

Abstract
Ischemia/reperfusion injury is associated with contractile dysfunction and increased cardiomyocyte death. Overexpression of the hematopoietic lineage substrate-1-associated protein X-1 (HAX-1) has been shown to protect from cellular injury but the function of endogenous HAX-1 remains obscure due to early lethality of the knockout mouse. Herein we generated a cardiac-specific and inducible HAX-1 deficient model, which uncovered an unexpected role of HAX-1 in regulation of sarco/endoplasmic reticulum Ca-ATPase (SERCA2a) in ischemia/reperfusion injury. Although ablation of HAX-1 in the adult heart elicited no morphological alterations under non-stress conditions, it diminished contractile recovery and increased infarct size upon ischemia/reperfusion injury. These detrimental effects were associated with increased loss of SERCA2a. Enhanced SERCA2a degradation was not due to alterations in calpain and calpastatin levels or calpain activity. Conversely, HAX-1 overexpression improved contractile recovery and maintained SERCA2a levels. The regulatory effects of HAX-1 on SERCA2a degradation were observed at multiple levels, including intact hearts, isolated cardiomyocytes and sarcoplasmic reticulum microsomes. Mechanistically, HAX-1 ablation elicited increased production of reactive oxygen species at the sarco/endoplasic reticulum compartment, resulting in SERCA2a oxidation and a predisposition to its proteolysis. This effect may be mediated by NAPDH oxidase 4 (NOX4), a novel binding partner of HAX-1. Accordingly, NOX inhibition with apocynin abrogated the effects of HAX-1 ablation in hearts subjected to ischemia/reperfusion injury. Taken together, our findings reveal a role of HAX-1 in the regulation of oxidative stress and SERCA2a degradation, implicating its importance in calcium homeostasis and cell survival pathways.
Keywords: heart, HAX-1, SERCA2a, NOX4, oxidative modification, ischemia reperfusion, proteolysis
1. Introduction
Ischemia reperfusion (I/R) injury and the resulting myocardial infarction are a leading cause of heart failure and death in the U.S. and worldwide [1]. One of the major characteristics of the stressed human or experimental heart is diminished contractile parameters, associated with depressed Ca2+-transport by the sarco/endoplasmic reticulum (SR/ER) Ca2+ ATPase (SERCA2a) and its regulator phospholamban (PLN)[2,3]. The resultant reduction of SR Ca2+ content adversely affects Ca2+ homeostasis causing aberrant SR/ER, mitochondrial, and cytosolic signaling that leads to cell death. Decades of research have suggested that targeting the SERCA2a/PLN activity may restore contractile function and benefit the stressed heart. PLN exists in a complex with SERCA2a, inhibiting its function and phosphorylation of PLN during β-agonist stimulation relieves its inhibitory effects [4]. Recent evidence indicates there are several other binding partners of PLN and SERCA2a, which modulate the function of SR Ca2+-transport through a much larger regulatory complex [5,6]. Among these partners, the HS-associated protein X-1 (HAX-1) has been found to directly interact with PLN [7]. In cardiomyocytes, HAX-1 also localizes to SR, where it increases inhibition of SERCA2a by PLN and depressing contractility [8].
HAX-1 is an approximately 35 kDa protein, which was originally found to form a complex with HS-1 (hematopoietic lineage cell-specific protein-1) in lymphocytes, and mediate lymphocyte differentiation. HAX-1 is ubiquitously expressed at the mitochondria with critical function in immune and neuronal cells [9]. Loss of HAX-1 protein as a result of human mutations causes severe neutropenia [10], through mitochondrial instability in neutrophils [9]. In the mouse, global genetic deletion of HAX-1 associates with a short life-span due to progressive loss of neuronal cells [9]. With respect to the heart, previous work has shown that HAX-1 overexpression protects from cell death and enhances recovery after ischemia/reperfusion (I/R) injury through regulation of inositol requiring enzyme-1 (IRE-1) ER stress signaling [11] and cyclophilin D dependent mitochondrial stability [12]. Interestingly, loss of SERCA2a activity by heterozygous deletion or pharmacological inhibition with thapsigargin can induce or exacerbate cell death through both of these pathways, suggesting a potential common link between them [13–15]. Furthermore, the decreases in SERCA2a protein levels and activity in human and experimental I/R [3,16–18] could serve as an upstream initiator of ER and mitochondrial stress signaling. Indeed, transgenic or viral over-expression of SERCA2a confers cardioprotection [19–21]. However, increased SERCA2a activity though PLN ablation corresponds to exacerbated injury during I/R [22], indicating a gap in our understanding of Ca2+ mediated cell death mechanisms.
The levels and activity of SERCA2a in cardiomyocytes can be modulated by various post-translation modifications (PTMs). Specifically, the small ubiquitin like modifier (SUMO) can be conjugated to SERCA2a, increasing expression and activity of the enzyme. Indeed, enhanced SUMOylation reverses the heart failure phenotype in small and large animal models indicating its therapeutic potential [23,24]. Additionally, tyrosine nitration and cysteine sulfonylation of SERCA2a are both associated with depressed function, whereas cysteine glutathionylation may elevate enzymatic activity. In particular, cysteine 674 is a critical site, which impacts vascular and cardiac function [25–30]. SERCA2a activity can be also modulated through proteolytic degradation, associated with increased activity of the Ca2+ sensitive proteases, calpain 1 and 2 after ischemia/reperfusion injury [16]. Yet, there is no clear evidence that this enzyme can serve as a specific substrate of calpain. Interestingly, previous studies suggested that HAX-1 may also down-regulate SERCA2a protein levels in HEK 293 cells [31]. However, these studies in non-muscle cell lines may not reflect findings in striated muscle.
Since global HAX-1 ablation results in early lethality, we generated a cardiac specific and inducible knockout mouse model to further characterize the role of endogenous HAX-1 in cardioprotection. Our findings demonstrate that HAX-1 deficiency results in diminished contractile recovery and increased infarct after I/R injury associated with increased SERCA2a degradation. The underlying mechanisms include increases in SERCA2a oxidation and reactive oxygen production at the ER/SR through direct interaction of HAX-1 with NOX4. Thus, a regulatory complex of SERCA2a/PLN/HAX-1/NOX4 may be a nodal point in the redox control of the heart and dysregulation may serve as a precipitating event in numerous stress pathways.
2. Materials and Methods
2.1. Human myocardial tissue
The current investigation conforms to the principles outlined in the Declaration of Helsinki. Briefly, failing heart samples were acquired from seven patients (4 females, 2 males, and 1 with gender that cannot be tracked), whose ages ranged from 48 to 69 years. Cardiac dysfunction was caused by ischemic heart disease (IHD), idiopathic dilated cardiomyopathy (IDC), and congestive heart failure (CHF). The average ejection fraction of the patients was 20 ± 3%, which can be defined as heart failure with reduced ejection fraction (HFrEF). All heart samples were obtained from explanted hearts at the time of cardiac transplantation. As controls, seven non-failing hearts (5 females and 2 males in the age range of 52–61 years) were obtained from donors who had normal cardiac function and died from neurological diseases or road traffic accidents, as previously described [32].
2.2. Mouse models
The HAXiKO mouse model (C57BL/6J: HAXflox/flox × αMHC-mer/cre/mer) was developed by crossing a floxed HAX-1 mouse [33] with a mouse expressing the mer/cre/mer driven by the myosin heavy chain promoter (αMHC-mer/cre/mer or CRE). Wild type (WT) mice were litter mates of the HAXiKO lacking the mer/cre/mer transgene (HAXflox/flox). Beginning at 8 weeks of age, mice of all backgrounds were treated with tamoxifen (40 mg/kg) for 14 days to induce cre recombinase activity and this resulted in full ablation of HAX-1 in the heart. Experiments were conducted 2-4 weeks after termination of tamoxifen treatment (12-14 weeks old) and were performed according to the National Institutes of Health Publication No. 8523: Guide for the Care and Use of Laboratory Animals. To assess potential adverse effects of tamoxifen/Cre on cardiac function, we performed echocardiography experiments at baseline, after completion of the tamoxifen treatment and at two weeks post-treatment. There were no differences in ejection fraction or LV dimensions between the HAXiKOs, MerCreMer and WT mice.
2.3. Quantitative real-time PCR assay
Total RNA was extracted and purified from heart tissue with miRNeasy Mini Kit (QIAGEN). The first-strand cDNA were generated from total RNA (1 μg) with reverse transcriptase kit (Invitrogen). PCR was then performed with Bio-Rad real-time thermal cycler by using the following specific primer sequences: Human HAX-1: (Forward) 5′- CTA CAG TAA CCC GAC ACG AAG -3′, (Reverse) 5′- AAT GGG TGA GAG GTG GAA AG-3′, and Human: GAPDH (Forward) 5′-GTC AAG GCT GAG AAC GGG AA-3′, (Reverse) 5′- AAA TGA GCC CCA GCC TTC TC-3′. The values were normalized to those obtained with GAPDH.
2.4. Global ischemia/reperfusion injury ex vivo
The cellular and functional responses to ischemia/reperfusion were assessed, using an isolated perfused heart model, as previously described [11]. Briefly, hearts were mounted on a Langendorff apparatus, and perfused with Krebs–Henseleit (KH) buffer. Temperature was maintained constant at 37 °C by water-jacketed glassware for the heart chamber, buffer reservoirs, and perfusion lines. In addition, an overhead light source was used to ensure maintenance of temperature during ischemia, which was monitored by a thermometer placed close to the perfused heart in the glass chamber. A water-filled balloon made of plastic film was inserted into the left ventricle and adjusted to achieve a left ventricular end-diastolic pressure (LVEDP) of 5 to 10 mmHg. The distal end of the catheter was connected to a Heart Performance Analyzer (Micro-Med) via a pressure transducer. Hearts were paced at 400 bpm except during ischemia, and pacing was reinitiated 2 minutes after reperfusion. After a 20-minute equilibration period, hearts were subjected to 40 minutes of no flow global ischemia, followed by 60 minutes of reperfusion. Maximum rate of contraction (+dP/dt), and maximum rate of relaxation (−dP/dt) were monitored during this process. In some experiments, 200 μM apocynin [34,35] was added directly into KH buffer and hearts were perfused with this solution during the ischemia/reperfusion protocol.
