

Abstract
We previously developed a Chlamydia trachomatis nanovaccine (PPM) by encapsulating a chlamydial M278 peptide within poly(lactic acid)-poly(ethylene glycol) biodegradable nanoparticles that immunopotentiated Chlamydia-specific immune effector responses in mice. Herein, we investigated the mechanistic interactions of PPM with mouse bone marrow-derived dendritic cells (DCs) for its uptake, trafficking, and T-cell activation. Our results reveal that PPM triggered enhanced expression of effector cytokines and chemokines, surface activation markers (Cd1d2, Fcgr1), pathogen-sensing receptors (TLR2, Nod1), co-stimulatory (CD40, CD80, CD86) and MHC class I and II molecules. Co-culturing of PPM-primed DCs with T cells from C. muridarium vaccinated mice yielded an increase in Chlamydia-specific immune effector responses including CD3+ lymphoproliferation, CD3+CD4+ IFN-γ-secreting cells along with CD3+CD4+ memory (CD44highand CD62high) and effector (CD44high and CD62low) phenotypes. Intracellular trafficking analyses revealed an intense expression and colocalization of PPM predominantly in endosomes. PPM also upregulated the transcriptional and protein expression of the endocytic mediator, caveolin-1 in DCs. More importantly, the specific inhibition of caveolin-1 led to decreased expression of PPM-induced cytokines and co-stimulatory molecules. Our investigation shows that PPM provided enhancement of uptake, probably by exploiting the caveolin-mediated endocytosis pathway, endosomal processing, and MHC II presentation to immunopotentiate Chlamydia-specific immune effector responses mediated by CD4+ T cells.
Keywords: Chlamydia muridarum, Nanovaccine, Caveolin, Dendritic cells, Endocytosis, PLA-PEG nanoparticles
1. Introduction
Biodegradable polymeric nanoparticles are appealing as nanodelivery systems for bioactive macromolecules, including peptides and proteins, and they are easily fabricated to evoke immune responses, by direct stimulation of antigen presenting cells (APCs) or delivering antigens to specific subcellular compartments [1, 2]. Recently, the novelty of vaccine adjuvanation mimicking various properties of actual pathogens without causing disease is gaining considerable attraction in the field of vaccinology. Nanoparticulate adjuvants are similar in size to bacteria and viruses [3–5] hence, vaccine delivery systems are simulating various properties of pathogens associated molecular patterns (PAMPs) to increase the vaccines’ immunogenicity. Vaccines based on nanoparticulate concepts like viral-like particles are available for influenza (Inflexal), hepatitis b (Engerix b) and the human papilloma (Gardasil and Cervarix) [3] viruses.
Dendritic cells (DCs) are specialized in a variety of endocytic uptake mechanisms and are key players in antigen presentation for eliciting specific T cell responses. Nanoparticulate-based vaccines can enter DCs using cellular endocytosis mechanisms, in particular, pinocytosis [6]. These mechanisms are classified either as clathrin-dependent, caveolae-dependent, and clathrin- and caveolae-independent endocytosis depending on the proteins that are endocytosed by these trafficking pathways [7, 8]. Caveolae-dependent endocytosis is likewise a common cellular entry pathway exploited by 20–100 nm size nanoparticles [9]. Caveolae-coated vesicles avoid fusion with lysosomes [10, 11] and thus enhance the concentrations of targeted therapeutics in endosomes or caveosomes, thus resulting in an improved therapeutic effect. The caveosomes containing targeted therapeutics move through microtubules to the endoplasmic reticulum [6, 12] with subsequent entry into the cytoplasm and nucleus. MHC class II presents antigens to CD4+ T cells involving endosomes, while MHC class I processes endoplasmic reticulum-derived antigens for the presentation of antigens to CD8 T cells [13]. Thus, many pathogens including viruses and bacteria exploit the caveolae pathway to evade lysosomal degradation [14].
Studies have reported that size, charge, and shape of nanoparticles play significant roles in antigen uptake. Nanoparticles having a comparable size to pathogens can be easily recognized and are consequently taken up efficiently by APCs for induction of immune responses. DCs preferentially uptake virus-sized particles (20 – 200 nm), while macrophages, on the other hand, uptake larger particles (0.5 – 5 μm) [15]. Higher uptake by macrophages of smaller poly lactide particles (200 – 600 nm) in comparison to larger ones (2–8 μm) has been described [16, 17]. It is, therefore, possible that APCs have evolved to efficiently process antigens with dimensions similar to those of pathogens including viruses and bacteria. Consequently, formulating nanoparticulate-based vaccines may provide enhancing the uptake, processing, and presentation of antigens by professional APCs [3].
Despite Chlamydia trachomatis being the most reported bacterial sexually transmitted infection globally with a plethora of disease manifestations, there is no approved vaccine to reduce the public health and economic burden of its infections. A noteworthy reason, perhaps, for the hurdle in developing a vaccine against Chlamydia is the lack of an adequate adjuvant delivery system that can provide a prolonged targeted delivery and potentiation. As such, the development of a vaccine against Chlamydia is continuously a top priority for researchers. We have focused on biodegradable polymeric nanoparticles as nanodelivery vaccine targets for Chlamydia [1, 18–20] because of their nanoparticulate, self-adjuvanting and slow-releasing attributes. We have previously developed a PLA-PEG-encapsulated M278 (named PPM) Chlamydia nanovaccine that immunopotentiated Chlamydia-specific Th1/Th2 immune effector responses in immunized mice [1]. Here, our goal is to gain a deeper understanding of how PPM triggers immunopotentiation by investigating its mechanistic interaction with mouse bone marrow-derived DCs for its uptake, intracellular trafficking, and T cell activation. First, we quantified the transcriptional and protein expressions of effector cytokines/chemokines and molecular signatures (surface activation and pathogen-sensing receptors, co-stimulatory and MHC molecules) that may be involved in cellular activation and maturation. Second, antigen presentation and induction of specific T cells immune effector responses (cytokines, lymphoproliferation, intracytoplasmic IFN-γ secretion and memory and effector T cell phenotypes) were studied by co-culturing antigen-primed DCs with T cells from both naïve and C. muridarum-vaccinated mice. Third, evidence of antigen processing was evaluated by intracellular trafficking and colocalization studies in subcellular compartments. Fourth, we quantified gene transcripts encoding mediators of the endocytic trafficking pathways (clathrin and caveolin-1) and their intracellular protein expression to evaluate their roles in antigen uptake. Finally, we tested the efficacy of specific inhibition of clathrin and caveolin-1 on the uptake, and the concomitant expression of immune effectors. Collectively, our investigation shows that PPM immunopotentiation of immune effector responses is mediated by endosomal processing for a prolonged presentation and cell activation, and probably exploitation of the caveolin-mediated endocytosis pathway for uptake and an MHC class II dependent presentation to trigger robust Chlamydia-specific immune effectors mediated by CD4+ T cells.
2. Material and Methods
2.1. Materials
PEG-b-PLA diblock polymer (Polyethylene glycol; MW 10,000 and Polylactic acid, MW 5,000) was purchased from Polysciences Incorporation (Warrington, PA). Polyvinyl alcohol (PVA), ethyl acetate, mitomycin-C, filipin III and chlorpromazine hydrochloride were obtained from Sigma-Aldrich (St Louis, MO). ELISA MAX™ Deluxe kits, rGM-CSF, anti-MHC-I (107650) and anti-MHC-II (114712) antibodies were purchased from Biolegend (San Diego, CA, USA). RPMI 1640 with GlutaMax™ and HEPES, heat-inactivated fetal bovine serum (FBS), goat serum, ACK lysing reagent, antibiotic-antimycotic, anti-fade solution, superscript vilo cDNA synthesis kit, TaqMan PreAmp master mix and TaqMan gene expression assays were all purchased from Life Technologies (Grand Island, NY). Chlamydia muridarum expressed as inclusion forming units (IFU)/mL was purchased from Virusys Corporation (Taneytown, MD). Anti-CD 90.2 magnetic beads were purchased from Miltenyi Biotech (Auburn, CA), and anti-PE-CD 90.2 antibody was purchased from eBioscience (San Diego, CA). Opti-EIA sets were purchased from BD-Biosciences (San Jose, CA, USA). RNeasy kit, Reaction Ready First Strand cDNA Synthesis, and mouse dendritic cells antigen presentation (PAMM-406Z) RT2 Profiler™ PCR array were all purchased from Qiagen (Valencia, CA). Anti-C. trachomatis MOMP polyclonal antibody (20C-CR2104GP) was obtained from Fitzgerald (Acton, MA). Anti-EEA1 (G4) (sc-137130) and anti-calregulin (F4) (sc-373863) antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX). Anti-rab7 (bs-6703R), anti-caveolin-1 (bs-1453R) and anti-LAMP-1(bs-1970R) antibodies were all purchased from Bioss Antibodies Inc. (Woburn, MA). Alexa-fluor 594, Alexa-fluor 546 and Alexa-fluor 488 labeled secondary antibodies and Cell-Trace CFSE (Carboxyfluorescein succinimidyl ester) cell proliferation assay kit (C34554) were all purchased from Thermo Fisher Scientific (Rockford, IL).
