5 ± 3 | 42.65 ± 2.85 | 45.45 ± 4.15 | 51 ± 3.8 |
| MCV (fL) | 50.7 ± 0.76 | 49.45 ± 0.75 | 49.5 ± 0.1 | 51.4 ± 0.9 |
| MCH (pg) | 14.8 ± 0.1 | 14.95 ± 0.05 | 14.65± 0.15 | 15 ± 0.5 |
| MCHC (%) | 29.13 ± 0.83 | 30.2 ± 0.6 | 29.6 ± 0.3 | 29.25 ± 0.45 |
| Platelets (10 3/μL) | 951.6 ± 86.3 | 865 ± 25 | 976.5 ± 81.5 | 854 ± 47 |
|
B. | ||||
| Control | 7 | 1 | 2 | |
| BUN (mg/dL) | 27.3 ± 2.66 | 29.5 ± 2.5 | 22 ± 2 | 21.5 ± 1.5 |
| Creatinine (mg/dL) | 0.33 ± 0.02 | 0.35 ± 0.12 | 0.31 ± 0.02 | 0.34± 0.1 |
| ALT (μ/L) | 54.3 ± 6.33 | 257 ± 27 | 95 ± 41 | 147.5 ± 86.5 |
| AST (μ/L) | 98.6 ± 0.05 | 573.5± 37.4 | 172 ± 11.2 | 416 ± 21.2 |
| Total Protein (g/dL) | 5.55 ± 0.15 | 5.5 ± 0.3 | 5.1 ± 0.1 | 5.75 ± 0.35 |
| Glucose (mg/dL) | 168 ± 3.3 | 309 ± 4 | 285 ± 24.5 | 288.5 ± 26.5 |
(A) Blood cytology analysis of C57BL/6 mice (n = 3 mice/group) treated with thiosemicarbazone 7 (750nmol/100μL), σ2 ligands 1 and 2 (1500nmol/100 μL) and vehicle (control) for 2 weeks. The differences in complete blood count laboratory values between the two groups are not statistically significant, p > 0.05.
(B) Biochemical analysis of C57BL/6 mice (n = 3 mice/group) treated with thiosemicarbazone 7 (750nmol/100μL), σ2 ligands 1 and 2 (1500nmol/100 μL) and vehicle (control) for 2 weeks. The differences in serum chemistries between the groups are not statistically significant, p > 0.05.
Figure 4.
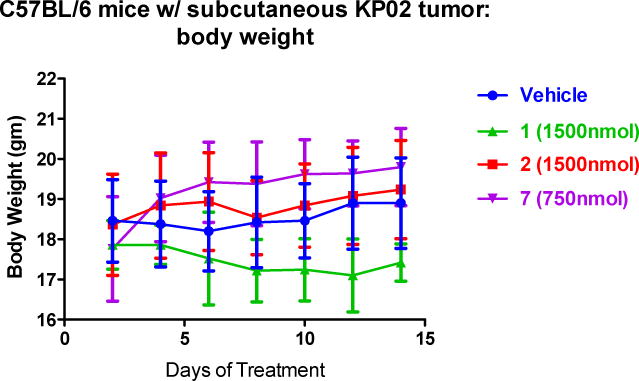
C57BL/6 mice tolerated the drugs well without signs of weight loss. Data represent Means ± SEM, n = 7-10 per group, p > 0.05.
3. Conclusion
Based on the encouraging activity profiles of the multifunctional thiosemicarbazones 1 and 2 that target the σ2 receptor and the P-gp efflux pump and have Iron chelating properties, a deconstruction approach was applied to investigate the contribution of each target hit to the overall action of these molecules. Because of the urgent need of novel treatments for pancreatic cancers, lead compounds 1 and 2 and the deconstructed analogues (7-11, 13 and 14) were investigated in a panel of pancreatic tumor cells. Among the compounds, N,N-dimethyl-thiosemicarbazone-bearing derivatives (1, 2, 7, 9) showed potent activity in most of the pancreatic cell lines studied, also in the absence of the σ2-targeting moiety (7 and 9), so that the iron chelating structural portion (N,N-dimethyl-thiosemicarbazone) appeared to be mainly responsible for the cytotoxic activity in these cells (likely because it confers a good balance between activity and cell permeability). Nevertheless, we showed that the death pathways engaged by these compounds (1, 2, 7 and 9) are not governed by the common N,N-dimethyl-thiosemicarbazones portion since: i) ROS increase was shown by 1, 2, and 9 but not by 7; ii) caspase-3 activity was only increased by σ2-targeting thiosemicarbazone 2; iii) mitochondrial superoxide production was only detected with the two σ2-targeted thiosemicarbazones 1 and 2. Our deconstructive approach showed that while σ2 receptor targeting is not necessary for the in vitro cytotoxicity of these multifunctional thiosemicarbazones (except for Panc-1 cells), the presence of the N-butyl linked basic moieties triggers alternative death pathways in comparison to those compounds that only contain metal chelating features, thus allowing for differential sensitization levels of cell death, which we believe could be useful for developing tailored treatments. Importantly, besides a contribution to the cytotoxic activity (as in Panc-1 cells), σ2 receptor binding may be beneficial for the targeted delivery to σ2 receptors overexepressing tumors, according to a promising recent approach [39]. In vivo administration of the multifunctional σ2 ligands 1, 2 or the metal chelator 7 markedly slowed the growth rate in a mouse stroma dense model of pancreatic cancer (C57BL/6 bearing KP02 tumors), demonstrating that these compounds reach their target and exert antitumor activity. While mice treated with the iron chlelator 7 revealed foci of pulmonary metastases of the implanted tumor and a mild chronic nephropathy, mice treated with multifunctional compounds 1 and 2 better tolerated the treatment without showing signs of off-target toxicity.
Despite the suggested role that P-gp may play in the resistance of pancreatic cancer to gemcitabine, the absence of the efflux pump in KP02 cells did not allow us to evaluate our compounds in the context of P-gp expression.
Overall, the results from our deconstructive approach from in vitro and in vivo experiments suggest that while the multifunctional drug composition is not necessary for the antitumor activity of these thiosemicarbazones, σ2-targeting may allow for a more specific targeted delivery to tumor foci in vivo (σ2 overexpressing tumors) with less toxic off-targets effects, together with the activation of alternative tumor cell death mechanisms. Such multifunctional molecules endowed with potent in vivo antitumor activity and reduced off-site toxicity are certainly worth studying further in the oncology field.
