

Abstract
Background
Lysosomal storage disease (LSD) is a rare inherited disease group. Consecutively there are few data on cardiac changes in mucopolysaccharidosis (MPS), Anderson Fabry disease (AFD), and other LSD (oLSD) including Pompe disease (PD) and Danon disease (DD), I-cell disease ICD and mucolipidosis III (ML III).
Methods
Between 1994 and 2011, we identified 39 patients with LSD: 25 with MPS, 8 with AFD, and 6 with oLSD including PD (1), ML III (2), DD (1), and ICD (2) at our institution fulfilling the inclusion criteria of at least one echocardiogram and ECG.
Results
Median age was 11.4 years (range: 2–27), 22 were females (56%). Normal echocardiograms were present in 12 patients (31%): 4 with MPS (16%), 7 AFD (88%), and 1 oLSD (17%). Valvular heart disease was present in 23 patients (59%) occurring more often in MPS (76%) and oLSD (67%) than in AFD (0%) (p < 0.001). The most common ECG abnormality was a short PR interval in 10 of 35 patients (29%) occurring in all LSD groups. Median follow-up was 5.8 (0.2–22.2) years showing diminished 5-year survival compared to an age-matched group. However, no patient died due to a cardiac cause and no cardiovascular intervention was necessary.
Conclusion
Echocardiographically detectable cardiovascular involvement in children with LSD is mostly confined to MPS and oLSD. Valve thickening in echo and a short PR interval in the ECG are the most frequent abnormalities. Routine repeat assessment is recommended in LSD. However, significant cardiac disease necessitating cardiac intervention is rare during a short follow-up.
Keywords: Lysosomal storage disease, Anderson Fabry, Mucopolysaccharidosis, Cardiovascular involvement
1. Introduction
Lysosomal storage disease (LSD) is a heterogeneous group of more than 40 different disorders due to genetic defects in a lysosomal acid hydrolase, causing progressive lysosomal accumulation of substrates specific for each disorder involving multiple organs; the severity of disease depends on residual enzyme activity. LSD shows an accumulation of various glycosaminoglycans, glycoproteins, or glycolipids within the lysosomes of various tissues [1]. LSD can affect the heart and constitute an important distinct and treatable cause of cardiomyopathy in children, accounting for approximately 5% of pediatric cardiomyopathies [2]. The most common LSDs in children are Anderson–Fabry disease (AFD), mucopolysaccharidosis (MPS) and Gaucher disease [3]. In adults, AFD is the most prevalent LSD; cardiac involvement in AFD in adults can mimic hypertrophic cardiomyopathy [4]. In MPS pronounced cardiovascular involvement can be a cause of death apart from upper airway obstruction 5, 6, 7. There are only rare studies comparing cardiac involvement between the various types of LSD. Most of these studies focus on one large group of the disorders such as MPS [7].
The aim of this study was first to describe and compare echocardiographic findings of clinical symptoms and ECG in children with LSD in a single institution, and second to analyze the possible impact of cardiac disease on survival.
2. Methods
2.1. Patients
The echocardiography database of the Children's University Hospital Zurich was searched from January 1994 to April 30, 2011. All patients with a diagnosis of MPS, AFD, mucolipidosis II and III (ML), Pompe disease (PD), or Danon (DD) disease were reviewed to meet our inclusion criteria of a biochemical or genetic diagnosis of the respective LSD. If the patient had more than one echocardiographic exam, the last exam was analyzed.
To segregate possible differences the study population was divided into 3 groups: MPS, AFD and other LSD group consisting of DD, PD and ML patients (oLSD). There were 40 patients fulfilling the inclusion criteria; as one patient refuses any research participation, 39 patients were included in the study. The local ethical committee approved the study according to institutional requirements.
2.2. Echocardiographic examination
A complete two-dimensional and Doppler echocardiographic exam was performed in each patient according to the criteria of the American Society of Echocardiography 8, 9. Left ventricular (LV) ejection fraction was determined using biplane Simpson's method. Left ventricular hypertrophy was defined as the Z-score of the left ventricular mass index (LVMI) being > + 2 standard deviations (SD) [10]. Dilatation of the LV or left atrium (LA) was defined as a Z-score > + 2SD 8, 9. BSA was calculated using the Mosteller formula [11]. Diastolic function was evaluated and analyzed as previously described including left ventricular inflow pattern, Doppler tissue imaging (including the E/e′ ratio), the isovolumic relaxation time and the pulmonary venous flow reversal velocity 12, 13.
The severity of valvular regurgitation and stenosis was determined according to ASE-guidelines [14]. Pulmonary hypertension was measured non-invasively and defined as an estimated systolic pulmonary artery pressure of > 35 mm Hg.
2.3. Electrocardiogram and 24 hour ECG
An ECG was available for review in 36 patients (92%). All ECGs were analyzed for heart rate, PR interval, QRS duration, QTc duration, QRS axis, and the presence of preexcitation or AV block. A 24-hour ECG was available in 12 patients (31%).
2.4. Follow-up
Follow-up for survival analysis was obtained in 38 patients (97%) from their last echocardiographic examination until April 30, 2011 by a clinical examination at our institution. One patient was lost to follow-up after referral to another center, where his follow-up data were not further accessible after he reached adulthood.
