

Abstract
In the United States, lung cancer is the second most common cancer in men and women. In 2017, 222,500 new cases and 155,870 deaths from lung cancer are estimated to have occurred. A tyrosine kinase receptor, epidermal growth factor receptor (EGFR) is over expressed or mutated in non-small cell lung cancer (NSCLC) resulting in increased cell proliferation and survival. Tyrosine kinase inhibitors (TKIs) are currently being used as therapy for NSCLC patients, however, they have limited efficacy in NSCLC patients due to acquisition of resistance. This study investigates the role of epithelial-mesenchymal transition (EMT) in the development of resistance against TKIs in NSCLC. Currently, the role of p120-catenin, Kaiso factor and PRMT-1 in reversal of EMT in T790M mutated and TKI-resistant NSCLC cells is a new line of study. In this investigation we found upregulation of cytoplasmic p120-catenin, and it was co-localized with Kaiso factor. In the nucleus, binding of p120-catenin to Kaiso factor initiates transcription by activating EMT-transcription factors such as Snail, Slug, Twist, and ZEB1. PRMT-1 was also found to be upregulated, which induces methylation of Twist and repression of E-cadherin activity, thus promoting EMT. We confirmed that TKI-resistant cells have mesenchymal cell type characteristics based on their cell morphology and gene or protein expression of EMT related proteins. EMT proteins, Vimentin and N-cadherin, displayed increased expression, whereas E-cadherin expression was downregulated. Finally, we found that the knockdown of p120-catenin and PRMT-1 by siRNA or use of a PRMT-1 inhibitor Furamidine increased Erlotinib sensitivity and could reverse EMT to overcome TKI resistance.
1. INTRODUCTION
In non-small cell lung cancer (NSCLC), the tyrosine kinase activity of growth factor receptors is dysregulated by various oncogenic mechanisms, such as gene mutations in the kinase domain of epidermal growth factor receptor (EGFR). This leads to enhanced kinase activity which signals cell survival pathways and promotes extensive cell proliferation, resulting in tumor progression[1]. A kinase domain mutation in EGFR leads to partial or fully ligand independent activation of tyrosine kinase activity of EGFR. The TK domain of the EGFR gene has a sensitizing L858R mutation (single point mutation in exon 21) that constitutes 40% all EGFR mutations[2,3]. This L858R mutation causes decreased affinity for ATP, which allows the ATP binding site to become available to TKIs. EGFR activating mutations, including exon 19 deletions and exon 21 L858R substitutions, constitute about 45% and 40% of EGFR mutations, respectively, and patients with these mutations generally have promising responses to EGFR TKIs[1,3]. There are numerous resistance mechanisms elucidated for the acquisition of TKI resistance, and one of the important mechanisms is the T790M mutation in EGFR, which is found in about 50% of the cases at the time of EGFR TKI resistance acquisition[1]. The T790M mutation in the EGFR kinase domain changes the conformation of the ATP binding pocket, increasing its affinity for ATP thus, reducing the binding of TKIs.
There are three different generations of TKIs developed against EGFR. First generation TKIs, such as Erlotinib and Gefitinib bind reversibly to the kinase domain of EGFR. However, NSCLC cells with wild type (WT) EGFR may undergo epithelial-mesenchymal transition (EMT) during TKI treatment and become resistant to first generation TKIs[2]. Therefore, second generation TKIs, such as Afatinib, Dacomitinib, and Neratinib were developed to overcome TKI resistance by binding irreversibly to the kinase domain of EGFR. However, second generation TKIs have minimal utility due to dose-limiting toxicity. Third generation TKIs, such as Osimertinib (AZD9291) and Rociletinib are T790M mutant-selective treatment options that spare WT EGFR[4]. Osimertinib is currently approved by the FDA as a breakthrough therapy that shows meaningful results.
EMT is a reversible biological process where epithelial cells lose cell adhesion and undergo changes to gain mesenchymal characteristics. The EMT process is regulated by key EMT mediators and EMT transcriptional factors (EMT-TFs) such as Snail, Slug, Twist, and ZEB1. E-cadherin, a cell adhesion protein in epithelial cells is repressed by these EMT-TFs. EMT results in a switch from E-cadherin to N-cadherin, which causes increased expression of Vimentin, a mesenchymal marker[2]. After acquisition of EMT, cells acquire enhanced migratory and invasive abilities, in addition to stem cell like characteristics and therapeutic resistance. The acquisition of cancer stem cell (CSC) characteristics by EMT positive cells induces tumor heterogeneity and thus these CSC biomarkers could be used for the development of new cancer therapies, potentially preventing tumor recurrence and drug resistance[5].