4. Experimental Section
4.1. Chemistry
Both column chromatography and flash column chromatography were performed with 60 Å pore size silica gel as the stationary phase (1:30 w/w, 63−200 μm particle size, from ICN, and 1:15 w/w, 15−40 μm particle size, from Merck, respectively). Melting points were determined in open capillaries on a Gallenkamp electrothermal apparatus. High-performance liquid chromatography (HPLC) on an Agilent Infinity 1260 system equipped with diode array with a multiwavelenght UV/vis detector set at λ = 230 nm, 254 nm and 280 nm, through a Phenomenex Gemini RP-18 column (250 × 4.6 mm, 5 μm particle size) was performed on target compounds confirming ≥ 95% purity. 1H NMR spectra were recorded on a Mercury Varian 300 MHz or on a 500-vnmrs500 Agilent spectrometer (499.801 MHz). The following data were reported: chemical shift (δ) in parts per million (ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), integration, and coupling constant(s) in hertz. 13C NMR (125 MHz) were recorded on a 500-vnmrs500 Agilent spectrometer (499.801 MHz) on novel final compounds: chemical shifts in ppm were reported. Recording of mass spectra was done on an Agilent 1100 series LCMSD trap system VL mass spectrometer; only significant m/z peaks, with their percentage of relative intensity in parentheses, are reported. High resolution mass spectroscopy (HRMS) was performed on a Agilent 6530 Accurate-Mass Q-TOF LC/MS spectrometer. For final compounds 10 and 11, 13C NMR and HRMS spectra were recorded on their hydrochloride salts. Chemicals were from Aldrich, TCI and Alpha Aesar and were used without any further purification.
4.2. 2-(4-Chlorobutyl)-6,7-dimethoxy-3,4-dihydroisoquinolin-1(2H)-one (4)
To a suspension of NaH (0.123 g, 5.12 mmol) in dry DMF (2 mL) a solution of 3 (0.425 g, 2.05 mmol) in dry DMF (3 mL) was added in a dropwise manner. The mixture was stirred at 0 °C for 15 min. Then a solution of 1-bromo-4-chlorobutane (0.3 ml, 2.66 mmol) in dry DMF (2 mL) was added and the resulting mixture was stirred for 4 h at room temperature. After cooling at 0 °C, the reaction mixture was quenched with water and the solvent concentrated under reduced pressure. The residue was dissolved in H2O and extracted with CH2Cl2 (3 ×10 mL). The collected organic layers were dried over Na2SO4 and the solvent was evaporated under reduced pressure to afford the crude as a yellow oil. Purification through column chromatography with CH2Cl2 as eluent afforded the title compounds as colorless oil (0.426 g, 70% yield). GC-MS m/z: 297 (M+, 20), 220 (100).
4.3. 1-(4-(6,7-Dimethoxy-1-oxo-3,4-dihydroisoquinolin-2(1H)-yl)butyl)indoline-2,3-dione (6)
A solution of 4 (0.426 g, 1.43 mmol) in CH3CN (10 mL) was added with K2CO3 (2.87 mmol, 0.396 g) and isatin 5 (0.192 g, 1.3 mmol). The resulting mixture was refluxed overnight under stirring. After the removal of the solvent under reduced pressure the residue was taken up with H2O and extracted with AcOEt (3 × 10 mL). The collected organic layers were dried (Na2SO4) and evaporated under reduced pressure to afford a crude oil, which was purified by column chromatography (CH2Cl2/AcOEt 1:1) to give the title compound (0.28 g, 50% yield).
1H NMR (500 MHz, CDCl3) δ 1.72-1.81 (m, 4H, CH2CH2CH2N), 2.92 (t, 2H, J = 6.85 Hz, NCH2CH2Ar), 3.53 (t, 2H, J = 6.85 Hz, NCH2CH2Ar), 3.64 (t, 2H, J = 6.85 Hz, NCH2CH2CH2CH2N), 3.81 (t, 2H, J = 6.85 Hz, NCH2CH2CH2CH2N), 3.90 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 6.64 (s, 1H, aromatic), 7.00 (d, 1H, J = 7.83 Hz, aromatic), 7.10 (t, 1 H, J = 7.34 Hz, aromatic), 7.54-7.61 (m, 3H, aromatic).
4.4. General procedure for the synthesis of final compounds 7-9
To a solution of isatins 5 or 6 (0.68 mmol) in Ethanol (10 mL) 4,4-dimethyl-3-thiosemicarbazide (0.68 mmol) was added and the resulting mixture was refluxed overnight. Upon cooling, precipitation of the final product was achieved.
4.4.1. (Z)-N,N-Dimethyl-2-(2-oxoindolin-3-ylidene)hydrazinecarbothioamide (7)
Crystallization from H2O/EtOH afforded the title compound as orange crystals (0.084g, 50% yield); mp = 250-250 °C; 1H NMR (500 MHz, DMSO-d6) δ 3.35 (s, 6H, CH3), 6.92-6.94 (m, 1H, aromatic), 7.06-7.09 (m, 1H, aromatic), 7.32-7.35 (m, 1H, aromatic), 7.51-7.53 (m, 1 H, aromatic), 11.28 (s, 1H, isatin NH), 13.41 (s, 1H, NHCS); 13C NMR (500 MHz, DMSO-d6) 43.75; 43.83; 117.96; 119.45; 124.34; 129.54; 131.31; 134.22; 141.23; 169.51; 177.83. LC-MS (ESI+) m/z: 271 [M+Na]+; LC-MS-MS 271: 226; LC-MS (ESI−) m/z 247 [M-H]−; LC-MS-MS 247: 204, 174; QTOF (m/z) Calcd for C11H12N4OS [M+Na]+: 271.0630, found: 271.0621. Compound was > 98% pure by HPLC analysis performed with MeOH/H2O, 80 : 20 v/v, at a flow rate 0.8 mL min−1.