2.5. Statistical analysis
Categorical variables were compared using chi-square analysis (two-sided exact significance). Continuous variables were expressed as mean ± 1 SD or as median with range and were compared using the Kruskal-Wallis test. All statistical analysis was two-tailed with a p-value of < 0.05 to indicate statistical significance. Overall survival was analyzed using Kaplan–Meier curves using the log-rank test. Expected survival of an age and sex matched US-population was computed using the R package survival [15]. Statistical analyses were performed using IBM SPSS Statistics version 20 (SPSS Inc., Chicago, IL, USA).
3. Results
A detailed summary of the exact type of LSD, the number of patients and the age at the echocardiographic exam is shown in Table 2. MPS was the most common LSD occurring in 25 patients (64%), whereas 8 patients (21%, 7 female) had AFD. The remaining 6 patients (15%) had oLSD. The largest groups among the MPS were MPS I (Hurler disease, 7 patients) and MPS IVa (6 patients).
Table 2.
Detailed summary of all our patients with the different LSD.
| Group | Diagnosis | No. pts | Age at most recent echo (years) |
|---|---|---|---|
| MPS | MPS I: Hurler | 7 | 2, 2, 4, 6, 10, 11, 13 |
| MPS I: Scheie | 1 | 16 | |
| MPS II: Hunter | 4 | 13, 15, 16, 19 | |
| MPS IIIa/b | 4 | 7, 11, 16, 19 | |
| MPS IVa | 6 | 3, 9, 11, 11, 12, 13 | |
| MPS VI | 2 | 9, 10, 17 | |
| Total | 25 | ||
| AFD | AFD | 8 | 6, 7, 8, 10, 11, 13, 13, 17 |
| oLSD | Pompe disease (PD) | 1 | 4 |
| Danon disease (DD) | 1 | 15 | |
| ML II: I-cell disease (ICD) | 2 | 2, 4 | |
| ML III | 2 | 12, 27 | |
| Total | 6 |
MPS = mucopolysaccharidosis; AFD = Anderson Fabry disease; ML = mucolipidosis; PD = Pompe disease; DD = Danon disease; ICD = I-cell disease.
3.1. Clinical characteristics
Clinical characteristics are shown in Table 3. There was no significant difference in median age, median body weight and gender between the 3 groups. A heart murmur was present in 49% of patients. Most patients were in NYHA class I. One patient of the oLSD group (20%) was in NYHA class III. Signs of heart failure were observed in 3 patients with MPS (1 with MPS, 2 with MPS VI) and in the patient with DD.
Table 3.
Clinical characteristics of the patients.
| All patients N = 39 | MPS N = 25 | AFD N = 8 | oLSD N = 6 | p value | |
|---|---|---|---|---|---|
| Median age (y) | 11.4 (2.0, 27.6) |
11.5 (2.0, 19.5) |
10.8 (6.2, 17.1) |
8.6 (2.9, 27.6) |
0.82 |
| Median body | 26.0 | 26.0 | 33.3 | 24.4 | 0.48 |
| Weight (kg) | (5.3, 73.4) | (11.0, 58.0) | (20.0, 73.4) | (5.3, 60.3) | |
| Female gender | 22 (56%) | 11 (44%) | 7 (88%) | 4 (67%) | 0.09 |
| Heart murmur | 19 (49%) | 13 (52%) | 2 (25%) | 4 (67%) | 0.33 |
| NYHA class I | 28/32 (88%) | 17/20 (85%) | 7/7 (100%) | 4/5 (80%) | 0.59 |
| NYHA class II or higher | 4/32 (13%) | 3/20 (15%) | 0 | 1/5 (20%) | 0.59 |
| Heart failure | 4 (10%) | 3 (12%) | 0 | 1 (17%) | 0.60 |
MPS = mucopolysaccharidosis; AFD = Anderson Fabry disease; oLSD = other lysosomal storage disease.
In 7 patients (18%) functional class could not be assessed because of orthopedic problems. In the remaining 32 patients, there was no statistically significant difference in NYHA classification between the 3 groups.
3.2. Echocardiographic findings
A completely normal echocardiographic exam was present in 4 of 25 patients with MPS (16%), 7 of 8 patients with AFD (88%) and in the patient with M. Pompe (17% of oLSD). Echocardiographically detectable changes are summarized in Table 4, Table 5. LV dilatation was rare and LV hypertrophy was found in MPS patients (20%) and oLSD patients (33%), but not in AFP patients (p = 0.56). The patient with DD had massive left ventricular hypertrophy with a left ventricular muscle mass index of 533 g/m2 (Fig. 1). All echocardiographic findings are shown in Table 4, Table 5.
Table 4.
