

Abstract
Accumulating evidence indicates that long noncoding RNAs (lncRNAs) are involved in diseases such as cancer. However, little is known about the role of lncRNAs in gastrointestinal stromal tumors (GIST). In the present study, we explored the biological function of the lncRNA coiled-coil domain-containing 26 (CCDC26) in imatinib resistance of GIST. We found that human GIST-882 cells with lower CCDC26 expression were less sensitive to imatinib compared with GIST-T1 cells with higher CCDC26 expression. CCDC26 expression decreased in a time-dependent manner in the presence of imatinib. Moreover, small interfering RNA (siRNA) knockdown of CCDC26 increased GIST cell sensitivity to imatinib. The RNA pull-down experiment showed that CCDC26 can interact with c-KIT and that CCDC26 knockdown can upregulate c-KIT expression. We also found that inhibiting c-KIT induced resistance to imatinib. Lastly, we proved that inhibiting c-KIT can reverse CCDC26 knockdown-mediated imatinib resistance in GIST. We suggest that CCDC26 knockdown can induce imatinib resistance in GIST cells by downregulating c-KIT expression. Our results provide a novel insight into imatinib resistance in GIST.
Keywords: Long noncoding RNA, GIST, c-KIT, CCDC26, chemoresistance
Introduction
Gastrointestinal stromal tumors (GIST), a type of mesenchymal tumor, are the most common mesenchymal tumors in the gastrointestinal tract [1]. GIST are identified immunohistochemically via the CD34 and KIT proteins [2]. Most (~75%) GIST carry gain-of-function KIT mutations in exons 9, 11, 13, or 17, and about 5% of GIST harbor platelet-derived growth factor receptor alpha (PDGFRA) mutations in exons 12, 14, or 18 [3,4]. The PDGFRA and KIT mutation types are related to drug resistance in GIST [5,6].
Imatinib is a rationally designed oral signal transduction inhibitor that antagonizes several mutant forms of type III tyrosine kinases such as KIT, PDGFRA, and BCR/ABL [7]. Imatinib has significant success in improving the clinical outcome of patients with GIST. Imatinib has become the first-line therapy for patients with metastatic or end-stage GIST, where disease control ranges 70-85%, and median progression-free survival and median overall survival is 29 months and 57 months, respectively [8]. However, almost 50% of imatinib-treated patients with GIST acquire secondary resistance in the first 2 years [9]. Despite the initial efficacy, drug resistance limits the long-term effect of imatinib. Hence, there is an urgent need to explore the potential mechanism underlying imatinib resistance in GIST to enhance its effect and improve patient survival.
Long noncoding RNAs (lncRNAs) are functionally defined as transcripts that are >200 nucleotides in length with no protein-coding potential [10]. Accumulating studies have demonstrated that lncRNAs are involved in serial steps of cancer development. LncRNAs can interact with DNA, RNA, protein, and/or their combinations, and play a vital role in chromatin organization and transcriptional and post-transcriptional regulation [11,12]. Abnormal lncRNA expression correlates with tumor initiation, growth, and metastasis [13]. The lncRNA coiled-coil domain-containing 26 (CCDC26), located on chromosome 8q24.21, was reported as a retinoic acid-dependent modulator in glioblastoma [14]. Several studies have demonstrated that CCDC26 polymorphism is related to glioma risk [15-17]. CCDC26 also controls myeloid leukemia cell growth by regulating KIT expression [18]. Moreover, CCDC26 is a novel oncogene in pancreatic cancer and is responsible for cancer cell growth and apoptosis by regulating proliferating cell nuclear antigen (PCNA) and Bcl-2 expression [19]. However, the biological function of CCDC26 in GIST remains unclear.
In the present study, we investigated the function of CCDC26 in GIST and reveal its role and potential mechanisms in imatinib resistance.
Materials and methods
Cell culture
The human GIST cell lines GIST-882 and GIST-T1 were purchased from the Chinese Academy of Science Cell Bank (Shanghai, China). Both cell lines were cultured in RPMI 1640 (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Gibco) at 37°C in 5% CO2.
Cell transfection
c-KIT, CCDC26, and negative control small interfering RNAs (siRNAs) were synthesized by GenePharma (Shanghai, China). Transfection was conducted using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Cell viability assay
The GIST cells (4000 cells/well) were seeded in 96-well plates. After the cells had adhered completely to the well, the culture medium was replaced with medium containing 1% FBS for 24 h to synchronize the cells. Then, the cells were cultured under the different treatment conditions at 37°C in a 5% CO2 incubator for 48 h. Cell viability was measured using Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) at 2 h according to the manufacturer’s instructions. An MRX II microplate reader (Dynex, Chantilly, VA, USA) was used to measure the optical density (OD) value at 450 nm.
