

Abstract
Development of resistance to doxorubicin-based chemotherapy limits curative effect in breast cancer (BC). N-acetyltransferase 10 (NAT10), a nucleolar protein involved in histone acetylation, is overexpressed in several cancers. We investigated whether NAT10 is involved in doxorubicin resistance in BC and explored the potential mechanisms. Remodelin, a NAT10 inhibitor, and a NAT10 small interfering RNA (siRNA) were used to inhibit NAT10; both remodelin and the NAT10 siRNA reduced cell viability and attenuated doxorubicin resistance in four BC cell lines. Remodelin and doxorubicin synergistically reduced cell viability, though knockdown of NAT10 and remodelin did not exert a synergistic effect in doxorubicin-treated cells. Remodelin upregulated E-cadherin and downregulated vimentin, canonical markers of the epithelial-mesenchymal transition (EMT), whereas doxorubicin had the opposite effects. Moreover, both remodelin and knockdown of NAT10 reversed the doxorubicin-induced EMT. Finally, when the EMT was blocked using a siRNA targeting Twist, remodelin could not alleviate doxorubicin resistance. Collectively, these findings demonstrate that inhibition of NAT10 attenuates doxorubicin resistance by reversing the EMT in BC. This represents a novel mechanism of doxorubicin resistance in BC and indicates remodelin may have potential clinical value to increase the efficacy of doxorubicin-based chemotherapy in BC.
Keywords: NAT10, doxorubicin, epithelial mesenchymal transition, remodelin, drug resistance
Introduction
Breast cancer (BC) is a leading cause of death among women worldwide [1]. Surgery and chemotherapy are the main treatments; chemotherapy can be administered before (neoadjuvant) or after surgery, with or without other interventions [2]. Although the goal of chemotherapy is to eliminate tumor cells and prevent metastasis, intrinsic and acquired resistance limit the efficacy of conventional chemotherapeutic agents [3]. A lack of a response to chemotherapeutic drugs remains a major problem in BC. The anthracycline doxorubicin is recommended as the first-line chemotherapeutic agent for treatment of BC resistant to endocrine therapy or advanced stage disease [4,5]. The antitumor activity of doxorubicin is mediated via initiation of DNA damage [5]. Although doxorubicin is still considered to be one of the most effective agents for BC, dose-related development of resistance limits its efficacy [6]. Therefore, it is essential to identify the mechanisms underlying resistance to doxorubicin to improve survival outcomes in BC.
NAT10 (N-acetyltransferase 10) is a 872 amino acid nucleolar protein containing an acetyltransferase domain and lysine-rich C-terminus. NAT10 enhances telomerase activity by stimulating human telomerase reverse transcriptase (hTERT) transcription [7] and is involved in several important biological processes, such as DNA damage responses, activation of rRNA transcription, cytokinesis and acetylation of microtubules [8-10]. NAT10 has been reported to participate in the development of several human cancers, including colorectal cancer and hepatocellular carcinoma [11-14]. Although dysregulation of NAT10 may play an important role in carcinogenesis, little is known about the role of NAT10 in BC or the effect of NAT10 on drug resistance in BC.
Remodelin, a specific inhibitor of NAT10 that can mediate nuclear shape rescue in laminopathic cells via microtubule reorganization, was identified by Larrieu et al. in 2014 [15]. Remodelin was recently reported to alleviate defects associated with prelamin A and improve the health of aged vascular smooth muscle cells (VSMCs) [16]. Remodelin also inhibits hepatocellular carcinoma cell invasion and migration under hypoxic conditions by blocking the epithelial-mesenchymal transition (EMT) [11]. However, the effect of remodelin has not been investigated in BC.
In the present study, we investigated the relationships between NAT10, doxorubicin and the EMT to explore the mechanisms underlying doxorubicin resistance in BC.
Methods and materials
Cell culture and reagents
Four human BC cell lines, Bcap-37, MCF-7, HCC1937 and MDA-MB-231, were originally purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultivated as instructed by the ATCC in a humidified incubator with 5% CO2 at 37°C. Bcap-37 and HCC193 cells were cultured in RPMI-1640 (Gibco, Grand Island, NY, USA) supplemented with 10% FBS; MCF-7 cells, in MEM (Gibco, Grand Island, NY, USA) supplemented with 10% FBS; MDA-MB-231 cells, in L15 (Gibco, Grand Island, NY, USA) supplemented with 10% FBS. Remodelin and doxorubicin were purchased from Selleckchem (S7641 and S1208; Houston, TX, USA).