E-cadherin forms a complex with intracellular proteins such as β-catenin and p120-catenin to stabilize itself on the cell membrane. p120-catenin is a multifunctional protein that binds to E-cadherin on the cell membrane, and its dissociation leads to E-cadherin degradation[6]. p120-catenin also acts as a transcription regulator by shuttling inside the nucleus and binding to the transcription repressor, Kaiso factor[6]. This results in activation of Wnt/β-catenin signaling pathway, which activates EMT-TF’s such as Snail, Slug, Twist, and ZEB1[7]. Upregulation of the EMT regulator protein arginine methyl transferase 1 (PRMT-1) results in methylation of Twist, a protein which also regulates E-cadherin degradation[8]. In this study, we determined the role of p120-catenin as a novel EMT regulator and show its colocalization with Kaiso factor in inherently TKI-resistant NSCLC cells with T790M mutations, in comparison to EGFR mutated TKI-sensitive cells with L858R mutation. This study also investigates how p120-catenin/Kaiso factor, PRMT-1, and their downstream pathways promote the EMT process in these cells, as well as their role in EMT mediated TKI resistance, which has been minimally studied. Finally, we attempted to reverse the EMT process by increasing sensitivity to Erlotinib in TKI-resistant cells by knockdown or inhibition of PRMT-1, or knockdown of p120-catenin.
2. Materials and Methods
2.1. Tyrosine Kinase Inhibitors and Growth Factor Ligands
Erlotinib hydrochloride (N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy) quinazolin-4-amine; C22H23N3O4•HCl) was obtained from LC laboratories (Woburn, MA). Erlotinib was reconstituted in Dimethyl Sulfoxide (DMSO) at a concentration of 20mM and stored in aliquots of 20μl at −20°C. Epidermal Growth Factor (EGF) was obtained from Peprotech (Rocky Hill, NJ) and reconstituted in PBS to obtain a concentration of 15ng/μl, then stored in aliquots of 100μl at −20°C.
2.2. Antibodies
Rabbit monoclonal antibodies for E-Cadherin, Claudin, Vimentin, N-Cadherin, Slug, and Snail were purchased from Cell Signaling (Danvers, MA) as part of the EMT antibody sampler kit. Rabbit monoclonal antibody for PRMT-1 was also obtained from Cell Signaling (Danvers, MA). A rabbit polyclonal Kaiso factor antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). Mouse monoclonal antibody for β-Actin was purchased from Sigma Aldrich (St. Louis, MO). Mouse monoclonal antibodies for p120-catenin and Twist were purchased from Thermo Fisher Scientific (St. Louis, MO). Anti-rabbit IgG and anti-mouse IgG HRP-linked secondary antibodies were obtained from Cell Signaling (Danvers, MA). All primary antibodies were diluted per manufacturer’s instruction in TBST with 1% BSA. Secondary antibodies were prepared at a dilution of 1:1000 in TBST with 1% blocking grade milk from Bio-Rad (Hercules, CA). The E-Cadherin antibody conjugated with Alexa-Fluor 647 for use in flow cytometry was purchased from BD Biosciences (Franklin Lakes, NJ). ABCB1 antibody conjugated with APC and control IgG were purchased from Miltenyi Biotec (Cambridge, MA).
2.3. Cell Lines
H2170 and H3255 NSCLC cell lines were purchased from American Type Culture Collection (ATCC) (Rockville, MD). H1975 was obtained from National Institute of Health (NIH) and cultured per their instructions. The H2170 cell line has WT EGFR status, whereas H3255 (L858R) and H1975 (L858R and T790M) cell lines have mutated EGFR. Parental H2170 cells were cultured in increasing drug concentrations of Erlotinib to obtain EGFR TKI-resistant cells as described earlier[9].
2.4. Immunoblotting
H1975 and H3255 cells were seeded in 35 mm petri-dishes. Once cells reached 90% confluency, serum-free media (RPMI with 0.5% BSA) was added for 24 hours. Cells were then treated with 10μM Erlotinib in serum-free media for 24 hours, followed by EGF (15ng/ml for 2.5 minutes in serum-free media). Cell lysates were prepared, electrophoresed, and transferred onto nitrocellulose membranes as previously described[2]. Membranes were then probed with appropriate antibodies, and protein expression was visualized using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) and exposure to autoradiography film (Scrip, Inc., Boling Brook, IL). Modulations in protein expression were calculated by densitometric analysis using NIH ImageJ software.
2.5. Real-Time PCR
All primers used in qPCR experiments (Table I) were purchased from Integrated DNA Technologies (Coralville, Iowa). H3255 and H1975 cells were cultured in 35mm petri dishes as described previously[2]. Total RNA was collected using PureLink RNA mini kit from Life Technologies. RNA was quantified, and qPCR protocols were conducted as described previously[2]. Cycle threshold (Ct) 2(−Δ(ΔCt)) values were recorded and normalized using GAPDH as an endogenous control to evaluate the fold changes.