4.4.2. (Z)-2-(2-Oxoindolin-3-ylidene)hydrazinecarbothioamide (8)
Crystallization from H2O/EtOH afforded the title compound as orange crystals (0.07g, 50% yield); mp = 253 – 255 °C; 1H NMR (500 MHz, DMSO-d6) δ 6.90-6.92 (m, 1H, aromatic), 7.06-7.09 (m, 1H, aromatic), 7.32-7.36 (m, 1H, aromatic), 7.63-7.65 (m, 1 H, aromatic), 8.67 (s, 1H, NHH), 9.03 (s, 1H, NHH), 11.19 (s, 1H, isatin NH), 12.46 (s, 1H, NHCS); 13C NMR (500 MHz, DMSO-d6) 117.66; 119.51; 124.54; 129.44; 131.25; 134.65; 141.32; 168.72; 180.82. LC-MS (ESI+) m/z: 243 [M+Na]+; LC-MS-MS 243: 226, 185; LC-MS (ESI−) m/z 219 [M-H]−; LC-MS-MS 219: 160; QTOF (m/z) Calcd for C9H8N4OS [M+Na]+: 243.0317, found: 243.0307. Compound was > 98% pure by HPLC analysis performed with MeOH/H2O, 80 : 20 v/v, at a flow rate 0.8 mL min −1.
4.4.3. (Z)-2-(1-(4-(6,7-Dimethoxy-1-oxo-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-2-oxoindolin-3-ylidene)-N,N-dimethylhydrazinecarbothioamide (9)
Crystallization from H2O/EtOH afforded the title compound as yellow crystals (0.173 g, 50% yield); mp = 201-202 °C; 1H NMR (500 MHz, CDCl3) δ 1.67-1.83 (m, 4H, CH2CH2CH2N), 2.88 (t, 2H, J = 6.85 Hz, NCH2CH2Ar), 3.48-3.54 (m, 8H, CH2NCH2CH2Ar and N(CH3)2), 3.58-3.64 (m, 2H, CH2NCH2CH2Ar), 3.80-3.87 (m, 2H, NCH2CH2CH2CH2N), 3.92 (s, 3 H, OCH3), 3.93 (s, 3H, OCH3), 6.63 (s, 1H, aromatic), 6.96 (d, 1H, J = 7.34 Hz, aromatic), 7.12 (t, 1 H, J = 7.34 Hz, aromatic), 7.33 (d, 1 H, J = 7.34 Hz, aromatic), 7.60 (s, 1H, aromatic), 7.83 (t, 1H, J = 7.34 Hz, aromatic), 13.62 (s, 1H, NH); 13C NMR (500 MHz, CDCl3) 24.74; 25.01; 27.72; 39.44; 46.17; 46.35; 56.04; 56.08; 109.23; 109.33; 110.44; 119.96; 121.66; 121.85; 123.27; 130.76; 131.54; 134.77; 142.04; 147.98; 151.88; 161.78; 164.62; 180.13. QTOF (m/z) Calcd for C26H31N5SO4 [M+Na]+: 532.1994, found: 532.1988. Compound was > 98% pure by HPLC analysis performed with MeOH/H2O, 80 : 20 v/v, at a flow rate 0.8 mL min −1.
4.5. General procedure for the synthesis of 10-11
A solution of 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline or 1-cyclohexylpiperazine (2.90 mmol) in CH3CN was added with K2CO3 (0.49 g, 3.50 mmol) and 1-bromobutane (0.38 ml, 3.50 mmol). The resulting mixture was refluxed overnight under stirring. After the removal of the solvent under reduced pressure the residue was taken up with H2O and extracted with CH2Cl2 (3 × 10 mL). The collected organic layers were dried (Na2SO4) and evaporated under reduced pressure to afford a crude oil which were purified by column chromatography (CH2Cl2/MeOH 95:5) to give the title compounds which were transformed into the corresponding hydrocloride salts, recrystallized from MeOH/Et2O.
4.5.1. N-Butyl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (10)
was obtained as white crystals (0.5 g, 90% yield); mp = 191-193 °C. 1H NMR (500 MHz, MeOH-d4) δ 1.03 (t, 3H, J = 7.3 Hz, NCH2CH2CH2CH3), 1.44-1.49 (m, 2H, NCH2CH2CH2CH3), 1.78-1.84 (m, 2H, NCH2CH2CH2CH3), 3.10-3.12 (m, 2H, NCH2CH2CH2CH3), 3.23-3.26 (m, 2 H, ArCH2CH2NCH2), 3.29-3.31 (m, 2H, ArCH2CH2NCH2), 3.80 (s, 6H, 2 OCH3), 4.86 (s, 2H, ArCH2NCH2CH2CH2CH3), 6.77 (s, 1H, aromatic), 6.82 (s, 1 H, aromatic); 13C NMR (500 MHz, MeOH-d4) 13.83; 18.75; 23.73; 25.74; 51.43; 53.67; 56.10; 59.56; 111.41; 111.63; 116.76; 130.45; 146.71; 146.92; GC-MS m/z: 248 (M+, 0.5), 206 (100); QTOF (m/z) Calcd for C15H23NO2 [M+H]+: 250.1807, found: 250.1800. Compound was > 98% pure by HPLC analysis performed with CH3CN/HCOONH4 (20 mM, pH = 5) 75 : 25 v/v, at a flow rate 1.0 mL min −1.
4.5.2. 1-Butyl-4-cyclohexyl-piperazine (11)
was obtained as white crystals (0.5 g, 77% yield); mp = 250-252 °C. 1H NMR (500 MHz, MeOH-d4) δ 1.01 (t, 3H, J = 7.34 Hz, NCH2CH2CH2CH3), 1.19-2.20 (m, 14H, cyclohexyl CH2 and NCH2CH2CH2CH3), 3.20-3.66 (m, 11H, piperazine CH and CH2 and NCH2CH2CH2CH3); 13C NMR (500 MHz, MeOH-d4) 13.82; 18.90; 24.32; 25.74; 25.65; 28.86; 50.10; 52.43; 53.82; 59.46; GC-MS m/z: 224 (M+, 30), 181 (100); QTOF (m/z) Calcd for C14H29N2 [M+H]+: 225.2331, found: 225.2324. Compound was > 98% pure by HPLC analysis performed with CH3CN/HCOONH4 (20 mM, pH = 5) 75 : 25 v/v, at a flow rate 1.0 mL min −1.
4.6. Biology
4.6.1. Materials
[3H]-DTG (50 Ci/mmol) and CulturePlate 96/wells plates were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA, USA). DTG was purchased from Tocris Cookson Ltd, UK. (+)-Pentazocine and calcein-AM were obtained from Sigma-Aldrich-RBI s.r.l. (Milan, Italy). Male Dunkin guinea-pigs (200-250 g) and Wistar Hannover rats (250-300 g) were from Harlan, Italy. Cell culture reagents were purchased from EuroClone (Milan, Italy). Protease inhibitor cocktail, was obtained from Sigma-Aldrich (Milan, Italy). Anti-P-Glycoprotein antibody produced in mouse (C219) was purchased from Calbiochem (Merck-Millipore, Germany). Anti-β-actin, secondary peroxidase antibodies and all reagents for western blotting were purchased from Life Technologies Italia (Monza, Italy).