Cell viability assay
MDA-MB-231 cells were seeded into 96-well microplates at 8,000 cells per well and the other three cell lines at 4,000 cells per well, cultured in media containing 1% FBS for 24 h to synchronize the cells, then cultured with different concentrations of doxorubicin (0.00 to 1.0 μg/mL), remodelin (0.00 to 10.0 μM), or both for 48 h. The CCK-8 assay (Dojindo, Kumamoto, Japan) was performed according to the manufacturer’s instructions. OD450 values were determined using a MRX II microplate reader (Dynex, Chantilly, VA, USA).
Cell proliferation assay
Cell proliferation was assessed by 5-ethynyl-2’-deoxyuridine (EdU) staining using the Click-iTEdU Imaging Kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions.Cells were treated with doxorubicin (0, 0.0625, 0.125, 0.25, 0.5, 1.0 μg/mL) or both doxorubicin and remodelin (0, 0.625, 1.25, 2.5, 5, 10 μM). for 48 h, then incubated with 10 µM EdU for 2 h at 37°C. The cells were fixed in 3.7% formaldehyde for 15 min at room temperature, permeabilized in 0.5% Triton X-100 for 20 min at room temperature, washed twice with Tris-buffered saline (TBS) and 0.1% Tween 20 (TBS-T) containing 5% bovine serum albumin (BSA), and incubated with 0.5 mL Click-iT® reaction cocktail for 30 min in the dark., followed by 1 mL of Hoechst 33,342 (1:2000) for 30 min. Three random fields of view per slide were captured using a fluorescence microscope (Olympus, Tokyo, Japan) and the numbers of proliferating cells (EdU-positive) were counted.
Cell transfection and RNA interference
Human NAT10 and Twist siRNAs were obtained from Shanghai GeneChem Co., Ltd. (Shanghai, China). The NAT10 siRNA (100 nM) sequence and the Twist siRNA (100 nM) sequence were purchased by Santa Cruz Biotechnology (Santa Cruz, Dallas, TX, USA). The siRNAs were transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Knockdown efficiency was determined by Western blotting.
Western blot analysis
Western blot analysis was performed following standard methods [17] using anti-E-cadherin (1:1000; Danvers MA, USA), anti-vimentin (1:1000; CST), anti-Twist (1:1000; CST), anti-GAPDH (1:1000; Cell Signaling technology, Danvers, MA, USA) and anti-NAT10 (1:1000; Abcam, Cambridge, UK). Secondary antibodies conjugated to horseradish peroxidase (1:2000) were obtained from Abcam. The grey values were quantified using Quantity One software (Bio-Rad, Drive Hercules, CA, USA).
Immunofluorescent staining
Cells were seeded onto coverslips, fixed in 4% formaldehyde for 30 min, blocked in 3% BSA for 30 min, followed by permeabilization with 0.1% Triton-X 100. Cells were then stained with primary E-cadherin or vimentin antibodies (1:100; Cell Signaling Technology, Danvers, MA, USA), followed by FITC-conjugated secondary antibody (Abcam, Cambridge, USA) and DAPI (1:1000). The cells were observed by confocal immunofluorescence microscopy (Zeiss, Oberkochen, Germany).
Statistical analysis
Data are presented as the mean ± SD. Statistical analyses were performed using. Differences between two groups were examined using the Student’s t-test and SPSS software (Version 18.0; SPSS Inc, Chicago, IL, USA) multiple groups, one-way analysis of variance (ANOVA). Significance was defined as P < 0.05.