2.5. Mouse myocyte isolation and viral infection
Mouse myocytes from were isolated as described [12]. Briefly, adult mouse hearts were excised following mouse anesthesia with sodium pentobarbital (70 mg/kg, i.p.) and cannulated on a Langendorff system. Ca-free Tyrode solution (113 nmol/L NaCl, 4.7 mmol/L KCl, 0.6 mmol/L KH2PO4, 0.6 mmol/L Na2HPO4, 1.2 mmol/L MgSO4⋅7H2O, 12 mmol/L NaHCO3, 10 mmol/L KHCO3, 5 mmol/L Hepes, 3 0 mmol/L taurine, 10 mmol/L 2,3-butanedione monoxime, and 5.5 mmol/L glucose, pH 7.4) was used to perfuse the heart for 3 min at 37 °C. The perfusion was then switched to a digestion solution containing 0.25 g/L liberase blendzyme (Roche). Following digestion, the left ventricular tissue was excised, minced, and dissociated into a cell suspension. Calcium was serially added to the cellular suspension until final calcium concentration in the Tyrode solution was 1.2 mmol/L. Cardiomyocytes were pelleted by gravity, resuspended in plating media [DMEM with 5% (vol/vol) FBS, 10 mmol/L 2,3-butanedione monoxime, 100 units/mL penicillin streptomycin and 2 mL of l-glutamine] and placed on a six-well plate containing laminin-coated coverslips for 1 h. Supplementing the media with 5% (vol/vol) FBS allows optimal cardiomyocyte attachment. Once the cells attached to the coverslip, the medium was replaced with infection medium (plating medium containing no FBS). Adenoviruses (Ad.GFP, Ad.Trx1) were applied at 500 multiplicity of infection (MOI) or virus particles per cell. After 3 h of incubation at 37 °C, medium was removed and replaced with culture medium, which is DMEM containing 5 mg/L ITS (bovine insulin, human transferrin and sodium selenite; Sigma), 100 units/mL penicillin streptomycin, 2 mmol/L l-glutamine, 4 mmol/L NaHCO3, 10 mmol/L Hepes, 0.2% BSA, and 25 μmol/L blebbistatin (Cayman Chemical). The cell phenotype and morphology remained similar among GFP and Trx1 adenoviral-infected groups after 24 h of infection.
2.6. Western blot analysis
Hearts were snap frozen in liquid nitrogen and homogenized in 1× Cell Lysis Buffer (Cell Signaling Technology) supplemented with 1mM PMSF and complete protease inhibitor cocktail (Roche Applied Science). For each protein, equal amounts of samples (5–120 μg) from each heart were analyzed by SDS-PAGE, as previously described. After transfer to membranes, immunoblotting analysis was performed with the corresponding primary antibodies HAX-1 from BD Biosciences; SERCA2a (custom-made commercially, Affinity Bioreagents), Calpain 1, Calpain 2, and Calpastatin (Cell Signalling), PLN, and Calsequestrin (Thermo). This was followed by incubation with Licor fluorescent secondary antibodies at a dilution of 1:10,000. Visualization was achieved using Licor Odyssey imager. The intensities of bands were determined by the Licor Odyssey software. For each protein, the densitometric values from pre-I/R WT controls were arbitrarily converted to 100%, and the values of samples from the other groups were normalized accordingly and expressed as fold changes. Calsequestrin (CSQ) was used as an internal standard.
2.7. Calpain activity measurement
The activity calpain were assessed using a fluorometric assay, according to the manufacturer’s instructions (Biovision). 100μg of protein from each heart were diluted to 85μl of Extraction buffer provided by the manufacturer, followed by addition of 10μl of 10X Reaction Buffer. The reaction was started by addition of calpain fluorescent substrate and carried out at 37°C for 1 hour in the dark. The caspase-3 or calpain activity was quantified by a fluorometer (excitation: 400nm, emission: 505 nm)
2.8. In vitro SERCA2a degradation
SR microsomes were isolated as previously described [36]. Briefly, isolated hearts were homogenized in buffer containing 10 mM imidazole (pH 7.0), 300 mM sucrose, 1 mM EDTA, 1 mM or 10 mM DTT, as indicated in the text, and 0.3 mM phenylmethylsulfonyl fluoride. Homogenates were centrifuged at 8,000 g for 20 minutes. The supernatant was removed and combined with NaCl to a final concentration of 600 mM, and centrifuged at 100,000 g for 60 min. The pellet was resuspended in homogenization buffer with 600 mM NaCl and centrifuged again at 100,000 g for 60 min. The pellet was resuspended in homogenization buffer without DTT. For H2O2 treatment, samples were incubated with 100 μM final concentration of urea-peroxide for 20 min on ice, which was neutralized by addition of 1 mM DTT. SR microsomal protein content was quantified via the Bradford method [36]. To assess SERCA2a degradation, the final reaction mixture included 400 ng/μl SR microsomal protein and 5 mM CaCl2 in homogenization buffer. The samples were incubated at 37 C and the reaction was initiated with the addition of purified Calpain 1 (Abcam) to a final concentration of 10 ng/μl. The reaction was terminated at indicated time points with 2x SDS sample buffer. SERCA2a degradation was assessed via Western blot quantification.
2.9. Free cysteine labeling
For biotin labeling, cardiac homogenates were diluted to 0.5 mg protein/ml in labeling buffer containing 20 mM Tris (pH 6.8), 1 mM EDTA, and 1% Triton-X100. Biotin maleimide (Sigma) was added to a final concentration of 100 μM, and samples were incubated in the dark for 1 hr at room temperature. The reaction was terminated with addition of DTT to a final concentration of 5 mM. Samples were mixed with 2x SDS buffer for SDS-PAGE separation and Western blotting using the indicated primary antibodies (SERCA2a and PLN). Membranes were then incubated with fluorescent secondary antibodies and streptavidin (to detect biotin) (Licor) and simultaneously imaged using a Licor Odyssey imager. The overlaying biotin signal corresponding to SERCA2a or PLN was then quantified with densitometry.
2.10. Neonatal rat myocyte isolation and HyPer ROS measurements
Primary cultures of ventricular cardiomyocytes were prepared from 1-day-old Hsd:WI Wistar rats (Envigo International Holdings), as described (Matsui et al. Circ Res 2007). The left ventricle was isolated and the tissue was digested with collagenase type IV(Sigma), 0.1% trypsin (Life Technologies), and 15 μg/ml DNase I (Sigma) to obtain a single cell suspension. Cardiomyocyte-rich fraction was obtained by centrifugation through a discontinuous Percoll gradient. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 medium supplemented with 5% horse serum, 4 μg/ml transferrin, 0.7 ng/ml sodium selenite (Life Technologies, Inc.), 2 g/liter bovine serum albumin (fraction V), 3 mM pyruvic acid, 15 mmol/liter HEPES, 100 μmol/liter ascorbic acid, 100 μg/ml ampicillin, 5 μg/ml linoleic acid, and 100 μmol/liter 5-bromo-2′-deoxyuridine (Sigma). Generation of replication-defective human adenovirus type 5 (devoid of E1) harboring cDNA of wild-cyto-Hyper, mito-HyPer, and ER-Hyper has been previously described [37]. Mitochondrial and ER-specific H2O2 production were evaluated through the expression of compartment-specific HyPer protein, using confocal microscopy. To detect HyPer probes, two excitation wave lengths (405nm and 488nm) are used with a single detection peak between 510-550nm. Images are captured as grayscale. Pseudocolor is applied to each channel for visualization. In this case, red and green colors were chosen as they were easy to display. Ratio is obtained as 488nm/405nm.
2.11. Cardiac morphology
Mice were anesthetized and the hearts were removed from the chest cavity. The tissues were rinsed in Dulbecco’s phosphate buffered saline, blotted with filter paper, and then weighed. The skeletal muscle of the lower hind limbs was dissected out, and the length of the left tibia was measured. Hearts were fixed in 10% formalin and sectioned for histological examination of hematoxylin and eosin (H&E), Masson’s trichrome stained ventricles, and wheat germ agglutinin (WGA) stained sarcolemma were carried out by the Department of Pathology at the Cincinnati Children Hospital Medical Center [38].
2.12. In vitro binding assays
GST-PLN, GST-HAX, and GST recombinant proteins were generated as previously described [7]. HEK293 cells were transfected with a FLAG-NOX4 plasmid (OriGene) and 24hrs after transfections the cells were harvested and lysed in 50mM Tris–HCl (pH 8.0), 150 mM NaCl, 1% NP-40, supplemented with protease inhibitors (Sigma). Cell lysates were then incubated with equivalent amounts of GST-PLN, GST-HAX, and GST-protein bound to glutathione matrices for 16 h at 4 °C. The beads were washed at 4 °C three times in lysis buffer and were subsequently resuspended in 2 × SDS Laemmli sample buffer. Samples were analyzed by SDS-PAGE, transferred to nitrocellulose membrane (Schleicher and Schuell Bioscience GmbH) and probed with a FLAG antibody (Sigma-Aldrich) and an anti-rabbit peroxidase-conjugated secondary antibody (Bio-Rad).
2.13. Co-immunoprecipitations
Hearts were homogenized with 1X cell lysis buffer (Cell Signaling Technology), supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail. The homogenate was centrifuged at 10,000 rpm for 30 minutes at 4°C. The centrifuged homogenate was diluted to 1g/L (1mL of final volume) and incubated with monoclonal anti-HAX-1 (BD Biosciences), monoclonal anti-NOX4 (abcam), monoclonal anti-PLN (Thermo Scientific), or IgG antibody (Santa Cruz Biotech) at 4°C overnight with rotation. 100μL of protein G PLUS agarose beads (Santa Cruz Biotech) were added into the mixture and incubated for an additional 5 hours. Agarose beads were sedimented and washed 6 times with the cell lysis buffer. Beads-bound proteins were dissociated in 2X SDS at room temperature for 30 minutes with vortexing every 5 minute-intervals. The identification of the associated proteins was detected by western blots. Pre-immunoprecipitated WT heart homogenate was used as positive control (input), and immunoprecipitate with anti-IgG PLUS agarose was used as negative control.
