

Abstract
Genetically modified mice are widely used as experimental models to study human heart function and diseases. However, the fast rate of normal mouse heart at 400–600 bpm limits its capacity of assessing kinetic parameters that are important for the physiology and pathophysiology of human heart that beats at a much slower rate (75–180 bpm). To extend the value of mouse models, we established a protocol to study ex vivo mouse working hearts at a human-like heart rate. In the presence of 300 μM Lidocaine to lower pacemaker and conductive activities and prevent arrhythmia, a stable rate of 120–130 bpm at 37°C is achieved for ex vivo mouse working hearts. The negative effects of decreased heart rate on force-frequency dependence and lidocaine as a myocardial depressant on intracellular calcium can be compensated by using a higher but still physiological level of calcium (2.75 mM) in the perfusion media. Multiple parameters were studied to compare the function at the human-like heart rate with that of ex vivo mouse working hearts at the standard rate of 480 bpm. The results showed that the conditions for slower heart rate in the presence of lidocaine did not have depressing effect on left ventricular pressure development, systolic and diastolic velocities and stroke volume with maintained positive inotropic and lusitropic responses to β-adrenergic stimulation. Compared with that at 480 bpm, the human-like heart rate increased ventricular filling and end diastolic volume with enhanced Frank-Starling responses. Coronary perfusion was increased from longer relaxation time and interval between beats whereas cardiac efficiency was significantly improved. Although the intrinsic differences between mouse and human heart remain, this methodology for ex vivo mouse hearts to work at human-like heart rate extends the value of using genetically modified mouse models to study cardiac function and human heart diseases.
Keywords: ex vivo mouse working heart, cardiac function, heart rate, left ventricular contractile parameters, lidocaine
Graphical Abstract
This paper report a protocol to extend the study of ex vivo mouse working heart function by measurements at a human-like heart rate.
1. Introduction
Genetically modified mice are widely-used as animal models to study human heart function and diseases. Despite having plausible similarities in cardiac anatomy, development and physiology [1, 2], the mouse and human hearts, however, have differences that need to be considered when interpreting studies of mouse hearts to understand human cardiac function. The adult mouse heart is small (~100–200 mg in weight) and fast beating (400–800 beats per min, bpm), whereas the adult human heart weighs 250–300 g, works at 60–70 bpm at rest and up to 180–200 bpm during exercise [2]. Implicating a difference in calcium homeostasis, the hearts of large, but not small, mammals display a positive force-frequency effect [3] while cardiomyocytes of small mammals with high heart rates have greater sarcoplasmic reticulum (SR) Ca2+-ATPase activity [4] and plasma membrane Na/Ca exchanger density [5]. The contractile proteins, especially myosin isoenzymes, are also different in mouse and human hearts [6–8]. Although some of these differences are intrinsic and unavoidable, mouse hearts remain a highly valuable experimental system in biomedical research and continuing technical improvement can maximize their application and impacts.
Isolated ex vivo working heart [9], is a powerful technique with many unique advantages over in vivo echocardiography [10] or Langendorff retrograde perfused heart [11] in the study of cardiac function. The working heart preparation is in a physiologically ejecting mode with in vivo-like dynamic changes in the left ventricular pressure and volume. The development of ventricular pressure and volume can be directly measured in real time using aortic pressure transducer and pressure-volume (P-V) catheter inserted into the left ventricular chamber. Temperature, preload, afterload, and heart rate are precisely controlled and readily adjustable. Left ventricular stroke volume and cardiac output can be directly and accurately measured. Systemic neurohumoral interferences are excluded. Drugs and other treatments such as pH, electrolytes and oxygen in the perfusant can be readily added or removed. Cardiac efficiency can be calculated from cardiac output, left ventricular (LV) systolic and kinetic integrals, and oxygen consumption measured from coronary effluent. Electrocardiograph (ECG) can also be recorded in ex vivo working hearts [12–14].
Ex vivo mouse working hearts are normally studied at heart rate of 400–600 bpm, mimicking their physiological rate in vivo. However, this fast heart rate imposes a major limitation to its use in the study of human cardiac function and diseases. For example, LV development pressure (LVDevP), ±dP/dt and cardiac power (the product of LVDevP and cardiac output) decline markedly when heart rate increases, concurring with an increase in LV end diastolic pressure (LVEDP) [13]. The high beating rate of mouse heart also produces shorter systolic and diastolic time durations with kinetic parameters significantly different from that of human hearts. Fast heart rate reduces ventricular end-diastolic volume, restricting the evaluation of Frank-Starling response in mouse working hearts. Taking the advantage that the heart rate of ex vivo working heart can be technically controlled, our present study developed a protocol to study mouse working hearts at a human-like heart rate in order to overcome some of the above limitations and extend its application in kinetic studies and the investigations of human heart diseases and failure.
2. Materials and Methods
2.1. Animals
Wild type male and female C57BL/6 mice of 4–5 months old were used in this study. All animal procedures were approved by the Institutional Animal Care and Use Committee of Wayne State University and were conducted in accordance with the Guiding Principles in the Care and Use of Animals, under the guidelines of the Council of the American Physiological Society.
2.2. Setup of ex vivo mouse working heart preparations
Preparation of isolated mouse working hearts was done using the protocol described in our previous studies [14]. Briefly, mouse was heparinized (100 U, i.p.) 30 min before anesthesia with pentobarbital (100 mg/kg, i.p.). After opening the chest, the heart was excised rapidly together with lung, trachea and thoracic aorta. The heart was submerged in Kreb’s buffer at room temperature and carefully dissected with the large vessels attached for aortic cannulation. Within 2 min after the opening of the chest, Langendorff retrograde perfusion of the heart using Kreb’s solution at 37°C was established from the aortic cannula connected to an 80 mmHg perfusion system (Radnoti, US).
The Kreb’s perfusion medium was modified from Krebs-Henseleit bicarbonate buffer, containing 118 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 2.25 mM MgSO4, 2.25 mM or 2.75 mM CaCl2, 0.32 mM EGTA, 2 mM pyruvate, and 15 mM D-glucose, equilibrated with 95% O2–5% CO2. NaHCO3 was added to adjust the pH to 7.4 at 37°C. The perfusion medium was filtered through a 0.45-μm membrane and was not circulated for reuse.