Results
Remodelin attenuates doxorubicin resistance in BC cells
Remodelin is an inhibitor of NAT10. To explore the role of NAT10 in doxorubicin resistance in BC, we first evaluated the effect of remodelin on BC cell viability in the presence of different concentrations of doxorubicin (Figure 1A). Combined doxorubicin and remodelin treatment reduced cell viability significantly more compared to cells treated with doxorubicin alone. EDU assays confirmed that both doxorubicin and remodelin decreased BC cell proliferation more compared to cells treated with doxorubicin alone (Figure 1B and 1C), suggesting remodelin attenuates doxorubicin resistance in BC cells. Western blotting demonstrated remodelin reduced the expression of NAT10, confirming remodelin is an effective inhibitor of NAT10 (Figure 1D).
Figure 1.
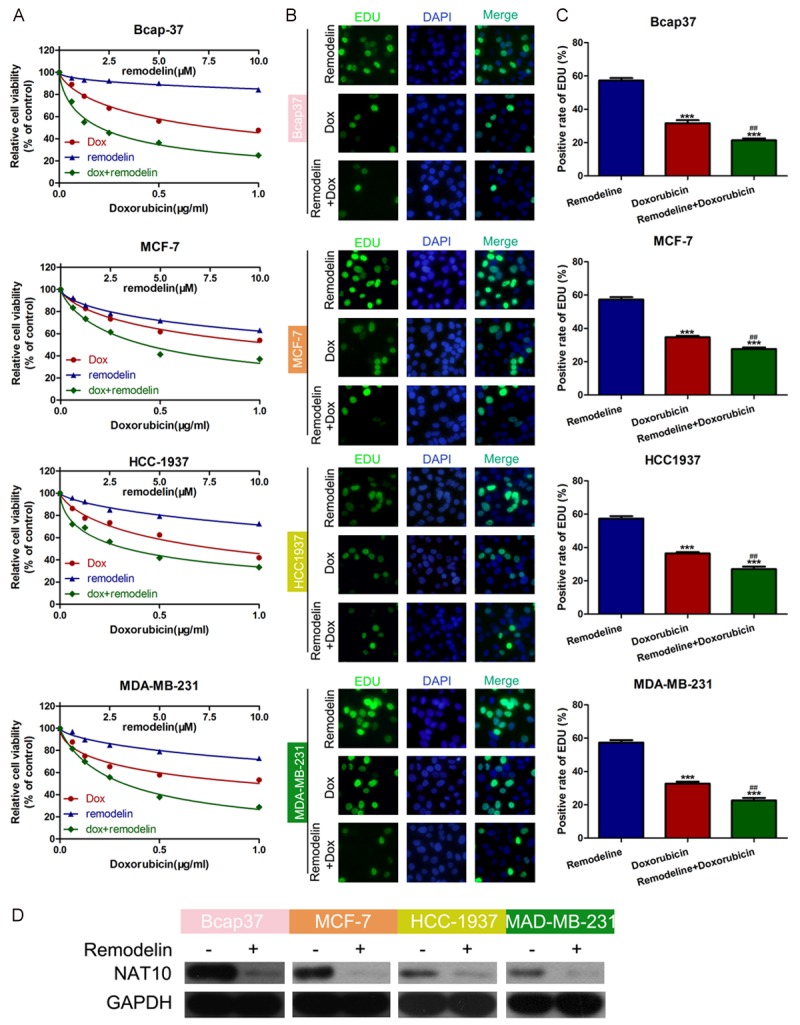
Remodelin attenuates doxorubicin resistance in BC cells. A. BC cell lines were treated with different concentrations of doxorubicin (0, 0.0625, 0.125, 0.25, 0.5, 1.0 μg/mL) and/or remodelin (0.00, 2.50, 5.0, 7.50, 10.0 μM). Cell viability was examined using the CCK-8 assay. B, C. BC cell lines were treated with doxorubicin (0, 0.0625, 0.125, 0.25, 0.5, 1.0 μg/mL) and/or remodelin (0, 0.625, 1.25, 2.5, 5, 10 μM). Cell proliferation was examined using the EDU assay; the numbers of EDU positive blotting confirmed remodelin effectively downregulated NAT10 protein expression. D. Western Bot was used to detect the interference efficiency of NAT10.