Cell proliferation assay
GIST cell proliferation was assayed using a Click-iT EdU Imaging Kit (Invitrogen) according to the manufacturer’s protocol. GIST cells treated with CCDC26 siRNA, c-KIT siRNA, or Negative siRNA were plated at 1×106 cells per well. Hoechst® 33342 solution was used to stain the nuclei. 5-Ethynyl-2’-deoxyuridine (EdU)-stained cells were mounted and imaged using fluorescence microscopy.
Apoptosis assay
GIST cell apoptosis was assayed using an annexin V/propidium iodide (PI) kit (Invitrogen). GIST cells (1×106) were treated with CCDC26 siRNA or c-KIT siRNA for 48 h, harvested, and washed with phosphate-buffered saline (PBS) three times. The cells were resuspended in binding buffer and stained with annexin V and PI according to the manufacturer’s protocol, and detected and quantified using fluorescence-activated cell sorting (FACS). The cells in the Q2 and Q3 quadrants were considered apoptotic cells.
Western blotting
Proteins were collected in radioimmunoprecipation assay (RIPA) lysis buffer (Beyotime, Shanghai, China) supplemented with a phenylmethanesulfonyl fluoride (PMSF) inhibitor (Beyotime), and quantitatively analyzed using a bicinchoninic acid (BCA) kit (Thermo Scientific, Waltham, MA, USA). Total protein (30 μg) was run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinylidene fluoride (PVDF) membrane, and blocked in 5% non-fat dry milk in Tris-buffered saline (TBS)-Tween (PBS and 0.05% Tween 20). The primary antibodies against c-KIT (1:1000) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:1000), and the secondary antibody (1:2000), were from Abcam (Cambridge, MA, USA). Anti-GAPDH antibody was used as the loading control. After incubation with the antibodies, the samples were visualized on film using an enhanced chemiluminescence (ECL) kit (GE Healthcare, Diegem, Belgium).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s instructions. Total RNA (0.5 μg) was reversed-transcribed to complementary DNA (cDNA). SYBR Green PCR Master mix (Takara, Shiga, Japan) was used to determine the CCDC26 and c-KIT RNA levels. All reactions were performed in triplicate. The relative expression of CCDC26 and c-KIT was normalized to the internal reference GAPDH. Data were analyzed using the comparative threshold cycle (2ΔΔCt) method.
RNA pull-down assay
The biotin-labeled CCDC26 was transcribed using Biotin RNA Labeling Mix (Roche, Pleasanton, CA, USA) and T7 RNA polymerase (Roche), treated with RNase-free DNase I (Roche), and purified using an RNeasy Mini Kit (Qiagen, Hilden, Germany). Proteins extracted from the GIST cells were mixed with biotinylated RNA. Washed Dynabeads (50 μL, Invitrogen) were added to each binding reaction and washed. The associated proteins were resolved by SDS-PAGE.
Statistical analysis
SPSS 18.0 software (SPSS Inc., Chicago, IL, USA) was used for the statistical analysis. The experimental data are expressed as mean ± SD and were assessed using a two-tailed Student t-test. Statistical significance was accepted if P<0.05.
Results
The GIST cell line with low CCDC26 expression was more resistant to imatinib
To investigate the function of CCDC26 on the sensitivity of GIST cells to imatinib, we first examined CCDC26 expression in two GIST cell lines using qRT-PCR, and found that GIST-T1 cells had higher CCDC26 expression than GIST-882 cells (Figure 1A). Then, we used the CCK-8 assay to detect the viability of the two GIST cell lines under different concentrations of imatinib, and found that GIST-882 cells had better viability than the GIST-T1 cells (Figure 1B). We further examined CCDC26 expression at different time points in the two cell lines cultured with their respective median inhibitory concentrations (IC50) of imatinib. CCDC26 expression in both cell lines decreased in a time-dependent manner in the presence of imatinib (Figure 1C, 1D).
Figure 1.