TABLE I.
LIST OF THE PRIMERS USED FOR Q-PCR WITH THEIR SEQUENCE
| Gene | Sequence | Melting Point | Number of DNA Bases |
| E-Cadherin | F: GAACAGCACGTACACAGCCCT | 59.8°C | 21 |
| R: GCAGAAGTGTCCCTGTTCCAG | 58.0°C | 21 | |
| N-Cadherin | F: GACGGTTCGCCATCCAGAC | 58.4°C | 19 |
| R: TCGATTGGTTTGACCACGG | 55.4°C | 19 | |
| p120-catenin | F: CGGCATACGTCATCCCCATT | 60.25°C | 20 |
| R: TCTTCCCTCAGCCCTCAAGT | 60.18°C | 20 | |
| GAPDH | F: TTGCCAATGACCCCTTCA | 54.4°C | 18 |
| R: CGCCCCACTTGATTTTGGA | 55.8°C | 19 |
2.6. Immunofluorescence
H1975 and H3255 cells (30,000 each) were plated in 8-well glass chamber slides. Cells were fixed, permeabilized, blocked, and incubated with primary antibodies at 4°C overnight as described previously[2]. Following overnight incubation, primary antibodies were removed, and cells were incubated for 1 hour at room temperature in the dark with anti-mouse/rabbit IgG secondary antibodies conjugated with DyLight™ 488 [1:250] (Thermo Fisher Scientific) or Cy3 (Jackson Immuno Research Laboratories Inc). Nuclei were stained using Hoechst dye [1:2500] (Life Technologies, Carlsbad, CA) diluted in PBS with 1% BSA. Cells were observed and imaged using the Olympus Fv10i Fluoview confocal microscope. Average fluorescence intensity was quantified using Olympus Fluoview image analysis software.
2.7. Flow Cytometry
H1975, H3255, H2170-P (parental), and H2170-ER (Erlotinib resistant) (500,000 cells each) were seeded in 100mm petri dishes to adhere and grow for 48 hours. After growth medium was removed, cells were washed with PBS, then treated with Accutase Cell Detachment solution (Affymetrix, eBioscience) in an incubator for 5 minutes or until cells detached. Cells were filtered through a 75μm filter (Fisher Scientific) and collected in a 15ml tube, pelleted using a centrifuge, then re-suspended in FACS buffer (PBS with 2mM EDTA and 0.5% BSA). The cells were washed, pelleted again, then probed directly with a fluorophore labeled antibody for 10 minutes on ice to determine surface marker staining. For intracellular staining, cells were fixed using 4% paraformaldehyde diluted in PBS buffer for 15 minutes, and permeabilized using 0.1% Tween 20 in FACS buffer for 15 minutes. After incubation with the antibody, cells were washed with FACS buffer to remove the excess antibody, then finally resuspended in FACS buffer for analysis using a BD FACS CALIBUR flow cytometer.
2.8. siRNA Transfection
24 hours before transfection, cells were seeded in media containing 10% FBS without antibiotics. Cells were then transfected with control siRNA (Cell Signaling), PRMT-1 siRNA (Dharmacon), or p120-catenin siRNA (Santa Cruz Biotechnology) using Dharmafect-2 transfection reagent, (GE Life Sciences) per manufacturer’s protocol. Cell lysates were collected for immunoblotting after 24 and 48 hours. For MTT cell viability assays, the transfection media was removed after 24 hours followed by addition of drug media.
2.9. MTT Cell Viability Assay
H1975 and H3255 cells were seeded at 5,500 cells per well in a 96-well plate to adhere and grow for 24 hours. Cells were then transfected with siRNA using Dharmafect-2 (GE Life Sciences) for 24 hours, followed by 10 μM Erlotinib for 24 hours. For treatment with a PRMT-1 inhibitor Furamidine (Tocris Bioscience), cells were incubated at 37°C in media containing 20–25μM Furamidine for 24 hours, followed by Erlotinib for an additional 24 hours. Finally, MTT cell viability assays were performed as described previously[2].