Knockdown of NAT10 confirms remodelin attenuates doxorubicin resistance in BC cells by inhibiting NAT10
To further confirm the ability of NAT10 to confer doxorubicin resistance in BC, we used a siRNA to knockdown NAT10. The CCK-8 assay showed that, similarly to remodelin treatment, knockdown of NAT10 attenuated doxorubicin resistance in BC cell lines compared to negative control-transfected cells. However, cell viability was similar in cells transfected with the NAT10 siRNA alone and cells treated with the NAT10 siRNA and remodelin (Figure 2A), confirming remodelin attenuates doxorubicin resistance in BC cells by inhibiting NAT10. The knockdown efficiency of the NAT10 siRNA was confirmed by Western blotting (Figure 2B).
Figure 2.
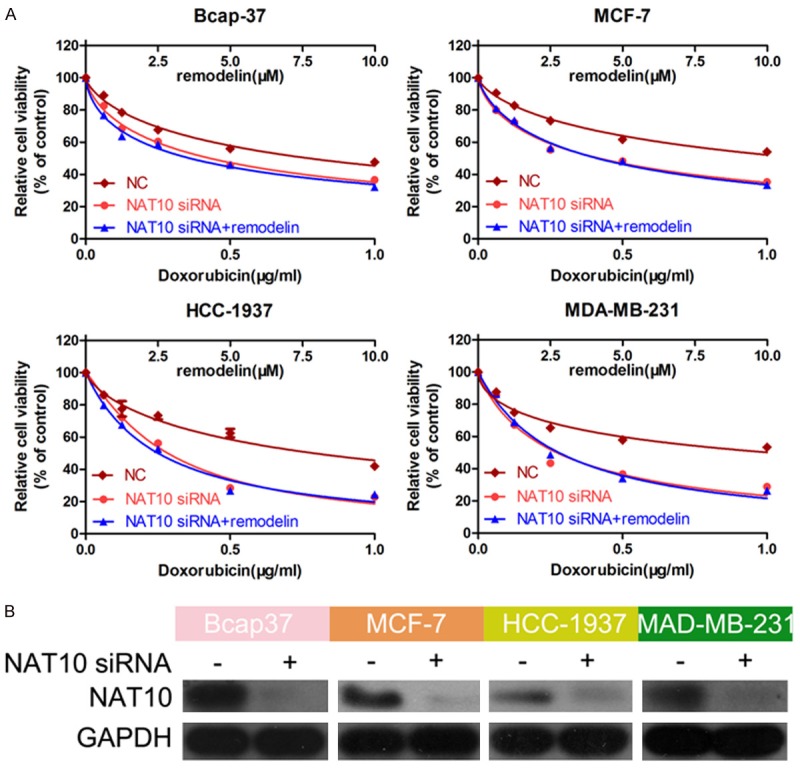
Knockdown of NAT10 confirms remodelin attenuates doxorubicin resistance in BC cells by inhibiting NAT10. A. BC cells were treated with different concentrations of doxorubicin (0, 0.0625, 0.125, 0.25, 0.5, 1.0 μg/mL), then transfected with the NAT10 siRNA, negative control siRNA, or NAT10 siRNA and treated with remodelin. Cell viability was examined using the CCK-8 assay. B. Western blotting was used to confirm the knockdown efficiency of the NAT10 siRNA; GAPDH was used as the internal control.
Remodelin reverses the EMT in BC cells
The EMT is closely associated with chemoresistance in cancer. Thus, we hypothesized remodelin may attenuate doxorubicin resistance in BC by reversing the EMT. Western blotting (Figure 3A) and immunofluorescent staining (Figure 3B) revealed remodelin significantly upregulated the epithelial marker E-cadherin and downregulated the mesenchymal marker vimentin in BC cells, indicating remodelin reverses the EMT in BC cells.
Figure 3.