The pulmonary vein and pulmonary artery were then cannulated for switching the heart to working mode. A 1.2F P-V catheter with 3.5 mm electrode spacing (Transonic Scisense Inc., Canada) was inserted into the left ventricular chamber through a track made at the apex using a 30g needle to record the intraventricular pressure and volume. A pair of lab-made pacing electrodes was placed on the surface of right atrium to provide electrical stimulation from an isolated stimulator (A365, World Precision Instrument, USA). An electrode made from a needle of 7-0 surgical suture was placed pericardial at the apex and connected to an ECG recording system (AD Instrument) together with a reference electrode placed inside the pulmonary vein-left atria cannula. A pressure transducer was place in the path of aortic cannula to record aortic pressure. A pair of copper wires with one wire attached with an iron clip was placed under the outlet of aortic flow to record the aortic output in calibrated drops in real time. Another pair was placed under the outlet of pulmonary flow to record coronary flow. The preload was set as the hydraulic height between the surface of the perfusant in the reservoir connected to the pulmonary vein and the left atrium. The afterload was set as the hydraulic height between the outlet of the aortic outflow track and the left ventricle. The signals of aortic pressure, LV pressure and volume, ECG, aortic output and coronary flow were digitized via an AD interface (16 Channels Powerlab, AD Instrument) and continuously collected using Chart5 computer software (AD Instrument) for later analysis.
After all cannulation and electrodes were setup, the heart was switched to ejection mode by turning on the left ventricular preload flow. Preload was set at 10 mmHg and afterload at 55 mmHg as the baseline condition. After functional stabilization was achieved, LV end systolic pressure-volume relationship (ESPVR) was measured by pressing the aortic outflow tubing to temporarily increase resistance and recording functional changes during the returning of afterload to normal after the tubing pressing was released. Left ventricular end diastolic pressure-volume relationship (EDPVR) was measured by lowering preload from 10 to 5mmHg. Frank-starling relationship was tested by altering preload between 5–20 mmHg.
2.3. Modified steps for the study of mouse working heart at human-like heart rate
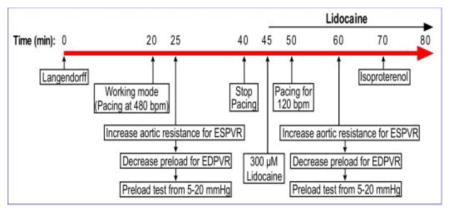
Illustrated in Fig. 1A, we modified the mouse working heart protocol to explore conditions for the study of cardiac function at slower and human-like heart rate. Functional measurements were first performed at the standard heart rate of 480 bpm [14]. Cardiac function was then measured at 420 bpm and 360 bpm pacing, and at the intrinsic sinus rate of ~230 bpm (37°C) without external pacing. A series of accumulated doses of lidocaine (VETONE) at 100, 200, and 300 μM were added to the preload reservoir to prevent arrhythmia and to decrease the intrinsic heart rate by inhibiting atrial rate with minor decreasing of A-V conduction [15]. Each dosage was maintained for 5 min to obtain stable functional measurements. Pacing was tested at various frequencies to identify a condition that produced a stable human-like rate of ventricular beating.
Figure 1. Testing the function of ex vivo mouse working heart at human-like lower heart rate.
A. Protocol steps to test ex vivo mouse working heart function at slower human-like heart rate. B. Representative LV pressure (LVP) and volume (LVV) recordings and P-V loops at 10 mmHg preload and 55 mmHg afterload demonstrate the relationship between the slower heart rate and higher LVP max, lower LVP min, larger end diastolic volume and higher stroke volume.
Considering that high doses of lidocaine has a cardiac depressant property by affecting intracellular calcium [16] and lower heart rate diminishes the effect of positive force-frequency relationship [17], we tested a compensation by increasing the Ca2+ concentration in the perfusion media. CaCl2 was increased from 2.25 mM to 2.75 mM, a concentration near the higher end of the physiological level in plasma. At the end of the protocol, 2 nM isoproterenol (ISO) was applied at 300 μM lidocaine to test β-adrenergic response.
2.4. Data analysis
All quantitative data are presented as mean ± SEM. Student’s t test was performed for comparisons between means and ANOVA was used for comparison between curves. P<0.05 was used to establish the level of significance.
3. Results
3.1. Increasing Ca2+ in perfusion media compensates for ex vivo mouse working heart function at decreased heart rates
Perfused with the Kreb’s solution normally used in working heart studies containing 2.25 mM CaCl2 (Fig. 1), LV stroke volume of ex vivo mouse working heart increased when heart rate was decreased from 480 to 360 bpm but then decreased at heart rate of ~260 bpm with decreased systolic and diastolic velocities (Table 1). Countering the decrease in intracellular Ca2+ at slower heart rate based on the positive force-frequency relationship of mammalian cardiomyocytes [17], the increase of Ca2+ concentration in Kreb’s solution to an upper physiological level of 2.75 mM effectively compensated for the reduction of contractility at slower heart rates (Fig. 2).
Table 1.
Effects of decreasing heart rate on the performance of ex vivo mouse working heart
| Heart Rate (bpm) | LVP max (mmHg) | LVP min (mmHg) | +dP/dt max (mmHg) | −dP/dt max (mmHg) | Stroke Volume (μL/mg) |
|---|---|---|---|---|---|
| 480 | 73.38±0.71 | 5.33±0.39 | 3527.64±109.04 | −3011.12±75.40 | 0.173±0.006 |
| 420 | 75.86±1.17 | 4.38±0.33* | 3669.39±99.90* | −3159.48±68.37* | 0.202±0.005* |
| 360 | 79.65±1.55# | 4.44±0.30* | 3745.55±103.68* | −3288.97±73.71 | 0.218±0.006* |
| 263.8±11.1 (No pacing) | 79.21±2.71* | 5.23±0.49#& | 2925.12±115.46*#& | −2446.35±97.65*#& | 0.188±0.009& |
Cardiac function increased when the paced heart rate was decreased from 480 to 420 and 360 bpm as shown by increased LVP max, +/−dP/dt and stroke volume, and decreased LVP min. At heart rate of ~260 bpm in the absence of external pacing, the contractile functions decreased, reflecting a negative impact of this further lowered heart rate on the force-frequency relationship of cardiac muscle. The test was done at 10 mmHg preload and 55 mmHg afterload.
Values are presented as mean ± SE. N = 7 hearts.
P<0.05 vs. 480;
P<0.05 vs. 420;
P<0.05 vs. 360 bpm in paired Student’s t test.
Fig. 2. Physiologically higher extracellular Ca2+ compensates for cardiac function at decreased heart rate of ex vivo mouse working heart.