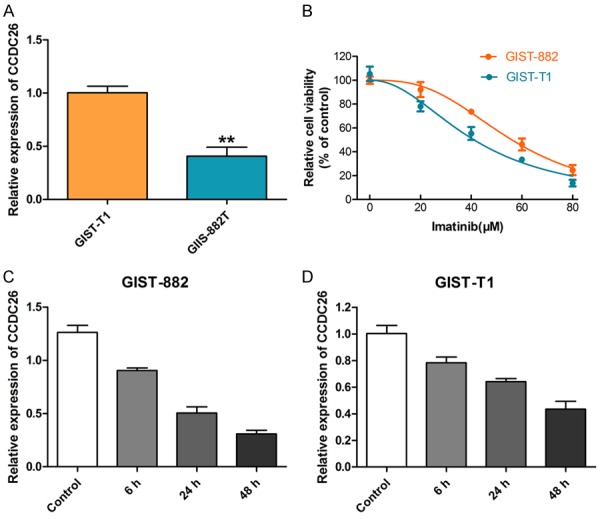
CCDC26 expression in GIST cells and its relationship with imatinib sensitivity. A. Real-time PCR detection of CCDC26 expression in GIST cell lines. B. CCK-8 assay of GIST cell viability in the presence of imatinib (0, 20, 40, 60, 80 μM). C, D. GIST cell viability in the presence of IC50 of imatinib (GIST-882, 56.06 μM; GIST-T1, 41.08 μM) for 0 h, 6 h, 24 h, and 48 h (**P<0.01).
CCDC26 knockdown enhanced imatinib resistance in GIST cells
We used siRNA knockdown of CCDC26 to explore the function of CCDC26 in GIST. Cells in which CCDC26 had been knocked down had better viability than the Negative siRNA in the presence of imatinib (Figure 2A, 2B). The EdU assay showed that cells with CCDC26 knockdown had a higher proliferation rate than the control (Figure 2C, 2D). Moreover, cells treated with both CCDC26 siRNA and imatinib had a lower rate of apoptosis compared to cells treated with imatinib and Negative siRNA (Figure 2E). These results indicate that negative regulation of CCDC26 correlates with decreased sensitivity of GIST cells to imatinib.
Figure 2.
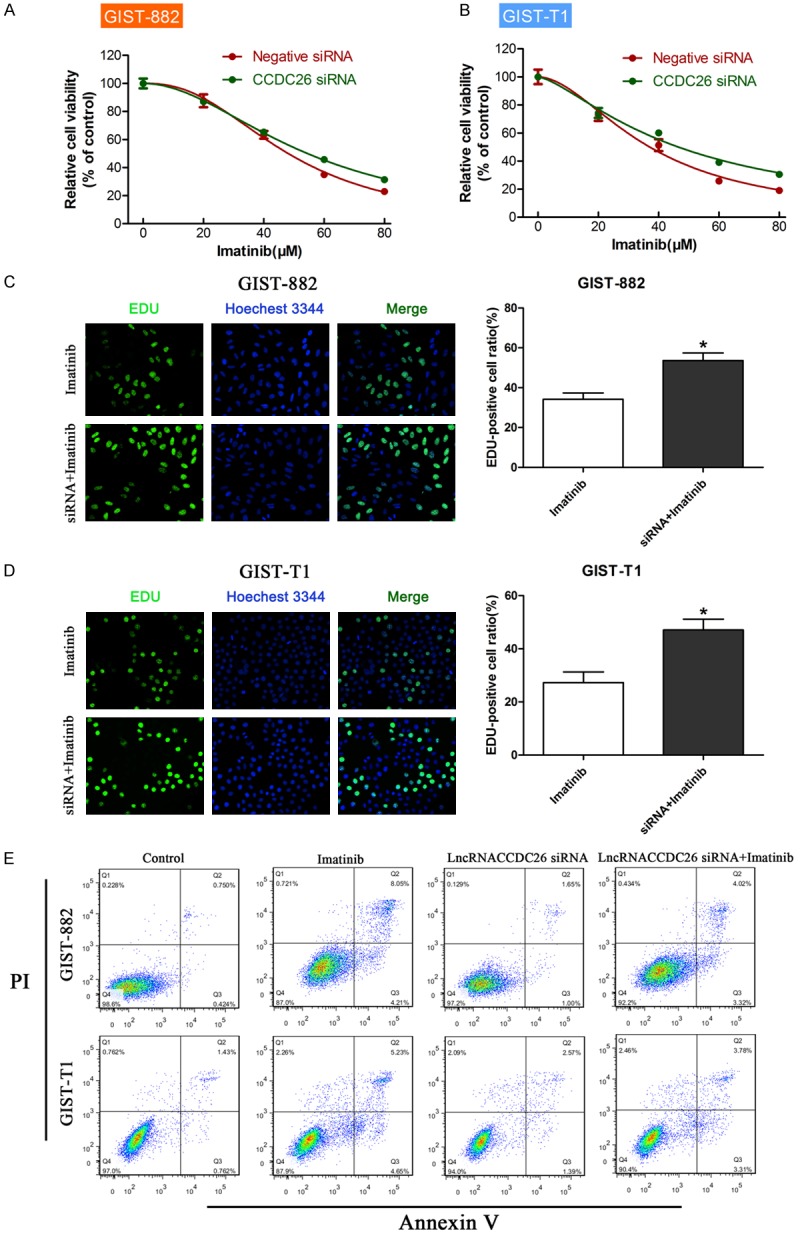
Effect of CCDC26 on GIST cell viability, proliferation, and apoptosis in vitro. A, B. CCK-8 assay of the viability of GIST cells treated with CCDC26 siRNA or control siRNA in the presence of imatinib (0, 20, 40, 60, 80 μM). C, D. EdU assay of cell proliferation rate under IC50 of imatinib in GIST cells treated with CCDC26 siRNA or control siRNA. The number of EdU-positive cells was counted. E. Flow cytometry analysis of apoptosis in GIST cells transfected with control siRNA or CCDC26 siRNA (*P<0.05).