2.14. Immuno-fluorescence staining
Cardiomyocytes were isolated as described above, and plated in a laminin-coated Lab-Tek chamber slides (Thermo Scientific). Cardiomyocytes were fixed with 1.2mmol/L Ca-Tyrode solution containing 4% Paraformaldehyde (Electron Microscopy Sciences) for 30 minutes at room temperature, and permeabilized with saline solution containing 0.1% Triton-X at 37°C for 15 minutes. Non-specific binding was limited by 4 hours incubation with blocking solution containing 2% goat serum. Cells were stained with monoclonal anti-HAX-1(BD Bioscience) and polyclonal anti-NOX-4 (Novus) antibodies, subsequently followed by incubation with Alex flour 488 goat anti-rabbit (Invitrogen) and DyLight 594 goat anti-rat (Abcam) in blocking solution. Images were obtained using Zeiss confocal microscopy.
2.15. Statistical Analysis
Data were expressed as the mean ± SEM. Comparisons between the means of two groups were performed by unpaired Student’s t test. Multiple groups were analyzed by using two-way ANOVA followed by Tukey’s multiple comparisons. Results were considered statistically significant at P < 0.05.
3. Results
3.1. HAX-1 expression is decreased in heart failure
The levels of the anti-apoptotic protein HAX-1 have been shown to decrease following ischemia reperfusion injury in mouse hearts [11]. In the present study, we used real-time PCR and found a significant down-regulation of HAX-1 mRNA in failing human left ventricular samples, compared to non-failing donor controls (Fig. 1A/B). These changes in mRNA were coupled with similar decreases in protein levels of HAX-1 (Fig. 1C/D). Interestingly, a strong correlation between HAX-1 protein levels and SERCA2a protein levels in human hearts is apparent, suggesting a potential cross-talk (Supplemental Fig. 1A). Similar decreases in HAX-1 levels were also observed in a more controlled experimental heart failure model, after transverse aortic constriction (Supplemental Fig. 1B). These observations suggest that decreases in HAX-1 expression may be a common pathological feature of cardiac stress, which can occur in human hearts.
Figure 1. HAX-1 mRNA and protein levels decrease in human failing hearts.
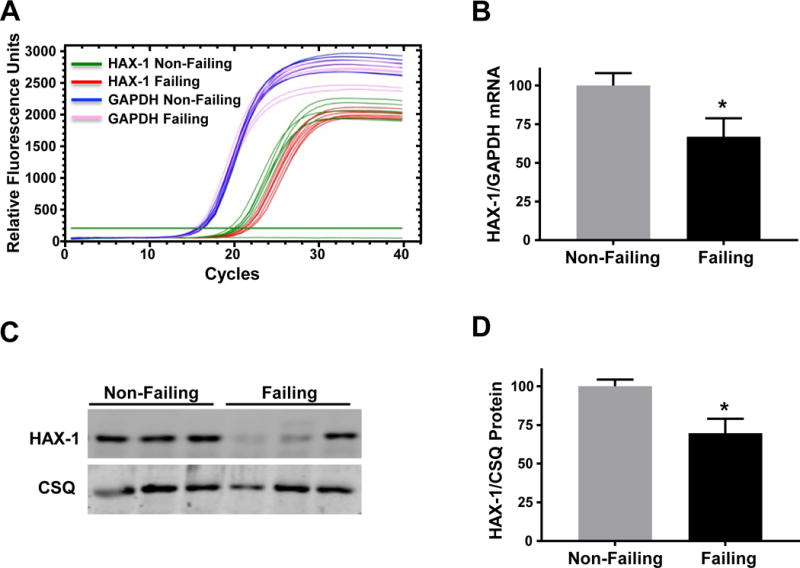
(A) Total mRNA was isolated and purified from human failing hearts (HFrEF) and non-failing hearts. Representative real-time PCR curves illustrating HAX-1 and glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA expression in non-failing and failing heart samples. (B) Quantitative analysis of HAX-1 expression levels relative to the non-failing control group after normalization to GAPDH levels. (C) Representative Western blots illustrating HAX-1 and calsequestrin (CSQ) protein expression along with (D) quantitative analysis of HAX-1 protein expression levels in failing hearts relative to the non-failing control group after normalization to CSQ. Data are presented as mean ± SEM (n = 7 hearts per group; P < 0.05: * vs non-failing group).
3.2. Inducible HAX-1 ablation does alter cardiac structure and morphology
Since global ablation of HAX-1 results in early lethality [33], the full role of endogenous HAX-1 in the heart has not been previously determined. Thus, we generated the HAXflox/flox × αMHC-mer/cre/mer mouse which is a cardiac-specific and inducible HAX-1 knockout model (HAXiKO) (Fig. 2A). HAX-1 has been identified as a critical regulator of cell death in many tissues [10,39–42] and cardiac overexpression of HAX-1 protects from ischemia/reperfusion injury (I/R)[11], and as such anticipated that HAX-1 ablation may cause cell death and cardiac dysfunction in response to the stress of aging. However, deletion of HAX-1 in the adult heart did not elicit any significant changes in heart and cell size, as determined by heart weight to tibia length and cardiomyocyte cross-sectional area at 12 months (Fig. 2B-D) compared to WT controls (HAXflox/flox). Additionally, hematoxylin and eosin staining (H&E) did not reveal any gross morphological changes between WT and HAXiKO hearts (Fig. 2E). Moreover, Masson’s trichrome stained cardiac sections showed no appreciable fibrosis in either WT or HAXiKO (Fig. 2E). These findings suggest that loss of the anti-apoptotic protein HAX-1 in the adult heart does not appear to have detrimental consequences in the absence of stress conditions.
Figure 2. HAX-1 ablation causes no morphological changes up to 12 months of age.
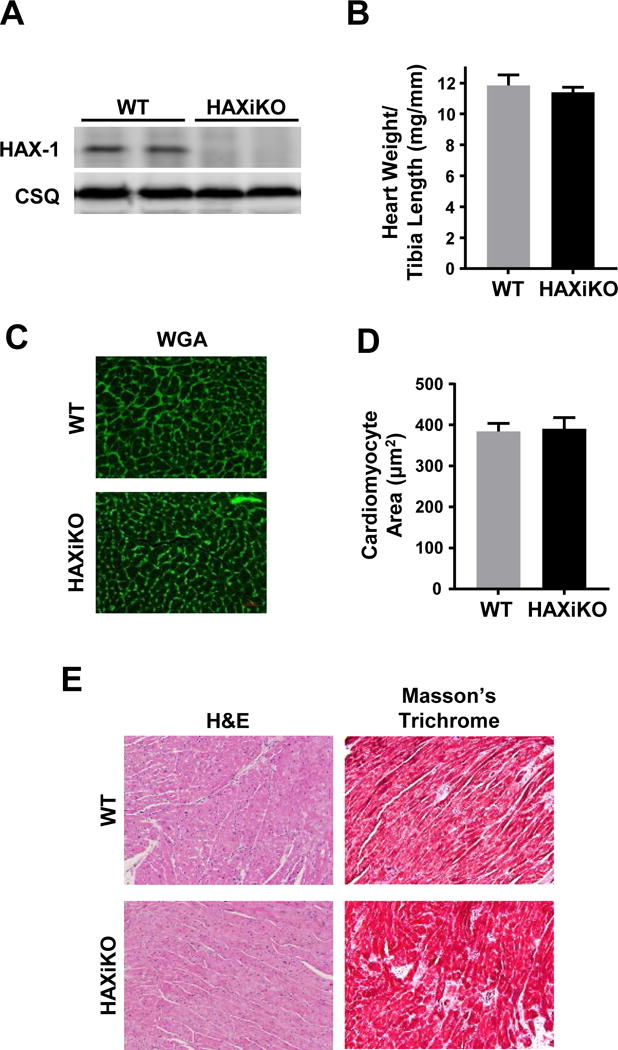
(A) Representative Western blots indicated full cardiac ablation at 3 months. (B) Hearts from 12 month old WT and HAXiKO mice were excised, weighed, and compared to tibia length. The hearts were then fixed and sectioned for histological analysis. (C) Representative images of wheat germ agglutinin (WGA) staining and (D) corresponding quantification of cardiomyocyte cross-sectional area. (E) Representative images for hematoxylin and eosin (H&E) and Masson’s Trichrome staining. Data are presented as mean ± SEM (n = 3-4).
3.3. HAX-1 deficient hearts exhibit attenuated contractile recovery and decreased SERCA2a levels upon ischemia/reperfusion injury
To elucidate the role of endogenous HAX-1 under stress conditions, hearts from HAXiKO and WT mice were subjected to 40 minutes of no flow ischemia followed by 60 minutes of reperfusion, using the ex vivo Langendorff perfusion system (Fig. 3). While basal function was enhanced by HAX-1 ablation (Supplemental Fig. 2), functional recovery during reperfusion, measured by the rates of contraction (+dP/dt) and relaxation (−dP/dt), was significantly decreased in HAX-1 null hearts (Fig. 3A/B) leading to depressed left ventricular developed pressure (LVDP) and higher left ventricular end diastolic pressure (EDP) (Fig. 3C/D). This reduction in contractile recovery was accompanied with an increase in myocardial infarct size, compared with WT hearts (45% HAXiKO, 33% WT; Fig. 3E). In parallel experiments, we did not observe any difference in functional recovery or infarct size between WT (HAXflox/flox) and CRE (αMHC-mer/cre/mer) mice (Supplemental Fig. 3), indicating that the effects in Fig. 3 were due to HAX-1 deletion and not Cre recombinase activity. The detrimental effects of HAX-1 ablation were also associated with increased apoptosis reflected by enhanced caspase-3 cleavage (Fig. 3F). Thus, loss of HAX-1 exacerbates cell death and decreases heart contractile recovery upon I/R injury.
Figure 3. HAX-1 ablation reduces functional recovery during ischemia/reperfusion.