Increasing of Ca2+ in perfusion medium from 2.25 mM to 2.75 mM significantly increased both systolic and diastolic functions as shown by the higher LVP max, lower LVPmin (A), higher ±dP/dt (B) and stroke volume (C) of ex vivo mouse working hearts at heart rate of ~260 bpm, compensating the decrease from lowering heart rate from 480 bpm at 2.25 mM Ca2+. The test was done at 10 mmHg preload and 55 mmHg afterload. N = 6 hearts. Values are presented as mean ± SE. *P<0.05 vs. 2.25 mM Ca2+ control in Student’s t test.
3.2. 300 μM Lidocaine showed no significant negative impact on the function of ex vivo mouse working heart when perfused with 2.75 mM Ca2+
Lidocaine is known to have a myocardial depressant property. Therefore, we examined its dose effect on the pumping function of ex vivo mouse working hearts. When administered at accumulated dosage from 100 to 300 μM, the non-paced intrinsic heart rate gradually decreased to 120–130 bpm (Table 2). Plausibly, cardiac function had no significant change at up to 300 μM lidocaine with LVP, systolic and diastolic velocities and stroke volume similar to that of control (Table 2).
Table 2.
Lidocaine reduced heart rate with no significant negative impact on cardiac function
| Control | 100 μM Lidocaine | 200 μM Lidocaine | 300 μM Lidocaine | |
|---|---|---|---|---|
| Heart Rate (bpm) | 251.99±25.06 | 215.99±31.50 | 198.83±23.84* | 133.70±8.86*#& |
| LVP max (mmHg) | 86.34±2.98 | 86.12±2.28 | 88.25±2.52 | 85.17±2.97 |
| LVP min (mmHg) | 3.65±0.26 | 3.23±0.20 | 2.97±0.15 | 2.74±0.22 |
| +dP/dt max (mmHg/s) | 4021.66±182.27 | 3999.58±172.06 | 4112.55±149.27 | 3705.04±176.84 |
| −dP/dt max (mmHg/s) | −3003.18±133.86 | −2969.2±167.56 | −2951.42±99.70 | −2592.51±91.036 |
| Stroke Volume (mL/mg) | 0.253±0.011 | 0.261±0.0139 | 0.269±0.015 | 0.271±0.015 |
The heart rate responses to 100, 200 and 300 μM lidocaine at 10 mmHg preload and 55 mmHg afterload showed a dose-dependent gradual decrease of heart rate to ~120 bpm with no significant change in LVPmax, LVPmin, systolic (+dP/dt) and diastolic (−dP/dt) velocities, and stroke volume. N = 6 hearts.
Values are presented as mean ± SE.
P<0.05 vs. Control;
P<0.05 vs. 100 μM;
P<0.05 vs. 200 μM in Student’s t test.
3.3. Atrial pacing in the presence of 300 μM lidocaine produces a stable ventricular pumping rate of 120 bmp
By inhibiting plasma membrane sodium channels using lidocaine to lower the activities of pace maker and conductive cells, ex vivo mouse working heart preparation can function at decreased heart rate at 37°C to mimic a physiological heart rate of human heart (Fig. 1). The effect of lidocaine also prevented ventricular arrhythmia that occurred very frequently when supraventricular pacemaker is absent, avoiding a major problem that would preclude reliable functional measurements (data not shown).
While the intrinsic heart rate in 300 μM lidocaine at 37°C falls in a human-like range of 120–130 bpm, it naturally has beat-to-beat variations. Since constant and stable heart rate is critical to accurate and reproducible measurement of contractile and kinetic parameters of cardiac function, we established a pacing condition for ex vivo mouse working heart to pump at constantly 120 bpm. Shown in Fig. 3A, atrial pacing at 240 per min in the presence of 300 μM lidocaine highly reproducibly produced LV beats of 120 bpm. Aligning the LVP trace with ECG, the data showed that a 2:1 A-V blockage was the mechanism that helped to achieve a supraventricular-originated stable human-like heart rate. Fig. 3B further shows that LVP waves of mouse ex vivo working hearts paced at 480 bpm at 2.25 mM Ca2+ without lidocaine and 120 bpm 2.75 mM Ca2+ with 300 μM lidocaine are similar, indicating normal ventricular pumping despite the constant 2:1 A-V blockage.
Figure 3. LVP trace of mouse working hearts at a human-like heart rate of 120 bpm.

A. The representative LVP and ECG traces of mouse ex vivo working hearts under 5 mmHg preload and 55 mmHg afterload under 240 per min atrium pacing at and 2.75 mM Ca2+ in 300 μM lidocaine produced 120 bpm ventricular pumping via a 2:1 A-V blockage. B. Comparison between LVP traces of mouse working hearts at 480 bpm at 2.25 mM Ca2+ without lidocaine and 120 bpm at 2.75 mM Ca2+ with 300 μM lidocaine, the LVP waves are similar while the lower heart rate produced an increased peak LVP.
3.4. 300 μM lidocaine reduces heart rate without negative effect on cardiac contractile function
To comprehensively evaluate the impact of 300 μM lidocaine on the function of ex vivo mouse working hearts, the data in Fig. 4 showed that perfused with 2.75 mM Ca2+, the lower heart rate of ~230 bpm or 120 bpm did not change LVP development as compared with at 480 bpm (Fig. 4A) and both decreased LV systolic and diastolic velocities (Fig. 4B), increased stroke volume (Fig. 4C), increased the time to develop peak pressure (TPT) and 50% of peak pressure (TP50) and the time for 50% (TR50) and 75% (TR75) relaxation (Fig. 4D) and elongated the total ejection time (ET) including both rapid and reduced ET (Fig. 4E). These changes were independent of the absence or presence of lidocaine, and therefore, were considered as primary effects of the decreased heart rates. The only difference unique to the presence of 300 μM lidocaine was the a longer TP50. These results demonstrate that the employment of 300 μM lidocaine to reduce ventricular beating rate of ex vivo mouse working hearts to the human-like 120 bpm does cause any prohibitive effect on cardiac contractile function, validating the value of this approach in extending functional characterizations of mouse models of human heart diseases.
Figure 4. Lidocaine reduced heart rate with preserved cardiac contractile function.