Echocardiographic findings.
| All patients N = 39 | MPS N = 25 | AFD N = 8 | oLSD N = 6 | p value | |
|---|---|---|---|---|---|
| Normal echo | 12 (31%) | 4 (16%) | 7 (88%) | 1 (17%) | 0.001 |
| Mean LVEDD (mm) | 38.5 ± 6.7 | 37.7 ± 5.7 | 39.8 ± 3.9 | 40.3 ± 12.2 | 0.62 |
| LV dilatation | 6 (15%) | 3 (12%) | 0 | 3 (50%) | 0.56 |
| Mean EF (%) | 58.0 ± 7.2 | 59.2 ± 4.2 | 57.5 ± 3.6 | 53.8 ± 16.1 | 0.47 |
| Median | 68.8 | 70.6 | 62.1 | 78.5 | 0.19 |
| LVMMI (g/m2) | (31.1, 533.0) | (31.1, 117.1) | (50.4, 80.1) | (56.0, 533.0) | |
| LVH | 7 (18%) | 5 (20%) | 0 | 2 (33%) | 0.30 |
| Mean LA size (mm) | 25.0 ± 5.1 | 24.7 ± 4.1 | 25.1 ± 5.4 | 25.6 ± 7.7 | 0.91 |
| LA dilatation | 8 (21%) | 5 (20%) | 0 | 3 (50%) | 0.30 |
| Mean dp | 20.4 ± 7.0 | 22.9 ± 9.0 | 18.7 ± 3.1 | 16.3 ± 3.5 | 0.31 |
| RV/RA | (n = 18) | (n = 9) | (n = 6) | (n = 3) | |
| PHT dpRV/RA > 25 mm Hg | 2/18 (11%) | 2/9 (22%) | 0/6 | 0/5 | 0.49 |
| Diastolic dysfunction | 5/24 (21%) | 4/13 (31%) | 1 (13%) | 0/3 | 0.60 |
MPS = mucopolysaccharidosis; AFD = Anderson Fabry disease; oLSD = other lysosomal storage disease; LVEDD = left ventricular end-diastolic diameter; LV = left ventricular; EF = ejection fraction; LVMMI = left ventricular muscle mass index; LVH = left ventricular hypertrophy; LA = left atrial; PHT = pulmonary hypertension.
Table 5.
Echocardiographic findings in valvular heart disease.
| All patients N = 39 | MPS N = 25 | AFD N = 8 | oLSD N = 6 | p value | |
|---|---|---|---|---|---|
| Any VHD | 23 (59%) | 19 (76%) | 0 | 4 (67%) | < 0.001 |
| Abnormal MV | 21 (54%) | 17 (68%) | 0 | 4 (67%) | 0.001 |
| ≥ Mild MS | 4 (10%) | 3 (12%) | 0 | 1 (17%) | 0.60 |
| ≥ Mild MR | 17 (44%) | 13 (52%) | 0 | 4 (67%) | 0.02 |
| Abnormal AV | 19 (49%) | 16 (64%) | 0 | 3 (50%) | 0.006 |
| ≥ Mild AS | 3 (8%) | 3 (12%) | 0 | 0 | 0.41 |
| ≥ Mild AR | 9 (23%) | 7 (28%) | 0 | 2 (33%) | 0.26 |
| Abnormal TV | 3 (8%) | 1 (4%) | 0 | 2 (33%) | 0.06 |
| Abnormal PV | 5 (13%) | 3 (12%) | 0 | 2 (33%) | 0.15 |
MPS = mucopolysaccharidosis; AFD = Anderson Fabry disease; oLSD = other lysosomal storage disease; VHD = valvular heart disease; MV = mitral valve; MS = mitral stenosis; MR = mitral regurgitation; AV = aortic valve; AR = aortic regurgitation; AS = aortic stenosis; TV = tricuspidal valve; PV = pulmonary valve.
Fig. 1.


15-Year old patient with Danon disease: parasternal long axis view with massive hypertrophy (panel a) measuring 39 mm. The 4-chamber view (Fig. 1b) shows the relation to the size of the right ventricle. In panel c, the ECG of this patient is shown with marked signs of left ventricular hypertrophy.
LA = left atrium; LV = left ventricle; AO = aorta; RA = right atrium.
Significant pulmonary hypertension was only seen in MPS (2/9, 22%). None of AFD and oLSD had non-invasively measured significantly elevated pulmonary artery pressures except for the patient with DD, who had an additional intracavitary pressure gradient in the right ventricle. There was no significant difference in the presence of diastolic dysfunction between the groups.
Valvular heart disease was frequent in MPS (76%) and oLSD (67%) as shown in Table 5. There was no difference between the presence of mitral or aortic valvular heart disease. In MPS, mitral valve (p = 0.001) and aortic valve abnormalities (p = 0.006) were significantly more commonly seen than in oLSD or in AFD.
Fig. 2, Fig. 3 show typical mitral valve abnormalities in a 18 years old woman with MPS I and a 14 years old boy with MPS II.
Fig. 2.

Example of an 18-year old woman with mucopolysaccharidosis. This shows the apical 4-chamber view (apex down) with the arrow pointing to the thickened mitral valve. No progression or regression of valvular changes after 2 years of enzyme replacement therapy was observed. LA = left atrium; LV = left ventricle; RV = right ventricle.
Fig. 3.
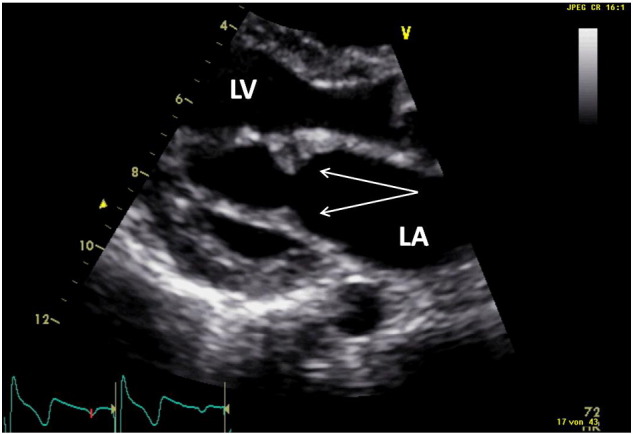
14-Year old patient with mucopolysaccharidosis Type II (hunter disease).
