

Abstract
Objective
Primary Sclerosing Cholangitis (PSC) is a chronic cholestatic liver disease that is characterized by severe peri-biliary tract inflammation and fibrosis, elevated oxidative stress and hepatocellular injury. A hallmark of PSC patients is the concurrent diagnosis of Inflammatory Bowel Disease occurring in approximately 70%–80% of PSC patients (PSC/IBD). The objective of this study was to determine the impact of end stage PSC/IBD on cellular antioxidant responses and the formation of protein carbonylation.
Methods
Using hepatic tissue and whole cell extracts isolated from age-matched healthy humans and patients diagnosed with end stage PSC/IBD, overall inflammation, oxidative stress, and protein carbonylation were assessed by Western blotting, and immunohistochemistry.
Results
Increased immunohistochemical staining for CD3+ (lymphocyte), CD68 (Kupffer cell) and myeloperoxidase (neutrophil) colocalized with the extensive Picrosirius red stained fibrosis confirming the inflammatory aspect of PSC. Importantly, the increased inflammation also colocalized with elevated periportal post-translational modification by the reactive aldehydes 4-HNE, MDA and acrolein. 4-HNE, MDA and acrolein IHC all displayed a significant component in hepatocytes adjacent to fibrotic regions. Furthermore, acrolein was also elevated within the nuclei of periportal inflammatory cells whereas MDA staining was increased in hepatocytes across the lobule. Prussian Blue staining, when compared to the positive controls (ALD, NASH), did not display any evidence of iron accumulation in PSC/IBD livers. Western analysis of PSC/IBD anti-oxidant responses revealed elevated expression of SOD2, GSTπ as well as upregulation of Akt Ser473 phosphorylation. In contrast, expression of GSTμ, GSTA4, catalase, Gpx1 and Hsp70 were suppressed. These data were further supported by a significant decrease in measured GST activity. Dysregulation of anti-oxidant responses in the periportal region of the liver was supported by elevated SOD2 and GSTπ IHC signals in periportal hepatocytes and cholangiocytes. Expression of the Nrf2-regulated proteins HO-1, NAD(P)H quinone reductase (NQO1) and Gpx1 was primarily localized to macrophages. In contrast, catalase staining decreased within periportal hepatocytes and was not evident within cholangiocytes.
Conclusions
Results herein provide additional evidence that cholestasis induces significant increases in periportal oxidative stress and suggest that there are significant differences in the cellular and subcellular generation of reactive aldehydes formed during cholestatic liver injury. Furthermore, these data suggest that anti-oxidant responses are dysregulated during end-stage PSC/IBD supporting pathological data. This work was funded by NIH 5R37AA009300-22 D.R.P.
Keywords: Aldehyde, Cholestasis, Oxidative Stress, cholangiocyte, Lipid peroxidation
Background
The orphan disease, Primary Sclerosing Cholangitis (PSC) is a progressive cholestatic liver disease of unknown etiology characterized by biliary inflammation, fibrosis, and stricturing of the intra and/or extra-hepatic bile ducts (Eaton et al., 2013). The incidence rate is ~1:100,000 person-years with no medical therapies currently available. Long-term disease progression leads to biliary obstruction, repeated bouts of cholangitis, and secondary biliary cirrhosis with a median time of survival following diagnosis of 12–18 years (Eaton et al., 2013). In the absence of liver transplant (LTx), patients carry a 10–20% lifetime risk of hepatobiliary malignancy. Furthermore, 20–40% develop recurrent disease even after LTx (Hirschfield et al., 2013; Kugelmas et al., 2003; Tamura et al., 2007). A primary risk factor for PSC is inflammatory bowel disease (IBD) which is present in at least 70% of PSC patients but has been reported in upwards of 80% making PSC/IBD the primary subtype of PSC (Jiang and Karlsen, 2017; Lunder et al., 2016; Weismuller et al., 2017).
Oxidative stress is a major contributing factor in regulating cellular protein post-translational modifications. In particular, the formation of reactive oxygen species (ROS) during chronic inflammation is hypothesized to be central to the progression of chronic liver diseases (Albano, 2006). ROS are produced by the mitochondrial respiratory chain, the cytochrome P450 system, auto-oxidation of heme proteins, the NADPH oxidase complex, xanthine oxidase, oxidative enzymes and other cellular systems (Aubert et al., 2011). While ROS are important in signal transduction, cellular physiology, and are involved in critical metabolic pathways, a high concentration of ROS can result in hepatocellular damage, apoptosis and necrosis (Leung and Nieto, 2013). ROS can also lead to a free radical chain reaction with unsaturated fatty acids generating toxic electrophilic α/β unsaturated aldehydes, a process called lipid peroxidation. Post-translational modification of proteins by products of lipid peroxidation has been implicated as a contributing factor in the progression of chronic liver disease (Osna et al., 2016; Sutti et al., 2014). We have recently demonstrated that elevated protein carbonylation is present in both end-stage alcoholic liver disease (ALD) as well as end-stage non-alcoholic liver disease (NASH) both of which possess elevated steatosis (Shearn et al., 2015b; Shearn et al., 2017).
Biliary cholestasis is implicitly connected to inflammation and oxidative stress and therefore hepatocellular damage (Bell et al., 2015; Salem et al., 2003; Tan et al., 2015; Vendemiale et al., 2002). In the liver, an important marker for increased oxidative stress is elevated levels of hepatic lipid peroxidation that generates electrophilic α/β unsaturated fatty acids such as 4-hydroxynonenal (4-HNE), acrolein and malondialdehyde (MDA). Due to a strong periportal localization, cholestatic inflammation and oxidative stress is unique compared to other well characterized diseases such as alcoholism (Kawamura et al., 2000; Osna et al., 2016; Shearn et al., 2015b). In cell culture, hydrophobic bile acids induce lipid peroxidation in macrophages (Ljubuncic et al., 1996). Elevated products of lipid peroxidation are present in the serum of patients diagnosed with Primary Biliary Cholangitis (Aboutwerat et al., 2003) and PSC patients, where elevated levels of lipid peroxides corresponded to decreased serum anti-oxidant capacity (Salem et al., 2003). In support of these data, when combined with ciprofloxacin, addition of the anti-oxidant N-acetylcysteine (NAC) significantly decreased hepatic enzymes and inflammation (Alkaline phosphatase, Gamma Glutmyl Transferase (GGT), % neutrophils) in 2 patients with partial biliary obstruction suggesting a neutrophil/oxidative stress component in cholestasis (Ozdil et al., 2010). In Primary Biliary Cholangitis (PBC), markers of protein carbonylation are upregulated corresponding to downregulation of Nrf2 (Wasik et al., 2017). PBC however, at least in part, originates from autoantibodies that cause chronic inflammation and hepatic injury (Trivedi et al., 2017). Even though both PBC and PSC are cholangiopathies, and an autoimmune component to PSC has been described, the fact that 70–80% of PSC patients also have a co-diagnosis of IBD making PSC clearly multifactorial and indicating that its origin is unclear (Jiang and Karlsen, 2017). The impact of PSC/IBD on protein lipid peroxidation and anti-oxidant responses within the liver has not been evaluated.