CCDC26 interacted with c-KIT and regulated its expression
Many lncRNAs play a role by interacting with proteins, and we therefore wondered whether CCDC26 has a possible protein partner, which might reveal the molecular mechanisms by which CCDC26 exerts its effects. Considering the vital role of c-KIT in imatinib resistance, we hypothesized that c-KIT may be a partner of CCDC26.
We performed an RNA pull-down experiment using CCDC26-specific DNA probes to pull down c-KIT and analyzed the interaction between CCDC26 and c-KIT via western blotting. The group treated with CCDC26 probes (Treated) showed c-Kit enrichment in the final eluted fluid, and the groups with the control probes (Unrelated) and the blank (None) showed c-KIT enrichment in the filtrated fluid (Figure 3A). This result indicates that CCDC26 can interact with c-KIT. Next, we explored the effect of CCDC26 on c-KIT expression, and found that CCDC26 knockdown upregulated c-KIT mRNA levels in the GIST cells (Figure 3B).
Figure 3.
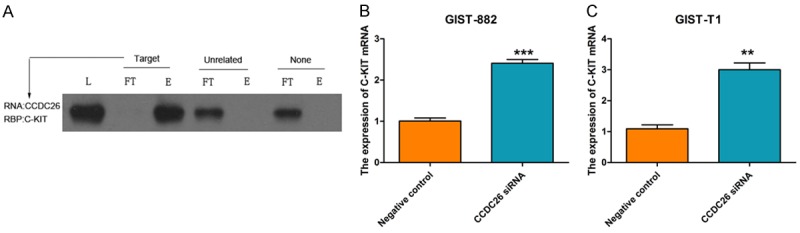
The relationship between CCDC26 and c-KIT. A. Western blot determination of c-KIT expression levels following RNA pull-down assay in CCDC26 RNA group (Target), control RNA group (Untreated), and blank group (None). (L, total protein; FT, flow-through; E, eluate). B, C. The effect of CCDC26 siRNA on c-KIT mRNA expression in GIST cells; GAPDH was the internal control (**P<0.01, ***P<0.001).
c-KIT inhibition enhanced GIST cell sensitivity to imatinib
We explored the role of c-KIT on GIST cell viability, proliferation, and apoptosis via siRNA transfection. Figure 4A and 4B show that c-KIT siRNA transfection reduced cell viability markedly in the presence of imatinib, and markedly reduced the cell proliferation rate (Figure 4C, 4D). Figure 4E shows that there were ~11.42% and 11.87% apoptotic GIST-882 and GIST-T1 cells, respectively, in the control group. However, there were ~19.37% and ~20.1% apoptotic GIST-882 and GIST-T1 cells, respectively, in the c-KIT siRNA group. Hence, our results indicate that c-KIT knockdown can increase imatinib sensitivity, playing an opposite role to that of CCDC26 knockdown.
Figure 4.