2.2. Preparation of nanoparticles and encapsulation efficiency
A recombinant peptide (M278) derived from the major outer membrane protein (MOMP) of C. trachomatis was cloned and encapsulated in PLA-PEG [poly(lactic acid)-poly (ethylene glycol)] biodegradable nanoparticles using a modified water/oil/water double emulsion evaporation technique to obtain PLA-PEG-M278 (PPM) as reported [1]. An equivalent volume of PBS as used for M278 was similarly encapsulated in PLA-PEG to obtain PLA-PEG-PBS (PPP) to serve as a negative control. All lyophilized nanoparticles were stored at −80°C in a sealed container until used. Encapsulation efficiency was calculated as reported [1], which was observed to be 60–65%.
2.3. Generation of mouse primary bone marrow-derived dendritic cells
Female 6–8 weeks old BALB/c mice were purchased from Charles River Laboratory (Raleigh, NC). The animal studies were performed following a protocol approved by the Alabama State University Institutional Animal Care and Use Committee (IACUC). Mice were housed under standard pathogen-free and controlled environmental conditions provided with food and water ad libitum. Femurs were collected from mice after euthanasia. Isolation and differentiation of bone marrow cells was conducted using previously published protocols [21, 22] with some modifications. Briefly, bone marrow cells were washed, RBCs lysed using ACK lysing reagent, and the resulting cell suspensions were filtered through a sterile 40-μm nylon mesh. Cells were centrifuged at 250g for 5 minutes and resuspended in differentiation media (RPMI 1640 supplemented with 20 ng/mL rGM-CSF, 50 μM mercaptoethanol, 10% FBS and antibiotic-antimycotic) at a cell density of 1 × 106 and transferred into sterile 100 mm2 petri dishes. Cultures were incubated at 37°C in a 5% CO2 humidified atmosphere with a change of media on days 2, 4 and 6, and harvesting of dendritic cells (DCs) on day 8 for subsequent experiments.
2.4. Dose-response and time-kinetics studies
For dose-response studies, DCs were plated at 0.5 × 106/mL in polypropylene round-bottom tubes and stimulated for 24 hours with M278 and PPM at concentrations ranging from 1.25, 2.5, 5 and 10 μg/mL. Next, time-kinetics studies were performed to determine the optimal time for maturation of DCs by stimulating them with 2.5 μg/mL of either M278 or PPM for 4, 24, 48 and 72 hours. As controls, PPP was used at an equivalent weight concentration as that of PPM, while unstimulated DCs served as the negative control. All dose-response and time-kinetics cell cultures were incubated at 37°C in a 5% CO2 humidified atmosphere. Cell-free supernatants were collected by centrifugation at 450g for 5 minutes at 4°C, and stored at −80°C until used. For some experiments, cell pellets were collected for flow cytometry or to extract RNA for TaqMan qPCR or PCR array studies as described below.
2.5. Quantification of cytokines by ELISA
Concentrations of cytokines (IL-12p40, IL-6, and IL-10) in cell-free supernatants were quantified in triplicate using BD-Biosciences Opti-EIA sets [1]. IFN-γ and IL-2 were similarly quantified in triplicate using Biolegend Max Deluxe kits. Each experiment was repeated at least 2–3 times.
2.6. TaqMan qPCR and PCR array
TaqMan qPCR was employed as described [1, 23] to quantify the transcription levels of CD80, CD86, CD40, MHC-I, MHC-II, caveolin-1(Cav-1), clathrin (Cltc), Nos2, Nod1, Nod2, TLR1, and TLR2. TaqMan qPCR was conducted using FAM (6-carboxyfluorescein)-labeled TaqMan gene expression assays on an Applied Biosystems ViiA™ 7 real-time PCR system according to the manufacturer’s instructions. The relative changes in gene expression levels were calculated using the ΔΔCT, where all values were normalized with respect to the GAPDH “housekeeping” gene mRNA levels. Each real-time PCR assay was performed in triplicate, and the results are expressed as the means ± standard deviations.
PCR array was performed as previously reported [23] using the mouse dendritic cells antigen presentation RT2 Profiler™ PCR array. Ninety-six-well plates containing gene-specific primer sets for 84 relevant pathway genes, housekeeping genes, and negative controls were used. After performing thermal cycling (according to the manufacturer’s protocol), real-time amplification data were gathered by using the ABI ViiA™ 7 system. Gene expression was normalized to internal controls (housekeeping genes) to quantify fold changes in gene expression between test and control samples by the ΔΔCT method. Each RNA sample was run in triplicate for data analyses using the RT2 Profiler™ PCR array software. Only those PCR array genes that were statistically significantly perturbed are presented in this study.
2.7. Phenotypic analyses of cell surface markers
DCs (0.5 × 106) were either left unstimulated or stimulated for 24 hours with bare M278 and PPM (each at 2.5 μg/mL) or an equivalent weight of PPP as used for PPM to quantify changes in expressions of CD80, CD86, CD40, MHC-I, and MHC-II as markers of differentiation and activation. DCs were washed, and Fc surface receptors were blocked for 15 minutes at 4°C using a purified mouse anti-CD16/CD32 receptor blocking antibody (BD Bioscience) diluted in fluorescent-activated cell sorting (FACS) buffer [phosphate-buffered saline (PBS) 0.1% NaN3, 1.0% FBS]. DCs were washed and stained with fluorochrome-conjugated antibodies against CD11c-APC-Cy7 (BD: 561241), MHC-I-A647 (BD: 562832) MHC-II-PE (BD: 558593), CD86-FITC (BD: 553691), CD80-PECy-7 (BD: 562504), CD40-BV421 (BD: 562846) each at 0.250 μg/100 μL. Unstimulated control cells (medium) were kept as background controls. Cells were then washed and fixed with 1% paraformaldehyde (PFA) solution for 20 minutes at 4°C. Data were acquired on a BD LSR II flow cytometer (BD Bioscience) with at least 1 × 105 events for each sample and analyzed using FlowJo software (Tree Star Inc., Ashland, OR, USA) [19].
2.8. Mice vaccination
Two groups of female 6 to 8 weeks old BALB/c mice at 8 mice/group were used for isolation of purified T cells. One group received a single intranasal (10 μL/nostril) vaccination of 1 ×105 IFUs of live C. muridarum elementary bodies (EBs) per mouse. The second control group received a similar intranasal vaccination but with SPG (sucrose-phosphate-glutamic acid) buffer (storage buffer of C. muridarum). All mice were sacrificed on day 42 to collect spleens for isolation of purified T cells as described below.
2.9. DCs prime and T cells co-cultures
T cells were isolated and purified from splenocytes of immunized mice as previously described [1]. In brief, splenocytes isolated from groups of naïve and EB-vaccinated mice were incubated with anti-CD 90.2-conjugated MACS magnetic beads, and total T cells were isolated by positive selection over magnetic MACS columns. The purity of T cells was higher than 95% as assessed by flow cytometry employing PE-conjugated anti-CD 90.2 antibodies at 0.25 μg/106 cells (data not shown). DCs were primed with either bare M278 and PPM (each at 2.5 μg/mL) or an equivalent weight of PPP as used for PPM for 24 hours at 37°C in a 5% CO2 humidified incubator to serve as antigen presenting cells (APCs). Primed DCs and T cells were co-cultured in polypropylene round-bottom tubes at a 1:5 ratio (1 × 106 DCs and 5 × 106 T cells) and incubated for 48 and 120 hours depending on the specific experiment. Cell-free supernatants, cell pellets or RNA samples were collected and used immediately or stored at −80°C until used.
2.10. Intracytoplasmic staining, memory and effector T cells phenotypes
For these studies, co-cultures were setup as described above and incubated for 120 hours for intracellular IFN-γ secretion and to quantify memory and effector T cells phenotypes (CD62L and CD44). To detect the intracellular presence of IFN-γ, the protein transport inhibitor, Brefeldin A, which blocks intracellular protein transport was added to co-cultures during the last remaining 4 hours of the 120 hours incubation period using BD cytofix/cytoperm kit (BD:554714) according to the manufacturer’s instructions. Briefly, cells were harvested after incubation, washed and Fc surface receptors were blocked as described above. Co-cultures were stained with fluorochrome-conjugated antibodies against CD3 using CD3-APC-Cy7. Permeabilization and fixation of cells were conducted using the BD cytofix/cytoperm kit according to the manufacturer’s instructions; cells were washed, and intracellular staining for IFN-γ secretion was performed using IFN-γ-APC (BD: 562018). For quantification of memory and effector T cell phenotypes, co-cultures were stained with fluorochrome-conjugated antibodies against CD3, CD62L, and CD44, respectively using CD3-APC-Cy7 (BD:560590), CD4-PerCP-Cy5.5 (BD:550954), CD62L-APC (BD:553152) and CD44-PE (BD:553134). All co-cultures were washed and fixed with 1% PFA and data were acquired and analyzed as described above.
2.11. T lymphocyte proliferation assay
Purified T cells isolated from splenocytes of naïve and EBs-vaccinated mice were labeled with 5 μM CFSE in PBS by incubating labeled cells for 20 minutes at 37°C in a 5% CO2 humidified atmosphere. After incubation, RPMI 1640 culture medium was added to remove unbound CFSE by incubating cells at 37°C for 5 minutes followed by washing three times with RPMI 1640 medium. Labeled T cells (1 × 106/mL) were co-cultured with DCs (1 × 105cells/mL) previously primed for 24 hours with M278, PPM or PPP. Co-cultures were incubated for 120 hours after which they were harvested and stained for CD3 cells using CD3-APC-Cy7 (BD: 560590). Cells were washed, fixed and data were acquired on a BD LSR II flow cytometer and analyzed for CD3+ CFSE+ T cells.