3. Results
3.1. Validation of Occurrence of EMT in TKI-resistant H1975 Cells
The expression of various key EMT regulators and EMT-TFs were analyzed in TKI-sensitive H3255 and TKI-resistant H1975 cells by immunoblotting (Fig. 1A, 1B). Results indicate that Slug, Snail, and Twist were all upregulated in H1975 cells between 1.6 to 4.7 fold (Fig. 1A, 1B), while several epithelial proteins such as E-cadherin, ZO-1, and Claudin were downregulated 3.5 to 8.6 fold (Fig. 1A, 1B). Additionally, Vimentin, a key mesenchymal marker, showed higher protein expression (4.3 fold) in H1975 cells than in H3255 cells. EMT regulators such as p120-catenin and PRMT-1 were also upregulated (2.4 fold), which promote EMT through upregulation of EMT-TFs (Fig. 1A, 1B). To further validate the occurrence of EMT, the expression of key EMT-related biomarkers in H1975 cells were checked and compared to TKI-sensitive H3255 cells using real-time PCR. These results confirmed high levels of mesenchymal markers Vimentin (10.7 fold) and N-cadherin (4.9 fold) were expressed in H1975 cells as compared to H3255 cells (p<0.01) (Fig. 1C). H1975 also showed higher expression of ZEB-1 (3.0 fold) and p120-catenin (3.1 fold) (Fig. 1C).
Fig. 1.
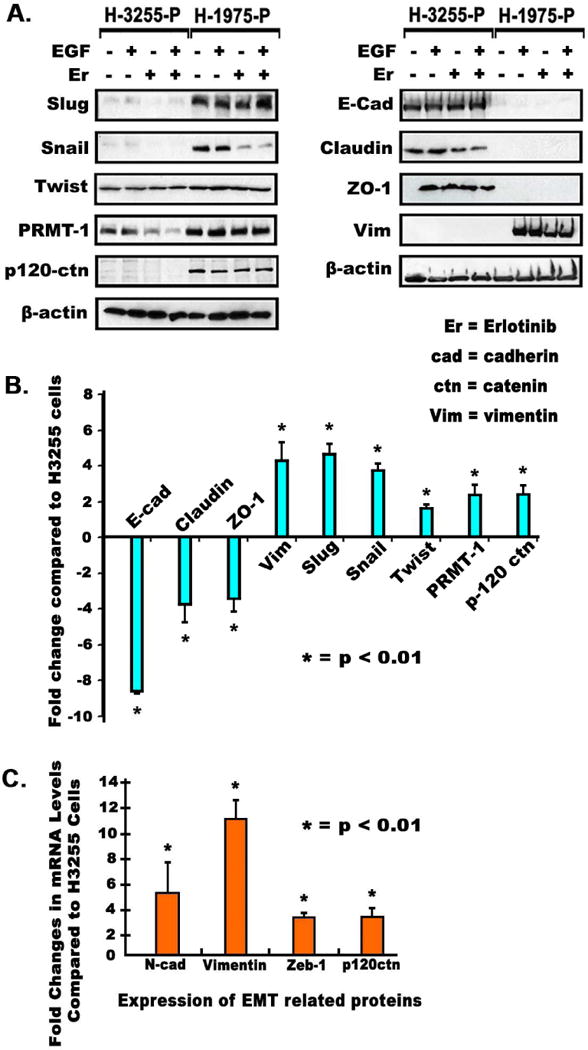
(A) 2.5×105 H1975 and H3255 cells were seeded in 35 mm dishes, grown for 24 hours, starved for 24 hours and then treated with Erlotinib for 24 hours after which immunoblotting was performed as described earlier[2]. (B) Protein modulations were calculated by densitometric analysis using ImageJ software, with data shown as bar graphs. (C) Cells were plated as described above and total RNA was then collected and quantified using qPCR. cDNA was prepared using 1μg of total RNA, and then used to determine mRNA levels of EMT related genes by qPCR as described earlier[2].
3.2. Increase in Vimentin Filaments and Cytoplasmic p120-catenin and Loss of E-cadherin in TKI-resistant H1975 Cells
Immunofluorescence was used to analyze changes in cellular morphology and expression of p120-catenin, E-cadherin, and Vimentin in H1975 and H3255 cells. We stained the cells with adherens junction protein p120-catenin, an E-cadherin stabilizer to study if p120-catenin was located on the cell membrane or in the cytoplasm. The results indicated that p120-catenin was localized on the membrane of H3255 cells, however, p120-catenin had a higher expression in H1975 cells (1.8 fold, p<0.01) and was distributed mainly in the cytoplasm and the nucleus (Fig. 2A). Our results also clearly showed membranous staining of E-cadherin in H3255 cells which was 2.3 fold (p<0.01) higher compared to H1975 cells (Fig. 2B). The mesenchymal marker Vimentin was upregulated (4.6 fold) in H1975 cells, which were noticeably elongated and thin, exhibiting a typical mesenchymal phenotype (Fig. 2C).
Fig. 2.