In the present study we have examined hepatocellular lipid peroxidation and antioxidant responses in tissue obtained from end-stage PSC patients that possess a combined diagnosis of PSC/IBD as observed in the majority of PSC patients. Our hypothesis was that we would find elevated lipid peroxidation products in the areas of active/ongoing pathology. We find that hepatic protein carbonylation is elevated and that anti-oxidant responses are dysregulated in tissue obtained from end-stage PSC/IBD patients. When compared to previously published reports regarding PBC, PSC may possess distinct hepatocellular anti-oxidant responses further supporting the uniqueness of PSC.
Methods
Sample procurement
To determine the status of protein carbonylation and acetylation during cholestasis, paraffin embedded and frozen hepatic tissue from normal and end stage PSC/IBD patients (N=9 PSC/IBD, 8 Normal) procured during transplantation (ages 25–62, Male/Female) were obtained from the University of Minnesota Liver Tissue cell Distribution Center NIH Contract #HHSN276201200017C. Whole cell extracts (WCE) of each sample was prepared by dounce homogenization (10X) of tissue resuspended in 50mM tricine pH 8.0, 0.001M NaCl plus phosphatase and protease inhibitors (SIGMA ALDRICH, St Louis, MO) followed by sonication (3X15 seconds@ 4°C). To remove debris, samples were centrifuged at 14,000RPM (16,000g)(4°C) for 10 minutes. Supernatants were drawn off and immediately flash frozen in liquid N2.
Histological Evaluation
To detect fibrosis, formalin fixed slides normal and end stage PSC patients (N=9 PSC, 8 Normal) were stained with Picro-Sirius (PSR). For Prussian blue staining, PSC, Normal, NASH (Shearn et al., 2017) and ALD (Osna et al., 2016) (N=6 per condition), slides were stained according to established protocols. In addition, immunohistochemical staining for 4-HNE, rabbit polyclonal(Shearn et al., 2014), acrolein rabbit polyclonal (Cell Sciences, Newburyport, MA), malondialdehyde (MDA) rabbit polyclonal (ABCAM, Billerica, MA), myeloperoxidase (MPO) goat polyclonal (Millipore, Billerica, MA), Rabbit polyclonal CD3 (DAKO/Agilent Santa Clara, CA), Rat Anti CD68 polyclonal (DAKO), catalase rabbit polyclonal (SIGMA ALDRICH, St. Louis, MO), and mitochondrial superoxide dismutase (SOD2) goat polyclonal (ABCAM, Billerica, MA), GSTπ rabbit polyclonal (MBL International, Woburn, MA), Glutathione Peroxidase rabbit polyclonal (ABCAM), Heme oxygenase rabbit polyclonal (ENZO Life Sciences, Farmingdale, N.Y.), cytokeratin 7 (ck7) rabbit polyclonal (ABCAM) was completed using citrate pH 6.0 antigen retrieval and the Biocare heating system (Biocare, Medical, Pacheco, CA) @100°C for 10 min as previously described (Shearn et al., 2013a). Histologic images were captured on an Olympus BX51 microscope equipped with a four-megapixel Macrofire digital camera (Optronics; Goleta, CA) using the PictureFrame Application 2.3 (Optronics). All images were cropped and assembled using Photoshop CS2 (Adobe Systems, Inc.; Mountain View, CA).
Western blotting
Western blotting for SOD2 (ABCAM, Cambridge, MA), catalase (Sigma, Saint Louis, MO), Superoxide dismutase 1 (ABCAM, Cambridge, MA), Glutathione Peroxidase 1 (Gpx1) (ABCAM, Billerica, MA), Heat Shock Protein 70 (Stressgen/Enzo Lifesciences Farmingdale, NY), GSTA4 (Protein Tech, Chicago, IL), GSTπ (MBL International, Woburn, MA), GSTμ (ABCAM, Cambridge, MA), and anti-GAPDH (Millipore, Billerica, MA) was performed from 10μg of whole cell liver extracts as previously described (Shearn et al., 2014; Shearn et al., 2011; Shearn et al., 2013a; Shearn et al., 2013b). Quantification of expression of each protein was performed using ImageJ (NIH) and normalized to overall GAPDH expression.
Statistical Analysis
The data are presented as means ± Standard Error (SE). Comparisons between normal and PSC tissue was accomplished by Student’s T-tests. Statistical significance was set at P<0.05. Prism 5 for Windows (GraphPad Software, San Diego, CA) was used to perform all statistical tests.
Results
For this study, fresh frozen human hepatic tissue and formalin fixed tissue from normal and end stage PSC/IBD patients was obtained prior to transplant from the University of Minnesota Liver Tissue cell Distribution Center (NIH Contract #HHSN276201200017C). The mean age of patients was 45.67 years old. For each patient relevant hepatic parameters was provided (Model for End-stage Liver Disease (MELD) (Kamath et al., 2007), aspartate aminotransferase (AST), International Normalized Ratio of prothrombin coagulation (INR), serum bilirubin, alkaline phosphatase and albumin). As shown in Table S1, all patients possessed increased MELD scores indicative of severe hepatic dysfunction. Examining individual parameters, INR (normal approximately 1.0), total bilirubin (adult normal range 0.1 – 1.3 mg/dL), serum AST (normal range 12 – 39 U/L), alkaline phosphatase (normal range 39 – 117 U/L), were all elevated and serum albumin (normal range 3.5–5.7g/dL) levels mildly suppressed.
Cirrhosis is characterized by a marked increase in fibrosis. To examine the extent of fibrosis in the tissues that were procured, tissue sections obtained from human PSC patients were stained with Hematoxylin and Eosin (H&E) and Picrosirius red. From the H&E staining shown in Figure S1 Panels A, E, as expected, end-stage PSC/IBD hepatic tissue exhibited “onion skin” biliary ducts characteristic of PSC. Overall, significant cirrhosis was evident but there was no evidence of cholangiocarcinoma in tissue obtained for any of the patients. As shown in Figure S1 Panels B, C, tissue from healthy donors did not exhibit Picrosirius red staining characteristic of abnormal fibrotic networks within the periportal and centrilobular regions of the liver. There was however some mild steatosis present in some normal samples (Data not shown). In patients with PSC, significant picrosirius staining indicative of bridging fibrosis and cirrhosis was clearly evident (Figure S1 Panels F, G). To further characterize fibrosis in PSC/IBD, immunohistochemical staining of α-smooth muscle actin (αSMA) staining was performed. As shown in Figure S1 Panels D, H, increased staining of αSMA was also present in PSC/IBD supporting the PSR data.