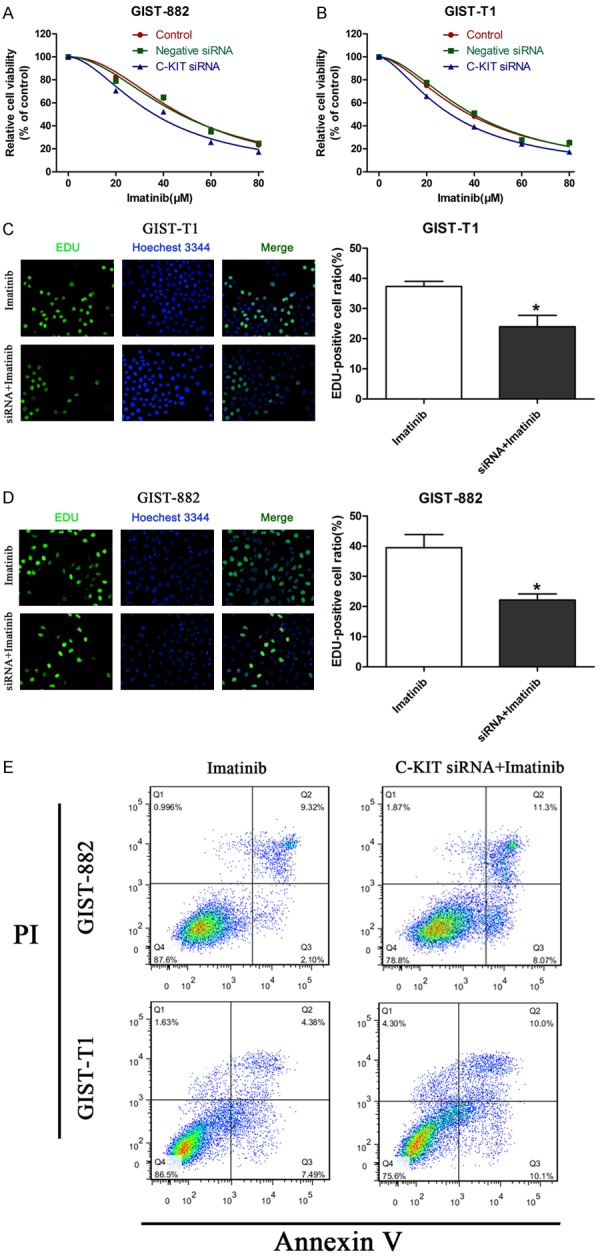
Effect of c-KIT on GIST cell viability, proliferation, and apoptosis in vitro. A, B. CCK-8 assay of the viability of GIST cells treated with c-KIT siRNA, Negative siRNA, or Control in the presence of imatinib (0, 20, 40, 60, 80 μM). C, D. EdU assay of cell proliferation rate under IC50 of imatinib in GIST cells treated with c-KIT siRNA or control siRNA. The number of EDU-positive cells was counted. E. Flow cytometry analysis of apoptosis in GIST cells transfected with control siRNA or CCDC26 siRNA (*P<0.05).
c-KIT knockdown reversed the imatinib resistance mediated by CCDC26 inhibition
To verify whether CCDC26 knockdown enhanced GIST cell resistance to imatinib by regulating c-KIT expression, we co-transfected GIST cells with siRNA against c-KIT and CCDC26, and examined the cell viability in the presence of imatinib. Western blotting confirmed the interference efficiency of the c-KIT siRNA (Figure 5A). The viability of GIST cells co-transfected with c-KIT siRNA and CCDC26 siRNA was the same as that of cells transfected with c-KIT siRNA alone, indicating that c-KIT knockdown can reverse CCDC26 inhibition-mediated imatinib resistance (Figure 5B, 5C).
Figure 5.
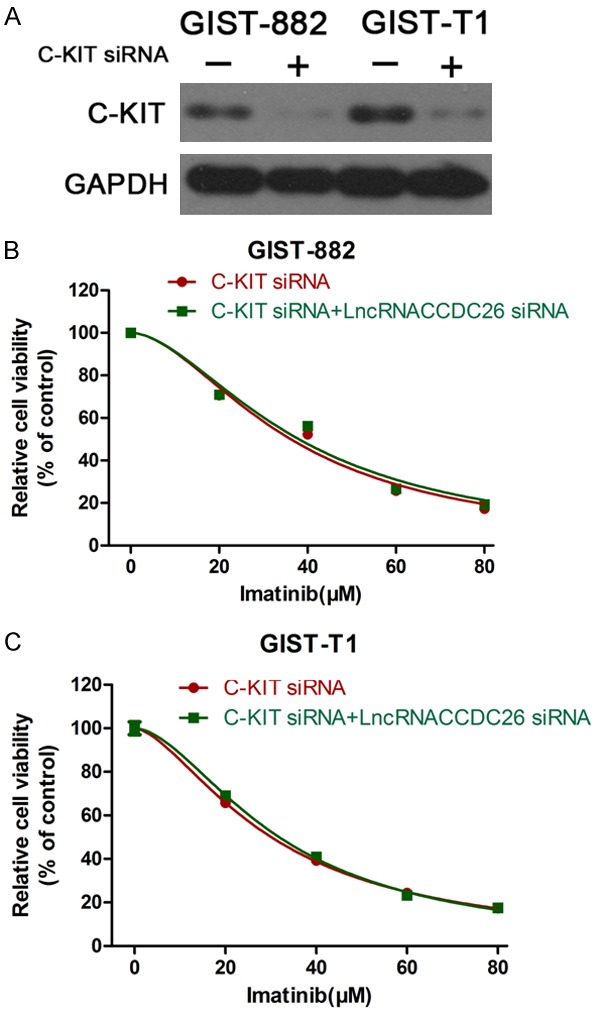
c-KIT knockdown reversed CCDC26 inhibition-mediated imatinib resistance. A. Western blotting validation of the interference efficiency of c-KIT siRNA. B, C. CCK-8 assay of the viability of GIST cells treated with c-KIT siRNA or both c-KIT siRNA and CCDC26 siRNA in the presence of imatinib (0, 20, 40, 60, 80 μM).