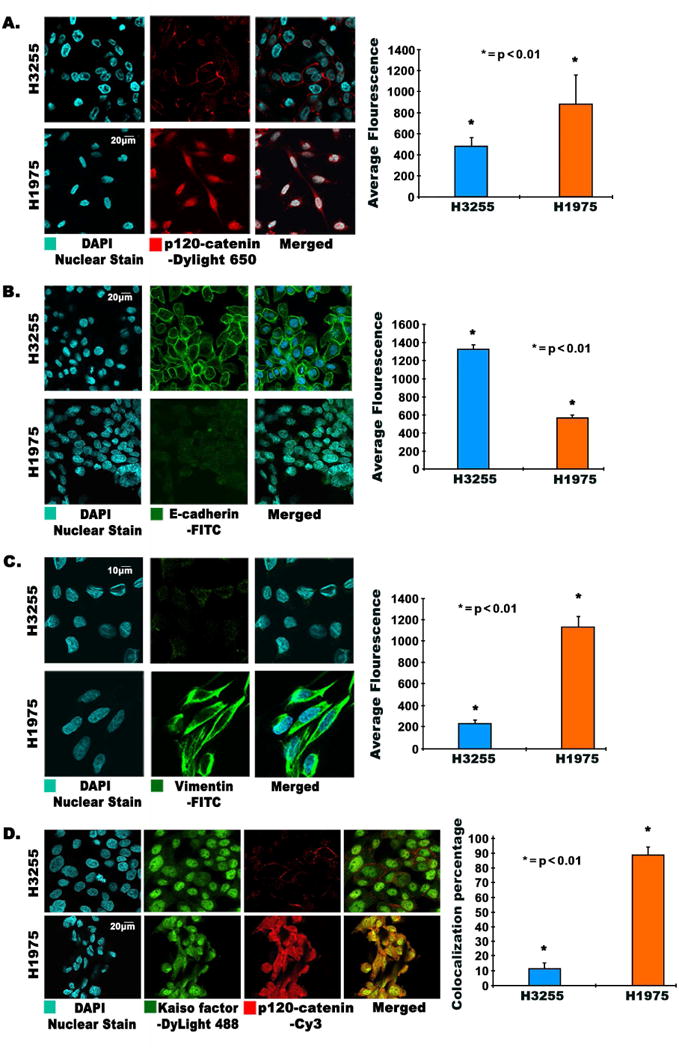
(A, B, C, D) Immunofluorescence images showing modulation and changes in morphology of EMT related proteins in TKI-resistant H1975 and H3255 cells. 3×104 cells per well were plated in an 8-well chamber slide, fixed, and probed for (A) p120-catenin, (B) E-cadherin, (C) Vimentin, and (D) colocalization of p120-catenin and Kaiso factor. Colocalization and immunofluorescence was conducted as described earlier[2]. Cells were probed with p120-catenin and Kaiso factor antibodies. DAPI was used as a nuclear stain; DyLight-488 conjugated secondary antibody was used for Kaiso factor, and Cy3 conjugated secondary antibody was used for p120-catenin. Images were captured using an Olympus fv10i confocal microscope. Fluorescence quantification was performed using the Olympus Fluoview image analysis software, and average fluorescence is graphically depicted showing statistically significant results.
3.3. Colocalization of p120-catenin and Kaiso Factor in TKI-resistant H1975 Cells
p120-catenin is able to shuttle inside the cell nucleus when it is released from the membrane. p120-catenin has an armadillo domain which can bind to Kaiso factor inside the nucleus, forming a complex. Since our studies revealed cytoplasmic localization of p120-catenin in H1975 cells, we wanted to check if p120-catenin was bound with Kaiso factor. Analysis of colocalization of Kaiso factor and p120-catenin by immunofluorescence indicated that p120-catenin and Kaiso factor were 10% colocalized in H3255 cells and 90% colocalized in H1975 cells (Fig. 2D). This demonstrates that p120-catenin and Kaiso factor were predominantly bound to each other in H1975 cells.
3.4 Effect of PRMT-1 and p120-catenin knockdown on Erlotinib efficacy in H1975 Cells
The results of the PRMT-1 knockdown experiment with siRNA showed that after 24 hours of transfection, PRMT-1 was inhibited by 3.4 fold and E-cadherin expression was increased by 2.0 fold (p<0.01) (Fig. 3A). The MTT cell viability assay results with PRMT-1 siRNA indicated that Erlotinib efficacy was increased by approximately 21% (p<0.01) (Fig. 3B) when compared to the viability of mock transfected cells in the presence of Erlotinib. We then examined the effect of PRMT-1 inhibitor Furamidine on Erlotinib-resistant H1975 cells. We found using an MTT assay that the PRMT-1 inhibitor, Furamidine, increased Erlotinib efficacy by 27% (p<0.01) when treated with 25μM Furamidine in H1975 cells (Fig. 3C). Differences in percentage viability of cells with different treatments were calculated with respect to the viability of cells in presence of Erlotinib alone (Fig. 3C). Next, we studied the effect of p120-catenin siRNA on Erlotinib efficacy. We found that p120-catenin knockdown (2 fold decrease, p<0.01) was successful and most efficient at 24 hours (Fig. 3D). In addition, E-cadherin levels were increased by 2.2 fold (p<0.01) at 24 hours after transfection (Fig. 3D). After p120-catenin knockdown, an MTT assay revealed that the efficacy of Erlotinib was increased by 40% (p<0.01) in p120-catenin downregulated H1975 cells, as compared to mock transfected cells in presence of Erlotinib (Fig. 3E).