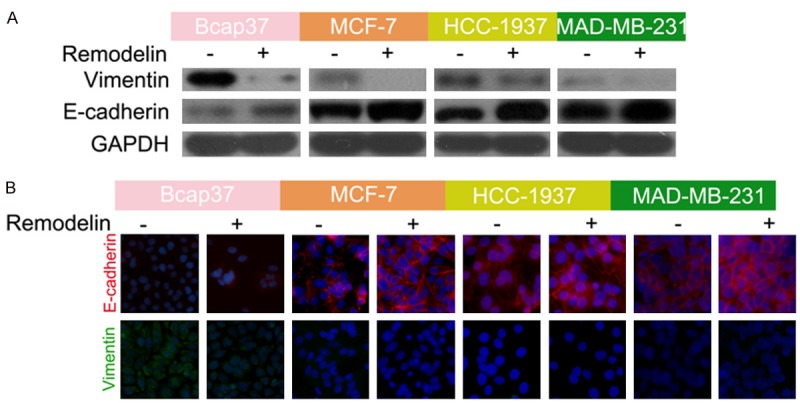
Remodelin reverses the EMT in BC cells. A. Western blotting of the effect of remodelin on E-cadherin and vimentin protein expression in BC cell lines. GAPDH was used as the internal control. B. Immunofluorescence assay of the effect of remodelin on E-cadherin (Red) and vimentin (Green) protein expression in BC cell lines; nuclei were stained using DAPI (blue).
Knockdown of NAT10 reverses the doxorubicin-induced EMT in BC cells
To further prove our hypothesis, next we examined the relationship between doxorubicin and the EMT. Doxorubicin significantly upregulated vimentin and downregulated E-cadherin in BC cell lines (Figure 4A and 4B). However, vimentin expression significantly decreased and E-cadherin expression increased in BC cells treated with both doxorubicin and remodelin, indicating remodelin reversed the doxorubicin-induced EMT in BC cells. Furthermore, NAT10 expression was significantly reduced by remodelin, but not by doxorubicin (Figure 4A and 4B). Knockdown of NAT10 using the siRNA also significantly reversed the doxorubicin-induced induced EMT (Figure 4C), confirming that inhibition of NAT10 reverses the EMT in BC cells.
Figure 4.
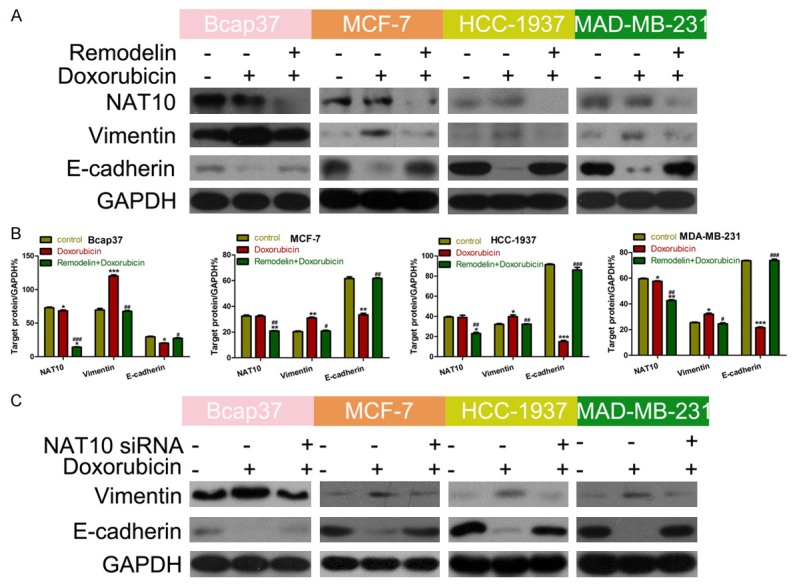
Knockdown of NAT10 reverses the doxorubicin-induced EMT in BC cells. A. Western blotting of the effect of remodelin and/or doxorubicin on E-cadherin, vimentin and NAT10 protein expression in BC cell lines. GAPDH was used as the internal control. B. Quantification of western blot grey values (*P < 0.05, **P < 0.01, ***P < 0.001 vs Control; #P <0.05, ##P < 0.01, ###P < 0.001 vs doxorubicin). C. Western blotting analysis the effect of transfected with NAT10 and/or doxorubicin on E-cadherin, vimentin in BC cell lines. GAPDH was used as the internal control.
To further explore the relationship between the EMT, doxorubicin resistance and remodelin, we knocked down Twist, a key molecule involved in the EMT, to block the EMT. The knockdown efficiency of the Twist siRNA was confirmed by western blotting (Figure 5A). After inhibition of the EMT using remodelin, the cell viability of cells treated with the Twist siRNA alone and Twist siRNA + remodelin were not significantly different in the presence of doxorubicin (Figure 5B), further confirming that remodelin attenuates doxorubicin resistance by reversing the EMT. Hence, these results prove our hypothesis that remodelin attenuates doxorubicin resistance in BC cells by reversing the EMT.