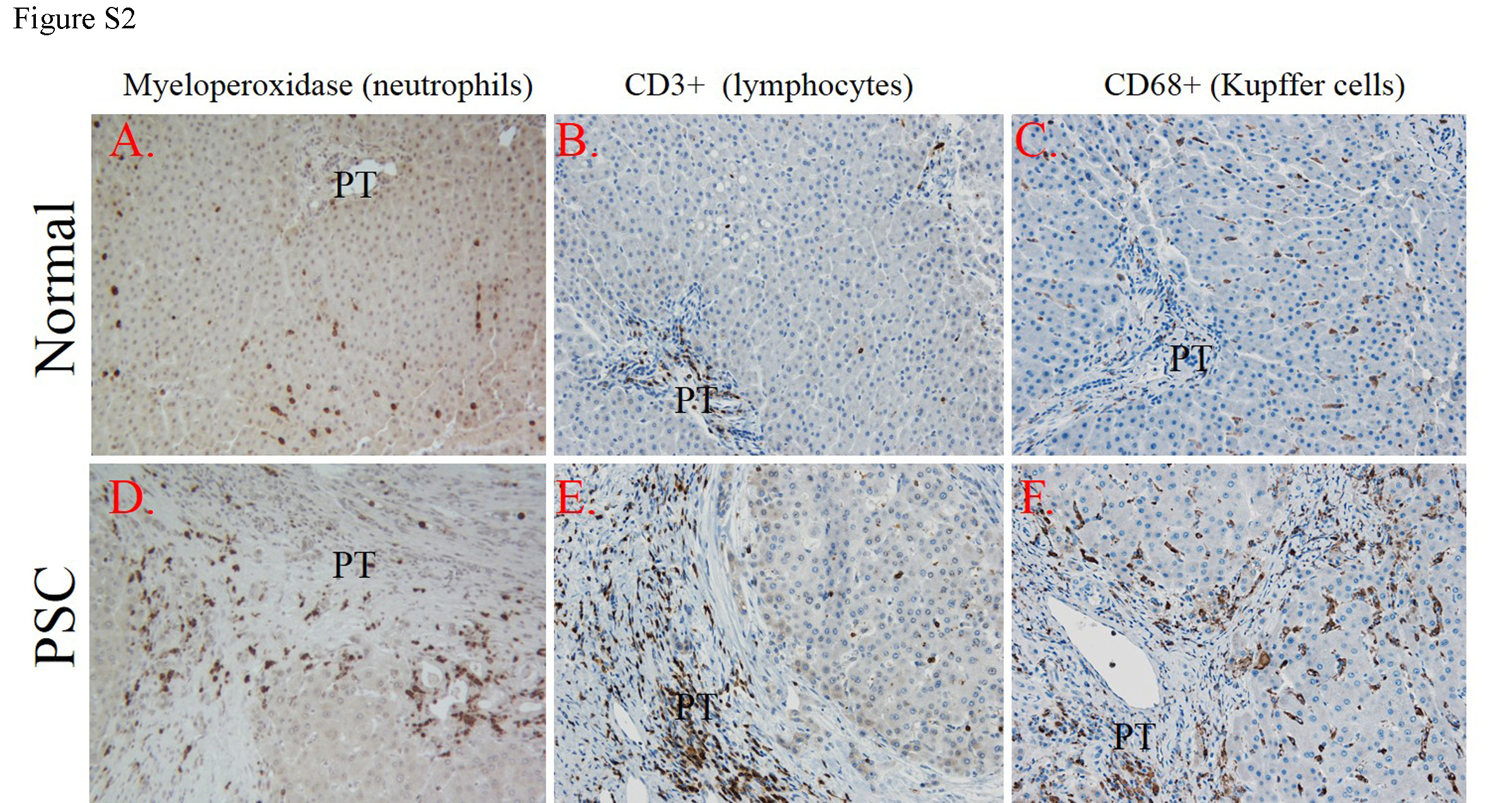
Primary Sclerosing cholangitis is characterized by significant periportal inflammation. Neutrophil infiltration and activation of the NADPH oxidase is a hallmark of PSC and can act as a significant source of reactive species that contribute to the formation of reactive aldehydes. To validate the increased neutrophil presence in tissue from these end-stage PSC patients, tissue sections were probed for myeloperoxidase (MPO). As shown in Figure S2 Panels A, D, in normal tissue, only modest numbers of MPO positive neutrophils were dispersed throughout the hepatic lobes. In PSC/IBD tissue, neutrophil numbers were significantly increased in the periportal region adjacent to the fibrosis. To further explore the inflammatory infiltrate in human PSC/IBD, tissue sections were subsequently stained for CD3+ lymphocytes and CD68+ Kupffer cells. In normal tissue, sparse numbers of CD3+ lymphocytes were randomly dispersed throughout the lobule. In PSC/IBD, periportal infiltration of CD3+ lymphocytes was very prominent with few CD3+ lymphocytes distributed in the centri-lobular region supporting the extreme hepatic localization of this disease (Figure S2 Panels B, E). Furthermore, although CD68+ cells were evenly dispersed throughout the lobule in normal tissue, in PSC/IBD, increased infiltrating CD68+ cells were evident in both the periportal region as well as in scattered foci across the lobule (Figure S2 Panels C, F).
Elevated products of lipid peroxidation and subsequent post-translational modification (protein carbonylation) are validated markers of oxidative stress during chronic liver disease (Osna et al., 2016). Pathophysiology of carbonylation has not been examined in end-stage patients diagnosed with PSC. Using IHC, tissue sections isolated from human end-stage patients were probed for protein carbonylation by the reactive aldehydes 4-HNE, acrolein and malondialdehyde (MDA). As shown in Figure 1A, in normal hepatic tissue, 4-HNE staining was not significant. Examining the tissue sections isolated from PSC/IBD patients revealed dramatically increased 4-HNE staining that primarily was in periportal hepatocytes that are adjacent to fibrotic tissue as well as in the cytosol of cholangiocytes. No staining was evident in inflammatory cells. Protein modification by the reactive aldehyde acrolein also is known to occur in models of chronic liver inflammation (Galligan et al., 2012; Shearn et al., 2016). From Figure 1A, acrolein staining was not significant in normal tissue but in end stage PSC tissue, periportal acrolein staining was prominent within fibrotic tissue, periportal inflammatory cells, and in the adjacent surrounding hepatocytes. In addition, scattered acrolein positive inflammatory cells with increased nuclear staining were present interspersed between hepatocytes as well as within fibrotic tissue. Significant elevation of MDA is present in serum isolated from PSC patients. Whether or not MDA is also elevated in the liver of PSC patients has not been investigated. As shown in Figure 1A, in normal tissue, minor MDA staining was present within punctate structures of hepatocytes around the central vein. In PSC/IBD tissue, numerous MDA positive cells were present along areas of damaged tissue forming a checkered pattern with high, moderate and low levels of staining. MDA positivity also was present within isolated hepatocytes across the hepatic lobe. MDA staining however was not punctuate, instead staining was throughout the hepatocyte with elevated staining along the plasma membrane as well as within fibrotic areas. These lipid peroxidation IHC results demonstrate that protein post-translational modification by 4-HNE, MDA and acrolein is elevated in PSC/IBD and that these modifications are occurring both in inflammatory cells and adjacent hepatocytes.