Fig. 3.
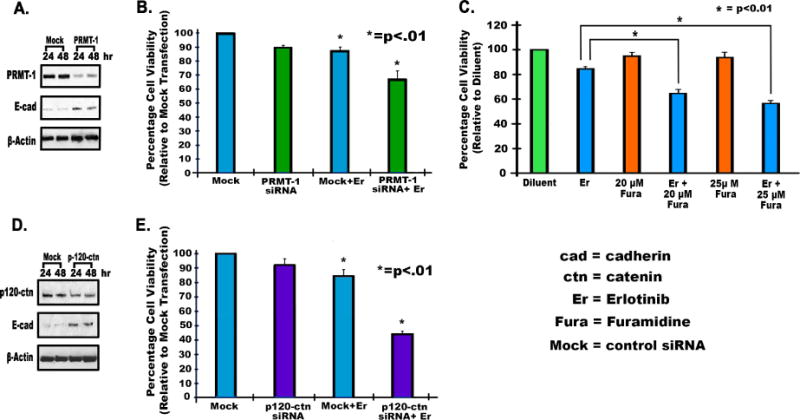
2.5×105 H1975 cells were seeded in antibiotic free media, and after 24 hours cells were transfected with mock siRNA, PRMT-1 siRNA, or p120-catenin siRNA, using Dharmafect-2 according to the manufacturer’s instructions. Cell lysates were collected after 24 and 48 hours. (A) Immunoblotting was performed after 24 and 48 hours to determine knockdown of PRMT-1 and upregulation of E-Cadherin. (B) 5500 cells per well were plated for an MTT assay as described above. To analyze Erlotinib efficacy after PRMT-1 knockdown, cells were treated with PRMT-1 siRNA for 24 hours followed by Erlotinib for 24 hours, after which an MTT assay was performed as described earlier[2], viability of transfected cells with Erlotinib and PRMT-1 siRNA was compared with mock and Erlotinib treatment. (C) To determine the effect of the PRMT-1 inhibitor Furamidine (20–25μM), H1975 cells were seeded in 96 well-plates (5500 per well) and treated with Furamidine for 24 hours and then Erlotinib (10μM) for 24 hr. MTT assays were done to study viability of treated cells. The differences in percentage viability of cells treated with Erlotinib alone were compared with different concentrations of Furamidine and Erlotinib treated cells. (D) p120-catenin siRNA was transfected as described above. Immunoblotting was performed after 24 and 48 hours to determine knockdown of p120-catenin and upregulation of E-Cadherin. (E) An MTT assay was then performed, as described above, for PRMT-1 siRNA to determine viability of transfected cells. The percentage viability of transfected cells with Erlotinib and p120-catenin siRNA was compared with mock and Erlotinib treatment.
3.5. Cancer Stem Cell Marker
We further investigated whether there was an association between acquisition of stem cell like properties and the EMT process. To determine this, flow cytometry was used to study the expression of CSC marker ABCB1 in EMT positive H1975 and H2170-ER cells which has WT EGFR[9]. Our results showed an increased expression of ABCB1 (1.8 and 1.5 fold, p<0.01) and decreased expression of E-cadherin (3.8 and 27.0 fold, p<0.001) (Fig. 4A, 4B), in H2170-ER and H1975 cells as compared to H2170-P and H3255 cells, respectively. This indicates that the EMT positive TKI-resistant cells had CSC-like properties.
Fig. 4.