Mouse ex vivo working heart was pace at right atrium and perfused with Kreb’s solution containing 2.75 mM Ca2+. Comparisons of LV functions at 480 bpm or ~230 bpm in the absence of lidocaine, and at 120 bpm in the presence of 300 μM lidocaine showed that LVPmax and LVPmin did not change between the three conditions (A). Independent of lidocaine, the slower heart rates of ~260 and 120 bpm both decreased LV systolic and diastolic velocities (B), increased stroke volume (C), increased the time to develop peak pressure (TPT) and 50% of peak pressure (TP50) and the time for 50% (TR50) and 75% (TR75) relaxation (D) and elongated the total ejection time (ET) including both rapid and reduced ET (E). The only difference unique to 120 bpm in the presence of 300 μM lidocaine was the a longer TP50 (D). N=5 of each condition. Values are presented as mean ± SE. *P<0.05 vs. 480 bpm; #P<0.05 vs. ~230 bpm. Statistical analysis was performed using Student’s t test.
3.5. Slower heart rate improves pumping functions of ex vivo mouse working heart
The comparison of LVP traces of ex vivo mouse working hearts at 480 and 120 bpm in Fig. 3B indicated that the slower human-like heart rate produced three notable differences that are further quantified: a) higher peak pressure development (Table 3), b) slightly increased duration of each contraction-relaxation cycle (Table 4), and c) longer intervals between beats (Fig. 3), which increased diastolic filling as shown by increased LV end diastolic volume (Fig. 1B and Table 3).
Table 3.
Improved contractile functions of mouse working heart at human-like heart rate
| Heart Rate (bpm) | LVP max (mmHg) | LVP min (mmHg) | Stroke Volume (mL/mg) | +dP/dt max (mmHg/s) | −dP/dt max (mmHg/s) | LVEDV (μL) | LVESV (μL) |
|---|---|---|---|---|---|---|---|
| 480 | 66.76±0.86 | 2.91±0.31 | 0.102±0.005 | 2869.07±139.04 | −2624.95±94.29 | 30.26±3.14 | 19.87±2.54 |
| 128.6±14.8* | 78.88±2.49* | 1.93±0.35 | 0.220±0.013* | 3448.76±177.78* | 2510.14±118.44 | 41.23±4.57 (P=0.091) | 23.93±2.75 |
At 5 mmHg preload and 55 mmHg afterload, mouse working hearts at human-like heart rate of 120–130 bpm in the presence of 300 μM lidocaine showed increased systolic function and stroke volume with increased diastolic filling in comparison to that at 480 bpm without lidocaine. N = 4 to 5 hearts.
Values are presented as Mean ± SE.
P<0.05 vs. 480 bpm control in Student’s t test.
Table 4.
Left ventricular systolic and diastolic time parameters (ms)
| Heart Rate (bpm) | T P50 | T PT | T R50 | T R75 | Ejection Time (ET) | Rapid ET | Reduced ET |
|---|---|---|---|---|---|---|---|
| 480 | 21.87±0.63 | 41±0.41 | 42.62±0.82 | 50.75±1.05 | 47.75±1.43 | 10±1.22 | 37.75±0.25 |
| 128.6±14.8 | 26±0.96* | 51.4±2.48* | 50.3±3.10 (P=0.07) | 60±3.36 (P=0.051) | 69.4±2.09* | 20.2±3.65* | 49.2±3.06* |
The time parameters of ex vivo mouse working hearts under 5 mmHg preload and 55 mmHg afterload showed elongated left ventricular systolic time (total and 50% time to peak pressure, TPT and TP50) and diastolic time (time for 50% and 75% relaxation, TR50 and TR75) at human-like heart rate of 120–130 bpm in the presence of 300 μM lidocaine as compared with that at 480 bpm without lidocaine. N = 4 to 5 hearts.
Values are presented as Mean ± SE.
P<0.05 vs. 480 bpm control in Student’s t test.
The human-like heart rate also produced larger stroke volume, faster systolic velocity, and a trend of larger LV end diastolic volume in mouse working hearts at 5 mmHg preload (Table 3). The systolic time and diastolic time of mouse working heart at heart rate of 120–130 bpm were elongated to increase ventricular ejection time (Table 4). Although there was no change in diastolic velocity (Table 3), the longer interval between beats (Fig. 3) allows increases in diastolic ventricular filling (Fig. 1B and Table 3).
Fig. 5A shows that the mouse working heart at human-like heart rate in the presence of 300 μM lidocaine did not change LV contractility with an ESPVR similar to that at 480 bpm without lidocaine. However, Fig. 5B shows that EDPVR at the slower heart rate was steeper than 480 bpm control due to increased end diastolic pressure at a larger end diastolic volume. This feature may reflect a higher myocardial stiffness and lower ventricular diastolic compliance. One molecular basis for this difference may be that cardiac muscle of large animals, such as human, with slower beating hearts has higher compliance and expresses more compliant N2BA titin with a relative N2BA:N2B ratio of ~0.4–1.2, whereas small animals, such as mouse, has lower compliance, expresses predominantly N2B titin with a N2BA:N2B ratio of ~0.05–0.25 [8, 18, 19]. It is worth noting that at the testing preload of 5–10 mmHg, mouse hearts working at 120–130 bpm had significantly larger LV end diastolic volume than the 480 bpm control. Our results suggest that the slower human-like heart rate facilitates ventricle filling, producing larger end diastolic volume and longer resting sarcomere length, which can increase the sensitivity in testing diastolic functions when mouse hearts are studied.
Figure 5. Mouse working hearts have preserved LV contractility with reduced compliance at slower human-like heart rate.

A. Representative P-V loops and the derived end systolic pressure volume relationship (ESPVR) showed similar LV contractility of mouse ex vivo working heart at heart rates of 480 and 120–130 bpm. B. Representative P-V loops and the derived end diastolic pressure volume relationship (EDPVR) showed decreased LV diastolic compliance in mouse ex vivo working heart at heart rate of 120–130 bpm as compared with that at 480 bpm. The slower heart rate allows higher LV filling and larger end diastolic volume corresponding to longer resting sarcomere length and lower end diastolic ventricular compliance. N = 4 to 5 hearts. Values are presented as mean ± SE. *P<0.05 vs. 480 bpm control in Student’s t test.