4.6.2. Compounds
σ2 Receptor ligands 1 [(Z)-2-(1-(4-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-2-oxoindolin-3-ylidene)-N,N-dimethylhydrazinecarbothioamide hydrochloride], 2 [(Z)-2-[1-[4-(4-Cyclohexylpiperazin-1-yl)butyl]-2-oxoindolin-3-ylidene]-N,N-dimethylhydrazinecarbothioamide di-hydrochloride], intermediate 3 [6,7-dimethoxy-3,4-dihydroisoquinolin-1(2H)-one], and σ1
fluorescent ligand 5-(dimethylamino)-2-(6-((5-(4-(4-methylpiperidin-1-yl)butyl)-5,6,7,8-tetrahydronaphthalen-2-yl)oxy)hexyl)isoindoline-1,3-dione were synthesized in our laboratories according to published methods [20,33,42]. Caspase-3 inhibitor Z-DEVD-FMK was purchased from Tocris Bioscience, α-tocopherol and N-Acetyl-L-cysteine from Sigma-Aldrich (St. Louis, MO). Compounds were dissolved in DMSO with final concentrations less than 0.3%.
4.6.3. Cell Culture
Human pancreas cancer cell lines BxPC3, AsPC1, MiaPaCa-2, and Panc1 were obtained from American Type Culture Collection (ATCC, Bethesda, MD). Murine pancreas adenocarcinoma Panc02 was a gift from Bryan Clary (Duke University). The mouse KCKO cell line isolated from a spontaneously developing pancreatic cancer overexpressing human MUC1 [41] was kindly provided by Dr. Pinku Mukherjee (University of North Carolina, Charlotte, NC). The mouse KP02 line was derived from pancreatic cancer tumor tissue obtained from p48-CRE/LSL-KrasG12D/p53flox/+ mice (backcrossed C57BL/6, n = 6). The MCF7σ1 was produced in our laboratory [43]. AsPC-1, BxPC-3 and Panc02 cells were cultured in RPMI-1940 medium with 10% fetal bovine serum (FBS). MIAPaCa-2 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% FBS and 2.5% horse serum. PANC-1 and MCF7σ1 cells were cultured in DMEM with 10% FBS. KCKO cells were cultured in RPMI-1940 medium with 10% FBS, 1% sodium pyruvate, 1% HEPES buffer, and 1% L-glutamine. KP02 cells were cultured in 1:1 mixture of DMEM and Ham’s F-12 Nutrient Mixture with 10% FBS. Penicillin (100 mg/mL) and streptomycin (100 mg/mL) were added to all media; cells were maintained in a humidified incubator at 37 °C with 5% CO2.
4.6.4. Competition σ2 Binding Assays
All the procedures for the binding assays were previously described. σ2 Receptor binding were carried out according to Berardi et al [44]. The specific radioligand and tissue sources was: [3H]-DTG in the presence of 1 μM (+)-pentazocine to mask σ1 receptors, rat liver membranes. The following compounds were used to define the specific binding reported in parentheses: DTG (85-96%). Concentrations required to inhibit 50% of radioligand specific binding (IC50) were determined by using six to nine different concentrations of the drug studied in at least three experiments with samples in duplicate. Scatchard parameters (Kd and Bmax) and apparent inhibition constants (Ki) values were determined by nonlinear curve fitting, using the Prism, version 3.0, GraphPad software [45].
4.6.5. σ1 Binding by Flow Cytometry studies
The procedure for σ1 binding by flow cytometry studies were carried out according to Abate et al 2016 [42]. MCF7σ1 cells were incubated with increasing concentrations (0.1, 1, 10, and 100 nmol/L and 1 and 10 μM) of (+)-pentazocine or PB212 [46–49] or thiosemicarbazones 1, 2 and 7, followed by 100 nmol/L of either σ1 fluorescent compound (σ1FC, 5-(dimethylamino)-2-(6-((5-(4-(4-methylpiperidin-1-yl)butyl)-5,6,7,8-tetrahydronaphthalen-2-yl)oxy)hexyl)isoindoline-1,3-dione) for 75 min at 37 °C. To mask σ2 receptors, 2-(3-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)propyl)-5-methoxy-3,4-dihydroisoquinolin-1(2H)-one F390 [28,42] (10 μM) was co-incubated. At the end of the incubation periods, cells were washed twice with PBS, detached with 200 mL of Cell Dissociation Solution (Sigma Chemical Co.) for 10 min at 37 °C, centrifuged at 13,000 g for 5 min and resuspended in 500 μL of PBS. The fluorescence was recorded using a Bio-Guava® easyCyte™ 5 Flow Cytometry System (Millipore, Billerica, MA), with a 530 nm band pass filter. For each analysis, 50,000 events were collected and analyzed with the InCyte software (Millipore).
4.6.6. Calcein-AM experiment
These experiments were carried out as already described [50]. MDCK-MDR1 cell line (50,000 cells per well) was seeded into black CulturePlate 96/wells plate with 100 μL medium and allowed to become confluent overnight. 100 μL of different concentrations of test compounds (0.1-100 μM) were solubilized in culture medium and added to each well. The 96/wells plate was incubated at 37 °C for 30 min. 100 μL of Calcein-AM, solved in Phosphate Buffered Saline (PBS), was added to each well to yield a final concentration of 2.5 μM, and the plate was incubated for 30 min. The plate was washed 3 times with 100 mL ice cold PBS. Saline buffer (100 μL) was added to each well and the plate was read by a PerkinElmer Victor3 spectrofluorimeter at excitation and emission wavelengths of 485 nm and 535 nm, respectively. In these experimental conditions, Calcein cell accumulation in the absence and in the presence of tested compounds was evaluated and fluorescence basal level was estimated by untreated cells. In treated wells the increase of fluorescence with respect to basal level was measured. EC50 values were determined by fitting the fluorescence increase percentage versus log[dose].