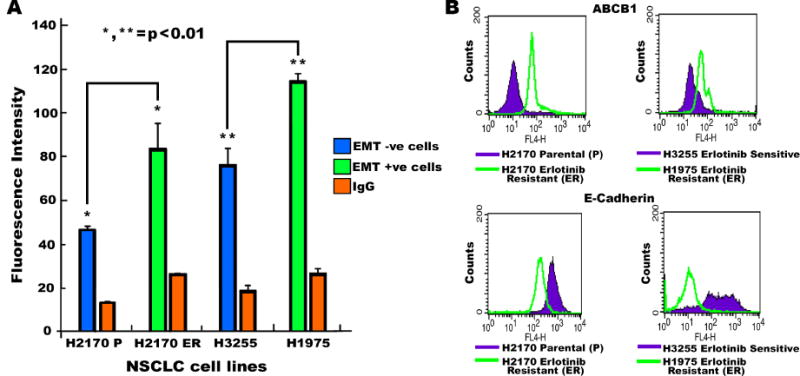
(A) 5×105 cells were plated and grown in 100mm dishes for 48 hours. Cells were then collected using Accutase media and probed with FITC-CD243 (ABCB1). The pellet was then washed and resuspended in flow cytometry buffer (PBS with 0.5% BSA and 5mM EDTA). An equal number of cells were analyzed, and a bar graph was generated in order to plot fluorescence intensity. (B) After analyzing equal numbers of cells by flow cytometry, a histogram for fluorescence intensity was plotted using CellQuest software. H2170-P and H2170-ER cells were compared. H3255 Erlotinib-sensitive and H1975-ER cells were also compared for expression of ABCB1 and E-cadherin.
4. Discussion
In this study, we found that TKI-resistant NSCLC cells with a T790M mutation undergo EMT, which contributes to TKI resistance in cancer cells. NSCLC cells with the T790M mutation have enhanced kinase activity especially in combination with L858R mutation as compared to WT EGFR. This affects downstream signaling pathways such as the Wnt and EGFR pathway leading to increased tumor cell survival[1,3]. Our results demonstrate that key EMT regulators such as PRMT-1, p120-catenin, and EMT-TFs such as Slug, Snail, Twist, and ZEB-1 were upregulated in H1975 cells. This resulted in lower expression of the epithelial proteins E-cadherin, Claudin, ZO-1, and higher expression of mesenchymal protein Vimentin in H1975 cells. Our previous and present studies show that upregulation of Vimentin and ZEB-1 and downregulation of Claudin occurs in TKI-resistant cells regardless of their EGFR status, which indicates that expression of these proteins plays an important role in TKI resistance[2]. PRMT-1 methylates Twist, and upregulation of PRMT-1 in H1975 cells may repress E-cadherin and promote expression of Vimentin[8]. We also demonstrated that knockdown of PRMT-1 and p120-catenin increased drug sensitivity, which indicates that these proteins may regulate the mechanism of EMT in TKI-resistant cells with the T790M mutation.
Our immunofluorescence studies revealed that H3255 control cells had an epithelioid and stratified morphology with high membranous E-cadherin, however, H1975 cells had an elongated and thin morphology which is indicative of an EMT positive cell phenotype. p120-catenin, an adherens junction protein, can dissociate from the adherens junction complex and shuttle into the cell nucleus[6]. Our studies showed that p120-catenin was upregulated, dissociated from the junction complex, and localized in the cytoplasm and nucleus of H1975 cells, in contrast to H3255 cells which mainly have membranous p120-catenin. Nuclear p120-catenin can also bind to Kaiso factor and interfere with its transcriptional repressor activity[6]. Results of our colocalization studies showed that p120-catenin and Kaiso factor were highly colocalized in H1975 cells, compared to H3255 cells. This may represent cytoplasmic localization and binding of these proteins, which disrupts Kaiso factor’s transcriptional repressor activity, resulting in activation of gene transcription and subsequent loss of E-cadherin.
Targeting important EMT regulators may be an attractive approach to reverse EMT and overcome TKI resistance in EGFR mutant NSCLC cells; hence we chose PRMT-1 and p120-catenin as key targets to reverse EMT. PRMT-1 or p120-catenin knockdown increased expression of E-cadherin, indicating possible reversal of EMT in H1975 cells. Cell viability assays in the presence of Erlotinib treatment in combination with the PRMT-1 inhibitor Furamidine or siRNA against PRMT-1/p120-catenin increased Erlotinib sensitivity. This increase in Erlotinib efficacy suggests that EMT, in addition to the T790M mutation, may be another important mechanism of acquired resistance. H1975 mutant cells may acquire EMT through amplifying PRMT-1 and presence of cytoplasmic p120-catenin. PRMT-1 methylates Twist which represses the activity of E-cadherin. Cytoplasmic p120-catenin may interact with the transcriptional repressor Kaiso factor and form a complex. The resulting complex may dissociate Kaiso factor from the promoter region and interfere with its transcriptional repressor activity resulting in activation of certain genes, which include ZEB-1, Twist, Slug and Snail.