LA = left atrium; LV = left ventricle.
3.3. ECG findings
An ECG was available in 36 of 39 patients. The findings are summarized in Table 6. There was no significant difference in age at ECG, heart rate, PR interval, QRS duration and QTc duration or ventricular arrhythmias between the groups (p = ns). There were no patients with ventricular tachycardias. ECG abnormalities included one 2nd degree AV block in one MPS; no higher degree AV blockage was found in our patient cohort. The patient with DD was diagnosed with a WPW syndrome. Other ECG parameters did not show any differences between the groups.
Table 6.
ECG findings.
| All patients N = 36 | MPS N = 23 | AFD N = 8 | oLSD N = 5 | p value | |
|---|---|---|---|---|---|
| Age (median) | 10.9 (0.3, 28.3) | 11.2 (2.1, 20.0) | 11.1 (6.3, 17.5) | 4.8 (0.3, 28.3) | 0.82 |
| Heart rate, mean (bpm) | 87.9 ± 25.2 | 88.9 ± 21.5 | 77.0 ± 9.1 | 100.6 ± 49.0 | 0.27 |
| PR interval, mean (ms) | 134.2 ± 42.3 | 141.2 ± 48.4 | 125.8 ± 18.8 | 110.5 ± 31.0 | 0.34 |
| PR interval < 120 ms | 10/35 (29%) | 6 (26%) | 2 (25%) | 2/4 (50%) | 0.73 |
| QRS duration, median (ms) | 81 (56, 170) | 82 (64, 108) | 79 (76, 86) | 84 (56, 170) | 0.76 |
| Median QTc duration (ms) | 423 (380, 489) | 425 (380, 452) | 409 (398, 432) | 432 (405, 489) | 0.14 |
| Mean QRS axis (°) | 68.0 ± 32.2 | 71.4 ± 37.6 | 63.6 ± 14.8 | 59.2 ± 27.3 | 0.39 |
| Preexcitation | 1 (3%) | 0 | 0 | 1 (20%) | 0.14 |
| 24 h-ECG | 12/39 (31%) | 4/25 (16%) | 6/8 (75%) | 2/6 (33%) | 0.006 |
| Isolated PVC's | 7/12 (58%) | 1/4 (25%) | 5/6 (83%) | 1/2 (50%) | 0.42 |
| AF | 1/12 (8%) | 0/4 | 0/6 | 1/2 (50%) | 0.17 |
| 2nd AVB | 1/12 (8%) | 1/4 (25%) | 0/6 | 0/2 | 0.50 |
MPS = mucopolysaccharidosis; AFD = Anderson Fabry disease; oLSD = other lysosomal storage disease; PVC's = premature ventricular contraction; AF = atrial fibrillation; 2nd AVB = second degree AV-block.
3.4. Treatment
Enzyme replacement therapy (ERT), was used in 13 patients (33%) suffering of MPS [9], AFD [3] or PD [1]. Bone marrow transplantation (BMT) was used in 6 patients (15%) with MPS I. There was no specific therapy for patients with ML or DD. No patient was under cardiac medication.
A summary of treatment for the LSD in these patients and the follow-up information is shown in Table 7.
Table 7.
Treatment and outcome.
| All patients N = 39 | MPS N = 25 | AFD N = 8 | oLSD N = 6 | p value | |
|---|---|---|---|---|---|
| BMT | 6 (15%) | 6 (24%) | – | – | 0.18 |
| ERT | 13 (33%) | 9 (36%) | 3 (38%) | 1 (17%) | 0.78 |
| No therapy | 22 (56%) | 12 (48%) | 5 (63%) | 5 (83%) | 0.26 |
| Death | 8/38 (21%) | 5/24 (21%) | 0 (0%) | 3 (33%) | 0.28* |
| Mean age death (y) | 12.0 ± 6.1 | 14.2 ± 5.2 | – | 8.3 ± 6.6 | 0.10 |
MPS = mucopolysaccharidosis; AFD = Anderson Fabry disease; oLSD = other lysosomal storage disease; BMT = bone marrow transplantation; ERT = enzyme replacement therapy; * log-rank test.
3.5. Follow-up
Median time of clinical follow-up after the first contact with the Children's University Hospital was 5.8 years (up to 22.2 years). Eight patients (21%) died during the follow-up period. Three deaths of the oLSD (50%) group had a diagnosis of ML II (2 deaths) and DD. One of each MPS group I, II, III, IIIa and VI died (5/24 (21%) patients with a regular follow-up). The patient with MPS I was under ERT. Although ventricular dilatation and/or abnormal function were present in 3 of the patients who died subsequently, none of them died directly from heart failure or sudden cardiac death. Causes of death were respiratory failure in 7 patients and septicemia in one patient. 30 patients (79%) were alive at the end of the follow-up period, including all patients who had undergone BMT and all patients of the AFD group. All findings about therapy and outcome are listed in Table 6. Five-year survival was 92% for MPS-, 100% for AFD- and 67% for oLSD-patients; there was no statistical significant difference between the groups (Fig. 4).