2.12. Intracellular trafficking and inhibition studies
Intracellular trafficking studies were conducted by plating DCs (2 ×104) in 8-well culture chamber slides followed by stimulation with bare M278, PPM or PPP (2.5 μg/mL). After 24 hours, DCs were washed, fixed with 2% PFA and blocked (buffer containing 0.20 % fish gelatin, 1 % BSA, 0.5 % saponin and 0.3 M glycine) for 1 hour at room temperature (RT). DCs were probed with antibodies against early to late endosome (anti-rab7), endoplasmic reticulum (anti-calregulin), lysosome (anti-LAMP1), early endosome (anti-EEA1), caveolin-1 (anti-caveolin 1), MHC-I (anti-MHC-I) and MHC-II (anti-MHC-II) (each used at 2 μg/mL) or M278 (anti-MOMP) (4 μg/mL) for 30 minutes at RT. After washing, cells were incubated with Alexa fluor labeled secondary antibodies: 594 (red), 546 (red) and 488 (green) (each at 1:400 dilution). DAPI (blue) at 1:1000 dilution was used to stain the nuclei. Colocalization of organelles with targeted M278 was visualized and imaged using a Nikon Eclipse Ti fluorescence Microscope (Nikon Instruments, Melville, NY).
For caveolin-1 and clathrin inhibition studies, DCs (2 ×106/well) were plated for 24 hours in 24-well tissue culture plates to permit attachment. Next, cells were pretreated for 30 minutes at 37°C in a 5% CO2 humidified atmosphere [24] with filipin III or chlorpromazine (each at 10 μg/mL) to inhibit the expression of caveolin-1 and clathrin, respectively. Cells were washed and stimulated with either 2.5 μg/mL of M278 and PPM or with PPP. Cell-free supernatants or RNA were collected after 24 hours to quantify cytokines or for TaqMan qPCR studies, respectively.
2.13. Statistical analysis
Data were analyzed by one- or two-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test, the two-tailed Mann-Whitney test or the one-tailed unpaired t-test with Welch correction using GraphPad Prism 5 Software. Significance was established at ***P < 0.001, **P < 0.01 and *P < 0.05.
3. Results
3.1. PPM enhances the expression of effector cytokines and chemokines
Immature DCs, in peripheral tissues or organs, capture and present foreign antigens to T cells in the lymphatic system; in contrast, mature DCs prime naive T cells to differentiate into specific T cell subsets that initiate adaptive immune responses. These processes involve expressions of cytokines and chemokines as well as costimulatory molecules to determine the functions of antigen-capturing to antigen-presenting DCs. It is well-established that the presence of a set of cytokines and chemokines determines the skewing of T cells as specialized Th1 responses to protect against Chlamydia infection [25, 26]. Accordingly, we assessed the maturation and activation of DCs after their exposure to stimulants by performing dose-response and time-kinetics studies and also to evaluate the release and stimulating properties of the PPM nanovaccine as compared to bare M278. Stimulation of DCs resulted in increased production of the Th1 pro-inflammatory cytokines, IL-12p40 and IL-6 and diminished level of the Th2 anti-inflammatory cytokine, IL-10 (15–30 fold less than IL-6 and IL-12p40), suggesting up-regulation of chiefly Th1 responses (Fig. 1A–C). Moreover, DCs stimulated with 10 μg/mL of either PPM or bare M278 resulted in comparable production levels of IL-12p40 and IL-6. However, bare M278 induced a typical protein dose-response with decreasing cytokine levels corresponding with decreasing stimulatory concentrations. Contrastingly, PPM continually induced enhanced secretion of IL-12p40 and IL-6 at all concentrations, suggesting potentiation for enhanced cell stimulation. The PPP control stimulated low production levels of IL-12p40 and IL-6 cytokine (Fig. 1A–C), suggesting its potentiating capacity.
Fig. 1. Differential transcriptional and protein expression of cytokines and chemokines induced by stimulated DCs.


For the dose-response studies, DCs were stimulated with various concentrations of PLA-PEG-M278 (PPM), M278 or PLA-PEG-PBS (PPP) and cell-free supernatants were collected after 24 hours to quantify the secretion of IL-12p40 (A), IL-6 (B) and IL-10 (C). For the time-kinetic studies, DCs were stimulated 4–72 hours with 2.5 μg/mL of stimulants to quantify IL-12p40 (D), IL-6 (E) and IL-10 (F). RNA samples were also collected after 24 hours to quantify mRNA gene transcripts of cytokines (G, H) and chemokines (I, J). Data were analyzed by two-way ANOVA followed by Tukey’s post-hoc test using GraphPad Prism 5 software. ***P < 0.001, **P < 0.01 and *P < 0.05.
Because all tested concentrations of PPM enhanced cytokines secretions, we selected 2.5 μg/mL for the time-kinetics and all subsequent experiments in this study to discern the potentiating capacity of PPM as compare to bare M278. The time-kinetics results further showed the increased stimulatory and slow releasing potential of PPM over the 72-hour time-frame for the enhanced secretion of cytokines as compared to bare M278 (Fig. 1D–F). Interestingly, the reversed secretion pattern of IL-10 as compared to IL-12p40 and IL-6 (Fig. 1A–F) may indicate their suppression by IL-10 evidently by their lower secretion levels during the earlier time-points but higher secretions during the late time-points, coinciding with the secretion patterns of IL-10 at similar time-points. Unstimulated DCs did not induce cytokines (data not shown). Together, these findings suggest that PPM induced enhanced secretion of Th1 cytokines as compared to bare M278. Moreover, the prolonged slow release of encapsulated M278 corroborates DCs activation for enhancements of IL-12p40 and IL-6 production levels over time.
Given that cytokines and chemokines produced by activated DCs prime T cells to migrate to infection sites for mounting adaptive immune responses, we additionally assessed whether PPM could enhance the mRNA transcriptional expression levels of cytokines and chemokines that are prerequisites for this process. Overall, PPM-stimulated DCs triggered higher expression of cytokines and chemokines as compared to bare M278 or PPP (Fig. 1G–J). The results presented in Fig. 1G–H, show that the mRNA gene transcripts of IL-12p40/IL-12β and IL-6 were significantly (P < 0.001) up-regulated by PPM stimulation, thus underscoring their up-regulated protein levels (Fig. 1A, B, D, E). Additionally, PPM significantly (P < 0.01) enhanced the expression of IFN-γ (Fig. 1H), which along with IL-12p40 and IL-6, are responsible for maturation of DCs and directing naïve T cells to become committed Th1 cells [27, 28]. PPM further enhanced the transcription of Csf2 (Fig. 1G), a cytokine also required for maturation of DCs, expression of costimulatory and MHC molecules, and for efficient antigen presentation to T cells [29]. The IL-10 gene transcript was highly expressed (Fig. 1H) as compared with those of other cytokines, which may suggest its anti-inflammatory effect on the IL-12p40/IL-12β and IL-6 lower secretions during the earlier time-points of the time-kinetics study (Fig. 1D–F). The mRNA gene transcripts of chemokines [Ccl12 (monocyte trafficking), Ccl5 (DCs and T cell interaction), Cxcl2 and Cxcl1 (neutrophil trafficking), Cxcl10 (Th1 promoting), and Ccl8 (Th2 responses)] [27, 30] were significantly (P < 0.001) enhanced by PPM stimulation, in contrast to those induced by M278 and PPP (Fig. 1I–J). Our results show that PPM increases the expression of effector cytokines and chemokines that are required for the maturation and activation of DCs to be efficient APCs for mounting adaptive immune responses and also for facilitating immune effector responses.
3.2. Recognition of PPM by select cell surface and pathogen-sensing receptors
We next investigated molecular signatures on DCs that may be involved in recognition and co-stimulation for induction of immune effector responses. We observed that PPM triggered significant (P < 0.001) transcriptional up-regulation of the surface binding markers, Cd1d2 and Fcgr1 (Fig. 2A). Of surprise, bare M278 did not up-regulate these markers; whereas PPP up-regulated Fcgr1 (Fig. 2A), which may suggest this receptors’ direct interaction with nanoparticles. Since Chlamydia is known to signal via the pathogen-sensing TLRs and Nod receptors pathways [31, 32], we hypothesized that PPM might signal via these same pathways. PPM significantly transcribed (P < 0.001) Nod1, Nod2, TLR1 and TLR2 in comparison to bare M278 (Fig. 2B–C). Expressions of Nod1 and TLR2 as induced by PPM were higher, suggesting a preferential signaling via these pathways. TLR1 and TLR2 transcripts as induced by PPP were surprisingly slightly higher than those of bare M278; thus indicative of PLA-PEG immune-potentiating actions.
Fig. 2. Enhanced expression of cell surface and pathogen-sensing receptors.


DCs were stimulated with PPM, M278 and PPP for 24 hours to quantify the mRNA gene transcripts of Cd1d2 and FcgR1 (A), pathogen-sensing receptors, TLR1, TLR2 (B) and Nod1 and Nod2 (C). Stimulated DCs were also subjected to flow cytometric analysis of CD80 (D), CD86 (E), CD40 (F), MHC-I (G) and MHC-II (H). Analyses were performed by gating on CD11c- APC-Cy7+ cells. The numbers included in D–H histograms are the % of positive cells for the respective molecules. Gene transcripts of co-stimulatory molecules were also quantified 24-hr post-stimulation of DCs (I).