Earlier studies show that both CSC properties and EMT contribute to tumor invasiveness, metastasis, and acquired resistance to TKIs in NSCLC[10]. Overexpression of ABCB1,a specific lung CSC marker, was found to be associated with both CSC properties and EMT, and was involved in acquired resistance to TKIs[11]. Studies have shown that EMT is closely associated with CSC characteristics in tumors, which facilitate the plasticity necessary for EMT. In our study, ABCB1 was found to be upregulated in EMT positive H2170-ER and H1975 cells when compared to WT H2170-P and the H3255 cells, indicating that they are enriched with CSC-like properties, as shown by expression of ABCB1. Additional studies are needed to determine the relationship between EMT and CSC formation and whether one process promotes the other.
In conclusion, the T790M mutation may mediate TKI resistance by activating EMT in addition to changing the conformation of the ATP binding pocket. Additionally, studies with CSC marker ABCB1 show that TKI-resistant cells undergoing EMT also possess CSC like characteristics. We found that T790M mutation mediated EMT is enhanced by cytoplasmic p120-catenin, which migrates to the nucleus and binds to Kaiso factor, relieving Kaiso factor mediated transcription repression. H1975 cells may undergo EMT due to another regulator, PRMT-1, which is upregulated in T790M mutated cells, and promotes EMT by repressing expression of E-cadherin. Our studies also showed that treatment of T790M mutant TKI-resistant NSCLC cells with p120-catenin and PRMT-1 siRNA can increase sensitivity of these cells towards Erlotinib. Therefore, we propose that targeting p120-catenin and PRMT-1 as anti-cancer treatments maybe a new potential strategy for NSCLC patients with acquired TKI resistance. Identifying and targeting molecules upstream of p120-catenin and PRMT-1 in T790M mutated cells may also be effective in overcoming EMT mediated TKI resistance in NSCLC.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number R21CA158965-01A1 (http://www.nih.gov) to Neelu Puri. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of potential conflict of interest
The authors have no conflicts of interest in this work.
References
- 1.Stewart EL, Tan SZ, Liu G, et al. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl Lung Cancer Res. 2015;4:67–81. doi: 10.3978/j.issn.2218-6751.2014.11.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rastogi I, Rajanna S, Webb A, et al. Mechanism of c-Met and EGFR tyrosine kinase inhibitor resistance through epithelial mesenchymal transition in non-small cell lung cancer. Biochem Biophys Res Commun. 2016;477:937–944. doi: 10.1016/j.bbrc.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jorge SE, Kobayashi SS, Costa DB. Epidermal growth factor receptor (EGFR) mutations in lung cancer: preclinical and clinical data. Braz J Med Biol Res. 2014;47:929–939. doi: 10.1590/1414-431X20144099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H. Three generations of epidermal growth factor receptor tyrosine kinase inhibitors developed to revolutionize the therapy of lung cancer. Drug Des Devel Ther. 2016;10:3867–3872. doi: 10.2147/DDDT.S119162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato R, Semba T, Saya H, et al. Concise Review: Stem Cells and Epithelial-Mesenchymal Transition in Cancer: Biological Implications and Therapeutic Targets, Stem Cells. 2016;34:1997–2007. doi: 10.1002/stem.2406. [DOI] [PubMed] [Google Scholar]
- 6.Kourtidis A, Ngok SP, Anastasiadis PZ. p120 catenin: an essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Prog Mol Biol Transl Sci. 2013;116:409–432. doi: 10.1016/B978-0-12-394311-8.00018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y, Lee JK, Ahn SH, et al. WNT signaling in glioblastoma and therapeutic opportunities. Lab Invest. 2016;96:137–150. doi: 10.1038/labinvest.2015.140. [DOI] [PubMed] [Google Scholar]
- 8.Avasarala S, Van Scoyk M, Karuppusamy Rathinam MK, et al. PRMT1 Is a Novel Regulator of Epithelial-Mesenchymal-Transition in Non-small Cell Lung Cancer. J Biol Chem. 2015;290:13479–13489. doi: 10.1074/jbc.M114.636050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fong JT, Jacobs RJ, Moravec DN, et al. Alternative signaling pathways as potential therapeutic targets for overcoming EGFR and c-Met inhibitor resistance in non-small cell lung cancer. PLoS One. 2013;8:e78398. doi: 10.1371/journal.pone.0078398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayachandran A, Dhungel B, Steel JC. Epithelial-to-mesenchymal plasticity of cancer stem cells: therapeutic targets in hepatocellular carcinoma. J Hematol Oncol. 2016;9:74. doi: 10.1186/s13045-016-0307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugano T, Seike M, Noro R, et al. Inhibition of ABCB1 Overcomes Cancer Stem Cell-like Properties and Acquired Resistance to MET Inhibitors in Non-Small Cell Lung Cancer. Mol Cancer Ther. 2015;14:2433–2440. doi: 10.1158/1535-7163.MCT-15-0050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.