3.6. Mouse working hearts have preserved Frank-Starling response at human-like heart rate
While ex vivo mouse working hearts produce higher stroke volume at 120–130 bpm than that at 480 bpm based on increased ventricular filling and end diastolic volume (Table 3 and Fig. 5B), Frank-Starling response was preserved as shown by the positive responses to increasing preload from 5 to 20 mmHg (Fig. 6A). Although the slower heart rate increased EDPVR (Fig. 5B), LVP min was not higher but significantly lower than that at 480 bpm under the preload up to 20 mmHg (Fig. 6B), indicating increased other than decreased diastolic function under physiological preloads. The already higher LVP max and systolic velocity at the slower human-like heart rate also retained trends of positive Frank-Starling response (Fig. 6C and 6D). Diastolic velocity was not changed at the slower heart rate (Table 3) and showed no positive response to increases in preload (Fig. 6E). Therefore, ex vivo mouse working hearts preserve Frank-Starling response at human-like heart rate mainly by improved ventricular filling (Fig. 1B) and Frank-Starling relationship-based stroke volume (Fig. 6A) since slower heart rate produces longer duration of the cardiac cycle and increases the intervals between beats.
Figure 6. Ex vivo mouse working hearts at human-like heart rate preserve Frank-Starling response.

In comparison with 480 bpm controls, LV stroke volume at preload from 5 to 20 mmHg demonstrates a preserved Frank-Starling response at heart rate of 120–130 bpm (A). Although LV EDPVR was higher at the slower heart rate (Fig. 5B), LVPmin showed less increase at heart rate of 120–130 bpm when preload was increased (B). The human-like heart rate increased LVPmax (C) and systolic velocity (D) but not diastolic velocity (E). N = 6 hearts. Values are presented as mean ± SE. *P<0.05 vs. 480 bpm control in Student’s t test.
3.7. Ex vivo mouse working hearts at human-like heart rate in the presence of 300 μM lidocaine preserves inotropic and lusitropic responses to β-adrenergic stimulation
A physiological level of isoproterenol (ISO) (2 nM) induced positive responses in ex vivo mouse working heart at human-like heart rate of ~130 bpm in the presence of 300 μM lidocaine (Table 5). The intrinsic heart rate increased to ~230 bpm following ISO treatment. LVP max and +/−dP/dt max also increased and LVP min reduced (Table 5), indicating preserved inotropic and lusitropic responses to β-adrenergic stimulation. Although stroke volume was not changed because the increased heart rate decreases ventricular filling, the significantly higher heart rate under ISO treatment resulted in significantly higher cardiac output.
Table 5.
Mouse ex vivo working hearts at human-like heart rate have preserved β-adrenergic responses
| Heart Rate (bpm) | LVPmax (mmHg) | LVP min (mmHg) | +dP/dt max (mmHg) | −dP/dt max (mmHg) | Stroke Volume (μL/mg) | |
|---|---|---|---|---|---|---|
| Control | 133.7±8.86 | 85.17±2.97 | 2.74±0.22 | 3705.04±176.84 | −2592.51±91.03 | 0.271±0.015 |
| ISO (2 nM) | 226.2±9.41* | 114.59±4.63* | 1.71±0.43* | 9717.48±599.23* | −4241.13±317.74* | 0.277±0.016 |
2 nM isoproterenol (ISO) increased heart rate in mouse ex vivo working hearts in the presence of 300 μM lidocaine. The preserved positive β-adrenergic responses were further shown by the increased LVPmax, +/−dP/dt and reduced LVPmin. Although the stroke volume was not changed due to the increased heart rate, cardiac output significantly increased in response to β-adrenergic stimulation as the function of increased heart rate (62.17±2.66 vs. 36.17±3.00 μL/mg, P<0.05). N = 6 hearts.
Values are presented as mean ± SE.
P<0.05 vs. untreated control in Student’s t test.
3.8. Ex vivo mouse working hearts at human-like heart rate have improved coronary perfusion and cardiac efficiency
Although the rate of coronary flow decreased in mouse hearts working at the slower heart rate as compared to that at 480 bpm, coronary flow per beat increased (Fig. 7A). This benefit may come from the elongated diastolic time and especially intervals between beats at the slower heart rate, permitting better myocardial perfusion.
Figure 7. Slower heart rate improves coronary perfusion and cardiac efficiency in ex vivo mouse working hearts.
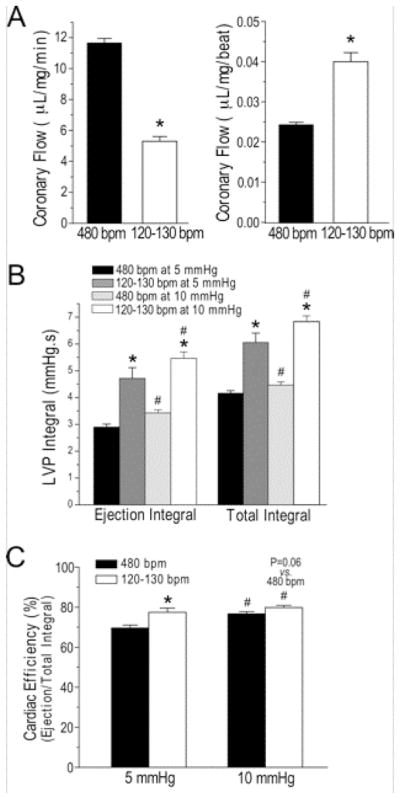
A. Although the rate coronary flow per min was decreased, coronary flow per beat increased in mouse working at human-like heart rate of 120–130 bpm as compared with that at 480 bpm. B. The increases of LVP ejection integral and total integral at preload of 5 and 10 mmHg were significantly higher in mouse ex vivo working hearts at 120–130 bpm than that at 480 bpm, indicating alleviated pumping function. C. Cardiac efficiency calculated by the ratio of ejection to total LVP integrals showed higher efficiency at the human-like lower heart rate as compared with that at 480 bpm. The difference was larger at 5 mmHg preload while the increase of preload improved cardiac efficiency. N = 4 to 6 hearts. Values are presented as mean ± SE. #P<0.05 vs. 5 mmHg; *P<0.05 vs. at 480 bpm in Student’s t test.
In the meantime, the longer systolic time elongates ventricular ejection time (Table 4). Together with the increased LVPmax (Table 1), the total integral and ejection integral of LVP, representing the total work and ejection work respectively, both increased significantly (Fig. 7B). The ratio of ejection integral vs. total integral of LVP indicates the efficiency of cardiac work [20]. Fig. 7C shows that ex vivo mouse working hearts at human-like heart rate of 120–130 bpm had a higher cardiac efficiency than that at 480 bpm. The difference is larger at 5 mmHg preload than that at 10 mmHg preload while the higher preload produced higher total and ejection cardia work (Fig. 7B) and higher cardiac efficiency (Fig. 7C), reflecting a positive effect of ventricular filling on pumping efficiency.