4.6.7. Cell Viability and ROS interference
Determination of cell growth was performed using the MTT assay at 48 h [51,21]. On day 1, 25,000 cells/well were seeded into 96-well plates in a volume of 100 μL. On day 2, the various drug concentrations (1 μM-100 μM) were added. In all the experiments, the various drug-solvents (EtOH, DMSO) were added in each control to evaluate a possible solvent cytotoxicity. After the established incubation time with drugs (48 h), MTT (0.5 mg/mL) was added to each well, and after 3-4 h incubation at 37 °C, the supernatant was removed. The formazan crystals were solubilized using 100 μl of DMSO/EtOH (1:1) and the absorbance values at 570 and 630 nm were determined on the microplate reader Victor 3 from PerkinElmer Life Sciences. The interference of ROS in cell viability was indirectly determined by MTT assay reported above at 24 h. On day 1, 25000 cells per well were seeded into 96-well plates in the presence or absence of α-tocopherol (100 μM). On day 2, the drugs (1 μM-100 μM) were added alone and in combination with α-tocopherol (100 μM). After incubation (24 h) with drugs, MTT assay was performed as above. The interference of ROS in cell viability was indirectly determined by MTT assay reported above at 48 h. On day 1, cells were plated at a density of 2 × 104 cells/well in opaque 96-well, clear-bottom plates 24 hours prior to treatment. On day 2, cells were treated with the drugs in the presence or absence of α-tocopherol (100 μM) or N-Acetyl-L-cysteine (100 μM). After incubation (48 h) with drugs, MTT assay was performed as above.
4.6.8. Western blotting
The experiment was carried out according to Niso et al. with minor modification [52]. All cells were washed twice with 10 ml phosphate-buffered saline (PBS), scraped in 1 ml PBS and centrifuged for 1 min at 11,000 g. Proteins were extracted from cells by homogenization in cold RIPA buffer (Life Technologies) containing 1× protease inhibitor cocktail and centrifuged at 14,000 g for 15 min at 4°C. The supernatant was recovered and the protein concentration was measured using the microLowry kit. 30 μg of protein extract was separated on 10% polyacrylamide gel (Life Technologies) and then transferred onto a polyvinylidene difluoride membrane (PVDF) by iBlot® Gel Transfer Device (Life Technologies). Membrane was blocked for 30 min at room temperature with blocking buffer (1% BSA, 0.05% Tween 20 in Tris-buffered saline, TBS). The membrane was then incubated overnight at 4°C with anti-P-Glycoprotein (1:500 mouse monoclonal) or for 1h at room temperature anti-β-actin (1:1000 mouse monoclonal) antibodies, diluted in blocking buffer. After incubation time, membrane was washed with washing buffer (0.05% Tween 20 in Tris-buffered saline, TBS) for three times and incubated with a secondary peroxidase antibody (1:2000 anti-mouse for P-glycoprotein and β-actin) for 1h at room temperature. After washing, the membrane was treated with the enhanced chemiluminescence (ECL, Life Technologies) according to the manufacturer’s instructions and the blot was visualized by UVITEC Cambridge (Life Technologies). The expression level was evaluated by densitometric analysis using UVITEC Cambridge software (Life Technologies) and β-actin expression level was used to normalize the sample values.
4.6.9. Detection of Caspase-3 activity in vitro
Caspase-3 activity was measured in KP02 cell lines with a Caspase-Glo® Assay Systems (Promega) according to protocol in which the reagent contain luminogenic caspase substrates that cleaved by activated caspase. Cells were seeded at a density of 1 × 104 in black 96-well, clear bottom plates for 24 hours before treatment with 25 μM of compounds in presence or absence of α-tocopherol (100 μM) or Z-DEVD-FMK (1μM) for 5 hours after treatment. The contents were then mixed using plate shaker for 30 seconds, and incubated at room temperature for 90 minutes. Luminescence signal was measured using multi-mode microplate reader (BioTek). Assay was performed in triplicates, and caspase activity was plotted compared cells treated with DMSO as a control.
4.6.10. Detection of Mitochondrial Superoxide by flow cytometry and ROS interference
MitoSOX™ Red reagent is a novel fluorogenic dye specifically targeted to mitochondria in live cells. Oxidation of MitoSOX™ Red reagent by superoxide produces red fluorescence. Mitochondrial superoxide is generated as a byproduct of oxidative phosphorylation. In an otherwise tightly coupled electron transport chain, approximately 1–3% of mitochondrial oxygen consumed is incompletely reduced; those “leaky” electrons can quickly interact with molecular oxygen to form superoxide anion, the predominant reactive oxygen species (ROS) in mitochondria [53–56]. MitoSOX™ Red reagent is readily oxidized by superoxide but not by other ROS- or reactive nitrogen species (RNS)–generating systems. KP02 cells were seeded into 12-well plates 24 hours before treatment with iron chelators (50 μM) for 2 hours at 37°C in presence or absence of α-tocopherol (100 μM) followed by staining with MitoSOX™ Red (5 μM). Two hours after red dye addition, the cells were washed twice with PBS and harvested with trypsin/EDTA buffer. The cells were washed twice with PBS before analysis with FACSCalibur (BD Bioscience, San Jose, CA). The oxidation product of MitoSOX™ Red by mitochondrial superoxide fluoresces with an emission maximus of 580 nm and was detected in the FL3 channel. Experiment was performed in triplicates.
4.6.11. In vivo assessment of tumor growth
Animal studies were performed according to the animal studies protocol (20130073) approved by the Washington University Institutional Animal Care Facility. In this pre-clinical model, we utilized the KP02 cell line. In vivo studies with mice were performed to compare the effect of σ2 ligands with iron chelator 7. Mice treated with vehicle alone (25% Cremophor in H2O) served as the control cohort. Female C57BL/6 mice (8 weeks old, National Cancer Institute Laboratories) were injected in the right flank with 200 μL of a single-cell suspension of KP02 cells in non-supplemented RPMI medium (2.5 × 105 cells per mouse). Treatment began when the mean tumor diameter was ~ 5-6 mm. Mice received daily intraperitoneal (i.p.) injections of the σ2 ligands 1 and 2 (750nmol), or 7 (750nmol) in 100 μL vehicle or vehicle alone (control) for 2 weeks. Tumors were measured three times weekly in two dimensions with a digital caliper, and tumor volumes were calculated by the standard formula of Tumor Volume = Length × Width2 × 0.5. All mice were euthanized when tumors reached a diameter of 15 mm or had ulcerated. The experiment was repeated using a double concentration of 1 and 2 following the same procedure described above. Several mice from each treatment cohort were assessed for pathologic evaluation (Digestive Diseases Research Core Center at Washington University School of Medicine, St. Louis, MO). Blood was collected for complete blood count (CBC) and biochemical analysis (AST, ALT, BUN, total protein, glucose and Cr). Organs were examined grossly and histologically.