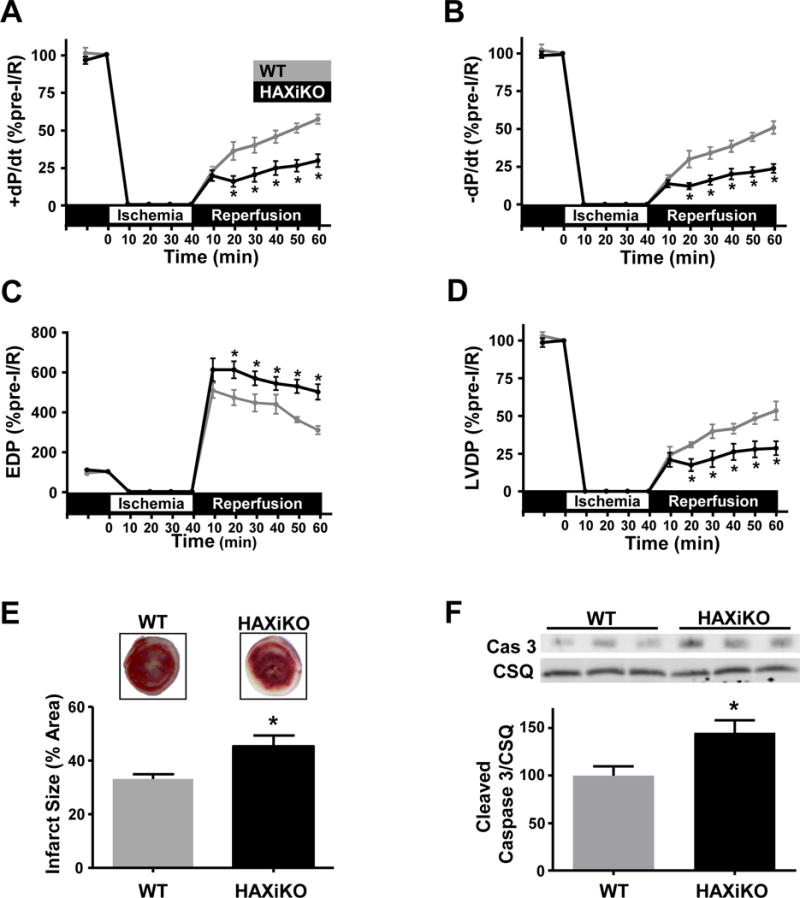
To determine the impact of HAX ablation on cardiac stress, isolated WT and HAXiKO hearts were subjected to 40 minutes of no-flow ischemia, followed by 60 minutes of reperfusion (I/R). HAX-1 ablation diminished functional recovery as assessed by (A) rate of contraction (+dP/dt), (B) rate of relaxation (-dP/dt), (C) end diastolic pressure (EDP), and (D) left ventricular developed pressure (LVDP) (n = 5). (E) Representative images of triphenyl tetrazolium chloride staining and quantification of infarct size after I/R in WT and HAXiKO hearts (n = 4). (F) Representative Western blots of cleaved caspase 3 from heart homogenates of WT and HAXiKO hearts after I/R. Calsequestrin was used as a loading control (n = 4). Data are presented as mean ± SEM (P < 0.05: * vs WT).
Previous studies in human clinical myocardial infarction and experimental-animal models of I/R have observed reduced SERCA2a levels and activity, which may contribute to decreased functional recovery and increased cell death [3,16,17]. Thus, we hypothesized that HAX-1 ablation may exacerbate decreases in SERCA2a expression, contributing to I/R injury, as HAX-1 has been previously observed to interact with SERCA2a besides PLN [7,31]. Indeed, the protein levels of SERCA2a were decreased in WT hearts post I/R (Fig. 4A). Interestingly, these decreases were parallel and time-dependent to those in HAX-1 levels (Supplemental Fig. 4). In addition, HAX-1 ablation resulted in further decreases in SERCA2a levels and the loss was almost double of that observed in WT hearts (Fig. 4A). This prompted us to examine the protein levels of SERCA2a in HAX-1 overexpressing hearts, which exhibit enhanced recovery and decreased cell death after I/R injury [11]. Indeed, HAX-1 overexpression fully prevented the loss of SERCA2a protein during I/R (Fig. 4B), which may contribute to the enhanced contractile recovery in that model [11]. The reduction in intensity of the full length 110kDa SERCA2a band was associated with the appearance of a lower molecular weight fragment, perhaps indicative of proteolytic cleavage (Fig. 4A/B).
Figure 4. HAX-1 regulates SERCA2a protein levels upon acute stress through calpain.
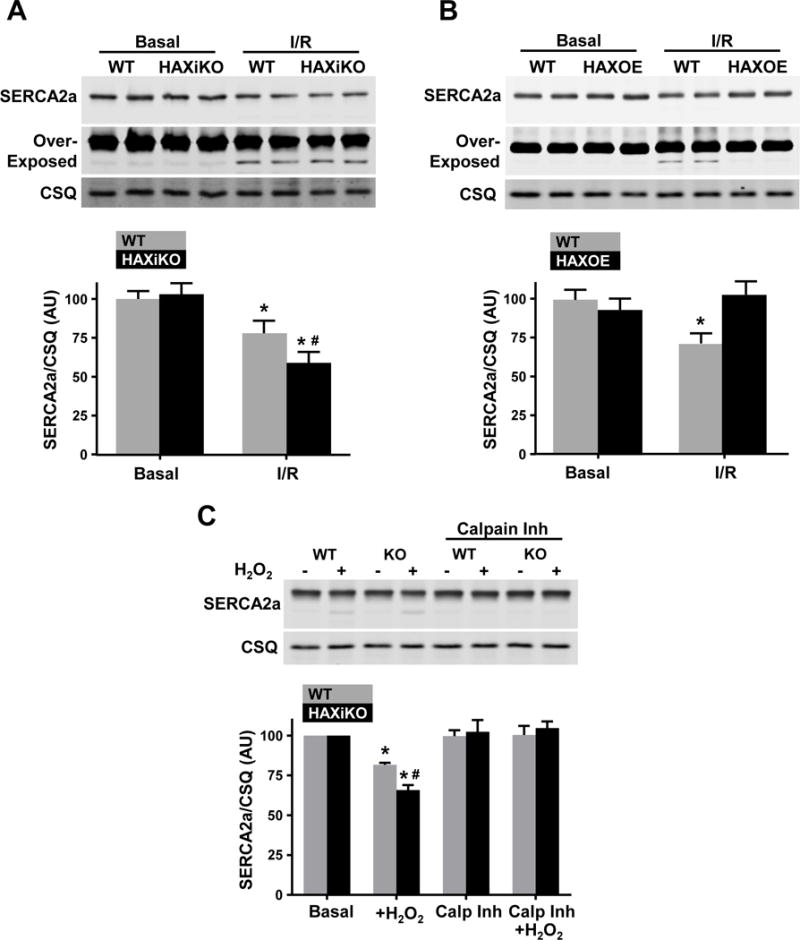
Quantitative Western blots of cardiac homogenates indicated that SERCA2a levels decrease upon I/R. The decreases are augmented by HAX-1 ablation but prevented by HAX-1 overexpression. Representative Western blots of SERCA2a and CSQ and corresponding quantification for (A) WT and HAXiKO hearts (n = 3-4) and (B) WT and HAXOE hearts (n = 3). Over-exposure of the SERCA2a blots shows a lower molecular weight fragment. (C) Cardiomyocytes were isolated from WT and HAXiKO hearts and exposed to H2O2 for 1 hr at 37 C with or without a 30 minute pretreatment with the calpain inhibitor (calp inh), MDL-28170. SERCA2a protein levels were assessed from these treated cells: Representative Western blot and corresponding quantification (n = 3). Data are presented as mean ± SEM (P < 0.05: * vs WT Basal, # vs WT I/R or H2O2).
Since a previous report suggested that the calpain family of Ca2+ sensitive proteases may be responsible for SERCA2a degradation during I/R [16], we investigated whether HAX-1 would affect the expression levels of calpain 1 and 2, the predominant cardiac isoforms. While expression of calpain 1 and 2 are both increased after I/R, no differences were observed between WT and HAXiKO hearts before and after I/R (Supplemental Fig. 5A/B). We then assessed the expression of calpastatin, the endogenous inhibitor of calpains, and found decreases upon I/R. However, the reduced levels of calpastatin were similar between WT and HAXiKO hearts (Supplemental Fig. 35A/B). In addition, we used a fluorometric assay to determine calpain activity (Supplemental Fig. 5C). There were increases after I/R but the activity was similar between WT and HAXiKO hearts. These findings suggest that changes in calpain expression or activation do not contribute to increased SERCA2a degradation in the HAX-1 ablated heart. Despite the lack of change with calpain expression and activity, we hypothesized that HAX-1 still regulated a cardiomyocyte degradation pathway. Thus, we isolated cardiomyocytes from WT and HAXiKO mice and treated them with 100 μM H2O2 to induce cellular stress, mimicking I/R in whole hearts. Indeed, SERCA2a levels were decreased (Fig. 4C), consistent with our observations in whole hearts (Fig. 4A). The decreases in SERCA2a levels were larger in HAXiKO cells, compared to WTs, upon H2O2 treatment (Fig. 4C). Importantly, blockade of calpain activity by its specific inhibitor MDL 28371 prevented loss of SERCA2a protein in WT and HAXiKO cardiomyocytes (Fig. 4C). Taken together, these results indicate that calpain mediates SERCA2a degradation and HAX-1 modulates downstream of calpain activation at the level of the cardiomyocyte.
3.4. HAX-1 modulates susceptibility of SERCA2a to oxidation dependent degradation
Induction of stress (I/R and H2O2) in intact tissue activates a complex network of response pathways, and as such, HAX-1 could be altering SERCA2a indirectly by effecting global cellular stress. To test if HAX-1 has a direct or indirect role in calpain mediated degradation of SERCA2a in the absence of stress signaling, SR microsomes were isolated from non-stressed WT and HAXiKO hearts under non-reducing conditions (-DTT) and were then exposed to purified calpain (-DTT). Consistent with findings in intact cardiomyocytes and hearts, HAX-1 deficient microsomes demonstrated increased susceptibility of SERCA2a to degradation by calpain in this purified in vitro setting (Fig. 5A). There was a 40% decrease in SERCA2a levels in HAXiKO, compared to a 25% decrease in WT (60 min of treatment with calpain) (Fig. 5A and 5B/-DTT). In parallel experiments, we isolated microsomes from HAXOE hearts and these exhibited protection against calpain degradation, compared to WT controls (Supplemental Fig. 6A). The loss of the full-length 110kDa band was accompanied by fragmentation of SERCA2a, observed in Western blots. At early time points, a series of bands between 90 and 70kDa was prominent, and these progressively diminished, while a doublet at ~35 kDa was observed, which became more intense with time (Supplemental Fig. 6B). These results indicate that calpain can directly cleave SERCA2a and at multiple sites. Furthermore, our findings suggest that HAX-1 alters the intrinsic susceptibility of SERCA2a to degradation prior to induction of stress and calpain activation.