4. Discussion
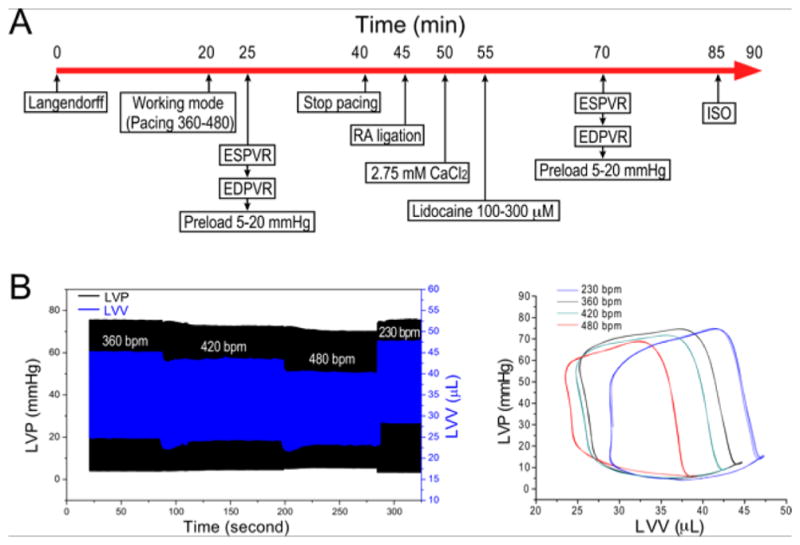
4.1. A practical protocol for the study of ex vivo mouse working hearts at human-like heart rate
Based on reproducible experimental data, our data presented the feasibility of a protocol to extend the study of ex vivo mouse working heart functions by including measurements at a human-like heart rate. Summarized in Fig. 8, the basic functional measurements including both mouse and human-like heart rate studies can be completed in 80 min. A properly prepared mouse working heart can normally work for over 2 hr without significant run-down in performance as shown in our over 1000 preparations represented in multiple publications over the past decade. Therefore, additional specific measurements, stress conditions and drug treatment can be readily integrated into the basic protocol when needed.
Figure 8. Basic protocol to extend the study of ex vivo mouse working heart function by including measurements at a human-like heart rate.

Protocol steps to test ex vivo mouse working heart function at both standard 480 bpm and a slower human-like heart rate of 120 bpm at 37°C is outlined. With both conditions included, the entire basic functional measurements can be completed in 80 min.
4.2. Lowering the pumping rate of ex vivo mouse working heart using lidocaine for cardiac contractility studies
Isolated ex vivo working heart preparations are preferably paced from RA to produce regular supraventricular heart beats for reproducible functional studies [12–14]. Intrinsic heart rate is normally determined by the spontaneous membrane depolarization of specialized pacemaker cells located in the sinoatrial node [21, 22]. Without electrical pacing and in the absence of neurohumoral stimulation, adult mouse sinus node produces a baseline heart rate around 250 bpm ex vivo at 37°C. In the presence of 300 μM lidocaine, ligating the RA to block sinus rhythm can lower ventricular beating rate to 120–130 bpm to provide a human-like range of heart rate for functional studies. 300 μM lidocaine also decreases the sinus-originated rate from ~250 bpm to a human-like rate of 120–130 bpm in ex vivo mouse working hearts. The negative effects of decreased heart rate on force-frequency dependence and lidocaine as a myocardial depressant on intracellular calcium can be compensated by using a higher but still physiological level of calcium (2.75 mM) in the perfusion media.
Since a constant heart rate is desired for accurate and reproducible functional measurements especially kinetic studies, we explored electrical pacing to precisely control the rhythm at a human-like heart rate. Lidocaine is known to lower heart rate through inhibiting A-V conduction in isolated rat heart [15, 23]. We found that when paced at RA at 240 per min a 2:1 A-V blockage produces a stable human-like ventricular beating of 120 bpm. The shapes of LVP waves (Fig. 3A) and P-V loops (Fig. 1B and Fig. 5) at 120 bpm generated by RA pacing at 240 per min are similar to that from sinus or RA pacing without A-V blockage, demonstrating normal supraventricular excitation via the ventricular conductive system to generate normal ventricular contraction for myocardial contractility and hemodynamic studies.
When the supraventricular heart rate decreases, lower level pacemaker activities increased the occurrence of ventricular arrhythmia in ex vivo mouse working hearts (data not shown), which was effectively prevented by lidocaine, a commonly used antiarrhythmic agent [24] that blocks the fast Na+ channels to increase membrane polarization of cardiomyocytes [25]. It is a safe drug clinically and has been used in cardioplegia solution to arrest the heart in myocardial revascularization and other surgical procedures [26, 27]. Lidocaine has also been used to improve functional recovery after prolonged cold static storage of isolated rat working hearts [28]. Consistently, we found that up to 300 μM lidocaine did not produce significant depressant effect on the function of ex vivo mouse working hearts.
Although the precise mechanisms of lidocaine’s action on plasma membrane ion-conducting pores are not fully understood [24], it closes fast Na+ channel in the atrial and ventricular cardiomyocytes and inhibits the phase 0 of action potential. Our results showed that lidocaine did not significantly prolong the contractile and relaxation time of the ventricular muscle and its effect on slowing down heart rate was based on increasing the interval between beats (Fig. 3). The partial A-V blockage produced by 300 μM lidocaine by inhibiting the excitability and conductivity of A-V node and His bundle allows for a 2:1 RA pacing to generate stable 120 bpm ventricular contractions that produce physiological pumping functions and provide a practical approach to study ex vivo mouse hearts at human-like heart rate.
4.3. Application and advantages of measuring mouse heart functions at a human-like beating rate
The protocol we developed in the present study provides a valuable approach to study ex vivo mouse working hearts at a human-like heart rate at 37°C with maintained left ventricular pressure development, systolic and diastolic velocities, preserved Frank-Starling response, and positive inotropic and lusitropic responses to β-adrenergic stimulation. The human-like heart rate produces increased ventricular filling and larger end diastolic volume to increase stroke volume based on Frank-Starling mechanism. The slower heart rate also results in increased coronary perfusion from longer relaxation and interval between beats with improved cardiac efficiency. This extended methodology can better characterize and further the value of genetically modified mouse models of human heart diseases.