Fig. 4.

Kaplan–Meier curve of the survival of the MPS, AFD and oLSD patients compared to an age and sex matched white American normal population.
MPS = mucopolysaccharidosis; AFD = Anderson Fabry disease; oLSD = other lysosomal storage disease.
4. Discussion
Cardiovascular changes are frequently found in a pediatric population with LSD. In our cohort only 31% of the patients had completely normal cardiac findings. A heart murmur (49%) and valvular heart disease (59%) mostly of mild degree were the most common abnormalities observed and they were almost exclusively confined to children with MPS and oLSD and not yet observed in children with AFD; this may be due to the rather young age and/or higher number of females in our AFD-group. Left ventricular hypertrophy and higher degree AV block were rare in this age group in all types of LSD.
Cardiac symptoms in this age group were rare. Despite significantly decreased survival, this was not due to cardiac disease and cardiac interventions were not necessary in any of these children and adolescents with MPS.
4.1. Differences in valvular heart disease between the lysosomal storage disorders
Valvular heart disease in LSD has been most commonly described in MPS, AFD, ML II and ML III (see Table 1). In our patients, we observed valvular heart disease in 76% of patients with MPS, in 67% of those with oLSD and in none of the patients with AFD. These findings are comparable to the literature. In a study on 28 patients with MPS, mitral valve thickening was described in 61% and aortic valve thickening in 36% [7]. In an article on cardiac manifestations of AFD in children and adolescents by Kampmann et al. [16], no heart valve changes were described. Heart valve changes in AFD can occur, they have been reported to occur in 14.6% in a registry; however, they are rarely hemodynamically significant even in adults with advanced disease 17, 18.
Table 1.
Typical cardiac findings of various lysosomal storage disorders according to the literature.
| LVH | Diastolic dysfunction | Valvular HD | AV block | Preexcitation | VPCs/VTs or SCD | PHT | |
|---|---|---|---|---|---|---|---|
| MPS 7, 19, 31 | +++ | ++ | ++ | ++ | – | – | ++ |
| AFD 4, 11, 28, 29, 34 | +++ | ++ | ++ | + | – | + | – |
| PD [47] | +++ | +++ | – | – | short PR | – | + |
| DD 24, 25 | +++ | +++ | – | – | short PR | +++ | – |
| ML II (ICD) [48] | +++ | ? | +++ | ? | – | +++ | ? |
| ML III [21] | ? | ? | +++ | ? | – | ? | ? |
| Sphingolipidosis: Gaucher [49] | + | + | + | ? | ? | ? | ? |
MPS = mucopolysaccharidosis; AFD = Anderson Fabry disease; ML = mucolipidosis; ICD = I-cell disease; + = rare, ++ = common; +++ = very common; – = not described; ? = unknown.
None of our patients needed valve surgery. In the literature, valve replacement has rarely been reported. In a group of children and young adults (21 months to 25 years) with MPS, Dangel et al. described valvular lesions and/or cardiomyopathy in 72% of patients [19]. The lesions were progressive but rarely led to cardiac symptoms. Only one boy with Hunter disease had to undergo successful mitral valve replacement. Both, aortic and mitral valve replacement was reported in a 14 year old female with Gaucher disease and mucolipidosis III 20, 21. Hopefully, in the current times of increasing options of enzyme replacement therapy in LSD, valve replacement in these patients will become even more rare.
4.2. Wall thickening of the left ventricle
Left ventricular wall thickening in AFD may mimic hypertrophic non-obstructive or obstructive cardiomyopathy in an adult population [22]. To our knowledge, however, obstructive hypertrophic cardiomyopathy due to AFD has not been described in children with AFD. In our pediatric AFD patients no significant left ventricular hypertrophy was detected. In AFD, onset of left ventricular wall thickening is earlier in males than females [23]. However, none of our male infants with AFD had left ventricular hypertrophy. Left ventricular wall thickening mimicking left ventricular “hypertrophy” predominated in the MPS group and oLSD. None of our patients with any LSD had a left ventricular outflow tract gradient. Left ventricular wall thickening can also occur in other LSD such as Danon disease and Pompe disease 24, 25, 26.
In our patients, the most impressive wall thickening was seen in the patients with Danon disease who died during follow-up. Lysosome-associated membrane protein-2 deficiency (LAMP-2 deficiency), also called Danon disease, is a rare X-linked lysosomal disorder characterized by impressive cardiomyopathy, vacuolar myopathy, and mental retardation. Danon disease may cause concentric left ventricular hypertrophy; in any male teenager with concentric LVH, especially in the presence of elevated serum hepatic enzymes and CK concentrations, and/or WPW syndrome with markedly increased voltage of the left ventricle. Rarely, left ventricular outflow tract obstruction has been described in Danon disease [27]. In patients with Danon disease timely molecular diagnosis and early consideration of heart transplantation are recommended [25].