Figure 5. HAX-1 Regulates SERCA2a degradation in SR microsomes and cardiomyocytes in a redox dependent manner.
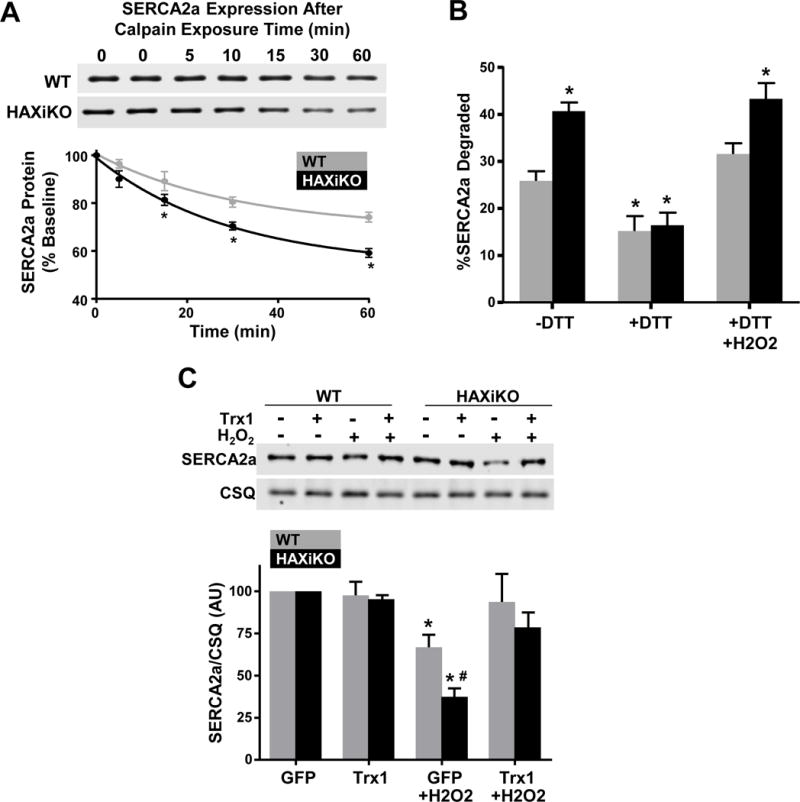
SR microsomes were isolated from WT and HAXiKO hearts and incubated with purified calpain 1 and 2 mM CaCl2 for indicated times at 37 C. (A) Representative Western blots and quantification of SERCA2a expression. (B) The extent of SERCA2a degradation in SR microsomes after 60 minutes of calpain exposure from WT and HAXiKO hearts is shown under: low reducing conditions (-DTT); high reducing condition (+DTT); and high reducing conditions followed by resuspension in non-reducing buffer and treated with H2O2 (+DTT/H2O2) (n = 3). (C) Cardiomyocytes were isolated from WT and HAXiKO hearts, infected with GFP or thioredoxin 1 (Trx1) adenoviruses, and exposed to H2O2 for 1 hr at 37 C. Representative Western blots of SERCA2a expression levels and corresponding quantification. Calsequestrin was used as a loading control (n = 3). Data are presented as mean ± SEM (P < 0.05: * vs WT GFP/-DTT, # vs WT GFP H2O2).
SERCA2a has several cysteine residues and some of them have been shown to be susceptible to oxidative modification under various experimental conditions [43]. Thus, we hypothesized that oxidative modification could serve as trigger of SERCA2a protein degradation and calpain may mediate degradation of SERCA2a in an oxidative sensitive manner. To determine this, we isolated microsomes under higher reducing conditions (10 mM DTT) to remove reversible oxidative cysteine modifications and performed the in vitro calpain assays, as described above. Indeed, SERCA2a proteolysis was significantly attenuated by 10 mM DTT and the extent of degradation was similar between WT and HAXiKO microsomes (Fig. 5B/+DTT). Accordingly, these protective effects of reducing conditions were abrogated, when microsomes were subsequently treated with H2O2 (Fig. 5B/+DTT+ H2O2).
To further confirm these findings in intact cells, we isolated cardiomyocytes from WT and HAXiKO hearts and infected them with an adenovirus encoding thioredoxin 1 (Trx1) to inhibit cysteine modification [44]. There were no effects of Trx1 on SERCA2a levels under basal conditions. Furthermore, SERCA2a degradation in GFP infected WT or HAXiKO cells after H2O2 exposure (Fig. 5C) was comparable to that observed in intact hearts and isolated cardiomyocytes (Fig 4A/C). However, SERCA2a integrity was fully protected by Trx1 in both WT and HAXiKO samples (Fig. 5C). Thus, the findings in microsomes and intact cardiomyocytes suggest that HAX-1 modulates SERCA2a degradation by calpain through an oxidative mechanism.
Finally, to determine whether the redox effects of HAX-1 on SERCA2a could be associated with direct oxidation of the enzyme on one (or more) of its cysteines, we utilized biotin maleimide to specifically label free cysteines from cardiac homogenates under basal conditions. Indeed, the biotin to SERCA2a ratio was lower in HAXiKO compared to WT, indicating that cysteine residues are modified to a greater extent in the HAXiKO even prior to stress induction (Fig. 6A/B). Interestingly, biotin labeling of PLN was not altered by HAX ablation (Fig. 6B/C). These results indicate an apparent specificity of HAX-1 induced redox modification for SERCA2a.
Figure 6. SERCA2a free cysteine labeling is altered by HAX-1.
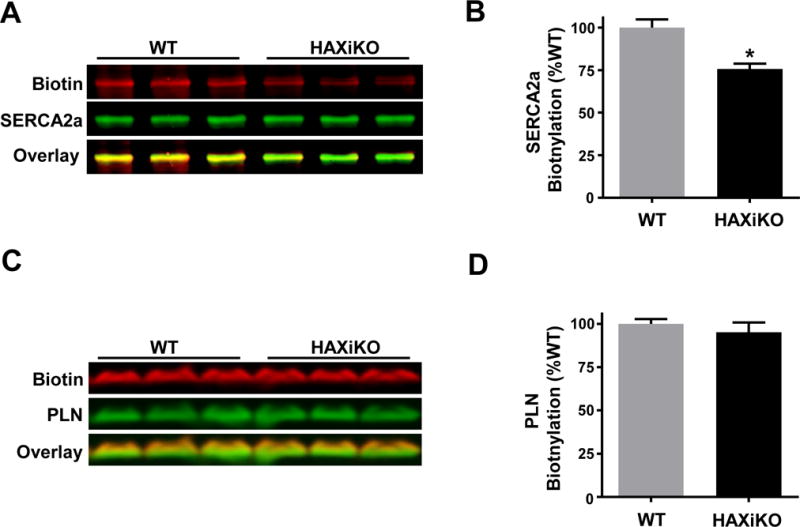
Homogenates from WT and HAXiKO hearts were treated with biotin maleimide to quantify labeling of free cysteines. Biotin and SERCA2a levels were simultaneously assessed by blotting with fluorescent streptavidin and SERCA2a polyclonal Ab/fluorescent secondary Ab after SDS-PAGE. (A) Representative blots for biotin, SERCA2a, and overlay; (B) corresponding quantification. Biotin and PLN levels were simultaneously assessed by blotting with fluorescent streptavidin and PLN antibody. (C) Representative blots for biotin, PLN, and overlay, and (D) corresponding quantification. Data are presented as mean ± SEM (P < 0.05: * vs WT).
3.5. HAX-1 regulates reactive oxygen species at the ER/SR
The findings above, led to the hypothesis that the modulation of SERCA2a oxidation by HAX-1 could be the result of increased cellular reactive oxygen production. However, the use of sense and antisense viral infection in neonatal rat ventricular myocytes (NRVM), indicated no impact on global H2O2 production, as assessed by Amplex Red (data not shown). We then assessed changes in local H2O2 production, using NRVMs that were infected with targeted HyPer sensors [37]. Cytosolic (Fig. 7A/B) and mitochondrial (Fig. 7C/D) targeted HyPer showed a slight increase in fluorescent signal in response to H2O2 treatment but there were no signal variations upon co-infection with LacZ or HAX sense/antisense adenoviruses. However, HAX-1 overexpression resulted in a basal decrease in ER-HyPer signal, whereas HAX-1 downregulation produced an increase in ER-HyPer signal in the presence of H2O2 treatment (Fig. 7E/F). LacZ infected cells also showed an increase in ER-HyPer signal after H2O2, but at a later time point (data not shown). These findings indicate that loss of HAX-1 increases ROS locally at the ER, suggesting that HAX-1 negatively regulates NOX4 at the SR/ER.
Figure 7. Oxidative stress in the ER is increased by downregulation of HAX-1.