Figure 1. Immunohistochemical analysis of protein carbonylation in human PSC/IBD.

A. Elevated protein carbonylation in PSC/IBD. Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically at 200X magnification of slides using polyclonal antibodies directed against 4-HNE, acrolein and MDA. B. Examination of protein carbonylation and inflammation in the portal triad in end stage PSC/IBD. Figures are representative of hepatic tissue isolated from four normal, and four PSC patients respectively (PT=portal triad).
Focusing on the biliary tract, to further explore the impact of aldehyde protein modification as well as the contribution of infiltrating inflammatory cells, sections were stained for 4-HNE, acrolein, MDA, CD68+, MPO and CD3+. Contrary to 4-HNE staining, acrolein staining was localized in punctate structures within cholangiocytes (Figure 1B-red arrows). In the periportal area, MDA positive staining was evident within connective tissue in and around inflammatory cells (Figure 1B yellow arrows). Within the bile duct, scattered cholangiocytes also exhibited the presence of MDA adducts (Figure 1B red arrows). Only scant 4-HNE IHC staining was observed. Comparing all three aldehyde IHC staining patterns with IHC staining patterns for MPO+ neutrophils and CD3+ lymphocytes revealed that in hepatocytes all three aldehyde modifications possessed neutrophils nearby (Figure 1B). Within fibrotic tissue, both MDA and acrolein staining occurred in or near CD3+ lymphocytes but not neutrophils. In contrast, CD68+ macrophages were interspersed among hepatocytes, were elevated in numbers near the inner edge in zone 1 and were present between cholangiocytes within the bile duct (Figure 1B Pink arrow). These results indicate that production of reactive aldehydes in PSC/IBD may be multifactorial and the most likely inflammatory cell source of these adductive molecules are macrophages.
By its ability to assist in the formation of toxic hydroxyl radicals via Fenton reactions, iron has been shown to contribute to the pathogenesis of non-alcoholic fatty liver disease (Datz et al., 2017). From the Fenton reaction, increased hydroxyl radicals can subsequently react with lipid to form lipid peroxides. In addition, recent data has indicated that elevated oxidative stress may contribute to oval cell proliferation and the ductular reaction during chronic liver disease (Gouw et al., 2011; Wang et al., 2014). To determine if an increase in hepatic iron concentrations may be a source of radicals contributing to the formation of products of lipid peroxidation, tissue sections were stained for iron accumulation using Prussian Blue. As a positive control, tissue sections isolated from end stage NASH and ALD patients were used (Datz et al., 2017; Harrison-Findik, 2007; Harrison-Findik, 2010; Shearn et al., 2015b). As shown in Figure 2 Panels A–D, contrary to ALD (6/6) and NASH (6/6), tissue obtained from PSC patients did not possess detectable iron (0/6). Examining the ductular reaction, cytokeratin 7 staining revealed significant oval cell proliferation in all three diseases but PSC and NASH also exhibited increased staining of adjacent hepatocyte-like cells (arrows)(Gouw et al., 2011). These data indicate that elevated reactive aldehydes that are present in PSC/IBD are not derived from radicals generated from the Fenton reaction.
Figure 2. Impact of PSC/IBD on hepatic iron accumulation and its correlation to oval cell proliferation. Panels.

A–D Tissue sections from normal, end-stage PSC/IBD, end-stage ALD and end-stage NASH were evaluated for iron accumulation using Prussian blue staining. Panels E–H. Cytokeratin 7 (CK7) staining of normal, PSC/IBD, ALD and NASH sections (Yellow arrows indicate hepatocyte-like cells that are CK7 positive). Figures are representative of hepatic tissue isolated from at least 4 normal, and 6 PSC/IBD, 6 ALD and 6 NASH patients respectively. All images are 200x magnification (PT=portal triad, CV-central vein).
Glutathione S-transferase (GST) isozymes A4, μ and π have all been proposed to possess catalytic glutathione conjugating activity towards lipid aldehydes (Berhane et al., 1994; Chen et al., 2016; Engle et al., 2004; Li et al., 2013; Ronis et al., 2015). To determine the impact of elevated inflammation and protein carbonylation on anti-oxidant responses, expression of the key anti-reactive aldehyde enzymes GSTA4, GSTπ, GSTμ as well as the anti-oxidant enzymes mitochondrial superoxide dismutase (SOD2), cytosolic superoxide dismutase (SOD1), catalase, glutathione peroxidase (Gpx1). In addition expression of the heat shock protein Hsp70 was examined (Figure 3A). From the quantification, examining all proteins, both SOD2 and GSTπ expression were significantly elevated in PSC/IBD. In contrast, catalase, GSTμ, GSTA4 and Gpx1 were all suppressed when compared to normal hepatic tissue. Expression of SOD1 exhibited no significant difference. In other chronic hepatic disorders, a decrease in hepatic GST activity also corresponds with elevated oxidative stress (Shearn et al., 2015a; Shearn et al., 2015b). These data indicate isoform specific dysregulation of GST’s. To determine the status of GST activity in end-stage PSC/IBD, an activity assay was performed using 1-chloro-dinitrobenzoic acid as a substrate and whole cell extracts. As shown in Figure 3B, GST activity is suppressed by approximately 30% in human PSC/IBD. Combined, these data support dysregulation of anti-oxidant responses in end-stage PSC/IBD which may be contributing to the observed elevation in reactive aldehyde production. Furthermore, both catalase and Gpx1 expression decreased in PSC/IBD supporting a possible defect in the ability of livers from PSC/IBD patients to detoxify hydrogen peroxide.
Figure 3. Dysregulation of oxidative stress responses in end-stage PSC/IBD.

(A) Western analysis of overall expression of SOD1, SOD2, catalase, Gpx1, NQO1, HO-1, Hsp70, GSTA4, GSTμ and GSTπ. Western blots were performed as described using 10μg of whole cell extracts prepared from normal, PSC/IBD hepatic tissue. Expression was normalized against GAPDH expression (B) Overall GST activity in whole cell extracts prepared from normal and PSC/IBD patients. Data are Means+/−SEM, N=6 patients per group, *p<0.05, ***p<0.001.