Figure 5.
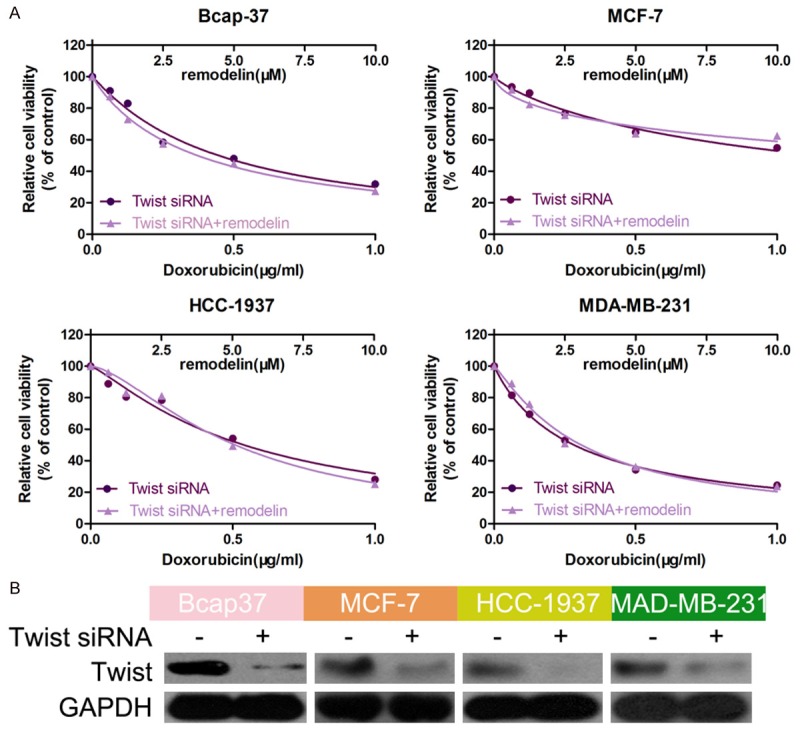
The effect of Remodelin was vanished in BC after knockdown of Twist. A. BC cell lines were transfected with a Twist siRNA and/or treated with remodelin in the presence of different concentrations of doxorubicin. Cell viability was examined using the CCK-8 assay. B. Western blotting was used to confirm the knockdown efficiency of the Twist siRNA; GAPDH was used as the internal control.
Discussion
Chemotherapy is widely used as an adjuvant therapy for BC; however, drug resistance limits the efficacy of chemotherapy [18,19]. This study demonstrates inhibition of NAT10 attenuates doxorubicin resistance in BC by reversing the EMT, and indicates the NAT10 inhibitor remodelin may have potential clinical value to reduce doxorubicin resistance in BC.
NAT10 activates telomerase activity and participates in several essential biological processes [8,9,20,21]. The role of NAT10 in cancer is poorly characterized. NAT10 can regulate the EMT and promote metastasis in HCC, and high expression of NAT10 is associated with poor patient survival [11,13]. In colorectal carcinoma, NAT10 is regulated by glycogen synthase kinase-3β (GSK-3β), involved in the Wnt signaling pathway and plays a critical role in p53 activation via acetylating p53 and counteracting mouse double minute2 (Mdm2) [12,14]. However, little is known about the role of NAT10 in drug resistance in BC. This study provides the first evidence that inhibition of NAT10 can relive doxorubicin resistance in BC.
The EMT is involved in drug resistance in cancer cells [22-24]. In BC, the EMT can induce drug resistance by promoting a stemness phenotype [25-28]. Furthermore, Ma et al. indicated that NAT10 could regulate the EMT in HCC [11]. Based on these previous reports, we hypothesized NAT10 may confer doxorubicin resistance in BC cells by regulating the EMT, and our in vitro experiments provided strong evidence to confirm this hypothesis.
The small molecule remodelin is an inhibitor of NAT10 that was first identified by Larrieu et al. in 2014 [15]. Remodelin can inhibit the invasion and migration of HCC cells under hypoxic conditions [11]. We demonstrated that remodelin could relieve doxorubicin resistance in BC cells. This finding indicates remodelin could be administered with doxorubicin to improve treatment efficacy in BC. Further studies are needed to fully explore the therapeutic value of remodelin in BC.