Another essential requirement for activation and maturation of DCs is the expression of co-stimulatory and the MHC molecules. Thus, we measured their protein expression levels by employing flow cytometry. Our analysis disclosed the enhancement of CD80, CD86 and CD40 expression by PPM over those of bare M278 (9.9, 16.1 and 21.6 %, respectively) (Fig. 2D–F). Similarly, enhanced expression of MHC-I and MHC-II molecules by PPM was observed over those of bare M278 (8.5 and 8.2%, respectively) (Fig. 2G–H). Additional experiments indicated that PPM likewise up-regulated the transcription of all co-stimulatory molecules (Fig. 2I). Except for MHC-I, the up-regulation of other molecules by PPP was lower than those of bare M278 (Fig. 2D–H). These findings demonstrate that the select up-regulated molecular signatures are probably mediators of PPM recognition to drive innate and adaptive immune effector responses.
3.3. Presentation of PPM to vaccinated T cells augments immune effector mediators
The specific interaction between PPM and DCs for presentation and recognition by T cells to trigger specific adaptive immune responses was tested by experiments using co-cultures of primed DCs and T cells (from naïve and C. muridarum-vaccinated mice). First, we determined whether antigen recognition by T cells would up-regulate effector mediators reported to clear Chlamydia infections such as MHC-II [31], Cd1d2 [33], IFN-γ and Nos2 [34–36]. All mediators were up-regulated by vaccinated co-cultures in comparison to naïve co-cultures, irrespective of the stimuli (Fig. 3A–D). However, up-regulation of mediators was higher for PPM-vaccinated T cells than for those of bare M278 and PPP. Similarly, IFN-γ (Fig. 3E) and IL-2 (Fig. 3F) cytokine secretion levels were significantly (P < 0.001) increased by PPM-vaccinated T cells as compared to bare M278. But, we also observed secretion of IFN-γ and IL-2 by PPM-naïve T cells, which may suggest some potentiation by the PLA-PEG nanoparticles. Nonetheless, PPM-vaccinated T cells, secreted significantly (P < 0.05) higher levels of IL-2 than did PPM-naïve T cells. The limited secretion of IL-10 (100 and 75 pg/mL, respectively for PPM and bare M278) underscored triggering of effective Chlamydia-specific Th1 immune responses. Unstimulated DCs and T cells co-cultures alone did not induce any significant cytokines (data not shown). These results show that PPM provides the enhancement of key immune effector responses that are essential for clearance of a chlamydial infection.
Fig. 3. PPM provides enhancement of Chlamydia-specific T cell immune effector responses.

DCs were primed with PPM, M278 or PPP for 24 hours before co-culturing with purified T cells from C. muridarum vaccinated or naïve mice for an additional 48 hours. RNA samples were collected to quantify the mRNA gene transcripts for MHC-II (A), Cd1d2 (B), Nos2 (C) and IFN-γ (D) or cell-free supernatants for quantifying IFN-γ (E) and IL-2 (F). Data were analyzed by two-way ANOVA followed by Tukey’s post-hoc test or the one-tailed unpaired t-test with Welch correction using GraphPad Prism 5 software. ***P < 0.001, **P < 0.01 and *P < 0.05.
We further focused on IFN-γ, whose elevated level is a Th1-mediated protective cytokine against a chlamydial infection, by quantifying its intracytoplasmic secretion by T cells. Our results, as shown in Fig. 4, confirm that presentation of PPM to naïve (Fig. 4A–C) or vaccinated (Fig. 4D–F) T cells stimulated higher percentages of CD3+ IFN-γ secreting T cells in comparison to those induced by bare M278 and PPP. PPM presentation led to 2.28% of CD3+ IFN-γ secreting T cells; in contrast, respectively to 1.77% and 1.28 % for bare M278 and PPP (Fig. 4A–C). An increase of these percentages was observed with PPM vaccinated T cells (2.39%) in comparison to those of bare M278 (1.67%) or PPP (1.49 %), suggesting an efficient presentation of PPM in stimulating IFN-γ secretion by CD3+ T cells. Further data analyses demonstrated that IFN-γ was secreted primarily by CD3+CD4+ and not by CD3+CD8+ (data not shown) T cells again with percentages in the order of magnitude: PPM > M278 > PPP. Naïve T cells (Fig. 4G–I) secreted less CD3+CD4+ IFN-γ in comparison to vaccinated T cells (Fig. 4J–L). This data confirms that PPM antigen presentation and recognition trigger T cell activation and induction of Chlamydia-specific IFN-γ secretion principally mediated via the MHC class II and CD4+ T cells pathways.
Fig. 4. Chlamydia-specific intracellular cytokine production from CD3+CD4+ IFN-γ secreting T cells.


DCs were primed with PPM, M278 or PPP before co-culturing with T cells from naïve and C. muridarum vaccinated mice. For intracellular IFN-γ secretion, cells were stained with CD3-APC-Cy7 and CD4-PerCP-Cy5.5 followed by staining with IFN-γ-APC. Analyses were performed by gating on CD3+ IFN-γ secreting cells for naïve ((A, B, C) and vaccinated co-cultures (D, E, F) and also for CD4+ IFN-γ secreting cells from naïve (G, H, I) and vaccinated (J, K, L) mice.
3.4. PPM provides enhanced T cell proliferative and memory and effector phenotypes
The increased percentages of CD3+ IFN-γ secreting cells suggested that PPM might overall enhance the proliferation of T cells for mounting cellular adaptive immune responses. Thus, naïve and vaccinated T cells were labeled with CFSE dye followed by co-culturing with primed DCs for 120 hours. Our results demonstrate that PPM triggered enhanced proliferation of naïve and vaccinated T cells as compared to proliferation induced by M278 and PPP (Fig. 5A–F). Naïve proliferating T cells were of the magnitude of 11.8% (PPM), 8.40% (M278) and 8.97% (PPP) as shown in Fig. 5A–C. These percentages increased and were evidently higher for PPM (17.5%) than for M278 (11.9%) or PPP (13.8 %) vaccinated proliferating T cells (Fig. 5D–F). These results show that priming of DCs with PPM efficiently enhances Chlamydia-specific T cell proliferative responses, which are essential for directing cellular adaptive immune responses.
Fig. 5. Chlamydia-specific proliferating total T cells, cell proliferation and memory and effector T cells phenotypes.


CSFE-labeled T cells from naïve (A, B, C) and vaccinated (D, E, F) mice were co-cultured with primed DCs and analyzed by flow cytometry for CD3+ CFSE+ T cells. Primed DCs were co-cultured with T cells from naïve and vaccinated mice and induction of memory, and effector T cell phenotypes were quantified by staining for CD3, CD4, CD62L, and CD44, respectively. Analyses were performed by gating on CD3+ CD4+ T cells of naïve (G, H, I) and vaccinated (J, K, L) co-cultures.
Induction of antigen-specific memory and effector T cells is key to an efficacious vaccine. Memory T cells rapidly proliferate to previously encountered antigen and is a measure of long-lived memory, which can convert into functional effector T cells against pathogens. Memory and effector T cells were quantified in co-cultures by targeting CD3+CD4+ (CD44 and CD62L) T cells using flow cytometry. Once more, PPM primed DCs were more efficient in antigen presentation for stimulating memory and effector T cells (Fig. 5G–L). Naïve T cells presented with PPM as compared with bare M278 or PPP (Fig. 5G–I), respectively exhibited 2.94%, 9.76% and 3.14 % of CD3+CD4+ memory T cells (CD44high and CD62L high). Vaccinated T cells presented with PPM or bare M278 had increased memory phenotypes, respectively of 20.6% and 15.2% (Fig. 5J–L).
Of significance, was the observed increase enhancement of the CD3+CD4+ effector T cells (CD44high and CD62Llow) by PPM presentation from 10.2% in naïve to 13.8% in vaccinated populations relative to those of bare M278 (14.8 % versus 10.9%) or PPP (21.8% versus 10%). The naïve CD3+CD4+ population (CD44low and CD62Lhigh) was higher with PPM presentation to naïve cells (65.3%); however, this percentage reduced (39.5%) probably due to the increase in the memory phenotype. A close look at the CD44low and CD62Lhigh phenotypes revealed that, overall, they remained high in bare M278 and PPP co-cultures most likely because of the presence of fewer memory cells (Fig. 5K–L). The increase in CD3+CD4+ memory and effector T cells underscores presentation of PPM in association with MHC class II for elicitation of Chlamydia-specific CD4+ adaptive immune effector responses.
3.5. PPM endosomal processing and presentation by MHC class II molecules
Given the observed augmentation of Chlamydia-specific CD4+ immune effectors triggered by PPM versus bare M278, we next looked at possible differences in their endocytic trafficking pathways by performing intracellular trafficking and colocalization studies employing immunofluorescence microscopy. We assessed the expression levels of subcellular organelles in stimulated DCs that may be involved in trafficking such as EE (EEA1, early endosome), LE (Rab7, late endosome), ER (endoplasmic reticulum) and LAMP-1 (lysosome). Colocalization of sub-cellular organelles with targeted M278 was confirmed by probing using an anti-chlamydial MOMP antibody. Our results demonstrate the release of encapsulated M278 as compared to bare M278 evidently by its intense intracellular expression (Fig. 6A–D). Apparently, early endosomes (Fig. 6A), late endosomes (Fig. 6B) and endoplasmic reticulum (Fig. 6C) were highly expressed and colocalized strongly with targeted M278 in the order of magnitude: PPM > M278 > PPP. Lysosome expression and colocalization were to a lesser extent and slightly higher for M278 than for PPM (Fig. 6D). These results confirmed that PPM is predominantly processed in the endosomal compartment and avoid degradation in lysosomes for prolonged presentation and cellular activation.