Discussion
Accumulating evidence has revealed that lncRNAs might perform a vital function in cancer by regulating a series of critical biological functions, including cell proliferation, apoptosis, and drug resistance [20,21]. Uncovering the mechanisms of lncRNAs in drug resistance would aid improvement of the long-term survival of patients with cancer [22]. In the present study, we investigated the relationship between CCDC26 expression and imatinib resistance in GIST. Our data first highlight the critical role of CCDC26 in the imatinib resistance of GIST.
LncRNAs play an essential role in many important biological processes, including epigenetic gene expression regulation and transcriptional and post-transcriptional regulation [23]. Aberrant lncRNA expression has been found widely in many cancers [24-27]. Researchers increasingly believe that these aberrant lncRNAs are specifically related to cancer development and progression [28,29]. Some well-researched lncRNAs, including H19, HOTAIR (HOX transcript antisense RNA), MALAT-1 (metastasis associated lung adenocarcinoma transcript 1 [non-protein coding]), and HULC (hepatocellular carcinoma upregulated lncRNA), have been proven to act as oncogenes or tumor suppressor genes and correlate with drug resistance [30-33]. However, little is known about the function of lncRNAs in GIST progression and drug resistance. To date, there has been no study on CCDC26 in GIST. In the present study, we first prove that CCDC26 knockdown can induce imatinib resistance in GIST. Our results provide a novel insight into the mechanism underlying imatinib resistance in GIST. Treatments aimed at upregulating CCDC26 expression in GIST may improve the effect of imatinib therapy and benefit long-term survival.
Imatinib is the first-line treatment for patients with metastatic and end-stage GIST, and has completely changed the management and prolonged the survival of patients with GIST [34]. However, imatinib resistance, including primary and secondary resistance, remains a major obstacle to better clinical outcomes [35]. In a phase III trial of imatinib, 12% of patients developed primary resistance to imatinib. Moreover, more than 40% of patients developed secondary resistance after a median follow-up of 25 months [36]. PDGFRA D842V and c-KIT exon 9 mutation, as well as the wild-type genotype, lead to primary resistance to imatinib [37,38]. During imatinib treatment, secondary resistance is often acquired because of secondary c-KIT or PDGFRA mutations that block drug binding [39,40]. c-KIT gene amplification also induces imatinib resistance in GIST [41]. However, the causes for imatinib resistance, especially secondary resistance, remain largely unknown. In our study, we prove that c-KIT knockdown can enhance sensitivity to imatinib, which provides evidence that c-KIT gene amplification can induce imatinib resistance in GIST. Moreover, we demonstrate that CCDC26 can pull down c-KIT in GIST and that CCDC26 can regulate c-KIT transcription. Finally, we prove that CCDC26 knockdown mediation of imatinib resistance may be due to c-KIT upregulation. These results consequently provide novel mechanisms for understanding imatinib resistance and novel clues for overcoming imatinib resistance in GIST.
In conclusion, our study provides key evidence that CCDC26 knockdown is functionally related to imatinib resistance in GIST. Moreover, we prove that CCDC26 can interact with c-KIT and regulate c-KIT transcription; CCDC26 knockdown-mediated imatinib resistance may be due to c-KIT upregulation. Understanding the precise function of CCDC26 in GIST imatinib resistance would improve the effect of imatinib and prolong the survival of patients with GIST.
Acknowledgements
The authors would like to thank the native English speaking scientists of Elixigen Company (Huntington Beach, California) for editing our manuscript. This work was sponsored by the National Science Foundation for Young Scholars of China [Grant number 81501380], the National Science Foundation of China [Grant number 81372364 and 81573007], The key research plan and social development project of Jiangsu Province, China [Grant number BE2016603], the National ministry of science and technology projects [Grant number 2016YFC0104105] and the Natural Science Foundation for Young Scholars of Jiangsu Province, China [Grant number BK20150110].
Disclosure of conflict of interest
None.