Mouse hearts at their fast physiological beating rates have only a small force-frequency reserve compared to that of human hearts [29]. Ventricular filling was limited at high heart rates, further impeding the enhancement of cardiac contractility and output. At the slower heart rate, LV end diastolic volume is larger, allowing the heart to work at the upper portion of the Frank-Starling curve. With sufficiently long interval between beats for full ventricular filling at the human-like heart rate, the preload response more accurately reflects the Frank-Starling length-tension relationship of the ventricular muscle. It is plausible to have sufficient filling time in ex vivo mouse working hearts for testing diastolic function and the effect of preload on Frank-Starling relationship in the study of mouse models of human heart disease. This added feature of mouse working heart at a human-like heart rate is also potentially valuable for studies of impaired diastolic function in heart failure with preserved ejection fraction (HFpEF) [30]. Our data in Fig. 5 demonstrated that the human-like slower heart rate notably increased the end diastolic volume of normal ex vivo mouse working heart, allowing the detection of diastolic dysfunctions in HFpEF models produced in mouse hearts.
The benefit of longer relaxed time of ventricular muscle at slower heart rate improves coronary perfusion. Coronary flow per beat was significantly increased as compared with that at 480 bpm (Fig. 7A). This effect increases oxygen and energy supply per cardiac cycle. In the meantime, the per minute rate of coronary flow was reduced at slower heart rate reflecting the decreased overall metabolic rate that produced less CO2 and thus less dilation of the coronary vessel [31–33]. As coronary perfusion is limited at high heart rate due to short diastolic time (Fig. 7), the slower human-like heart rate in ex vivo mouse working hearts is beneficial for studies of cardiac function while levitating the limitation of energetic supply.
The increased LVP development and elongated systolic, diastolic and ejection time result in increases of total ventricular work and ejection work as calculated from the ventricular pressure integrals (Fig. 7B). At the slower human-like heart rate, cardiac efficiency derived from the ratio of ejection to total LVP integrals was significantly increased from that at 480 bpm (Fig. 7C). This feature should increase the sensitivity of detecting impaired myocardial efficiency in mouse working heart models of human diseases and heart failure.
4.4. Remaining limitations of using mouse hearts to study human cardiac function and diseases
Despite the added value of studying mouse hearts at a human-like heart rate, there are remaining limitations. The slower heart rate is not what the mouse heart normally works at in vivo and adult mouse heart naturally expresses 100% αMHC, a myosin isoform with faster ATPase rate than the rate of βMHC that is the sole myosin isoenzyme in human heart [34]. This is an intrinsic issue for all studies of human heart diseases using mouse models. To consider its specific impact on ex vivo mouse working heart at slower heart rate, one precaution is that the higher ATPase rate of α myosin in mouse heart may limit the benefit of slower heart rate on slowing down contractile velocity and improving pumping efficiency [14].
The resting human heart works at 60–80 bpm. Our attempts to further lower the rate of mouse working heart was less effective. The optimal concentration of lidocaine to lower the ventricular rate of mouse heart to <100 bpm was not reproducible although continuous recording at 400 μM was able to obtain short periods of stable functional data (not shown). Lidocaine can also produce arrhythmia at high concentrations and at 400 μM it can significantly depress cardiac function (data not shown).
Despite these limitations from intrinsic differences between human and mouse hearts, the readily applicable protocol we presented allows informative functional characterizations of ex vivo mouse working hearts at human-like heart rate to extend the use of genetically modified mouse models of human heart diseases, which can help to maximize the value of these currently non-dispensable resources to facilitate cardiac function and mechanistic studies.
Highlights.
We report a protocol to study ex vivo mouse working heart at human-like heart rate
300 μM lidocaine effectively maintains stable heart rate of 120–130 bpm at 37°C
Cardiac depressant effect of lidocaine is compensated by 2.75 mM Ca2+ in perfusant
The protocol does not have depressing effect on contractile functions
The slow down of heart rate allows more sensitive studies of diastolic dysfunction
Acknowledgments
Sources of funding
This research was supported in part by a grant from the National Institutes of Health (HL127691) to JPJ.
Abbreviations
- HR
heart rate
- ISO
isoproterenol
- LV
left ventricle
- LVP
left ventricular pressure
- Lido
Lidocaine
- RA
right atrium
Footnotes
Disclosures
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaese S, Verheule S. Cardiac electrophysiology in mice: a matter of size. Frontiers in physiology. 2012;3:345. doi: 10.3389/fphys.2012.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wessels A, Sedmera D. Developmental anatomy of the heart: a tale of mice and man. Physiol Genomics. 2003;15(3):165–76. doi: 10.1152/physiolgenomics.00033.2003. [DOI] [PubMed] [Google Scholar]
- 3.Gattoni S, Roe AT, Frisk M, Louch WE, Niederer SA, Smith NP. The calcium-frequency response in the rat ventricular myocyte: an experimental and modelling study. J Physiol. 2016;594(15):4193–224. doi: 10.1113/JP272011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su Z, Li F, Spitzer KW, Yao A, Ritter M, Barry WH. Comparison of sarcoplasmic reticulum Ca2+-ATPase function in human, dog, rabbit, and mouse ventricular myocytes. J Mol Cell Cardiol. 2003;35(7):761–7. doi: 10.1016/s0022-2828(03)00119-6. [DOI] [PubMed] [Google Scholar]
- 5.Su Z, Bridge JH, Philipson KD, Spitzer KW, Barry WH. Quantitation of Na/Ca exchanger function in single ventricular myocytes. J Mol Cell Cardiol. 1999;31(5):1125–35. doi: 10.1006/jmcc.1999.0949. [DOI] [PubMed] [Google Scholar]
- 6.Harada K, Sugaya T, Murakami K, Yazaki Y, Komuro I. Angiotensin II type 1A receptor knockout mice display less left ventricular remodeling and improved survival after myocardial infarction. Circulation. 1999;100(20):2093–9. doi: 10.1161/01.cir.100.20.2093. [DOI] [PubMed] [Google Scholar]