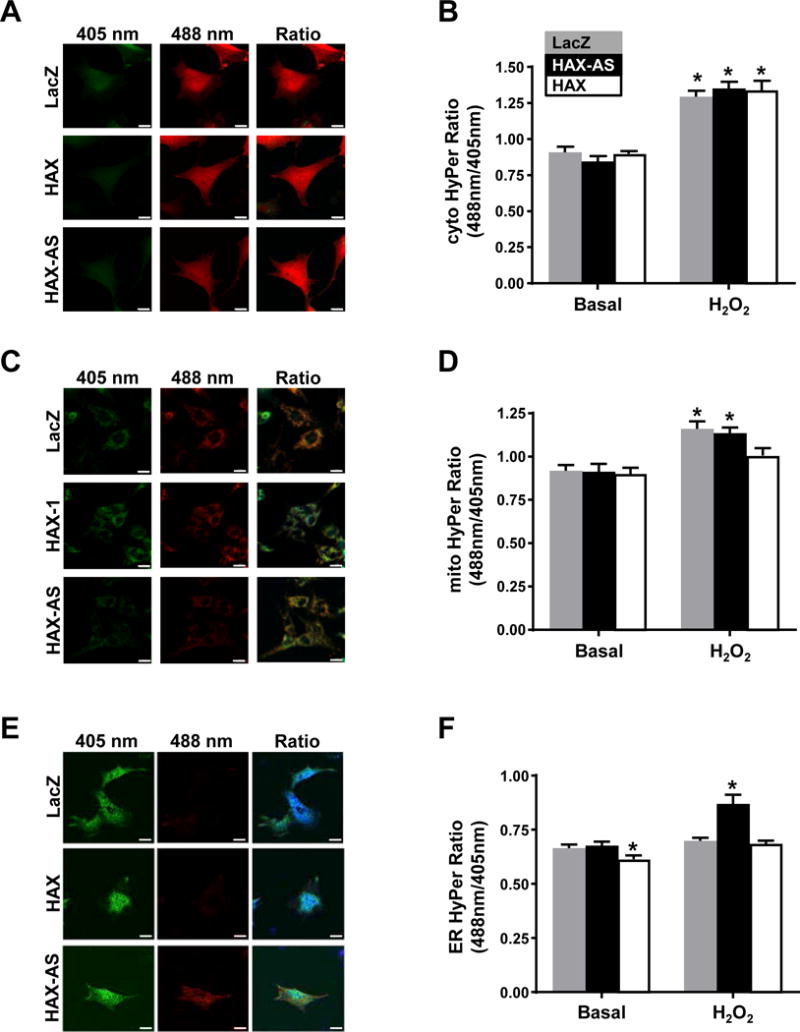
Rat neonatal cardiomyocytes were infected with Cyto-HyPer (A/B), Mito-HyPer (C/D), or ER-HyPer adenovirus (E/F) along with indicated HAX-1 adenovirus, Ad.HAX-1 (HAX-1) or Ad.HAX-1-AS (HAX-1-AS), or LacZ. Fluorescence was measured by live-cell imaging in confocal microscopy as a ratio of excitation at 488nm and 405nm. A time lapse experiment was conducted and 50μM H2O2 was added to the cells at 4:45sec and the compartment specific-H2O2 levels were measured every 20 sec. Representative images of Cyto-Hyper, Mito-HyPer and ER-HyPer fluorescence along with quantification of fluorescence ratio are shown. Scale bar = 20 μm (n = 3). Data are represented as mean ± SEM. (P < 0.05: * vs LacZ).
3.6. Inhibition of NOX4, a novel binding partner of HAX-1, attenuates I/R injury
NADPH oxidase 4 (NOX4) produces H2O2 at the ER [37] consistent with our observation in the NRVM experiments (Fig. 7). Previous studies have also indicated that SERCA2a may be oxidized by NOX4 in rat arterial smooth muscle and this is related to carotid artery injury [45]. Thus, we hypothesized that HAX-1 negatively regulates NOX4 to decrease SERCA2a oxidation and degradation. To establish a potential regulatory interaction, we used recombinant proteins and in vitro binding assays. Indeed, FLAG-NOX4 was able to bind to GST-HAX-1, and to a lesser extent GST-PLN (Fig. 8A). In addition, HAX-1 was identified as a binding partner of NOX4 in a yeast two hybrid screening of a human heart cDNA library, using the C-terminal cytosolic domain of NOX4 (amino acids 406-578) as bait [46]. Furthermore, co-immunoprecipitation experiments in mouse cardiac homogenates, using HAX-1 or NOX4 as bait, indicated reciprocal pull-down of each other partner (Fig. 8B). When PLN was used as bait, it pulled down both HAX-1 and NOX4 indicating that these proteins may exist as a complex in the mouse heart (Fig 8B). Accordingly, immunofluorescent staining of isolated mouse cardiomyocytes demonstrated extensive co-localization of HAX-1 and NOX4 (Fig 8C).
Figure 8. HAX-1 interacts with NOX4 and NOX inhibition rescues effects of HAX-1 ablation.
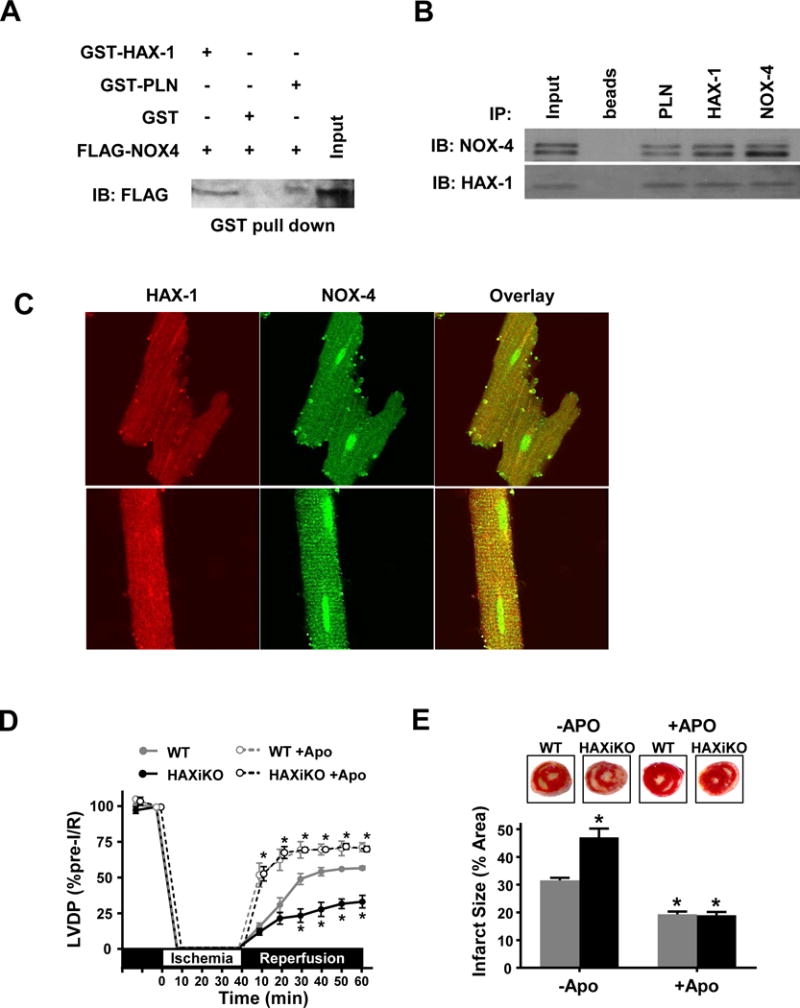
(A) Equivalent amounts of recombinant GST-HAX-1, GST-PLN, or GST protein were attached to glutathione matrices and incubated with FLAG-NOX4 samples separated by SDS-PAGE. Representative blot for FLAG-NOX4, indicating binding to GST-HAX-1. (B) Representative Western blots for co-immunoprecipitation (co-IP) experiments in WT cardiac homogenates using PLN, HAX-1, or NOX-4 as bait and probing for NOX4 and HAX-1 interactions. (C) Representative confocal images of WT isolated cardiomyocytes stained for HAX-1 (red), NOX4 (green) and overlay (yellow). NOX inhibition with apocynin (Apo) during 40 min of ex vivo no flow ischemia, followed by 60 min of reperfusion (D), improved contractile recovery as measured by LVDP (n = 5), (E) reduced infarct size (n = 4) and eliminated the effects of HAX-1 deletion. Data are offset along the x-axis for visualization and presented as mean ± SEM (P < 0.05: * vs WT).
To further confirm the role of HAX-1 in the regulation of NOX4, we performed the same ex vivo I/R protocol (Fig. 3) in the presence of the NOX inhibitor, apocynin. Indeed, apocynin significantly enhanced functional recovery as assessed by LVDP (Fig. 8D) and reduced infarct size (Fig. 8E). Consistent with these results, apocynin increased +dP/dt and -dP/dt and decreased EDP during reperfusion (Supplemental Fig. 7). Apocynin also normalized the differences between WT and HAXiKO hearts (Fig. 8D/E and Supplemental Fig. 7). Furthermore, the degree of functional recovery (75%) and infarct size (20%) observed in the presence of apocynin were similar to those exhibited by HAX overexpressing hearts [11]. Taken together, these results suggest that HAX-1 mediates cardioprotection through regulation of NOX activity.
4. Discussion
The current study demonstrates for the first time that HAX-1 regulates oxidation and degradation of SERCA2a, impacting cardiac function and survival under stress conditions. The underlying mechanisms included binding of HAX-1 to NOX4 and modulation of reactive oxygen species at the ER/SR compartment under basal conditions, a previously unidentified paradigm of cell death regulation by HAX-1. Inhibition of NOX4 eliminates the detrimental effects of HAX-1 ablation during I/R. While HAX-1 is widely viewed to confer cellular protection, its localization differs in various cell-types [9]. As such, the survival mechanisms mediated by HAX-1 may be different in the heart, where it shows significant ER/SR localization [7,8], in addition to its ubiquitous mitochondrial localization. This notion is supported by findings in HEK cells, indicating that redistribution of HAX-1 to ER through PLN co-expression enhanced survival [7]. Our previous studies indicated that the cardiac protective effects of HAX-1 involve both reduction in the ER stress signaling pathway [11] and downregulation of cyclophilin D in mitochondria, preserving mitochondrial transition pore (MTP) integrity [12]. Our current findings further support a pleiotropic role of HAX-1 in the heart, and indicate additional cardioprotective functions through NOX4 in the SR/ER compartment, resulting in inhibition of SERCA2a degradation under stress conditions. Intriguingly, the deleterious changes in ROS levels and Ca2+ homeostasis, associated with loss of HAX-1, could also trigger ER stress and loss of mitochondrial stability, as suggested by previous studies [11,12]. Thus, HAX-1 may represent a “fail-safe” operating on many different mechanistic levels along the same pathways.