4.3. Pulmonary hypertension
The etiology of pulmonary hypertension in patients with LSD is multifactorial: severe scoliosis, obstructive sleep apnea in MPS patients, and diastolic dysfunction are the main causes. Pulmonary hypertension was detected by echocardiography only in 2 patients, and it was not hemodynamically significant. In any patient with MPS and pulmonary hypertension, one has to think of obstructive sleep apnea as in these patients, partially degraded GAGs can accumulate also in the upper airways [27]. In our patients, sleep studies were not performed routinely.
4.4. Changes in AV conduction in lysosomal storage disorders
A short PR interval is considered typical of AFD and due to accelerated atrioventricular conduction; it is seen in 14 to 40% of patients 28, 29. Older patients with AFD may develop bundle branch block and progressive AV conduction abnormalities. In our patients, a short PR interval did not only occur in patients with AFD (in 25%) but also in MPS (26%) and oLSD (50%). Thus shortening of the PR interval is not specific for AFD.
Complete AV block can occur in MPS 30, 31 and may even cause sudden cardiac death in MPS as described by Hishitani et al. [31]. In the past, the incidence of sudden death in patients with MPS was reported to be as high as 11%, thus in these patients, careful surveillance with Holter ECG is needed [32]. In one of our patients with MPS, there was 2nd degree AV block in the 24 h-ECG during daily activities. This patient died 19 years old due to respiratory infection. Complete AV block in AFD has been described in middle-aged women [33], but to our knowledge this is a rarity in children.
4.5. Impact of therapies
Nowadays, the clinical course of many children with LSD is attenuated by treatments such as ERT and BMT in MPS I; the long-term effect of which has yet to be determined. Among our patients, 6 of 7 (85%) with MPS I (Hunter) had BMT, all 7 patients with MPS I were alive at the last follow- up.
Thirty-three percent of our patients had an ERT (3 AFD, 3 MPS I, 3 MPS II, 3 MPS VI, one PD). One 9 years old girl with MPS VI died due to respiratory failure and progressive hypoventilation subsequent to craniocervical compression myelopathy despite treatment with ERT. The safety and effectiveness of ERT for AFD, MPS I, MPS II and MPS VI, as well as for PD have been demonstrated in well-designed clinical trials, and the treatments are now commercially available throughout the world 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45. However, except for PD (Invasive ventilator-free survival, changes in LVMI) efficacy end points did not include cardiac function. The heart is one of the major organs affected in patients with AFD, almost all male patients with classic AFD will develop hypertrophic cardiomyopathy if untreated. Thus cardiac disorders including conduction disturbances, valve disease and heart failure have been well documented; ERT results in a dramatic improvement in cardiac symptoms in a substantial number of patients [46]. To further assess objective cardiac benefits including reduction of cardiovascular morbidity and mortality in MPS disorders, data from large registries will be of utmost importance, due to the rarity of the individual disorders.
Thus many of these patients will hopefully reach adulthood underlining the necessity that adult cardiologists are also getting familiar with these rare diseases, although we recommend that most of these patients are followed-up in specialized centers.
4.6. Limitations
In our patient group, follow-up time was limited and averaged only 5.8 years. However, many patients with LSD die prematurely and do not reach adulthood, which explains part of the short follow-up. In our study, 8 patients died, one was lost to follow-up.
Unfortunately, detailed analysis of diastolic function is not available in all patients, most often due to a fast heart rate or diminished echo quality due to body habitus. Also, no detailed analysis with modern techniques (speckle tracking) has been performed routinely in older studies.
LSD is a rare disease. So, although these children and adolescents are all patients of a tertiary referral center, this is still a small group. Thus, comparisons between the groups are difficult to interpret.
We do not have data on polysomnography in all patients with pulmonary hypertension. Therefore, the exact etiology of pulmonary hypertension cannot be determined.
5. Conclusions
Echocardiographically detectable cardiovascular involvement is frequent in children with LSD. However, cardiovascular findings are rarely hemodynamically significant and mostly do not necessitate any intervention. Valvular heart disease occurs mainly in patients with MPS and oLSD but is rare in AFD. A short PR interval is a characteristic ECG abnormality. Routine repeat evaluation with echocardiography and ECG is recommended in children with LSD.