Fig. 6. Intracellular trafficking and colocalization of targeted M278 in DCs.




DCs were stimulated for 24 hours with PPM, M278 and PPP followed by staining for the subcellular organelles EE (EEA1, early endosome) (A), LE (Rab7, late endosome) (B), ER (endoplasmic reticulum) (C) and lysosome (LAMP-1) (D). Colocalization with organelles was confirmed by probing for the expression of the targeted M278. DAPI (blue) was used to stain the nuclei. The top row (merge) indicate the overlay of images. Direct visualization and imaging were performed employing immunofluorescence microscopy.
Since we observed that augmentation of adaptive immune effectors was mediated primarily by MHC class II and CD4+ T cells, we considered it necessary to compare the expression and colocalization of both MHC-I and MHC-II by their direct visualization in cells. Our results revealed that MHC-I and MHC-II expressions were triggered by stimulants sequentially as: PPM > M278 > PPP (Fig. 7A–B). However, PPM provided enhanced expression of, and colocalization with, MHC-II than it did with MHC-I (Fig. 7A–B), thereby confirming that presentation of PPM is primarily mediated via the MHC-II pathway.
Fig. 7. Expression and colocalization of MHC class I and II molecules.


DCs were stimulated with PPM, M278 and PPP followed by staining for MHC class I (A) and II (B) molecules. Subcellular colocalizations with MHC class I and II were confirmed by probing for the expression of the targeted M278. DAPI (blue) was used to stain the nuclei. The top row (merge) indicate the overlay of images. Direct visualization and imaging were performed employing immunofluorescence microscopy.
3.6. Cellular entry and uptake of PPM
The mechanism(s) of antigen uptake determines the fate of antigen processing inside cells and presentation to T cells. Therefore, we sought to understand the mechanism mediating the uptake of PPM and possibly responsible for immunopotentiation of immune effectors. We selected caveolin-1 (Cav1) and clathrin (Cltc) because these endocytic mediators are reported to be involved in uptake of molecules (50–200 nm size) by DCs and macrophages by the process of pinocytosis [37, 38]. TaqMan qPCR was used to quantify the expression of Cav1 and Cltc 24 hours after stimulating DCs with PPM bare M278 or PPP. We observed significant up-regulation of Cav1 expression in PPM-stimulated DCs as compared to those stimulated with M278 and PPP (Fig 8A). Cav1 was also more expressed by PPP than M278 stimulation, indicating uptake of nanoparticles by this pathway. Expression of Cltc was low, but yet significantly (P < 0.05) induced by PPM than by M278 and PPP (Fig. 8A). Subsequently, since the expression of Cltc was weak, intracellular and colocalization studies were only performed to investigate Cav1 involvement in the uptake of PPM. PPM provided enhanced expression of Cav1 in comparison to expression levels induced by M278 or PPP (Fig. 8B), which is in agreement with the transcription data. We also observed extensive colocalization of Cav1 with targeted M278, which shows that Cav1 may be involved in the uptake of PPM.
Fig. 8. Expression of endocytic mediators and the specific-inhibition of caveolin-1 on immune effector responses.
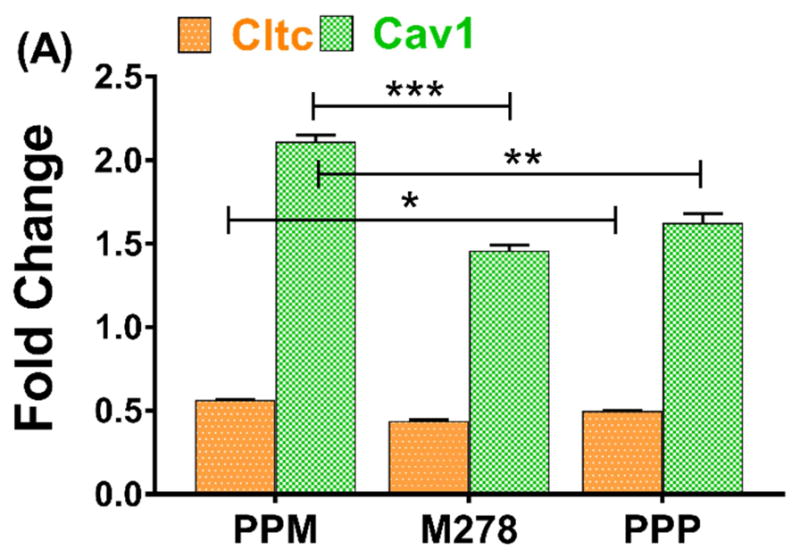


DCs were stimulated with PPM, M278, and PPP for 24 hours to quantify the gene transcripts expression of clathrin (Cltc) and caveolin-1 (Cav1) (A). Stimulated cells were stained for Cav1 expression and intracellular colocalization by probing for the expression of the targeted M278 (B). DAPI (blue) was used to stain the nuclei. The top row (merge) indicate the overlay of images. Direct visualization and imaging were performed employing immunofluorescence microscopy. Specific inhibition of Cav-1 by its inhibitor, filipin caused reduced expression of Cav1 (C) resulting in diminishing the expression of CD40 (D), CD86 (E), IL-6 (F) and IL-12p40 (G). Data analyses and asterisks are as described in Fig. 1.
Finally, we questioned whether or not Cav1 and Cltc endocytosis might be responsible for uptake of PPM. To address this issue, we inhibited the expression of Cav1 and Cltc using their respective inhibitors, filipin III and chlorpromazine hydrochloride, followed by stimulation of DCs with bare M278, PPM, and PPP. Inhibition of Cltc did not impact changes in its expression levels as compared with untreated DCs (data not shown) probably because of its low expression (Fig. 8A). Thus, we did not conduct further Cltc inhibition studies. On the other hand, Cav1 expression was significantly enhanced by all stimulants, particularly PPM (Fig. 8B) and inhibition of Cav 1 significantly (P < 0.001) inhibited its expression (Fig. 8C). Of major significance is that inhibition of Cav1 expression resulted in significant down-regulation of the expression of the costimulatory molecules, CD40 (Fig. 8B), CD86 (Fig. 8C), and the Th1 cytokines, IL-6 (Fig. 8D) and IL-12p40 (Fig. 8E). To note, inhibition of Cltc, neither inhibited nor up-regulated the expression of Cav1, suggesting that Cltc did not perturb the functions of Cav1. This exciting finding of PPM to provide high expression of Cav1 coupled with modulation of immune effectors following its specific inhibition suggest a possible caveolin-mediated endocytosis of PPM for mounting Chlamydia-specific CD4+ mediated immune effectors.
4. Discussion
Development of a vaccine against Chlamydia continues to be a top priority because of the global morbidity and socioeconomic burdens posed by this pathogen, such as pelvic inflammatory disease, infertility, and ectopic pregnancy. The absence of an approved Chlamydia vaccine may partly stem from ineffective delivery systems to provide the necessary protective immune effector responses. Recently, biodegradable nanoparticles are emerging as attractive alternative nanodelivery vaccine targets because of their unique nanoparticulate, self-adjuvanting and sustained release of antigens to the immune system [1, 19, 20, 39, 40]. We recently developed a PLA-PEG-M278 Chlamydia nanovaccine (PPM) that triggered intense immunopotentiation of specific Th1/Th2 adaptive immune effectors in immunized mice [1]. Here, we provide several mechanistic possibilities that could account for PPM immunopotentiation of immune effector responses as follows: (i) PPM provided a slow and prolonged targeted release of encapsulated M278 to DCs for the enhancement of transcribed and/or secreted effector cytokines and chemokines that are facilitators of cellular maturation, activation, and differentiation. (ii) PPM was recognized by select surface activation markers (Cd1d2, Fcgr1) and pathogen-sensing receptors (TLR2, Nod1) that bolstered activation of co-stimulatory and MHC molecules to enhance processing and T cell activation (iii) PPM primarily engaged the MHC class II pathway for targeted M278 presentation to mediate T cell specific immune effector responses (iv) PPM-primed DCs efficiently boosted targeted presentation to T cells from C. muridarium vaccinated mice by the increase of CD3+ lymphoproliferation, CD3+CD4+ IFN-γ secreting cells as well as CD3+CD4+ memory and effector phenotypes (v) PPM was delivered endosomally for processing for a prolonged presentation and cell activation, and (vi) PPM exploited the caveolin-mediated endocytosis pathway for possibly uptake and cell entry.
The self-adjuvanting and potentiating properties of PPM were apparent as it provided a slow and prolonged release of targeted encapsulated M278 that enhanced secretion of effector cytokines and chemokines that are facilitators of cellular maturation, activation and differentiation [27, 30]. Noteworthy was that low concentrations of PPM enhanced the secretion of cytokines/chemokines over time in comparison to bare M278. This significant finding is an attractive feature for a vaccine in administering a single rather than multiple booster doses. The diverse profile of secreted cytokines/chemokines was inclusive of IL-10, a known balancer of Th1/Th2 responses, whose secretion subsided over time coinciding with increasing levels of IL-6 and IL-12p40 that drive Th1 skewed immune responses. The significance of Th1-skewed immune responses for protection against chlamydial infection has been previously recognized. For example, Lu et al. [41], reported that IL-12 is required for chlamydial antigen-pulsed DCs to induce protection against a C. trachomatis infection, while Chen et al. [42] showed the development of C. muridarum-induced pathologies in IL-12 knockout mice. Also, Hook and colleagues [25] reported that human epithelial cells and DCs stimulated with C. trachomatis, respectively produced IL-18 and IL-12, which activated NK cells to secrete IFN-γ; a protagonist cyokine involved in inhibiting the replication of Chlamydia and inducing Th1 immune responses [25, 43].