4.6.12. Statistical analysis
Statistical analyses and data plotting were performed using GraphPad Prism software version 6.03 (San Diego, CA). Results were expressed as mean ± standard error of the mean of at least 3 biological replicates. EC50 values were calculated by curve fitting normalized viability versus drug concentration. Differences in viability, caspase-3 activity, and tumor volume were analyzed using two-way ANOVA to identify differences and confirmed with paired two tailed t-tests. Mann-Whitney test was used to compare the difference in CBC and biochemistry analyses. Kaplan-Meier survival analyses were used to assess differences between treatment groups and were compared using a log-rank test. A p-value < 0.05 was considered significant for all analyses.
Supplementary Material
Highlights.
Metal Chelation, σ2 and P-gp proteins interaction in multifunctional thiosemicarbazones.
Deconstruction of potent multifunctional thiosemicarbazones.
Multifunctional ligands reduce pancreatic cancer growth in mouse stroma dense model.
Diverse death pathways from compounds sharing N,N-dimethyl-thiosemicarbazone.
Different sensitization of cells useful for developing tailored treatments.
Acknowledgments
This work was funded in part by a National Institute of Health R01 grant (US NIH 5R01CA16376402) (W.G. Hawkins). This work was conducted under the Agreement regulating cultural and scientific cooperation between the University of Bari ALDO MORO (Italy) and Washington University School of Medicine in St. Louis (USA).
Abbreviations
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
- BUN
blood urea nitrogen
- Cr
Creatinine
- i.p
intraperitoneal
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Information: Synthesis of intermediate compound 12 and final compounds 13 and 14. Western blot for P-gp detection in KP02 cell. ROS involvement, caspase-3 activity and mitochondrial superoxide detection in AsPC1 and Panc02 cell lines. Flow cytometry curves for σ1 binding of references compounds and 1, 2 and 7.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;363:298. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 3.Yeo D, Huynh N, Beutler JA, Christophi C, Shulkes A, Baldwin GS, Nikfarjam M, He H. Glaucarubinone and gemcitabine synergistically reduce pancreatic cancer growth via down-regulation of P21-activated kinases. Cancer letters. 2014;346:264–272. doi: 10.1016/j.canlet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeo TP, Hruban RH, Leach SD, Wilentz RE, Sohn TA, Kern SE, Iacobuzio-Donahue CA, Maitra A, Goggins M, Canto MI, Abrams RA, Laheru D, Jaffee EM, Hidalgo M, Yeo CJ. Pancreatic cancer. Curr Probl Cancer. 2002;26:176–275. doi: 10.1067/mcn.2002.129579. [DOI] [PubMed] [Google Scholar]
- 5.Allison DC, Piantadosi S, Hruban RH, Dooley WC, Fishman EK, Yeo CJ, Lillemoe KD, Pitt HA, Lin P, Cameron JL. DNA content and other factors associated with ten-year survival after resection of pancreatic carcinoma. J Surg Oncol. 1998;67:151–159. doi: 10.1002/(sici)1096-9098(199803)67:3<151::aid-jso2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 6.Castellanos E, Berlin J, Cardin DB. Current treatment options for pancreatic carcinoma. Curr Oncol Rep. 2011;13:195–205. doi: 10.1007/s11912-011-0164-1. [DOI] [PubMed] [Google Scholar]
- 7.Colabufo NA, Berardi F, Abate C, Contino M, Niso M, Perrone R. Is the sigma2 receptor a histone binding protein? J Med Chem. 2006;49:4153–4158. doi: 10.1021/jm0600592. [DOI] [PubMed] [Google Scholar]
- 8.Abate C, Elenewski J, Niso M, Berardi F, Colabufo NA, Azzariti A, Perrone R, Glennon RA. Interaction of the sigma2 receptor ligand PB28 with the human nucleosome: Computational and experimental probes of interaction with the H2A/H2B dimer. ChemMedChem. 2010;5:268–273. doi: 10.1002/cmdc.200900402. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Zeng C, Chu W, Pan F, Rothfuss JM, Zhang F, Tu Z, Zhou D, Zeng D, Vangveravong S, Johnston F, Spitzer D, Chang KC, Hotchkiss RS, Hawkins WG, Wheeler KT, Mach RH. Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nat Commun. 2011;2:380. doi: 10.1038/ncomms1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abate C, Niso M, Infantino V, Menga A, Berardi F. Elements in support of the ‘non-identity’ of the PGRMC1 protein with the σ2 receptor. Eur J Pharmacol. 2015;758:16–23. doi: 10.1016/j.ejphar.2015.03.067. [DOI] [PubMed] [Google Scholar]
- 11.Chu UB, Mavlyutov TA, Chu ML, Yang H, Schulman A, Mesangeau C, McCurdy CR, Guo LW, Ruoho AE. The sigma-2 receptor and Progesterone Receptor Membrane Component 1 are different binding sites derived from independent genes. EBioMedicine. 2015;2:1806–1813. doi: 10.1016/j.ebiom.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pati ML, Groza D, Riganti C, Kopecka J, Niso M, Berardi F, Hager S, Heffeter P, Hirai M, Tsugawa H, Kabe Y, Suematsu M, Abate C. Sigma-2 receptor and progesterone receptor membrane component 1 (PGRMC1) are two different proteins: Proofs by fluorescent labeling and binding of sigma-2 receptor ligands to PGRMC1. Pharmacol Res. 2017;117:67–74. doi: 10.1016/j.phrs.2016.12.023. [DOI] [PubMed] [Google Scholar]
- 13.Alon A, Schmidt HR, Wood MD, Sahn JJ, Martin SF, Kruse AC. Identification of the gene that codes for the σ2 receptor. Proc Natl Acad Sci U S A. 2017;114:7160–7165. doi: 10.1073/pnas.1705154114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crawford KW, Bowen WD. Sigma-2 receptor agonists activate a novel apoptotic pathway and potentiate antineoplastic drugs in breast tumor cell lines. Cancer Res. 2002;62:313–322. [PubMed] [Google Scholar]