References
- 1.Lim KT, Tan KY. Current research and treatment for gastrointestinal stromal tumors. World J Gastroenterol. 2017;23:4856–4866. doi: 10.3748/wjg.v23.i27.4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirota S, Isozaki K. Pathology of gastrointestinal stromal tumors. Pathol Int. 2006;56:1–9. doi: 10.1111/j.1440-1827.2006.01924.x. [DOI] [PubMed] [Google Scholar]
- 3.Reichardt P, Demetri GD, Gelderblom H, Rutkowski P, Im SA, Gupta S, Kang YK, Schoffski P, Schuette J, Soulieres D, Blay JY, Goldstein D, Fly K, Huang X, Corsaro M, Lechuga MJ, Martini JF, Heinrich MC. Correlation of KIT and PDGFRA mutational status with clinical benefit in patients with gastrointestinal stromal tumor treated with sunitinib in a worldwide treatment-use trial. BMC Cancer. 2016;16:22. doi: 10.1186/s12885-016-2051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schoffski P, Wozniak A, Schoffski O, van Eycken L, Debiec-Rychter M. Overcoming cost implications of mutational analysis in patients with gastrointestinal stromal tumors: a pragmatic approach. Oncol Res Treat. 2016;39:811–816. doi: 10.1159/000453057. [DOI] [PubMed] [Google Scholar]
- 5.Kang W, Zhu C, Yu J, Ye X, Ma Z. KIT gene mutations in gastrointestinal stromal tumor. Front Biosci (Landmark Ed) 2015;20:919–926. doi: 10.2741/4346. [DOI] [PubMed] [Google Scholar]
- 6.Corless CL. Gastrointestinal stromal tumors: what do we know now? Mod Pathol. 2014;27(Suppl 1):S1–16. doi: 10.1038/modpathol.2013.173. [DOI] [PubMed] [Google Scholar]
- 7.Blay JY, Casali PG, Dei Tos AP, Le Cesne A, Reichardt P. Management of gastrointestinal stromal tumour: current practices and visions for the future. Oncology. 2015;89:1–13. doi: 10.1159/000374120. [DOI] [PubMed] [Google Scholar]
- 8.Ksienski D. Imatinib mesylate: past successes and future challenges in the treatment of gastrointestinal stromal tumors. Clin Med Insights Oncol. 2011;5:365–379. doi: 10.4137/CMO.S4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopes LF, Bacchi CE. Imatinib treatment for gastrointestinal stromal tumour (GIST) J Cell Mol Med. 2010;14:42–50. doi: 10.1111/j.1582-4934.2009.00983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khorkova O, Hsiao J, Wahlestedt C. Basic biology and therapeutic implications of lncRNA. Adv Drug Deliv Rev. 2015;87:15–24. doi: 10.1016/j.addr.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li XL, Wu ZQ, Fu XB, Han WD. lncRNAs: Insights into their function and mechanics in underlying disorders. Mutat Res Rev Mutat Res. 2014;762:1–21. doi: 10.1016/j.mrrev.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 12.Rinn JL. lncRNAs: linking RNA to chromatin. Cold Spring Harb Perspect Biol. 2014:6. doi: 10.1101/cshperspect.a018614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochim Biophys Acta. 2014;1839:1097–1109. doi: 10.1016/j.bbagrm.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 14.Yin W, Rossin A, Clifford JL, Gronemeyer H. Co-resistance to retinoic acid and TRAIL by insertion mutagenesis into RAM. Oncogene. 2006;25:3735–3744. doi: 10.1038/sj.onc.1209410. [DOI] [PubMed] [Google Scholar]
- 15.Wu Q, Peng Y, Zhao X. An updated and comprehensive meta-analysis of association between seven hot loci polymorphisms from eight GWAS and glioma risk. Mol Neurobiol. 2016;53:4397–4405. doi: 10.1007/s12035-015-9346-4. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Luo T, Ruan M, Liu P, Wang S, Zhu W. Association of the CCDC26 rs4295627 polymorphism with the risk of glioma: evidence from 7,290 cases and 11,630 controls. Mol Clin Oncol. 2016;4:878–882. doi: 10.3892/mco.2016.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu HW, Huang M, Wang JH, Sun XL, Ke YQ. CCDC26 rs4295627 polymorphism (8q24.21) and glioma risk: a meta-analysis. Genet Mol Res. 2015;14:12074–12084. doi: 10.4238/2015.October.5.20. [DOI] [PubMed] [Google Scholar]
- 18.Hirano T, Yoshikawa R, Harada H, Harada Y, Ishida A, Yamazaki T. Long noncoding RNA, CCDC26, controls myeloid leukemia cell growth through regulation of KIT expression. Mol Cancer. 2015;14:90. doi: 10.1186/s12943-015-0364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng W, Jiang A. Long noncoding RNA CCDC26 as a potential predictor biomarker contributes to tumorigenesis in pancreatic cancer. Biomed Pharmacother. 2016;83:712–717. doi: 10.1016/j.biopha.2016.06.059. [DOI] [PubMed] [Google Scholar]