Western blotting data was determined from whole cell lysates but it does not provide insight into the specific hepatocellular region where dysregulation is occurring. Not all antibodies that are validated for Western blotting are also valid for immunohistochemistry. We were able to perform immunohistochemical localization of SOD2, catalase, glutathione peroxidase (Gpx) GSTπ, as well as additional Nrf2 targets Heme oxygenase (HO-1) and NAD(P)H Quinone Dehydrogenase 1 (NQO1) was performed. From these IHC, in normal tissue, SOD2 expression is primarily centrilobular whereas in PSC tissue, SOD2 expression is significantly increased in hepatocytes that ringed damaged tissue in the periportal region (Figure 4). Western blotting indicated increased GSTπ expression in PSC/IBD, from the histochemical staining, in normal tissue, GSTπ expression was present in Kupffer cells across the lobule as well as cholangiocytes in the biliary tract. In PSC/IBD, GSTπ expression was elevated in hepatocytes, infiltrating inflammatory cells and within cholangiocytes in the periportal region. No significant differences in staining was evident in other regions of the lobule. Examining catalase, in normal tissue, catalase expression was primarily localized around the central vein. In PSC, staining for catalase was not significantly different when compared to control tissue. An immunohistochemical analysis of Glutathione peroxidase 1 expression has not been reported in human liver. In normal tissue, Gpx1 staining is most prominent within resident Kupffer cells. In PSC/IBD, Gpx1 staining is not noticeably different with the exception that there are more macrophages present in the periportal region. In murine models of chronic liver disease, HO-1 has been reported to be expressed primarily in macrophages with minor expression in hepatocytes (Bakhautdin et al., 2014; Jais et al., 2014). From HO-1 staining, HO-1 expression is also limited to macrophages in normal tissue. In PSC/IBD, expression was not markedly different with the exception that there is increased HO-1 staining of inflammatory infiltrates. NAD(P)H quinone oxidoreductase 1 (NQO1) is not normally significantly expressed in human hepatic tissue. In PBC, NQO1 expression is induced in the periportal region. We sought to determine if NQO1 induction also occurred in PSC/IBD. From Figure 4, in normal tissue, NQO1 expression was evident primarily in resident Kupffer cells. In tissue isolated from PSC/IBD patients, overall NQO1 staining was slightly increased with substantial increases in staining of periportal inflammatory infiltrates, in scattered hepatocytes adjacent to damaged tissue, and in the sinusoidal Kupffer cells.
Figure 4. Immunohistochemical analysis of antioxidant responses in end stage PSC/IBD.

A. Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against SOD2, Catalase, GSTπ, Gpx1, Heme Oxygenase 1, and NQO1 (PT=portal triad, CV-central vein). B. Colocalization of catalase and SOD2 using fluorescent microscopy. Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against catalase and SOD2 followed by FITC-conjugated anti-rabbit and Alexa Fluor 615-conjugated anti-goat secondary antibodies. Slides were examined using confocal microcopy (Green=SOD2, Red=Catalase, Blue= Hoechst 33342 nuclear staining). Figures are representative of hepatic tissue isolated from four normal and four PSC patients respectively.
To determine the specific effects of cholestasis, fibrosis and inflammation on both cholangiocytes as well as hepatocytes, dual fluorescent staining of tissue sections was used. As shown in Figure 4B, SOD2 (Green) is apparent in hepatocytes (Panel A) as well as in cholangiocytes (Panel B). Catalase expression (Red) is present within interlobular hepatocytes and to a small degree in a few of the cholangiocytes but is not evident in SOD2 positive hepatocyte located adjacent to the fibrosis. These immunohistochemical data strongly support dysregulation of anti-oxidant responses in hepatic tissue isolated from PSC patients which may be contributing to the observation of elevated protein carbonylation.
Elevated H2O2 concentrations occur during hepatic injury (Roskams et al., 2003). Exposure to high concentrations of H2O2 can contribute to cell proliferation due to H2O2-dependent inactivation of the lipid phosphatase PTEN and activation of its downstream target Akt (Cho et al., 2004; Covey et al., 2010; Lee et al., 2002). Elevated oxidative stress may be potentiating suppression of catalase by an Akt-dependent mechanism (Venkatesan et al., 2007). To determine if there is increased Akt activation in human PSC/IBD, pSer473Akt was examined in whole cell extract. From Fig 5, pSer473Akt is elevated in PSC, supporting the hypothesis of H2O2/Akt-dependent suppression of catalase expression.
Figure 5. Increased Akt activation in end-stage PSC/IBD.

Western analysis of total Akt and phospho-Ser473 Akt expression. Western blots were performed as described using 10μg of whole cell extracts prepared from normal, PSC/IBD hepatic tissue and a 7% SDS-PAGE gel. Expression was normalized against GAPDH expression. Data are Means+/−SEM, N=6 patients per group, ***p<0.001.
Discussion and Conclusions
Previous studies have shown that oxidative stress markers such as 4-HNE and MDA are elevated in murine models of cholestasis such as the bile duct ligation model as well as in Mdr2−/− mice (Fickert et al., 2006; Parola et al., 1996; Peres et al., 2000). In addition, elevated lipid peroxidation and decreased anti-oxidant capacity in the form of suppressed concentrations of serum GSH have been demonstrated in patients with cholestatic liver disease (Aboutwerat et al., 2003). The data presented in this article extends these previous observations into hepatic tissue isolated from patients diagnosed with PSC/IBD.
Our data demonstrate lipid peroxidation is strongly periportal in PSC/IBD with staining of all three aldehydes dramatically increasing in areas surrounding fibrotic tissue supporting the biliary origins of PSC. Yet there are subtle differences in both the cellular and subcellular localization of individual aldehydes. Comparing acrolein, acrolein staining is strongly evident in inflammatory cells whereas inflammatory cells clearly did not possess 4-HNE positivity. Furthermore, within cholangiocytes, 4-HNE staining was diffuse throughout the cells but acrolein strongly localized in punctate vesicles. Comparing 4-HNE and acrolein staining with a 3rd aldehyde MDA, there are more differences. In agreement with both 4-HNE and acrolein, MDA staining increased in areas of damaged tissue, yet MDA positivity also extended into scattered hepatocytes across the lobule indicating that there is elevated oxidative stress that does not colocalize with the observed increased periportal inflammation and suggesting different sources of reactive species. We hypothesize that as hepatocytes are slowly denied that ability to obtain nutrients as well as oxygen during cirrhosis, they are stressed and the MDA staining could be a marker of the aforementioned stress.
It has been established that in PBC, NAFLD as well as ALD, lipid aldehydes may be generated due to iron catalyzed production of hydroxyl radicals which are then capable of reacting with membrane lipids forming reactive lipid peroxides (Datz et al., 2017; Sorrentino et al., 2010). Surprisingly, although samples obtained from end stage human ALD as well as NASH have substantial hepatic accumulation of iron, liver sections from PSC/IBD patients have no evidence of accumulation of iron in the liver. Given that these patients possess a combinatorial diagnosis of PSC/IBD it may be that they are also iron deficient due to malabsorption. Unfortunately, data are not available regarding dietary status of these patients but malnutrition and iron deficiency is not uncommon in patients with cholestatic liver diseases (Kryskiewicz et al., 2012; Mattar et al., 2005). This suggests that lipid aldehyde production in PSC/IBD may be unique or that iron-mediated catalysis and the subsequent production of lipid aldehydes is not contributing to formation of lipid aldehydes.