- 15.Ostenfeld MS, Fehrenbacher N, Hoyer-Hansen M, Thomsen C, Farkas T, Jaattela M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005;65:975–983. doi: 10.1158/0008-5472.CAN-05-0269. [DOI] [PubMed] [Google Scholar]
- 16.Zeng C, Rothfuss J, Zhang J, Chu W, Vangveravong S, Tu Z, Pan F, Chang KC, Hotchkiss R, Mach RH. Sigma-2 ligands induce tumour cell death by multiple signalling pathways. Br J Cancer. 2012;106:693–701. doi: 10.1038/bjc.2011.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abate C, Perrone R, Berardi F. Classes of Sigma2 (σ2) receptor ligands: structure affinity relationship (SAfiR) studies and antiproliferative activity. Curr Pharm Des. 2012;18:938–949. doi: 10.2174/138161212799436485. [DOI] [PubMed] [Google Scholar]
- 18.Niso M, Abate C, Contino M, Ferorelli S, Azzariti A, Perrone R, Colabufo NA, Berardi F. Sigma-2 receptor agonists as possible antitumor agents in resistant tumors: hints for collateral sensitivity. ChemMedChem. 2013;8:2026–2035. doi: 10.1002/cmdc.201300291. [DOI] [PubMed] [Google Scholar]
- 19.Mir SU, Schwarze SR, Jin L, Zhang J, Friend W, Miriyala D, St Clair D, Craven RJ. Progesterone receptor membrane component 1/Sigma-2 receptor associates with MAP1LC3B and promotes autophagy. Autophagy. 2013;9:1566–1578. doi: 10.4161/auto.25889. [DOI] [PubMed] [Google Scholar]
- 20.Pati ML, Niso M, Ferorelli S, Abate C, Berardi F. Novel Metal Chelators Thiosemicarbazones with activity at the σ2 Receptors and P-glycoprotein: an Innovative Strategy for Resistant Tumors Treatment. RSC Adv. 2015;5:103131–103146. [Google Scholar]
- 21.Pati ML, Hornick JR, Niso M, Berardi F, Spitzer D, Abate C, Hawkins W. Sigma-2 receptor agonist derivatives of 1-Cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) induce cell death via mitochondrial superoxide production and caspase activation in pancreatic cancer. BMC Cancer. 2017;17:51. doi: 10.1186/s12885-016-3040-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kashiwagi H, McDunn JE, Jr, Simon PO, Goedegebuure PS, Xu J, Jones L, Chang K, Johnston F, Trinkaus K, Hotchkiss RS, Mach RH, Hawkins WG. Selective sigma-2 ligands preferentially bind to pancreatic adenocarcinomas: applications in diagnostic imaging and therapy. Mol Cancer. 2007;6:48. doi: 10.1186/1476-4598-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kashiwagi H, McDunn JE, Jr, Simon PO, Goedegebuure PS, Vangveravong S, Chang K, Hotchkiss RS, Mach RH, Hawkins WG. Sigma-2 receptor ligands potentiate conventional chemotherapies and improve survival in models of pancreatic adenocarcinoma. J Transl Med. 2009;7:24. doi: 10.1186/1479-5876-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hornick JR, Xu J, Vangveravong S, Tu Z, Mitchem JB, Spitzer D, Goedegebuure P, Mach RH, Hawkins WG. The novel sigma-2 receptor ligand SW43 stabilizes pancreas cancer progression in combination with gemcitabine. Mol Cancer. 2010;9:298. doi: 10.1186/1476-4598-9-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hornick JR, Vangveravong S, Spitzer D, Abate C, Berardi F, Goedegebuure P, Mach RH, Hawkins WG. Lysosomal membrane permeabilization is an early event in Sigma-2 receptor ligand mediated cell death in pancreatic cancer. J Exp Clin Cancer Res. 2012;31:41. doi: 10.1186/1756-9966-31-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pluchino KM, Hall MD, Goldsborough AS, Callaghan R, Gottesman MM. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist Updat. 2012;15:98–105. doi: 10.1016/j.drup.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao J, Yang J, Ramachandran V, Arumugam T, Deng D, Li Z, Xu L, Logsdon CD. TM4SF1 Promotes Gemcitabine Resistance of Pancreatic Cancer In Vitro and In Vivo. PLoS One. 2015;10:e0144969. doi: 10.1371/journal.pone.0144969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abate C, Selivanova SV, Müller A, Krämer SD, Schibli R, Marottoli R, Perrone R, Berardi F, Niso M, Ametamey SM. Development of 3,4-dihydroisoquinolin-1(2H)-one derivatives for the Positron Emission Tomography (PET) imaging of σ2 receptors. Eur J Med Chem. 2013;69:920–930. doi: 10.1016/j.ejmech.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 29.Mésangeau C, Amata E, Alsharif W, Seminerio MJ, Robson MJ, Matsumoto RR, Poupaert JH, McCurdy CR. Synthesis and pharmacological evaluation of indole-based sigma receptor ligands. Eur J Med Chem. 2011;46:5154–5161. doi: 10.1016/j.ejmech.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abate C, Mosier PD, Berardi F, Glennon RA. A structure-affinity and comparative molecular field analysis of sigma-2 (sigma2) receptor ligands. Cent Nerv Syst Agents Med Chem. 2009;9:246–57. doi: 10.2174/1871524910909030246. [DOI] [PubMed] [Google Scholar]
- 31.Cratteri P, Romanelli MN, Cruciani G, Bonaccini C, Melani F. GRIND-derived pharmacophore model for a series of alpha-tropanyl derivative ligands of the sigma-2 receptor. J Comput Aided Mol Des. 2004;18:361–374. doi: 10.1023/b:jcam.0000047815.22931.3b. [DOI] [PubMed] [Google Scholar]
- 32.Glennon RA. Pharmacophore identification for sigma-1 (sigma1) receptor binding: application of the “deconstruction-reconstruction-elaboration” approach. Mini Rev Med Chem. 2005;5:927–940. doi: 10.2174/138955705774329519. [DOI] [PubMed] [Google Scholar]
- 33.Abate C, Pati ML, Contino M, Colabufo NA, Perrone R, Niso M, Berardi F. From mixed sigma-2 receptor/P-glycoprotein targeting agents to selective P-glycoprotein modulators: small structural changes address the mechanism of interaction at the efflux pump. Eur J Med Chem. 2015;89:606–615. doi: 10.1016/j.ejmech.2014.10.082. [DOI] [PubMed] [Google Scholar]