Footnotes
Available online 13 November 2013
References
- 1.Wappner R.S. Lysosomal storage disorders. In: McMillan J.A., Feigin R.D., editors. Oski's pediatrics. Principles and practice. Lippincott Williams & Wilkins; Phildelphia: 2006. p. 2199. [Google Scholar]
- 2.Cox G.F. Diagnostic approaches to pediatric cardiomyopathy of metabolic genetic etiologies and their relation to therapy. Prog Pediatr Cardiol. 2007;24(1):15–25. doi: 10.1016/j.ppedcard.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meikle P.J., Hopwood J.J., Clague A.E., Carey W.F. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 4.Linhart A., Elliott P.M. The heart in Anderson–Fabry disease and other lysosomal storage disorders. Heart. 2007;93(4):528–535. doi: 10.1136/hrt.2005.063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hopwood J.J., Morris C.P. The mucopolysaccharidoses. Diagnosis, molecular genetics and treatment. Mol Biol Med. 1990;7(5):381–404. [PubMed] [Google Scholar]
- 6.Kurihara M., Kumagai K., Goto K., Imai M., Yagishita S. Severe type Hunter's syndrome. Polysomnographic and neuropathological study. Neuropediatrics. 1992;23(5):248–256. doi: 10.1055/s-2008-1071352. [DOI] [PubMed] [Google Scholar]
- 7.Leal G.N., de Paula A.C., Leone C., Kim C.A. Echocardiographic study of paediatric patients with mucopolysaccharidosis. Cardiol Young. 2010;20(3):254–261. doi: 10.1017/S104795110999062X. [DOI] [PubMed] [Google Scholar]
- 8.Lopez L., Colan S.D., Frommelt P. Recommendations for quantification methods during the performance of a pediatric echocardiogram: a report from the Pediatric Measurements Writing Group of the American Society of Echocardiography Pediatric and Congenital Heart Disease Council. J Am Soc Echocardiogr. 2010;23(5):465–495. doi: 10.1016/j.echo.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 9.Lang R.M., Bierig M., Devereux R.B. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;8:1454–1457. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Devereux R.B., Alonso D.R., Lutas E.M. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. Feb 15 1986;57(6):450–458. doi: 10.1016/0002-9149(86)90771-x. [DOI] [PubMed] [Google Scholar]; J Am Soc Echocardiogr. 2005;18(12):1440–1463. doi: 10.1016/j.echo.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Mosteller R.D. Simplified calculation of body surface area. N Engl J Med. 1987 Oct 22;317(17):1098. doi: 10.1056/NEJM198710223171717. [DOI] [PubMed] [Google Scholar]
- 12.McMahon C.J., Nagueh S.F., Pignatelli R.H. Characterization of left ventricular diastolic function by tissue Doppler imaging and clinical status in children with hypertrophic cardiomyopathy. Circulation. 2004;109(14):1756–1762. doi: 10.1161/01.CIR.0000124723.16433.31. [DOI] [PubMed] [Google Scholar]
- 13.Bu'Lock F.A., Mott M.G., Martin R.P. Left ventricular diastolic function in children measured by Doppler echocardiography: normal values and relation with growth. Br Heart J. 1995;73(4):334–339. doi: 10.1136/hrt.73.4.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zoghbi W.A., Enriquez-Sarano M., Foster E. Recommendations for evaluation of the severity of native valvular regurgitation with two-dimensional and Doppler echocardiography. J Am Soc Echocardiogr. 2003;16(7):777–802. doi: 10.1016/S0894-7317(03)00335-3. [DOI] [PubMed] [Google Scholar]
- 15.Therneau T and original Report by Lumley T (2009). survival: Survival analysis, including penalised likelihood. R package version 2.35–8. http://CRAN.R-project.org/package=survival.
- 16.Kampmann C., Wiethoff C.M., Whybra C., Baehner F.A., Mengel E., Beck M. Cardiac manifestations of Anderson–Fabry disease in children and adolescents. Acta Paediatr. 2008;97(4):463–469. doi: 10.1111/j.1651-2227.2008.00700.x. [DOI] [PubMed] [Google Scholar]
- 17.Linhart A., Kampmann C., Zamorano J.L. Cardiac manifestations of Anderson–Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;28(10):1228–1235. doi: 10.1093/eurheartj/ehm153. [DOI] [PubMed] [Google Scholar]
- 18.Weidemann F., Strotmann J.M., Niemann M. Heart valve involvement in Fabry cardiomyopathy. Ultrasound Med Biol. 2009;35(5):730–735. doi: 10.1016/j.ultrasmedbio.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Dangel J.H. Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders—clinical and echocardiographic findings in 64 patients. Eur J Pediatr. 1998;157(7):534–538. doi: 10.1007/s004310050872. [DOI] [PubMed] [Google Scholar]
- 20.Cindik N., Ozcay F., Süren D. Gaucher disease with communicating hydrocephalus and cardiac involvement. Clin Cardiol. 2010;33(1):E26–E30. doi: 10.1002/clc.20348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cripe L.H., Ware S.M., Hinton R.B. Replacement of the aortic valve in a patient with mucolipidosis III. Cardiol Young. 2009;19(6):641–643. doi: 10.1017/S1047951109991120. [DOI] [PubMed] [Google Scholar]
- 22.Elliott P., Baker R., Pasquale F. ACES study group. Prevalence of Anderson–Fabry disease in patients with hypertrophic cardiomyopathy: the European Anderson–Fabry Disease survey. Heart. 2011;97(23):1957–1960. doi: 10.1136/heartjnl-2011-300364. [DOI] [PubMed] [Google Scholar]
- 23.Kampmann C., Linhart A., Baehner F. Onset and progression of the Anderson–Fabry disease related cardiomyopathy. Int J Cardiol. 2008;130(3):367–373. doi: 10.1016/j.ijcard.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Balmer C., Ballhausen D., Bosshard N.U., Steinmann B., Boltshauser E., Bauersfeld U. Familial X-linked cardiomyopathy (Danon disease): diagnostic confirmation by mutation analysis of the LAMP2gene. Eur J Pediatr. 2005;164(8):509–514. doi: 10.1007/s00431-005-1678-z. [DOI] [PubMed] [Google Scholar]