In conclusion, we provide the first evidence that inhibition of NAT10 can reduce doxorubicin resistance in BC cells by reversing the EMT. Moreover, the NAT10 inhibitor remodelin may represent a potential anti-cancer drug, and could lead to synergistic treatment effect if administered with doxorubicin. However, more experiments are needed to understand the precise molecular mechanisms underlying doxorubicin resistance in BC.
Disclosure of conflict of interest
None.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Joerger M, Thurlimann B. Chemotherapy regimens in early breast cancer: major controversies and future outlook. Expert Rev Anticancer Ther. 2013;13:165–178. doi: 10.1586/era.12.172. [DOI] [PubMed] [Google Scholar]
- 3.Velaei K, Samadi N, Barazvan B, Soleimani Rad J. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast. 2016;30:92–100. doi: 10.1016/j.breast.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Lv L, Qiu K, Yu X, Chen C, Qin F, Shi Y, Ou J, Zhang T, Zhu H, Wu J, Liu C, Li G. Amphiphilic copolymeric micelles for doxorubicin and curcumin co-delivery to reverse multidrug resistance in breast cancer. J Biomed Nanotechnol. 2016;12:973–985. doi: 10.1166/jbn.2016.2231. [DOI] [PubMed] [Google Scholar]
- 5.Xiang S, Dauchy RT, Hauch A, Mao L, Yuan L, Wren MA, Belancio VP, Mondal D, Frasch T, Blask DE, Hill SM. Doxorubicin resistance in breast cancer is driven by light at night-induced disruption of the circadian melatonin signal. J Pineal Res. 2015;59:60–69. doi: 10.1111/jpi.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Teng Z, Wang H, Wang C, Liu Y, Tang Y, Wu J, Sun J, Wang H, Wang J, Lu G. Increasing the cytotoxicity of doxorubicin in breast cancer MCF-7 cells with multidrug resistance using a mesoporous silica nanoparticle drug delivery system. Int J Clin Exp Pathol. 2014;7:1337–1347. [PMC free article] [PubMed] [Google Scholar]
- 7.Shen Q, Zheng X, McNutt MA, Guang L, Sun Y, Wang J, Gong Y, Hou L, Zhang B. NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp Cell Res. 2009;315:1653–1667. doi: 10.1016/j.yexcr.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Ito S, Horikawa S, Suzuki T, Kawauchi H, Tanaka Y, Suzuki T, Suzuki T. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA) J Biol Chem. 2014;289:35724–35730. doi: 10.1074/jbc.C114.602698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu H, Ling Y, Gong Y, Sun Y, Hou L, Zhang B. DNA damage induces N-acetyltransferase NAT10 gene expression through transcriptional activation. Mol Cell Biochem. 2007;300:249–258. doi: 10.1007/s11010-006-9390-5. [DOI] [PubMed] [Google Scholar]
- 10.Chi YH, Haller K, Peloponese JM Jr, Jeang KT. Histone acetyltransferase hALP and nuclear membrane protein hsSUN1 function in decondensation of mitotic chromosomes. J Biol Chem. 2007;282:27447–27458. doi: 10.1074/jbc.M703098200. [DOI] [PubMed] [Google Scholar]
- 11.Ma R, Chen J, Jiang S, Lin S, Zhang X, Liang X. Up regulation of NAT10 promotes metastasis of hepatocellular carcinoma cells through epithelial-to-mesenchymal transition. Am J Transl Res. 2016;8:4215–4223. [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, Tan Y, Zhang C, Zhang Y, Zhang L, Ren P, Deng H, Luo J, Ke Y, Du X. NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Rep. 2016;17:349–366. doi: 10.15252/embr.201540505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Liu J, Yan S, Huang K, Bai Y, Zheng S. High expression of N-acetyltransferase 10: a novel independent prognostic marker of worse outcome in patients with hepatocellular carcinoma. Int J Clin Exp Pathol. 2015;8:14765–14771. [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Hou W, Wang HL, Liu HJ, Jia XY, Zheng XZ, Zou YX, Li X, Hou L, McNutt MA, Zhang B. GSK-3beta-regulated N-acetyltransferase 10 is involved in colorectal cancer invasion. Clin Cancer Res. 2014;20:4717–4729. doi: 10.1158/1078-0432.CCR-13-3477. [DOI] [PubMed] [Google Scholar]