Decreases in SERCA2a expression and activity have been observed in experimental and clinical ischemia reperfusion injury [18] and they were suggested to exacerbate cardiac injury. Indeed, heterozygous SERCA2a deficient mice exhibited depressed cardiac functional recovery and increased infarct upon I/R [3,47], while transgenic [19] or viral [20,21] overexpression of SERCA2a were shown to reduce the extent of cardiac injury. Furthermore, loss of SERCA2a function, resulting in elevated cytosolic Ca2+ and decreased SR Ca2+ load is a well-established upstream initiator of ER stress and mitochondrial dysfunction [13–15]. However, despite the frequency at which decreases in SERCA2a expression have been found to associate with various cardiac injuries and diseases, clear degradation mechanisms and pathways have yet to be established. One study showed that perfusion of hearts with the calpain inhibitor MDL28170 could block SERCA2a proteolysis in an ex vivo I/R model [16] and a correlation between calpain activation and SERCA2a degradation was observed in platelets [48] and bladder [49]. Evidence that calpain can directly cleave SERCA2a was provided by the current findings, which also showed that HAX-1 can modulate this process through oxidation of the enzyme that occurs prior to stress. In heart failure, increased calpain activity and SERCA2a oxidation present a plausible contributing factor for decreased SERCA2a levels in the chronically stressed heart. Notably, an apparent linear correlation between HAX-1 and SERCA2a levels was observed in human failing hearts (Supplemental Fig. 1A), further supporting this notion. Interestingly, HAX-1 ablation did not alter basal protein levels of SERCA2a and there was no observed remodeling in the long-term under non-stress conditions. This may be due to the localization of calpains, which are predominantly cytosolic under basal conditions, and stress induces their translocation to the membranes, including SR [50,51]. Accordingly, calpain overexpression exacerbated the effects of stress, but did not produce a cardiac phenotype under basal conditions [50]. Thus, activation and localization of calpains are likely critical determinants of SERCA2a degradation in addition to the oxidative state of the Ca2+ pump.
Oxidative modification of SERCA2a has been observed in cardiomyocytes [29,30] though this pathway is more extensively characterized in the vascular system [25–28], where NOX4 has been implicated in this oxidation process [45]. Moreover, ROS and NOX4 are known to play key roles in the regulation of neutrophils [51]. Importantly, human mutations resulting in loss of HAX-1 protein or activity are associated with increased ROS levels in the neutrophils of the human carriers [10]. In the heart, the NADPH oxidases NOX2 and NOX4 are major sources of ROS and have been demonstrated to play critical roles in physiology and disease, though their specific biochemical targets are not fully understood. NOX2 and NOX4 are upregulated in the failing myocardium, contributing to hypertrophic and fibrotic signaling [52,53]. Consistently, increased expression of NOX4 and the resulting ROS generation associates with aggravation of I/R induced injury [54]. We have shown previously that NOX4 is localized at intracellular membranes, including mitochondria, nucleus, and endoplasmic reticulum, and its protein expression and function is regulated by both transcriptional as well as post-translational mechanisms [37,46,52,55]. We here show that NOX4 physically interacts with HAX-1 in the ER/SR. Since the level of H2O2 in the ER/SR is significantly elevated when HAX-1 is down-regulated, we speculate that NOX4, a major ROS producing enzyme in the ER/SR, is negatively regulated by HAX-1. Although further investigation is required to address the potential role of NOX2 in SERCA2a modification, the observed ER/SR specificity of ROS modulation by HAX-1, coupled with the reported localization of NOX2 to plasma membrane [56], make this unlikely. The current findings suggest that basally elevated ROS in the SR leads to cysteine oxidation of SERCA2a, which in turn increases its susceptibility to degradation when calpain is activated during injury. Reversal of I/R injury and elimination of HAX-1 effects through pharmacological inhibition of NOX substantiates this idea. Intriguingly, PLN which has 3 cysteines residues did not display a change in oxidation parallel to that of SERCA2a. These 3 cysteines are in the transmembrane region, which may make PLN less susceptible to oxidation compared to SERCA2a which has over 20 cysteines outside the membrane that are conserved in mouse and human. Given the ubiquitous expression of various SERCA isoforms, NOX4, and calpains, this mechanism may be relevant in other tissues besides the heart.
In summary, the current study is the first to demonstrate a new role of HAX-1 in cardioprotection through regulation of SERCA2a levels. This appears to be mediated through interaction of HAX-1 with NOX4 and alteration of ROS levels in the micro-environment of the SERCA2a pump. Our findings not only suggest an important safeguarding role of HAX-1 in the SR Ca2+ transport, but also have wide-ranging implications in multiple tissues and diseases, given the powerful nature of NOX4 derived ROS.
Supplementary Material
Acknowledgments
This work was supported by: NIH grants HL-26057 and HL-64018 to EGK and NIH grant HL 125204 and AHA Postdoctoral Fellowship 13POST13860006 to PAB. We would like to thank Dr. James Ihle and Dr. Evan Parganas (St. Jude, Memphis TN) for graciously donating the floxed HAX-1 mouse.
Footnotes
Disclosures
None
References
- 1.Murphy E, Steenbergen C. Mechanisms Underlying Acute Protection From Cardiac Ischemia-Reperfusion Injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W, Terentyev D. Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch. 2014;466:1129–37. doi: 10.1007/s00424-014-1469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talukder MAHAH, Zweier JL, Periasamy M. Targeting calcium transport in ischaemic heart disease. Cardiovasc Res. 2009;84:345–52. doi: 10.1093/cvr/cvp264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–77. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 5.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–60. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haghighi K, Bidwell P, Kranias EG. Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J Mol Cell Cardiol. 2014;77:160–7. doi: 10.1016/j.yjmcc.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vafiadaki E, Sanoudou D, Arvanitis DA, Catino DH, Kranias EG, Kontrogianni-Konstantopoulos A. Phospholamban interacts with HAX-1, a mitochondrial protein with anti-apoptotic function. J Mol Biol. 2007;367:65–79. doi: 10.1016/j.jmb.2006.10.057. [DOI] [PubMed] [Google Scholar]
- 8.Zhao W, Waggoner JR, Zhang ZG, Lam CK, Han P, Qian J, Schroder PM, Mitton B, Kontrogianni-Konstantopoulos A, Robia SL, Kranias EG. The anti-apoptotic protein HAX-1 is a regulator of cardiac function. Proc Natl Acad Sci U S A. 2009;106:20776–81. doi: 10.1073/pnas.0906998106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fadeel B, Grzybowska E. HAX-1: A multifunctional protein with emerging roles in human disease. Biochim Biophys Acta - Gen Subj. 2009;1790:1139–1148. doi: 10.1016/j.bbagen.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schäffer AA, Rathinam C, Boztug K, Schwinzer B, Rezaei N, Bohn G, Melin M, Carlsson G, Fadeel B, Dahl N, Palmblad J, Henter JI, Zeidler C, Grimbacher B, Welte K. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease) Nat Genet. 2007;39:86–92. doi: 10.1038/ng1940. [DOI] [PubMed] [Google Scholar]
- 11.Lam CK, Zhao W, Cai W, Vafiadaki E, Florea SM, Ren X, Liu Y, Robbins N, Zhang Z, Zhou X, Jiang M, Rubinstein J, Jones WK, Kranias EG. Novel role of HAX-1 in ischemic injury protection involvement of heat shock protein 90. Circ Res. 2013;112:79–89. doi: 10.1161/CIRCRESAHA.112.279935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lam CK, Zhao W, Liu GS, Cai WF, Gardner G, Adly G, Kranias EG. HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc Natl Acad Sci U S A. 2015;112:E6466–75. doi: 10.1073/pnas.1508760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen K, Horn S, Niemann MT, Daniel PT, Schulze-Osthoff K, Fischer U. Inhibition of the ER Ca 2+ pump forces multidrug-resistant cells deficient in Bak and Bax into necrosis. J Cell Sci. 2009;122:4481–4491. doi: 10.1242/jcs.055772. [DOI] [PubMed] [Google Scholar]
- 15.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.French JP, Quindry JC, Falk DJ, Staib JL, Lee Y, Wang KKW, Powers SK. Ischemia-reperfusion-induced calpain activation and SERCA2a degradation are attenuated by exercise training and calpain inhibition. Am J Physiol Heart Circ Physiol. 2006;290:H128–36. doi: 10.1152/ajpheart.00739.2005. [DOI] [PubMed] [Google Scholar]
- 17.Prunier F, Kawase Y, Gianni D, Scapin C, Danik SB, Ellinor PT, Hajjar RJ, Del Monte F. Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation. 2008;118:614–24. doi: 10.1161/CIRCULATIONAHA.108.770883. [DOI] [PubMed] [Google Scholar]
- 18.Boštjančič E, Zidar N, Glavač D. MicroRNAs and cardiac sarcoplasmic reticulum calcium ATPase-2 in human myocardial infarction: expression and bioinformatic analysis. BMC Genomics. 2012;13:552. doi: 10.1186/1471-2164-13-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talukder MAH, Kalyanasundaram A, Zhao X, Zuo L, Bhupathy P, Babu GJ, Cardounel AJ, Periasamy M, Zweier JL. Expression of SERCA isoform with faster Ca2+ transport properties improves postischemic cardiac function and Ca2+ handling and decreases myocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293:H2418–28. doi: 10.1152/ajpheart.00663.2007. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Escoubet B, Prunier F, Amour J, Simonides WS, Vivien B, Lenoir C, Heimburger M, Choqueux C, Gellen B, Riou B, Michel JB, Franz WM, Mercadier JJ. Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation. 2004;109:1898–903. doi: 10.1161/01.CIR.0000124230.60028.42. [DOI] [PubMed] [Google Scholar]