- 25.Maron B.J., Roberts W.C., Arad M. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA. 2009;301(12):1253–1259. doi: 10.1001/jama.2009.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Z., Cui Q., Tian Z. Danon disease as a cause of concentric left ventricular hypertrophy in patients who underwent endomyocardial biopsy. Eur Heart J. 2012;33(5):649–656. doi: 10.1093/eurheartj/ehr420. [DOI] [PubMed] [Google Scholar]
- 27.John A., Fagondes S., Schwartz I. Sleep abnormalities in untreated patients with mucopolysaccharidosis type VI. Am J Med Genet A. 2011;155A(7):1546–1551. doi: 10.1002/ajmg.a.33902. [DOI] [PubMed] [Google Scholar]
- 28.O'Mahony C., Elliott P. Anderson–Fabry disease and the heart. Prog Cardiovasc Dis. 2010;52(4):326–335. doi: 10.1016/j.pcad.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Namdar M., Kampmann C., Steffel J. PQ interval in patients with Fabry disease. Am J Cardiol. 2010;105(5):753–756. doi: 10.1016/j.amjcard.2009.10.056. [DOI] [PubMed] [Google Scholar]
- 30.Toda Y., Takeuchi M., Morita K. Complete heart block during anesthetic management in a patient with mucopolysaccharidosis type VII. Anesthesiology. 2001;95(4):1035–1037. doi: 10.1097/00000542-200110000-00041. [DOI] [PubMed] [Google Scholar]
- 31.Hishitani T., Wakita S., Isoda T., Katori T., Ishizawa A., Okada R. Sudden death in Hunter syndrome caused by complete atrioventricular block. J Pediatr. 2000;136(2):268–269. doi: 10.1016/s0022-3476(00)70117-x. [DOI] [PubMed] [Google Scholar]
- 32.Krovetz J., Schiebler G. Cardiovascular manifestations of genetic mucopolysaccharidoses. Virth Defects. 1972;8:192. [Google Scholar]
- 33.Doi Y., Toda G., Yano K. Sisters with atypical Fabry's disease with complete atrioventricular block. Heart. 2003;89(1):e2. doi: 10.1136/heart.89.1.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spada M., Chiappa E., Ponzone A. Cardiac response to enzyme-replacement therapy in Gaucher's disease. N Engl J Med. 1998;339(16):1165–1166. doi: 10.1056/NEJM199810153391615. [DOI] [PubMed] [Google Scholar]
- 35.Schiffmann R., Kopp J.B., Austin H.A., III, Sabnis S., Moore D.F., Weibel T. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;6(285(21)):2743–2749. doi: 10.1001/jama.285.21.2743. [DOI] [PubMed] [Google Scholar]
- 36.Eng C.M., Banikazemi M., Gordon R.E. A phase 1/2 clinical trial of enzyme replacement in Fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet. 2001;68(3):711–722. doi: 10.1086/318809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eng C.M., Guffon N., Wilcox W.R. Safety and efficacy of recombinant human alpha-galactosidase A—replacement therapy in Fabry's disease. N Engl J Med. 2001 Jul 5;345(1):9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 38.Wilcox W.R., Banikazemi M., Guffon N. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75(1):65–74. doi: 10.1086/422366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banikazemi M., Bultas J., Waldek S. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;16(146(2)):77–86. doi: 10.7326/0003-4819-146-2-200701160-00148. [DOI] [PubMed] [Google Scholar]
- 40.Kakkis E.D., Muenzer J., Tiller G.E. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med. 2001;18(344(3)):182–188. doi: 10.1056/NEJM200101183440304. [DOI] [PubMed] [Google Scholar]
- 41.Wraith J.E., Clarke L.A., Beck M. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-l-iduronidase (laronidase) J Pediatr. 2004;144(5):581–588. doi: 10.1016/j.jpeds.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 42.Harmatz P., Giugliani R., Schwartz I.V. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux–Lamy syndrome) J Pediatr. 2004;144(5):574–580. doi: 10.1016/j.jpeds.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 43.Muenzer J., Gucsavas-Calikoglu M., McCandless S.E., Schuetz T.J., Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome) Mol Genet Metab. 2007;90(3):329–337. doi: 10.1016/j.ymgme.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 44.Kishnani P.S., Corzo D., Nicolino M. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;9(68(2)):99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 45.Harmatz P., Giugliani R., Schwartz I. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase Bor rhASB) and follow-on, open-label extension study. J Pediatr. 2006;148(4):533–539. doi: 10.1016/j.jpeds.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 46.Mehta A., Beck M., Sunder-Plassmann G., editors. In: Fabry disease: perspectives from 5 years of FOS. Oxford PharmaGenesis; Oxford: 2006. [PubMed] [Google Scholar]
- 47.Kishnani P.S., Howell R.R. Pompe disease in infants and children. J Pediatr. 2004;144(5 Suppl.):S35–S43. doi: 10.1016/j.jpeds.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 48.Satoh Y., Sakamoto K., Fujibayashi Y. Cardiac involvement in mucolipidosis. Importance of non-invasive studies for detection of cardiac abnormalities. Jpn Heart J. 1983;24(1):149–159. doi: 10.1536/ihj.24.149. [DOI] [PubMed] [Google Scholar]
- 49.Rosengarten D., Abrahamov A., Nir A. Outcome of ten years' echocardiographic follow-up in children with Gaucher disease. Eur J Pediatr. 2007;166(6):549-5. doi: 10.1007/s00431-006-0276-z. [DOI] [PubMed] [Google Scholar]