Both Cd1d2 and Fcgr1 were up-regulated by PPM, thus suggesting their participatory role in cell surface binding and activation of downstream effectors. Apparently, an up-regulated CD1d expression may aid in the clearance of Chlamydia by host innate immune responses. Alternatively, low expression of CD1 can promote Chlamydia survival as observed in human urethral epithelial cells where Chlamydia evaded killing via down-regulating CD1d through proteasomal degradation [44]. Similarly, an increase in bacterial burden was reported in Cd1d knockout mice [45, 46]. Blocking of CD1d functions reduced the levels of cytokines and chemokines necessary for activating NKT cells to recruit early inflammatory cells and the homing of activated DCs to the genital tract [46]. Fcgr1 a potentiator of humoral and cellular immune responses was up-regulated by PPM and also by the PPP control, which may indicate this receptors’ preference for interacting with nanoparticulate targets. Supposedly, IFN-γ activation of Fcgr1 receptors increases the uptake concentration of antigens in APCs for effective presentation, thus significantly lowering the T cell activating adjuvant dose by several folds [47, 48]. Moreover, mice lacking the Fcgr1 receptor lose multiple cellular functions including antigen presentation and impaired protection from bacterial infections [49]. Fc receptor functions also can promote the acceleration of elevated Th1 responses for clearance of chlamydiae as seen in FcR−/− mice that showed a delayed and reduced frequency of Chlamydia-specific Th1 cells and anti-chlamydial antibodies augmented Th1 activation by FcR+/+ but not FcR−/− APCs [48]. Both Cd1d2 and Fcgr1 recognition underscores the significance of our nanovaccine in targeting critical cell surface mediators that are involved in the clearance of Chlamydia infections.
Extracellular and intracellular signaling pathways leading to host cell inflammation and innate immunity to Chlamydia include those mediated by the pathogen-sensing TLRs and Nod proteins [32, 50–52]. Our results show that recognition of PPM most likely occurs by binding to TLR2 and Nod1 for activation of downstream immune effectors. In epithelial cells, TLR2-dependent signaling contributes to local immune responses via induction of inflammatory mediators [51, 52]. Others have reported that TLR2−/− mice infected with C. muridarum have more severe disease and persistent infection with blunted IFN-γ and T cell proliferation [53]. The role of Nod1/Nod2 proteins in host defense against C. pneumonia lung infections is well documented [52] but alternatively with discordant data for C. trachomatis vaginal infections [54], perhaps, because of tissue-specific recognition of Nod1/Nod2. The above observations suggest that APCs similarly recognize our nanovaccine as viable infective Chlamydia for triggering innate and adaptive immune effectors, which have significant implications for a vaccine candidate.
PPM stimulated the expression of both MHC class I and II molecules that are required for presentation and T cells recognition. However, MHC-II was more up-regulated and efficient in the presentation of PPM to vaccinated T cells as underscored by the significant production of IFN-γ and CD3+CD4+ IFN-γ secreting cells, a finding that is congruent with our previous report whereby PPM boosted the secretion of IFN-γ by T cells from immunized mice [1]. IFN-γ is a Th1-mediated protective cytokine against chlamydial infection as mice deficient in IL-12 or IFN-γ have exacerbated bacteria shedding from the genital tract [55–57]. The role of CD4+ IFN-γ secreting cells in protection against Chlamydia is unequivocally substantiated [58, 59]. For example, T cells and IFN-γ triggered up-regulation of epithelial iNOS genes and NO production, thus destroying Chlamydia [60]. In a more recent study, in vitro killing of C. muridarum by neutrophils was supported by an IFN-γ-dependent antibody-mediated mechanism [61], giving further credence to the importance of IFN-γ in Chlamydia clearance. The observed PPM-induced expression of MHC-I indicates simultaneous activation of CD4+ and CD8+ T cells or cross-presentation, which coincidentally may help explain the up-regulated Cd1d2 mediator that is known to activate CD8+ cells and is involved in the clearance of Chlamydia [44–46]. Moreover, both CD4+ and CD8+ T cells reportedly may provide anti-chlamydial immunity in mice [62, 63].
We additionally observed an increase in Chlamydia-specific CD3+CD4+-mediated memory (CD44highand CD62high) and effector (CD44high and CD62low) phenotypes in this study, which have significant implications for our nanovaccine. Induction of antigen-specific memory and effector T cells are indispensable for a vaccine’s efficacy because of the rapidity by which conversion of memory cells occurs into functional effector T cells against pathogens. Experiments conducted in Chlamydia-infected mice showed that effector T cells differentiation takes place in uterus-draining lymph nodes for recruitment to the genital mucosa to mediate bacterial clearance [64–67]. Recently it was discovered that tissue-resident memory T cells (Trm) are necessary for protection against a C. trachomatis genital challenge infection mediated by CD4+ memory T cells [67]. The definitive role of Trm in mediating protective immunity against Chlamydia is a new concept that requires additional investigations. Nevertheless, our finding substantiates PPM triggering of IFN-γ and memory and effector T cells that are crucial in protective immunity against Chlamydia. In this context, the potentiating property of PLA-PEG as a nanodelivery system can be linked to it facilitating the uptake of antigens by APCs along with enhancing IFN-γ production [68] as observed herein by the up-regulated expression of Fcrg1, a mediator of antigen uptake.
Our intracellular trafficking study indicates with surety that processing of PPM occurred predominantly endosomally and not lysosomally, suggesting its avoidance of lysosomal degradation. Avoidance of lysosomal degradation may address the prolonged activation of cells for the enhanced production of immune effectors whereas; the rapid decline and lower expression of effectors triggered by bare M278 could be due to rapid lysosomal degradation. Indeed, PPM up-regulation of FcgR1, which functionally concentrates antigens in APCs may also provide a mechanism for PPM enhancement of immune responses. Another finding of significance in the present study was the high expression and colocalization of the endocytic mediator caveolin-1, suggesting its possible role in the cellular uptake mechanisms of PPM. However, more importantly, the ensuing modulation of immune effectors following the specific inhibition of caveolin-1 suggests that cellular uptake of PPM maybe via the caveolin-endocytosis pathway. Also, its endosomal processing may indicate the formation of caveosomes for sustained activation thereby enhancing the concentration of our targeted nanovaccine in endosomes/caveosomes [11] for prolonged triggering of immune effector responses.
Currently, there is emerging consensus amongst vaccinologists to develop vaccines that mimic actual pathogens in the form of particulate vaccines. As previously reported, our PPM nanoparticles are spherical with a diameter size of ~73–100 nm [1]. Interestingly, they are very similar in size to the developmental stage of Chlamydia EBs of ~0.2–0.3 μm that undergo endocytosis upon contact with epithelial cells. The endocytic vesicles are modified by the EBs to prevent their entry into the endocytic-lysosomal pathway and alternatively engage trafficking on intermediate cytoskeletal filaments to the endoplasmic reticulum/Golgi activity center. Our results are supportive of other studies revealing that C. trachomatis employs the caveolin-endocytosis pathway for its uptake and internalization [69–72]. Seemingly the cellular uptake and entry of PPM in our study, thus, mimic the natural infection course of Chlamydia. The caveolin-endocytic uptake route is believed to be beneficial for enhancing the concentration of targeting antigens and improving the vaccine’s therapeutic effect. Similar observations have been made by others where caveolae-vesicles induce the intracellular migration of biomaterials with a diameter size of 50–80 nm [15]. Nanoparticles with sizes mimicking those of pathogens (20–200 nm) are also efficiently taken up by DCs [15, 73] and the caveosomes containing targeted nanoparticulate-vaccines are transported with microtubules to the endoplasmic reticulum [12, 74] to stimulate enhanced immune responses.
In conclusion, our data show that in comparison to bare M278, the PLA-PEG delivery of targeted M278 enhances its stimulatory outcomes regarding recognition, uptake, processing, and presentation that are mediated by multiple pathways to activate innate and adaptive immune effectors. Specifically, immunopotentiation by our nanovaccine is mediated by processing endosomally and avoiding lysosomal degradation for prolonged presentation and cell activation, and most-likely exploitation of the caveolin-mediated endocytosis pathway for uptake with an MHC class II-dependent presentation to trigger robust Chlamydia-specific CD4+ mediated immune effector responses. Moreover, this study provides ‘proof of feasibility’ in supporting PLA-PEG as an efficient nanodelivery system and PPM as a Chlamydia nanovaccine candidate given its triggering of key cellular immune effector responses.