- 21.del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK, Hajjar RJ. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci U S A. 2004;101:5622–7. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cross HR, Kranias EG, Murphy E, Steenbergen C. Ablation of PLB exacerbates ischemic injury to a lesser extent in female than male mice: protective role of NO. Am J Physiol - Hear Circ Physiol. 2003;284:H683–H690. doi: 10.1152/ajpheart.00567.2002. [DOI] [PubMed] [Google Scholar]
- 23.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tilemann L, Lee A, Ishikawa K, Aguero J, Rapti K, Santos-Gallego C, Kohlbrenner E, Fish KM, Kho C, Hajjar RJ. SUMO-1 Gene Transfer Improves Cardiac Function in a Large-Animal Model of Heart Failure. Sci Transl Med. 2013;5:211ra159–211ra159. doi: 10.1126/scitranslmed.3006487. [DOI] [PubMed] [Google Scholar]
- 25.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schöneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–7. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 26.Evangelista AM, Thompson MD, Weisbrod RM, Pimental DR, Tong X, Bolotina VM, Cohen RA. Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid Redox Signal. 2012;17:1099–108. doi: 10.1089/ars.2011.4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong X, Ying J, Pimentel DR, Trucillo M, Adachi T, Cohen RA. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol. 2008;44:361–9. doi: 10.1016/j.yjmcc.2007.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ying J, Tong X, Pimentel DR, Weisbrod RM, Trucillo MP, Adachi T, Cohen RA. Cysteine-674 of the sarco/endoplasmic reticulum calcium ATPase is required for the inhibition of cell migration by nitric oxide. Arterioscler Thromb Vasc Biol. 2007;27:783–90. doi: 10.1161/01.ATV.0000258413.72747.23. [DOI] [PubMed] [Google Scholar]
- 29.Lancel S, Zhang J, Evangelista A, Trucillo MP, Tong X, Siwik DA, Cohen RA, Colucci WS. Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ Res. 2009;104:720–3. doi: 10.1161/CIRCRESAHA.108.188441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hobai IA, Buys ES, Morse JC, Edgecomb J, Weiss EH, Armoundas AA, Hou X, Khandelwal AR, Siwik DA, Brouckaert P, Cohen RA, Colucci WS. SERCA Cys674 sulphonylation and inhibition of L-type Ca2+ influx contribute to cardiac dysfunction in endotoxemic mice, independent of cGMP synthesis. Am J Physiol Heart Circ Physiol. 2013;305:H1189–200. doi: 10.1152/ajpheart.00392.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vafiadaki E, Arvanitis DA, Pagakis SN, Papalouka V, Sanoudou D, Kontrogianni-Konstantopoulos A, Kranias EG. The anti-apoptotic protein HAX-1 interacts with SERCA2 and regulates its protein levels to promote cell survival. Mol Biol Cell. 2009;20:306–18. doi: 10.1091/mbc.E08-06-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai WF, Liu GS, Lam CK, Florea S, Qian J, Zhao W, Pritchard T, Haghighi K, Lebeche D, Lu LJ, Deng J, Fan GC, Hajjar RJ, Kranias EG. Up-regulation of micro-RNA765 in human failing hearts is associated with post-transcriptional regulation of protein phosphatase inhibitor-1 and depressed contractility. Eur J Heart Fail. 2015;17:782–793. doi: 10.1002/ejhf.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chao JRR, Parganas E, Boyd K, Hong CY, Opferman JT, Ihle JN. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature. 2008;452:98–102. doi: 10.1038/nature06604. [DOI] [PubMed] [Google Scholar]
- 34.Duda M, Konior A, Klemenska E, Beręsewicz A. Preconditioning protects endothelium by preventing ET-1-induced activation of NADPH oxidase and xanthine oxidase in post-ischemic heart. J Mol Cell Cardiol. 2007;42:400–410. doi: 10.1016/j.yjmcc.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 35.Han Y, Zhao H, Tang H, Li X, Tan J, Zeng Q, Sun C. 20-Hydroxyeicosatetraenoic acid mediates isolated heart ischemia/reperfusion injury by increasing NADPH oxidase-derived reactive oxygen species production. Circ J. 2013;77:1807–16. doi: 10.1253/circj.cj-12-1211. [DOI] [PubMed] [Google Scholar]
- 36.Brittsan AG, Carr AN, Schmidt AG, Kranias EG. Maximal inhibition of SERCA2 Ca(2+) affinity by phospholamban in transgenic hearts overexpressing a non-phosphorylatable form of phospholamban. J Biol Chem. 2000;275:12129–35. doi: 10.1074/jbc.275.16.12129. [DOI] [PubMed] [Google Scholar]
- 37.Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M, Sadoshima J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2α/activating transcripti. Circ Res. 2013;113:1253–64. doi: 10.1161/CIRCRESAHA.113.301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pritchard TJ, Kawase Y, Haghighi K, Anjak A, Cai W, Jiang M, Nicolaou P, Pylar G, Karakikes I, Rapti K, Rubinstein J, Hajjar RJ, Kranias EG. Active Inhibitor-1 Maintains Protein Hyper-Phosphorylation in Aging Hearts and Halts Remodeling in Failing Hearts. PLoS One. 2013;8:e80717. doi: 10.1371/journal.pone.0080717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawaguchi Y, Nakajima K, Igarashi M, Morita T, Tanaka M, Suzuki M, Yokoyama A, Matsuda G, Kato K, Kanamori M, Hirai K. Interaction of Epstein-Barr virus nuclear antigen leader protein (EBNA-LP) with HS1-associated protein X-1: implication of cytoplasmic function of EBNA-LP. J Virol. 2000;74:10104–11. doi: 10.1128/jvi.74.21.10104-10111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuda G, Nakajima K, Kawaguchi Y, Yamanashi Y, Hirai K. Epstein-Barr virus (EBV) nuclear antigen leader protein (EBNA-LP) forms complexes with a cellular anti-apoptosis protein Bcl-2 or its EBV counterpart BHRF1 through HS1-associated protein X-1. Microbiol Immunol. 2003;47:91–9. doi: 10.1111/j.1348-0421.2003.tb02790.x. [DOI] [PubMed] [Google Scholar]
- 41.Cilenti L, Soundarapandian MM, Kyriazis GA, Stratico V, Singh S, Gupta S, Bonventre JV, Alnemri ES, Zervos AS. Regulation of HAX-1 Anti-apoptotic Protein by Omi/HtrA2 Protease during Cell Death. J Biol Chem. 2004;279:50295–50301. doi: 10.1074/jbc.M406006200. [DOI] [PubMed] [Google Scholar]
- 42.Yedavalli VSRK, Shih HM, Chiang YP, Lu CY, Chang LY, Chen MY, Chuang CY, Dayton AI, Jeang KT, Huang LM. Human Immunodeficiency Virus Type 1 Vpr Interacts with Antiapoptotic Mitochondrial Protein HAX-1. J Virol. 2005;79:13735–13746. doi: 10.1128/JVI.79.21.13735-13746.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharov VS, Dremina ES, Galeva NA, Williams TD, Schöneich C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem J. 2006;394:605–15. doi: 10.1042/BJ20051214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagarajan N, Oka S, Sadoshima J. Modulation of signaling mechanisms in the heart by thioredoxin 1. Free Radic Biol Med. 2016 doi: 10.1016/j.freeradbiomed.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong X, Hou X, Jourd’heuil D, Weisbrod RM, Cohen RA. Upregulation of Nox4 by TGF{beta}1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ Res. 2010;107:975–83. doi: 10.1161/CIRCRESAHA.110.221242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsushima S, Kuroda J, Zhai P, Liu T, Ikeda S, Nagarajan N, Oka S, Yokota T, Kinugawa S, Hsu CP, Li H, Tsutsui H, Sadoshima J. Tyrosine kinase FYN negatively regulates NOX4 in cardiac remodeling. J Clin Invest. 2016;126:3403–3416. doi: 10.1172/JCI85624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Talukder MAH, Kalyanasundaram A, Zuo L, Velayutham M, Nishijima Y, Periasamy M, Zweier JL. Is reduced SERCA2a expression detrimental or beneficial to postischemic cardiac function and injury? Evidence from heterozygous SERCA2a knockout mice. Am J Physiol Heart Circ Physiol. 2008;294:H1426–34. doi: 10.1152/ajpheart.01016.2007. [DOI] [PubMed] [Google Scholar]
- 48.Randriamboavonjy V, Pistrosch F, Bölck B, Schwinger RHG, Dixit M, Badenhoop K, Cohen RA, Busse R, Fleming I. Platelet sarcoplasmic endoplasmic reticulum Ca2+-ATPase and mu-calpain activity are altered in type 2 diabetes mellitus and restored by rosiglitazone. Circulation. 2008;117:52–60. doi: 10.1161/CIRCULATIONAHA.107.719807. [DOI] [PubMed] [Google Scholar]
- 49.Zhao Y, Levin SS, Wein AJ, Levin RM. Correlation of ischemia/reperfusion or partial outlet obstruction-induced spectrin proteolysis by calpain with contractile dysfunction in rabbit bladder. Urology. 1997;49:293–300. doi: 10.1016/S0090-4295(96)00452-9. [DOI] [PubMed] [Google Scholar]
- 50.Galvez AS, Diwan A, Odley AM, Hahn HS, Osinska H, Melendez JG, Robbins J, Lynch RA, Marreez Y, Dorn GW. Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ Res. 2007;100:1071–8. doi: 10.1161/01.RES.0000261938.28365.11. [DOI] [PubMed] [Google Scholar]
- 51.Pittermann E, Lachmann N, MacLean G, Emmrich S, Ackermann M, Göhring G, Schlegelberger B, Welte K, Schambach A, Heckl D, Orkin SH, Cantz T, Klusmann JH. Gene correction of HAX1 reversed Kostmann disease phenotype in patient-specific induced pluripotent stem cells. Blood Adv. 2017;1:903–914. doi: 10.1182/bloodadvances.2016003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by Hypertrophic Stimuli Promotes Apoptosis and Mitochondrial Dysfunction in Cardiac Myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsushima S, Kuroda J, Ago T, Zhai P, Ikeda Y, Oka S, Fong GH, Tian R, Sadoshima J. Broad Suppression of NADPH Oxidase Activity Exacerbates Ischemia/Reperfusion Injury Through Inadvertent Downregulation of Hypoxia-inducible Factor-1? and Upregulation of Peroxisome Proliferator-activated Receptor-? Circ Res. 2013;112:1135–1149. doi: 10.1161/CIRCRESAHA.111.300171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie LH, Tian B, Sadoshima J. Increased Oxidative Stress in the Nucleus Caused by Nox4 Mediates Oxidation of HDAC4 and Cardiac Hypertrophy. Circ Res. 2013;112:651–663. doi: 10.1161/CIRCRESAHA.112.279760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maejima Y, Kuroda J, Matsushima S, Ago T, Sadoshima J. Regulation of myocardial growth and death by NADPH oxidase. J Mol Cell Cardiol. 2011;50:408–16. doi: 10.1016/j.yjmcc.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.