In other chronic hepatic diseases such as nonalcoholic fatty liver disease and especially alcoholic liver disease, the induction of cytochrome P4502E1 plays a major role in the formation of reactive species and in the formation of lipid peroxidation (Aubert et al., 2011; Chen et al., 2016; Leung and Nieto, 2013). We do not see evidence of Cyp2E1 elevation in our PSC/IBD tissue (data not shown) and lipid peroxidation is periportal. Cytochrome p450’s are expressed around the central vein, suggesting that inflammation and/or cholestasis are the primary factor(s) in the production of oxidative stress contrary to ALD and NASH providing evidence for the uniqueness of cholestatic liver disease derived products of lipid peroxidation. Concurrently, periportal neutrophil infiltration correlates with increased protein modification by reactive aldehydes in our PSC tissues. Neutrophil infiltration has been demonstrated to positively contribute with both oxidative stress, lipid peroxidation and disease severity in patients with choledocholithiasis which frequently occurs in PSC (Damnjanovic et al., 2013). Furthermore, the extreme localization of lipid peroxidation evident in our samples also supports previous reports demonstrating a close link between fibrosis formed using bile duct ligations and lipid peroxidation (Galicia-Moreno et al., 2012).
In normal hepatic tissue, both catalase and SOD2 (although weak) expression is primarily localized in the centri-lobular region. From our histology, elevated SOD2 expression is evident in the periportal region in human PSC. Specifically, expression is particularly evident in hepatocytes immediately adjacent to fibrotic tissue. Expression of these two proteins however is not necessarily in the same cell as there are some cells which exhibit SOD2 positivity that do not appear to demonstrate changes in catalase. This would be indicative of elevated mitochondrial oxidative stress as well as peroxisomal stress within the cholangiocytes in PSC. In the literature, both catalase and SOD activity (not specifically SOD2) have been demonstrated to be increased (in children with obstructive cholestasis) and suppressed in patients with PSC (albeit in the serum)(Salem et al., 2003). Of minor interest, in our colocalization studies, catalase and SOD2 do not colocalize even within the same cell supporting previous observations that catalase is predominantly expressed in peroxisomes whereas SOD2 is mitochondrial specific (Karnati et al., 2013). Although we did not examine organelle specific hepatic GST activity, GSH, GSSG in individual cellular fractions due to procurement of frozen tissue, activity overall was suppressed. We predict that GST activity would also be impacted in both the mitochondrial (due to GSTA4 suppression) and cytosolic fractions (GSTμ and GSTπ). In PBC, immunohistochemical analysis of GSTπ expression trended down but was not significantly different further supporting distinct pathologies of PSC/IBD and PBC (Salunga et al., 2007). Furthermore, in patients with obstructive cholestasis which frequently occurs in PSC, GSH is also decreased supporting an environment of severe impairment of cellular redox capacity (Vendemiale et al., 2002). Since we do not see elevated CD3 positive cells or MPO staining immediately adjacent to damaged bile ducts but we do see CD68+ macrophages, we hypothesize that elevated reactive aldehyde production is due to direct exposure to high concentrations of bile acids and/or it is arising from infiltrating macrophages. This is supported in part by cell culture models where exposure to hydrophobic bile acids results in an increase in mitochondrial TBARS and generation of hydrogen peroxide which provides a plausible explanation the increase in SOD2 (Sokol et al., 1995). It does not provide evidence for suppression of catalase or Gpx1 expression. In agreement with our data, mitochondrial oxidative stress has previously been reported in both murine models of bile duct ligation and patients with cholestatic liver disease (Arduini et al., 2012; Shen et al., 2015). This suggests that cellular anti-oxidant responses are occurring within the cholangiocytes in PSC but are not sufficient when combined with the presence of decreased GST activity and indicates possible avenues for additional therapy in cholestatic liver disease.
In summary, this study is the first to perform comprehensive immunohistochemical analysis of the formation of products of lipid peroxidation and its impact on anti-oxidant responses in PSC/IBD. Based on immunohistochemical staining, oxidative stress and carbonylation present the highest increase in hepatocytes adjacent to fibrosis as well as in cholangiocytes tissue. In combination with protein expression data, this report validates cell specific dysregulation of anti-oxidant responses in PSC/IBD. Dysregulation is further supported with evidence that reactive aldehyde generation can be cell specific and that different aldehydes may originate from different sources e.g. bile and or inflammatory cells and iron-dependent formation of hydroxyl radicals is not the source of reactive aldehydes in PSC/IBD.
Supplementary Material
A. Paraffin embedded formalin fixed tissue sections were stained with Panels A, E: Hematoxylin &Eosin, Panels B, F: picrosirius red (PSR), Panels C, G: PSR polarize light exposure), Panels D, H: sections were probed using alpha smooth muscle actin rabbit polyclonal antibodies (αSMA) and DAB. All images are 200X and are representative of at least four normal and for PSC/IBD patients respectively.
{kind=link}
A. Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against myeloperoxidase, CD3+ lymphocytes and CD68+ macrophages. All images are 200X magnification and figures are representative of hepatic tissue sections isolated from at least four normal and four PSC/IBD patients respectively (PT=portal triad).
{kind=link}
Data was obtained from the University of Minnesota Liver Tissue cell Distribution Center NIH Contract #HHSN276201200017C., MELD (Model for End Stage Liver Disease), INR (International normalized ratio), AST (Aspartate Aminotransferase), Alk Phos (Alkaline Phosphatase). Data are Means+/−SEM, N=9.
Highlights.
Elevated cell specific peri-portal carbonylation occurs in PSC/IBD
PSC/IBD patients exhibit dysregulation of anti-oxidant responses.
SOD2 expression is upregulated in PSC/IBD
In PSC/IBD catalase expression is not evident in cholangiocytes
Patients with PSC/IBD possess elevated Akt activation.
Acknowledgments
Financial Support and Acknowledgements: This research was supported by the following grants from the National Institutes of Health; R37AA009300-22 DRP. The authors wish to thank E. Erin Smith, HTL(ASCP)CMQIHC of the University of Colorado Denver Cancer Center Research Histology Core for assistance in preparing histology slides. The UCDCCRHC is supported in part by NIH/NCRR Colorado CTSI Grant Number UL1 RR025780 and the University of Colorado Cancer Center Grant (P30 CA046934).