Acknowledgments
This research was supported by the National Institute of Allergy And Infectious Diseases of the National Institutes of Health (NIH) under Award Number R21AI111159, NIH-NIGMS-RISE (1R25GM106995-01) and the National Science Foundation (NSF)-CREST (HRD-1241701) grants. The authors would like to thank Yvonne Williams, LaShaundria Lucas and Juwana Smith-Henderson of CNBR for their excellent administrative assistance. Special thanks go to Golden Muse (www.golden-muse.com) for the illustration used in this manuscript, and Marion L Spell, the University of Alabama at Birmingham CFAR Flow Core Facility (An NIH funded program-P30 AI027767), for assistance with the FACS data. G. H. G. is a member of the Research Career of CONICET (Argentina).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dixit S, Singh SR, Yilma AN, Agee RD, 2nd, Taha M, Dennis VA. Poly(lactic acid)-poly(ethylene glycol) nanoparticles provide sustained delivery of a Chlamydia trachomatis recombinant MOMP peptide and potentiate systemic adaptive immune responses in mice. Nanomedicine. 2014;10:1311–21. doi: 10.1016/j.nano.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalkanidis M, Pietersz GA, Xiang SD, Mottram PL, Crimeen-Irwin B, Ardipradja K, et al. Methods for nano-particle based vaccine formulation and evaluation of their immunogenicity. Methods. 2006;40:20–9. doi: 10.1016/j.ymeth.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 3.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787–96. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 4.Jennings GT, Bachmann MF. Designing recombinant vaccines with viral properties: a rational approach to more effective vaccines. Curr Mol Med. 2007;7:143–55. doi: 10.2174/156652407780059140. [DOI] [PubMed] [Google Scholar]
- 5.Shi Y, Huang G. Recent developments of biodegradable and biocompatible materials based micro/nanoparticles for delivering macromolecular therapeutics. Crit Rev Ther Drug Carrier Syst. 2009;26:29–84. doi: 10.1615/critrevtherdrugcarriersyst.v26.i1.20. [DOI] [PubMed] [Google Scholar]
- 6.Pelkmans L, Puntener D, Helenius A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science. 2002;296:535–9. doi: 10.1126/science.1069784. [DOI] [PubMed] [Google Scholar]
- 7.Rappoport JZ. Focusing on clathrin-mediated endocytosis. Biochem J. 2008;412:415–23. doi: 10.1042/BJ20080474. [DOI] [PubMed] [Google Scholar]
- 8.Wang J, Byrne JD, Napier ME, DeSimone JM. More effective nanomedicines through particle design. Small (Weinheim an der Bergstrasse, Germany) 2011;7:1919–31. doi: 10.1002/smll.201100442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Tiruppathi C, Minshall RD, Malik AB. Size and dynamics of caveolae studied using nanoparticles in living endothelial cells. ACS Nano. 2009;3:4110–6. doi: 10.1021/nn9012274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu S, Olenyuk BZ, Okamoto CT, Hamm-Alvarez SF. Targeting receptor-mediated endocytotic pathways with nanoparticles: rationale and advances. Adv Drug Deliv Rev. 2013;65:121–38. doi: 10.1016/j.addr.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yameen B, Choi WI, Vilos C, Swami A, Shi J, Farokhzad OC. Insight into nanoparticle cellular uptake and intracellular targeting. J Control Release. 2014;190:485–99. doi: 10.1016/j.jconrel.2014.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- 13.Neefjes J, Ovaa H. A peptide’s perspective on antigen presentation to the immune system. Nat Chem Biol. 2013;9:769–75. doi: 10.1038/nchembio.1391. [DOI] [PubMed] [Google Scholar]
- 14.Kou L, Sun J, Zhai Y, He Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian Journal of Pharmaceutical Sciences. 2013;8:1–10. [Google Scholar]
- 15.Xiang SD, Scholzen A, Minigo G, David C, Apostolopoulos V, Mottram PL, et al. Pathogen recognition and development of particulate vaccines: does size matter? Methods. 2006;40:1–9. doi: 10.1016/j.ymeth.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 16.Kanchan V, Panda AK. Interactions of antigen-loaded polylactide particles with macrophages and their correlation with the immune response. Biomaterials. 2007;28:5344–57. doi: 10.1016/j.biomaterials.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 17.Zhao L, Seth A, Wibowo N, Zhao CX, Mitter N, Yu C, et al. Nanoparticle vaccines. Vaccine. 2014;32:327–37. doi: 10.1016/j.vaccine.2013.11.069. [DOI] [PubMed] [Google Scholar]
- 18.Cambridge CD, Singh SR, Waffo AB, Fairley SJ, Dennis VA. Formulation, characterization, and expression of a recombinant MOMP Chlamydia trachomatis DNA vaccine encapsulated in chitosan nanoparticles. International journal of nanomedicine. 2013;8:1759–71. doi: 10.2147/IJN.S42723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fairley SJ, Singh SR, Yilma AN, Waffo AB, Subbarayan P, Dixit S, et al. Chlamydia trachomatis recombinant MOMP encapsulated in PLGA nanoparticles triggers primarily T helper 1 cellular and antibody immune responses in mice: a desirable candidate nanovaccine. International journal of nanomedicine. 2013;8:2085–99. doi: 10.2147/IJN.S44155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taha MA, Singh SR, Dennis VA. Biodegradable PLGA85/15 nanoparticles as a delivery vehicle for Chlamydia trachomatis recombinant MOMP-187 peptide. Nanotechnology. 2012;23:325101. doi: 10.1088/0957-4484/23/32/325101. [DOI] [PubMed] [Google Scholar]
- 21.Inaba K, Swiggard WJ, Steinman RM, Romani N, Schuler G, Brinster C. Isolation of dendritic cells. Current protocols in immunology. 2009;Chapter 3(Unit 3):7. doi: 10.1002/0471142735.im0307s86. [DOI] [PubMed] [Google Scholar]
- 22.Marim FM, Silveira TN, Lima DS, Jr, Zamboni DS. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PloS one. 2010;5:e15263. doi: 10.1371/journal.pone.0015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gautam A, Dixit S, Embers M, Gautam R, Philipp MT, Singh SR, et al. Different patterns of expression and of IL-10 modulation of inflammatory mediators from macrophages of Lyme disease-resistant and -susceptible mice. PloS one. 2012;7:e43860. doi: 10.1371/journal.pone.0043860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voigt J, Christensen J, Shastri VP. Differential uptake of nanoparticles by endothelial cells through polyelectrolytes with affinity for caveolae. Proc Natl Acad Sci U S A. 2014;111:2942–7. doi: 10.1073/pnas.1322356111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hook CE, Matyszak MK, Gaston JS. Infection of epithelial and dendritic cells by Chlamydia trachomatis results in IL-18 and IL-12 production, leading to interferon-gamma production by human natural killer cells. FEMS immunology and medical microbiology. 2005;45:113–20. doi: 10.1016/j.femsim.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 26.Igietseme JU, Murdin A. Induction of protective immunity against Chlamydia trachomatis genital infection by a vaccine based on major outer membrane protein-lipophilic immune response-stimulating complexes. Infect Immun. 2000;68:6798–806. doi: 10.1128/iai.68.12.6798-6806.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebre MC, Burwell T, Vieira PL, Lora J, Coyle AJ, Kapsenberg ML, et al. Differential expression of inflammatory chemokines by Th1- and Th2-cell promoting dendritic cells: a role for different mature dendritic cell populations in attracting appropriate effector cells to peripheral sites of inflammation. Immunology and cell biology. 2005;83:525–35. doi: 10.1111/j.1440-1711.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 28.Vieira PL, de Jong EC, Wierenga EA, Kapsenberg ML, Kalinski P. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J Immunol. 2000;164:4507–12. doi: 10.4049/jimmunol.164.9.4507. [DOI] [PubMed] [Google Scholar]
- 29.Ozawa H, Ding W, Torii H, Hosoi J, Seiffert K, Campton K, et al. Granulocyte-macrophage colony-stimulating factor gene transfer to dendritic cells or epidermal cells augments their antigen-presenting function including induction of anti-tumor immunity. The Journal of investigative dermatology. 1999;113:999–1005. doi: 10.1046/j.1523-1747.1999.00769.x. [DOI] [PubMed] [Google Scholar]
- 30.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annual review of immunology. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 31.Morrison RP, Feilzer K, Tumas DB. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infect Immun. 1995;63:4661–8. doi: 10.1128/iai.63.12.4661-4668.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou Y, Lei W, He Z, Li Z. The role of NOD1 and NOD2 in host defense against chlamydial infection. FEMS Microbiol Lett. 2016:363. doi: 10.1093/femsle/fnw170. [DOI] [PubMed] [Google Scholar]
- 33.Dowds CM, Kornell SC, Blumberg RS, Zeissig S. Lipid antigens in immunity. Biological chemistry. 2014;395:61–81. doi: 10.1515/hsz-2013-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Igietseme JU, Perry LL, Ananaba GA, Uriri IM, Ojior OO, Kumar SN, et al. Chlamydial infection in inducible nitric oxide synthase knockout mice. Infect Immun. 1998;66:1282–6. doi: 10.1128/iai.66.4.1282-1286.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Donnell H, Pham OH, Li LX, Atif SM, Lee SJ, Ravesloot MM, et al. Toll-like receptor and inflammasome signals converge to amplify the innate bactericidal capacity of T helper 1 cells. Immunity. 2014;40:213–24. doi: 10.1016/j.immuni.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rottenberg ME, Gigliotti-Rothfuchs A, Wigzell H. The role of IFN-gamma in the outcome of chlamydial infection. Current opinion in immunology. 2002;14:444–51. doi: 10.1016/s0952-7915(02)00361-8. [DOI] [PubMed] [Google Scholar]