- 15.Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014;344:527–532. doi: 10.1126/science.1252651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cobb AM, Larrieu D, Warren DT, Liu Y, Srivastava S, Smith AJ, Bowater RP, Jackson SP, Shanahan CM. Prelamin A impairs 53BP1 nuclear entry by mislocalizing NUP153 and disrupting the Ran gradient. Aging Cell. 2016 doi: 10.1111/acel.12506. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Zheng C, Xu H, He W, Ruan Y, Ma J, Zheng J, Ye C, Li W. Inhibition of AMPK-related kinase 5 (ARK5) enhances cisplatin cytotoxicity in non-small cell lung cancer cells through regulation of epithelial-mesenchymal transition. Am J Transl Res. 2017;9:1708–1719. [PMC free article] [PubMed] [Google Scholar]
- 18.Moulder S. Intrinsic resistance to chemotherapy in breast cancer. Womens Health (Lond) 2010;6:821–830. doi: 10.2217/whe.10.60. [DOI] [PubMed] [Google Scholar]
- 19.Saeki T, Tsuruo T, Sato W, Nishikawsa K. Drug resistance in chemotherapy for breast cancer. Cancer Chemother Pharmacol. 2005;56(Suppl 1):84–89. doi: 10.1007/s00280-005-0106-4. [DOI] [PubMed] [Google Scholar]
- 20.Cai S, Liu X, Zhang C, Xing B, Du X. Autoacetylation of NAT10 is critical for its function in rRNA transcription activation. Biochem Biophys Res Commun. 2017;483:624–629. doi: 10.1016/j.bbrc.2016.12.092. [DOI] [PubMed] [Google Scholar]
- 21.Montgomery DC, Garlick JM, Kulkarni RA, Kennedy S, Allali-Hassani A, Kuo YM, Andrews AJ, Wu H, Vedadi M, Meier JL. Global profiling of acetyltransferase feedback regulation. J Am Chem Soc. 2016;138:6388–6391. doi: 10.1021/jacs.6b03036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chow AK, Ng L, Lam CS, Wong SK, Wan TM, Cheng NS, Yau TC, Poon RT, Pang RW. The Enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS One. 2013;8:e78675. doi: 10.1371/journal.pone.0078675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliveras-Ferraros C, Corominas-Faja B, Cufi S, Vazquez-Martin A, Martin-Castillo B, Iglesias JM, Lopez-Bonet E, Martin AG, Menendez JA. Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin) Cell Cycle. 2012;11:4020–4032. doi: 10.4161/cc.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X, Lingala S, Khoobyari S, Nolta J, Zern MA, Wu J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J Hepatol. 2011;55:838–845. doi: 10.1016/j.jhep.2010.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo M, Brooks M, Wicha MS. Epithelialmesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des. 2015;21:1301–1310. doi: 10.2174/1381612821666141211120604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliveras-Ferraros C, Vazquez-Martin A, Cuyas E, Corominas-Faja B, Rodriguez-Gallego E, Fernandez-Arroyo S, Martin-Castillo B, Joven J, Menendez JA. Acquired resistance to metformin in breast cancer cells triggers transcriptome reprogramming toward a degradome-related metastatic stem-like profile. Cell Cycle. 2014;13:1132–1144. doi: 10.4161/cc.27982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mallini P, Lennard T, Kirby J, Meeson A. Epithelial-to-mesenchymal transition: what is the impact on breast cancer stem cells and drug resistance. Cancer Treat Rev. 2014;40:341–348. doi: 10.1016/j.ctrv.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 28.Nicolini A, Ferrari P, Fini M, Borsari V, Fallahi P, Antonelli A, Berti P, Carpi A, Miccoli P. Stem cells: their role in breast cancer development and resistance to treatment. Curr Pharm Biotechnol. 2011;12:196–205. doi: 10.2174/138920111794295657. [DOI] [PubMed] [Google Scholar]