Abbreviations
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- AST
Aspartate Aminotransferase
- Cyp2E1
Cytochrome P4502E1
- GPX
Glutathione peroxidase
- GST
Glutathione S-Transferase
- HO-1
Heme Oxygenase 1
- 4-HNE
4-hydroxy-2-nonenal
- IBD
Inflammatory Bowel Disease
- INR
International Normalized Ratio
- LTx
liver transplant
- MDA
malondialdehyde
- MELD
Model for End-stage Liver Disease
- MPO
Myeloperoxidase
- NAFLD
Nonalcoholic fatty liver disease
- NASH
Nonalcoholic Steatohepatitis
- PBS
Primary Biliary Cholangitis
- PSC
Primary Sclerosing Cholangitis
- PSR
Picro-Sirius red
- ROS
Reactive oxidative species
- αSMA
Alpha Smooth Muscle Actin
- SOD2
mitochondrial superoxide dismutase
- WCE
whole cell extracts
Footnotes
Conflict of Interest
The authors have no conflict of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboutwerat A, et al. Oxidant stress is a significant feature of primary biliary cirrhosis. Biochim Biophys Acta. 2003;1637:142–50. doi: 10.1016/s0925-4439(02)00225-9. [DOI] [PubMed] [Google Scholar]
- Albano E. Alcohol, oxidative stress and free radical damage. The Proceedings of the Nutrition Society. 2006;65:278–90. doi: 10.1079/pns2006496. [DOI] [PubMed] [Google Scholar]
- Arduini A, et al. Mitochondrial dysfunction in cholestatic liver diseases. Front Biosci (Elite Ed) 2012;4:2233–52. doi: 10.2741/539. [DOI] [PubMed] [Google Scholar]
- Aubert J, et al. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clinics and research in hepatology and gastroenterology. 2011;35:630–7. doi: 10.1016/j.clinre.2011.04.015. [DOI] [PubMed] [Google Scholar]
- Bakhautdin B, et al. Protective role of HO-1 and carbon monoxide in ethanol-induced hepatocyte cell death and liver injury in mice. J Hepatol. 2014;61:1029–37. doi: 10.1016/j.jhep.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell LN, et al. Serum metabolic signatures of primary biliary cirrhosis and primary sclerosing cholangitis. Liver Int. 2015;35:263–74. doi: 10.1111/liv.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berhane K, et al. Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proc Natl Acad Sci U S A. 1994;91:1480–4. doi: 10.1073/pnas.91.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WY, et al. Acrolein Is a Pathogenic Mediator of Alcoholic Liver Disease and the Scavenger Hydralazine Is Protective in Mice. Cell Mol Gastroenterol Hepatol. 2016;2:685–700. doi: 10.1016/j.jcmgh.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SH, et al. Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS Lett. 2004;560:7–13. doi: 10.1016/s0014-5793(04)00112-7. [DOI] [PubMed] [Google Scholar]
- Covey TM, et al. Alkylation of the tumor suppressor PTEN activates Akt and beta-catenin signaling: a mechanism linking inflammation and oxidative stress with cancer. PLoS One. 2010;5:e13545. doi: 10.1371/journal.pone.0013545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damnjanovic Z, et al. Correlation of inflammation parameters and biochemical markers of cholestasis with the intensity of lipid peroxidation in patients with choledocholithiasis. Vojnosanit Pregl. 2013;70:170–6. doi: 10.2298/vsp1302170d. [DOI] [PubMed] [Google Scholar]
- Datz C, et al. Iron overload and non-alcoholic fatty liver disease. Minerva Endocrinol. 2017;42:173–183. doi: 10.23736/S0391-1977.16.02565-7. [DOI] [PubMed] [Google Scholar]
- Eaton JE, et al. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology. 2013;145:521–36. doi: 10.1053/j.gastro.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle MR, et al. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of mGsta4 null mouse. Toxicol Appl Pharmacol. 2004;194:296–308. doi: 10.1016/j.taap.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Fickert P, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465–81. doi: 10.1053/j.gastro.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Galicia-Moreno M, et al. Antifibrotic and antioxidant effects of N-acetylcysteine in an experimental cholestatic model. Eur J Gastroenterol Hepatol. 2012;24:179–85. doi: 10.1097/MEG.0b013e32834f3123. [DOI] [PubMed] [Google Scholar]
- Galligan JJ, et al. Protein carbonylation in a murine model for early alcoholic liver disease. Chem Res Toxicol. 2012;25:1012–21. doi: 10.1021/tx300002q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouw AS, et al. Ductular reactions in human liver: diversity at the interface. Hepatology. 2011;54:1853–63. doi: 10.1002/hep.24613. [DOI] [PubMed] [Google Scholar]
- Harrison-Findik DD. Role of alcohol in the regulation of iron metabolism. World J Gastroenterol. 2007;13:4925–30. doi: 10.3748/wjg.v13.i37.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison-Findik DD. Gender-related variations in iron metabolism and liver diseases. World J Hepatol. 2010;2:302–10. doi: 10.4254/wjh.v2.i8.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfield GM, et al. Primary sclerosing cholangitis. Lancet. 2013;382:1587–99. doi: 10.1016/S0140-6736(13)60096-3. [DOI] [PubMed] [Google Scholar]
- Jais A, et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell. 2014;158:25–40. doi: 10.1016/j.cell.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Karlsen TH. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat Rev Gastroenterol Hepatol. 2017;14:279–295. doi: 10.1038/nrgastro.2016.154. [DOI] [PubMed] [Google Scholar]
- Kamath PS, et al. The model for end-stage liver disease (MELD) Hepatology. 2007;45:797–805. doi: 10.1002/hep.21563. [DOI] [PubMed] [Google Scholar]
- Karnati S, et al. Mammalian SOD2 is exclusively located in mitochondria and not present in peroxisomes. Histochem Cell Biol. 2013;140:105–17. doi: 10.1007/s00418-013-1099-4. [DOI] [PubMed] [Google Scholar]
- Kawamura K, et al. Enhanced hepatic lipid peroxidation in patients with primary biliary cirrhosis. Am J Gastroenterol. 2000;95:3596–601. doi: 10.1111/j.1572-0241.2000.03300.x. [DOI] [PubMed] [Google Scholar]
- Kryskiewicz E, et al. Bone metabolism in cholestatic children before and after living-related liver transplantation--a long-term prospective study. J Clin Densitom. 2012;15:233–40. doi: 10.1016/j.jocd.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Kugelmas M, et al. Different immunosuppressive regimens and recurrence of primary sclerosing cholangitis after liver transplantation. Liver Transpl. 2003;9:727–32. doi: 10.1053/jlts.2003.50143. [DOI] [PubMed] [Google Scholar]
- Lee SR, et al. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–42. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. Journal of hepatology. 2013;58:395–8. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- Li T, et al. Glutathione S-transferase P1 correlated with oxidative stress in hepatocellular carcinoma. Int J Med Sci. 2013;10:683–90. doi: 10.7150/ijms.5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubuncic P, et al. Effect of deoxycholic acid and ursodeoxycholic acid on lipid peroxidation in cultured macrophages. Gut. 1996;39:475–8. doi: 10.1136/gut.39.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunder AK, et al. Prevalence of Sclerosing Cholangitis, Detected by Magnetic Resonance Cholangiography, in Patients with Long-term Inflammatory Bowel Disease. Gastroenterology. 2016 doi: 10.1053/j.gastro.2016.06.021. [DOI] [PubMed] [Google Scholar]