- 37.Kumari S, Mg S, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010;20:256–75. doi: 10.1038/cr.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tonigold M, Mailander V. Endocytosis and intracellular processing of nanoparticles in dendritic cells: routes to effective immunonanomedicines. Nanomedicine (London, England) 2016;11:2625–30. doi: 10.2217/nnm-2016-0195. [DOI] [PubMed] [Google Scholar]
- 39.Grossen P, Witzigmann D, Sieber S, Huwyler J. PEG-PCL-based nanomedicines: A biodegradable drug delivery system and its application. J Control Release. 2017;260:46–60. doi: 10.1016/j.jconrel.2017.05.028. [DOI] [PubMed] [Google Scholar]
- 40.Liu YW, Chen YH, Chen JW, Tsai PJ, Huang IH. Immunization with Recombinant TcdB-Encapsulated Nanocomplex Induces Protection against Clostridium difficile Challenge in a Mouse Model. Front Microbiol. 2017;8:1411. doi: 10.3389/fmicb.2017.01411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu H, Zhong G. Interleukin-12 production is required for chlamydial antigen-pulsed dendritic cells to induce protection against live Chlamydia trachomatis infection. Infect Immun. 1999;67:1763–9. doi: 10.1128/iai.67.4.1763-1769.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Lei L, Zhou Z, He J, Xu S, Lu C, et al. Contribution of interleukin-12 p35 (IL-12p35) and IL-12p40 to protective immunity and pathology in mice infected with Chlamydia muridarum. Infect Immun. 2013;81:2962–71. doi: 10.1128/IAI.00161-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasilevsky S, Greub G, Nardelli-Haefliger D, Baud D. Genital Chlamydia trachomatis: understanding the roles of innate and adaptive immunity in vaccine research. Clinical microbiology reviews. 2014;27:346–70. doi: 10.1128/CMR.00105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawana K, Quayle AJ, Ficarra M, Ibana JA, Shen L, Kawana Y, et al. CD1d degradation in Chlamydia trachomatis-infected epithelial cells is the result of both cellular and chlamydial proteasomal activity. The Journal of biological chemistry. 2007;282:7368–75. doi: 10.1074/jbc.M610754200. [DOI] [PubMed] [Google Scholar]
- 45.Bilenki L, Wang S, Yang J, Fan Y, Joyee AG, Yang X. NK T cell activation promotes Chlamydia trachomatis infection in vivo. J Immunol. 2005;175:3197–206. doi: 10.4049/jimmunol.175.5.3197. [DOI] [PubMed] [Google Scholar]
- 46.Jiang J, Karimi O, Ouburg S, Champion CI, Khurana A, Liu G, et al. Interruption of CXCL13-CXCR5 axis increases upper genital tract pathology and activation of NKT cells following chlamydial genital infection. PloS one. 2012;7:e47487. doi: 10.1371/journal.pone.0047487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wallace PK, Tsang KY, Goldstein J, Correale P, Jarry TM, Schlom J, et al. Exogenous antigen targeted to FcgammaRI on myeloid cells is presented in association with MHC class I. Journal of immunological methods. 2001;248:183–94. doi: 10.1016/s0022-1759(00)00351-3. [DOI] [PubMed] [Google Scholar]
- 48.Moore T, Ekworomadu CO, Eko FO, MacMillan L, Ramey K, Ananaba GA, et al. Fc receptor-mediated antibody regulation of T cell immunity against intracellular pathogens. The Journal of infectious diseases. 2003;188:617–24. doi: 10.1086/377134. [DOI] [PubMed] [Google Scholar]
- 49.Ioan-Facsinay A, de Kimpe SJ, Hellwig SM, van Lent PL, Hofhuis FM, van Ojik HH, et al. FcgammaRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity. 2002;16:391–402. doi: 10.1016/s1074-7613(02)00294-7. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Liu Q, Chen D, Guan J, Ma L, Zhong G, et al. Chlamydial Lipoproteins Stimulate Toll-Like Receptors 1/2 Mediated Inflammatory Responses through MyD88-Dependent Pathway. Front Microbiol. 2017;8:78. doi: 10.3389/fmicb.2017.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Massari P, Toussi DN, Tifrea DF, de la Maza LM. Toll-like receptor 2-dependent activity of native major outer membrane protein proteosomes of Chlamydia trachomatis. Infect Immun. 2013;81:303–10. doi: 10.1128/IAI.01062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shimada K, Chen S, Dempsey PW, Sorrentino R, Alsabeh R, Slepenkin AV, et al. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS pathogens. 2009;5:e1000379. doi: 10.1371/journal.ppat.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beckett EL, Phipps S, Starkey MR, Horvat JC, Beagley KW, Foster PS, et al. TLR2, but not TLR4, is required for effective host defence against Chlamydia respiratory tract infection in early life. PloS one. 2012;7:e39460. doi: 10.1371/journal.pone.0039460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Welter-Stahl L, Ojcius DM, Viala J, Girardin S, Liu W, Delarbre C, et al. Stimulation of the cytosolic receptor for peptidoglycan, Nod1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cellular microbiology. 2006;8:1047–57. doi: 10.1111/j.1462-5822.2006.00686.x. [DOI] [PubMed] [Google Scholar]
- 55.Naglak EK, Morrison SG, Morrison RP. IFNgamma is Required for Optimal Antibody-Mediated Immunity against Genital Chlamydia Infection. Infect Immun. 2016 doi: 10.1128/IAI.00749-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jayarapu K, Kerr M, Ofner S, Johnson RM. Chlamydia-specific CD4 T cell clones control Chlamydia muridarum replication in epithelial cells by nitric oxide-dependent and -independent mechanisms. J Immunol. 2010;185:6911–20. doi: 10.4049/jimmunol.1002596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gondek DC, Olive AJ, Stary G, Starnbach MN. CD4+ T cells are necessary and sufficient to confer protection against Chlamydia trachomatis infection in the murine upper genital tract. J Immunol. 2012;189:2441–9. doi: 10.4049/jimmunol.1103032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cotter TW, Ramsey KH, Miranpuri GS, Poulsen CE, Byrne GI. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infect Immun. 1997;65:2145–52. doi: 10.1128/iai.65.6.2145-2152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang S, Fan Y, Brunham RC, Yang X. IFN-gamma knockout mice show Th2-associated delayed-type hypersensitivity and the inflammatory cells fail to localize and control chlamydial infection. European journal of immunology. 1999;29:3782–92. doi: 10.1002/(SICI)1521-4141(199911)29:11<3782::AID-IMMU3782>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 60.Johnson RM, Brunham RC. Tissue-Resident T Cells as the Central Paradigm of Chlamydia Immunity. Infect Immun. 2016;84:868–73. doi: 10.1128/IAI.01378-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Naglak EK, Morrison SG, Morrison RP. Neutrophils Are Central to Antibody-Mediated Protection against Genital Chlamydia. Infect Immun. 2017:85. doi: 10.1128/IAI.00409-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Igietseme JU, Magee DM, Williams DM, Rank RG. Role for CD8+ T cells in antichlamydial immunity defined by Chlamydia-specific T-lymphocyte clones. Infect Immun. 1994;62:5195–7. doi: 10.1128/iai.62.11.5195-5197.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nogueira CV, Zhang X, Giovannone N, Sennott EL, Starnbach MN. Protective immunity against Chlamydia trachomatis can engage both CD4+ and CD8+ T cells and bridge the respiratory and genital mucosae. J Immunol. 2015;194:2319–29. doi: 10.4049/jimmunol.1402675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li LX, McSorley SJ. B cells enhance antigen-specific CD4 T cell priming and prevent bacteria dissemination following Chlamydia muridarum genital tract infection. PLoS pathogens. 2013;9:e1003707. doi: 10.1371/journal.ppat.1003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roan NR, Gierahn TM, Higgins DE, Starnbach MN. Monitoring the T cell response to genital tract infection. Proc Natl Acad Sci U S A. 2006;103:12069–74. doi: 10.1073/pnas.0603866103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olive AJ, Gondek DC, Starnbach MN. CXCR3 and CCR5 are both required for T cell-mediated protection against C. trachomatis infection in the murine genital mucosa. Mucosal immunology. 2011;4:208–16. doi: 10.1038/mi.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stary G, Olive A, Radovic-Moreno AF, Gondek D, Alvarez D, Basto PA, et al. VACCINES. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science. 2015;348:aaa8205. doi: 10.1126/science.aaa8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bramwell VW, Perrie Y. Particulate delivery systems for vaccines: what can we expect? The Journal of pharmacy and pharmacology. 2006;58:717–28. doi: 10.1211/jpp.58.6.0002. [DOI] [PubMed] [Google Scholar]
- 69.Norkin LC, Wolfrom SA, Stuart ES. Association of caveolin with Chlamydia trachomatis inclusions at early and late stages of infection. Experimental cell research. 2001;266:229–38. doi: 10.1006/excr.2001.5202. [DOI] [PubMed] [Google Scholar]
- 70.Stuart ES, Webley WC, Norkin LC. Lipid rafts, caveolae, caveolin-1, and entry by Chlamydiae into host cells. Experimental cell research. 2003;287:67–78. doi: 10.1016/s0014-4827(03)00059-4. [DOI] [PubMed] [Google Scholar]
- 71.Webley WC, Norkin LC, Stuart ES. Caveolin-2 associates with intracellular chlamydial inclusions independently of caveolin-1. BMC infectious diseases. 2004;4:23. doi: 10.1186/1471-2334-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rank RG. Models of Immunity. Chlamydia: American Society of Microbiology; 1999. [Google Scholar]
- 73.Gutjahr A, Phelip C, Coolen AL, Monge C, Boisgard AS, Paul S, et al. Biodegradable Polymeric Nanoparticles-Based Vaccine Adjuvants for Lymph Nodes Targeting. Vaccines. 2016:4. doi: 10.3390/vaccines4040034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pelkmans L, Kartenbeck J, Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nature cell biology. 2001;3:473–83. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]