- 7.Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86(4):386–90. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]
- 8.Milani-Nejad N, Janssen PM. Small and large animal models in cardiac contraction research: advantages and disadvantages. Pharmacol Ther. 2014;141(3):235–49. doi: 10.1016/j.pharmthera.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neely JR, Liebermeister H, Battersby EJ, Morgan HE. Effect of pressure development on oxygen consumption by isolated rat heart. Am J Physiol. 1967;212(4):804–14. doi: 10.1152/ajplegacy.1967.212.4.804. [DOI] [PubMed] [Google Scholar]
- 10.Scherrer-Crosbie M, Thibault HB. Echocardiography in translational research: of mice and men. J Am Soc Echocardiogr. 2008;21(10):1083–92. doi: 10.1016/j.echo.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skrzypiec-Spring M, Grotthus B, Szelag A, Schulz R. Isolated heart perfusion according to Langendorff---still viable in the new millennium. J Pharmacol Toxicol Methods. 2007;55(2):113–26. doi: 10.1016/j.vascn.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Gauthier NS, Matherne GP, Morrison RR, Headrick JP. Determination of function in the isolated working mouse heart: issues in experimental design. J Mol Cell Cardiol. 1998;30(3):453–61. doi: 10.1006/jmcc.1997.0610. [DOI] [PubMed] [Google Scholar]
- 13.Larsen TS, Belke DD, Sas R, Giles WR, Severson DL, Lopaschuk GD, Tyberg JV. The isolated working mouse heart: methodological considerations. Pflugers Arch. 1999;437(6):979–85. doi: 10.1007/s004240050870. [DOI] [PubMed] [Google Scholar]
- 14.Feng HZ, Biesiadecki BJ, Yu ZB, Hossain MM, Jin JP. Restricted N-terminal truncation of cardiac troponin T: a novel mechanism for functional adaptation to energetic crisis. J Physiol. 2008;586(14):3537–50. doi: 10.1113/jphysiol.2008.153577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komai H, Rusy BF. Effects of bupivacaine and lidocaine on AV conduction in the isolated rat heart: modification by hyperkalemia. Anesthesiology. 1981;55(3):281–5. doi: 10.1097/00000542-198109000-00017. [DOI] [PubMed] [Google Scholar]
- 16.Tsuboi M, Chiba S. Effects of lidocaine on isolated, blood-perfused ventricular contractility in the dog. Heart Vessels. 1999;14(6):289–94. doi: 10.1007/BF03257241. [DOI] [PubMed] [Google Scholar]
- 17.Endoh M. Force-frequency relationship in intact mammalian ventricular myocardium: physiological and pathophysiological relevance. Eur J Pharmacol. 2004;500(1–3):73–86. doi: 10.1016/j.ejphar.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 18.Cazorla O, Freiburg A, Helmes M, Centner T, McNabb M, Wu Y, Trombitas K, Labeit S, Granzier H. Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ Res. 2000;86(1):59–67. doi: 10.1161/01.res.86.1.59. [DOI] [PubMed] [Google Scholar]
- 19.Opitz CA, Linke WA. Plasticity of cardiac titin/connectin in heart development. J Muscle Res Cell Motil. 2005;26(6–8):333–42. doi: 10.1007/s10974-005-9040-7. [DOI] [PubMed] [Google Scholar]
- 20.Feng HZ, Jin JP. Coexistence of cardiac troponin T variants reduces heart efficiency. Am J Physiol Heart Circ Physiol. 2010;299(1):H97–H105. doi: 10.1152/ajpheart.01105.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiFrancesco D. Pacemaker mechanisms in cardiac tissue. Annual review of physiology. 1993;55:455–72. doi: 10.1146/annurev.ph.55.030193.002323. [DOI] [PubMed] [Google Scholar]
- 22.Irisawa H, Brown HF, Giles W. Cardiac pacemaking in the sinoatrial node. Physiological reviews. 1993;73(1):197–227. doi: 10.1152/physrev.1993.73.1.197. [DOI] [PubMed] [Google Scholar]
- 23.Krikava I, Jarkovsky J, Stourac P, Novakova M, Sevcik P. The effects of lidocaine on bupivacaine-induced cardiotoxicity in the isolated rat heart. Physiol Res. 2010;59(Suppl 1):S65–9. doi: 10.33549/physiolres.932014. [DOI] [PubMed] [Google Scholar]
- 24.Kambouris NG, Nuss HB, Johns DC, Marban E, Tomaselli GF, Balser JR. A revised view of cardiac sodium channel “blockade” in the long-QT syndrome. The Journal of clinical investigation. 2000;105(8):1133–40. doi: 10.1172/JCI9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hearse DJ, O’Brien K, Braimbridge MV. Protection of the myocardium during ischemic arrest. Dose-response curves for procaine and lignocaine in cardioplegic solutions. The Journal of thoracic and cardiovascular surgery. 1981;81(6):873–9. [PubMed] [Google Scholar]
- 26.Fiore AC, Naunheim KS, Taub J, Braun P, McBride LR, Pennington DG, Kaiser GC, Willman VL, Barner HB. Myocardial preservation using lidocaine blood cardioplegia. Ann Thorac Surg. 1990;50(5):771–5. doi: 10.1016/0003-4975(90)90683-w. [DOI] [PubMed] [Google Scholar]
- 27.Yammine M, Neely RC, Loberman D, Rajab TK, Grewal A, McGurk S, Fitzgerald D, Aranki SF. The Use of Lidocaine Containing Cardioplegia in Surgery for Adult Acquired Heart Disease. J Card Surg. 2015;30(9):677–84. doi: 10.1111/jocs.12597. [DOI] [PubMed] [Google Scholar]
- 28.Rudd DM, Dobson GP. Eight hours of cold static storage with adenosine and lidocaine (Adenocaine) heart preservation solutions: toward therapeutic suspended animation. J Thorac Cardiovasc Surg. 2011;142(6):1552–61. doi: 10.1016/j.jtcvs.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 29.Georgakopoulos D, Kass D. Minimal force-frequency modulation of inotropy and relaxation of in situ murine heart. J Physiol. 2001;534(Pt. 2):535–45. doi: 10.1111/j.1469-7793.2001.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma K, Kass DA. Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ Res. 2014;115(1):79–96. doi: 10.1161/CIRCRESAHA.115.302922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Case RB, Greenberg H. The response of canine coronary vascular resistance to local alterations in coronary arterial P CO2. Circ Res. 1976;39(4):558–66. doi: 10.1161/01.res.39.4.558. [DOI] [PubMed] [Google Scholar]
- 32.Feinberg H, Gerola A, Katz LN. Effect of changes in blood CO2 level on coronary flow and myocardial O2 consumption. Am J Physiol. 1960;199:349–54. doi: 10.1152/ajplegacy.1960.199.2.349. [DOI] [PubMed] [Google Scholar]
- 33.Kittle CF, Aoki H, Brown EB., Jr The Role of Ph and Co2 in the Distribution of Blood Flow. Surgery. 1965;57:139–54. [PubMed] [Google Scholar]
- 34.Rundell VL, Manaves V, Martin AF, de Tombe PP. Impact of beta-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am J Physiol Heart Circ Physiol. 2005;288(2):H896–903. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]