- Mattar RH, et al. Nutritional status and intestinal iron absorption in children with chronic hepatic disease with and without cholestasis. J Pediatr (Rio J) 2005;81:317–24. [PubMed] [Google Scholar]
- Osna NA, et al. Aberrant post-translational protein modifications in the pathogenesis of alcohol-induced liver injury. World J Gastroenterol. 2016;22:6192–6200. doi: 10.3748/wjg.v22.i27.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdil B, et al. Potential benefits of combined N-acetylcysteine and ciprofloxacin therapy in partial biliary obstruction. J Clin Pharmacol. 2010;50:1414–9. doi: 10.1177/0091270010361257. [DOI] [PubMed] [Google Scholar]
- Parola M, et al. On the role of lipid peroxidation in the pathogenesis of liver damage induced by long-standing cholestasis. Free Radic Biol Med. 1996;20:351–9. doi: 10.1016/0891-5849(96)02055-2. [DOI] [PubMed] [Google Scholar]
- Peres W, et al. The flavonoid quercetin ameliorates liver damage in rats with biliary obstruction. J Hepatol. 2000;33:742–50. doi: 10.1016/s0168-8278(00)80305-0. [DOI] [PubMed] [Google Scholar]
- Ronis MJ, et al. Increased 4-hydroxynonenal protein adducts in male GSTA4-4/PPAR-alpha double knockout mice enhance injury during early stages of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2015;308:G403–15. doi: 10.1152/ajpgi.00154.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskams T, et al. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am J Pathol. 2003;163:1301–11. doi: 10.1016/S0002-9440(10)63489-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem TA, et al. Study of antioxidant enzymes level and phagocytic activity in chronic liver disease patients. Egypt J Immunol. 2003;10:37–45. [PubMed] [Google Scholar]
- Salunga TL, et al. Oxidative stress-induced apoptosis of bile duct cells in primary biliary cirrhosis. J Autoimmun. 2007;29:78–86. doi: 10.1016/j.jaut.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Shearn CT, et al. Identification of 5′ AMP-activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J Biol Chem. 2014;289:15449–62. doi: 10.1074/jbc.M113.543942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, et al. Modification of Akt2 by 4-hydroxynonenal inhibits insulin-dependent Akt signaling in HepG2 cells. Biochemistry. 2011;50:3984–96. doi: 10.1021/bi200029w. [DOI] [PubMed] [Google Scholar]
- Shearn CT, et al. Deletion of GSTA4-4 results in increased mitochondrial post-translational modification of proteins by reactive aldehydes following chronic ethanol consumption in mice. Redox Biol. 2015a;7:68–77. doi: 10.1016/j.redox.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, et al. Liver-Specific Deletion of Phosphatase and Tensin Homolog Deleted on Chromosome 10 Significantly Ameliorates Chronic EtOH-Induced Increases in Hepatocellular Damage. PLoS One. 2016;11:e0154152. doi: 10.1371/journal.pone.0154152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, et al. Increased hepatocellular protein carbonylation in human end-stage alcoholic cirrhosis. Free Radic Biol Med. 2015b doi: 10.1016/j.freeradbiomed.2015.10.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, et al. Differential carbonylation of proteins in end-stage human fatty and nonfatty NASH. Free Radic Biol Med. 2017 doi: 10.1016/j.freeradbiomed.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, et al. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic Biol Med. 2013a;65:680–92. doi: 10.1016/j.freeradbiomed.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearn CT, et al. Increased dietary fat contributes to dysregulation of the LKB1/AMPK pathway and increased damage in a mouse model of early-stage ethanol-mediated steatosis. J Nutr Biochem. 2013b;24:1436–45. doi: 10.1016/j.jnutbio.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, et al. Epigallocatechin 3-gallate ameliorates bile duct ligation induced liver injury in mice by modulation of mitochondrial oxidative stress and inflammation. PLoS One. 2015;10:e0126278. doi: 10.1371/journal.pone.0126278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol RJ, et al. Generation of hydroperoxides in isolated rat hepatocytes and hepatic mitochondria exposed to hydrophobic bile acids. Gastroenterology. 1995;109:1249–56. doi: 10.1016/0016-5085(95)90585-5. [DOI] [PubMed] [Google Scholar]
- Sorrentino P, et al. Oxidative stress and steatosis are cofactors of liver injury in primary biliary cirrhosis. J Gastroenterol. 2010;45:1053–62. doi: 10.1007/s00535-010-0249-x. [DOI] [PubMed] [Google Scholar]
- Sutti S, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59:886–97. doi: 10.1002/hep.26749. [DOI] [PubMed] [Google Scholar]
- Tamura S, et al. Recurrence of primary sclerosing cholangitis after living donor liver transplantation. Liver Int. 2007;27:86–94. doi: 10.1111/j.1478-3231.2006.01395.x. [DOI] [PubMed] [Google Scholar]
- Tan M, et al. SIRT1/PGC-1alpha signaling protects hepatocytes against mitochondrial oxidative stress induced by bile acids. Free Radic Res. 2015;49:935–45. doi: 10.3109/10715762.2015.1016020. [DOI] [PubMed] [Google Scholar]
- Trivedi HD, et al. Primary biliary cholangitis: new treatments for an old disease. Frontline Gastroenterol. 2017;8:29–36. doi: 10.1136/flgastro-2016-100741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendemiale G, et al. Hepatic oxidative alterations in patients with extra-hepatic cholestasis. Effect of surgical drainage. J Hepatol. 2002;37:601–5. doi: 10.1016/s0168-8278(02)00234-9. [DOI] [PubMed] [Google Scholar]
- Venkatesan B, et al. Downregulation of catalase by reactive oxygen species via PI 3 kinase/Akt signaling in mesangial cells. J Cell Physiol. 2007;211:457–67. doi: 10.1002/jcp.20953. [DOI] [PubMed] [Google Scholar]
- Wang X, et al. Osteopontin induces ductular reaction contributing to liver fibrosis. Gut. 2014;63:1805–18. doi: 10.1136/gutjnl-2013-306373. [DOI] [PubMed] [Google Scholar]
- Wasik U, et al. Protection against oxidative stress mediated by the Nrf2/Keap1 axis is impaired in Primary Biliary Cholangitis. Sci Rep. 2017;7:44769. doi: 10.1038/srep44769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weismuller TJ, et al. Patient Age, Sex, and Inflammatory Bowel Disease Phenotype Associate With Course of Primary Sclerosing Cholangitis. Gastroenterology. 2017 doi: 10.1053/j.gastro.2017.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Paraffin embedded formalin fixed tissue sections were stained with Panels A, E: Hematoxylin &Eosin, Panels B, F: picrosirius red (PSR), Panels C, G: PSR polarize light exposure), Panels D, H: sections were probed using alpha smooth muscle actin rabbit polyclonal antibodies (αSMA) and DAB. All images are 200X and are representative of at least four normal and for PSC/IBD patients respectively.
A. Paraffin embedded formalin fixed tissue sections were analyzed immunohistochemically using polyclonal antibodies directed against myeloperoxidase, CD3+ lymphocytes and CD68+ macrophages. All images are 200X magnification and figures are representative of hepatic tissue sections isolated from at least four normal and four PSC/IBD patients respectively (PT=portal triad).
Data was obtained from the University of Minnesota Liver Tissue cell Distribution Center NIH Contract #HHSN276201200017C., MELD (Model for End Stage Liver Disease), INR (International normalized ratio), AST (Aspartate Aminotransferase), Alk Phos (Alkaline Phosphatase). Data are Means+/−SEM, N=9.