- 34.Yu Y, Rahmanto YS, Richardson DR. Bp44mT: an orally active iron chelator of the thiosemicarbazone class with potent anti-tumour efficacy. Br J Pharm. 2012;165:148–166. doi: 10.1111/j.1476-5381.2011.01526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowol CR, Trondl R, Heffeter P, Arion VB, Jakupec MA, Roller A, Galanski M, Berger W, Keppler BK. Impact of metal coordination on cytotoxicity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (triapine) and novel insights into terminal dimethylation. J Med Chem. 2009;52:5032–5043. doi: 10.1021/jm900528d. [DOI] [PubMed] [Google Scholar]
- 36.Yuan J, Lovejoy DB, Richardson DR. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood. 2004;104:1450–1458. doi: 10.1182/blood-2004-03-0868. [DOI] [PubMed] [Google Scholar]
- 37.Ohman KA, Hashim YM, Vangveravong S, Nywening TM, Cullinan DR, Goedegebuure SP, Liu J, Van Tine BA, Tiriac H, Tuveson DA, DeNardo DG, Spitzer D, Mach RH, Hawkins WG. Conjugation to the sigma-2 ligand SV119 overcomes uptake blockade and converts dm-Erastin into a potent pancreatic cancer therapeutic. Oncotarget. 2016;7:33529–33541. doi: 10.18632/oncotarget.9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vilner BJ, John CS, Bowen WD. Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res. 1995;55:408–413. [PubMed] [Google Scholar]
- 39.Hayashi T, Su TP. Sigma-1 receptor ligands: potential in the treatment of neuropsychiatric disorders. CNS Drugs. 2004;18:269–284. doi: 10.2165/00023210-200418050-00001. [DOI] [PubMed] [Google Scholar]
- 40.Bowen WD. Sigma receptors: Recent advances and new clinical potentials. Pharm Acta Helv. 2000;74:211–218. doi: 10.1016/s0031-6865(99)00034-5. [DOI] [PubMed] [Google Scholar]
- 41.Tinder TL, Subramani DB, Basu GD, Bradley JM, Schettini J, Million A, Skaar T, Mukherjee P. MUC1 enhances tumor progression and contributes toward immunosuppression in a mouse model of spontaneous pancreatic adenocarcinoma. J Immunol. 2008;181:3116–3125. doi: 10.4049/jimmunol.181.5.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abate C, Riganti C, Pati ML, Ghigo D, Berardi F, Mavlyutov T, Guo LW, Ruoho A. Development of sigma-1 (σ1) receptor fluorescent ligands as versatile tools to study σ1 receptors. Eur J Med Chem. 2016;108:577–585. doi: 10.1016/j.ejmech.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abate C, Ferorelli S, Niso M, Lovicario C, Infantino V, Convertini P, Perrone R, Berardi F. 2-Aminopyridine derivatives as potential σ(2) receptor antagonists. ChemMedChem. 2012;7:1847–1857. doi: 10.1002/cmdc.201200246. [DOI] [PubMed] [Google Scholar]
- 44.Berardi F, Abate C, Ferorelli S, Uricchio V, Colabufo NA, Niso M, Perrone R. Exploring the importance of piperazine N-atoms for sigma(2) receptor affinity and activity in a series of analogs of 1-cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) J Med Chem. 2009;52:7817–7828. doi: 10.1021/jm9007505. [DOI] [PubMed] [Google Scholar]
- 45.Prism Software, version 3.0 for Windows. GraphPad Software, Inc: San Diego, CA; 1998. [Google Scholar]
- 46.Berardi F, Ferorelli S, Abate C, Pedone MP, Colabufo NA, Contino M, Perrone R. Methyl substitution on the piperidine ring of N-[omega-(6-methoxynaphthalen-1-yl)alkyl] derivatives as a probe for selective binding and activity at the sigma(1) receptor. J Med Chem. 2005;48:8237–8244. doi: 10.1021/jm050654o. [DOI] [PubMed] [Google Scholar]
- 47.Skuza G, Szymańska M, Budziszewska B, Abate C, Berardi F. Effects of PB190 and PB212, new σ receptor ligands, on glucocorticoid receptor-mediated gene transcription in LMCAT cells. Pharmacol Rep. 2011;63:1564–1568. doi: 10.1016/s1734-1140(11)70722-8. [DOI] [PubMed] [Google Scholar]
- 48.Gasparre G, Abate C, Berardi F, Cassano G. The sigma-1 receptor antagonist PB212 reduces the Ca2+ -release through the inositol (1, 4, 5)-trisphosphate receptor in SK-N-SH cells. Eur J Pharmacol. 2012;684:59–63. doi: 10.1016/j.ejphar.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 49.Skuza G, Sadaj W, Kabziński M, Cassano G, Gasparre G, Abate C, Berardi F. The effects of new sigma (σ) receptor ligands, PB190 and PB212, in the models predictive of antidepressant activity. Pharmacol Rep. 2014;66:320–324. doi: 10.1016/j.pharep.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 50.Niso M, Pati ML, Berardi F, Abate C. Rigid versus flexible anilines or anilides confirm the bicyclic ring as the hydrophobic portion for optimal σ2 receptor binding and provide novel tools for the development of future σ2 receptor PET radiotracer. RSC Adv. 2016;6:88508–88518. [Google Scholar]
- 51.Colabufo NA, Berardi F, Contino M, Niso M, Abate C, Perrone R, Tortorella V. Antiproliferative and cytotoxic effects of some sigma2 agonists and sigma1 antagonists in tumour cell lines. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:106–113. doi: 10.1007/s00210-004-0961-2. [DOI] [PubMed] [Google Scholar]
- 52.Niso M, Riganti C, Pati ML, Ghigo D, Berardi F, Abate C. Novel and selective fluorescent σ2 -receptor ligand with a 3,4-dihydroisoquinolin-1-one scaffold: a tool to study σ2 receptors in living cells. Chembiochem. 2015;16:1078–1083. doi: 10.1002/cbic.201402712. [DOI] [PubMed] [Google Scholar]
- 53.Batandier C, Fontaine E, Kériel C, Leverve XM. Determination of mitochondrial reactive oxygen species: methodological aspects. J Cell Mol Med. 2002;6:175–187. doi: 10.1111/j.1582-4934.2002.tb00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem. 2004;279:4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- 55.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 56.Gauuan PJ, Trova MP, Gregor-Boros L, Bocckino SB, Crapo JD, Day BJ. Superoxide dismutase mimetics: synthesis and structure-activity relationship study of MnTBAP analogues. Bioorg Med Chem. 2002;10:3013–3021. doi: 10.1016/s0968-0896(02)00153-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.