- 20.Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 21.Cheetham SW, Gruhl F, Mattick JS, Dinger ME. Long noncoding RNAs and the genetics of cancer. Br J Cancer. 2013;108:2419–2425. doi: 10.1038/bjc.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malek E, Jagannathan S, Driscoll JJ. Correlation of long non-coding RNA expression with metastasis, drug resistance and clinical outcome in cancer. Oncotarget. 2014;5:8027–8038. doi: 10.18632/oncotarget.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651–669. doi: 10.1534/genetics.112.146704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, Zhang L, Zhang Y, Zhou F. Increased expression of LncRNA BANCR is associated with clinical progression and poor prognosis in gastric cancer. Biomed Pharmacother. 2015;72:109–112. doi: 10.1016/j.biopha.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Gu W, Gao T, Sun Y, Zheng X, Wang J, Ma J, Hu X, Li J, Hu M. LncRNA expression profile reveals the potential role of lncRNAs in gastric carcinogenesis. Cancer Biomark. 2015;15:249–258. doi: 10.3233/CBM-150460. [DOI] [PubMed] [Google Scholar]
- 26.Zheng HT, Shi DB, Wang YW, Li XX, Xu Y, Tripathi P, Gu WL, Cai GX, Cai SJ. High expression of lncRNA MALAT1 suggests a biomarker of poor prognosis in colorectal cancer. Int J Clin Exp Pathol. 2014;7:3174–3181. [PMC free article] [PubMed] [Google Scholar]
- 27.Shi D, Zheng H, Zhuo C, Peng J, Li D, Xu Y, Li X, Cai G, Cai S. Low expression of novel lncRNA RP11-462C24.1 suggests a biomarker of poor prognosis in colorectal cancer. Med Oncol. 2014;31:31. doi: 10.1007/s12032-014-0031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martens-Uzunova ES, Bottcher R, Croce CM, Jenster G, Visakorpi T, Calin GA. Long noncoding RNA in prostate, bladder, and kidney cancer. Eur Urol. 2014;65:1140–1151. doi: 10.1016/j.eururo.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Sun M, Nie FQ, Wang ZX, De W. Involvement of lncRNA dysregulation in gastric cancer. Histol Histopathol. 2016;31:33–39. doi: 10.14670/HH-11-655. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, Zhou Y, Huang T, Cheng AS, Yu J, Kang W, To KF. The interplay of LncRNA-H19 and its binding partners in physiological process and gastric carcinogenesis. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18020450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan J, Dang Y, Liu S, Zhang Y, Zhang G. LncRNA HOTAIR promotes cisplatin resistance in gastric cancer by targeting miR-126 to activate the PI3K/AKT/MRP1 genes. Tumour Biol. 2016 doi: 10.1007/s13277-016-5448-5. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 32.Huang JK, Ma L, Song WH, Lu BY, Huang YB, Dong HM, Ma XK, Zhu ZZ, Zhou R. LncRNA-MALAT1 promotes angiogenesis of thyroid cancer by modulating tumor-associated macrophage FGF2 protein secretion. J Cell Biochem. 2017;118:4821–4830. doi: 10.1002/jcb.26153. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Song X, Wang X, Hu J, Jiang L. Silencing of LncRNA HULC enhances chemotherapy induced apoptosis in human gastric cancer. J Med Biochem. 2016;35:137–143. doi: 10.1515/jomb-2015-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishida T, Blay JY, Hirota S, Kitagawa Y, Kang YK. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer. 2016;19:3–14. doi: 10.1007/s10120-015-0526-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reid T. Reintroduction of imatinib in GIST. J Gastrointest Cancer. 2013;44:385–392. doi: 10.1007/s12029-013-9532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang YK, Ryu MH, Yoo C, Ryoo BY, Kim HJ, Lee JJ, Nam BH, Ramaiya N, Jagannathan J, Demetri GD. Resumption of imatinib to control metastatic or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib (RIGHT): a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2013;14:1175–1182. doi: 10.1016/S1470-2045(13)70453-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, Debiec-Rychter M. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:4375–4384. doi: 10.1158/1078-0432.CCR-12-0625. [DOI] [PubMed] [Google Scholar]
- 38.Siehl J, Thiel E. C-kit, GIST, and imatinib. Recent Results Cancer Res. 2007;176:145–151. doi: 10.1007/978-3-540-46091-6_12. [DOI] [PubMed] [Google Scholar]
- 39.Zheng S, Huang KE, Pan YL, Zhou Y, Pan SD, Li X, Jia J, Zheng XL, Tao DY. KIT and BRAF heterogeneous mutations in gastrointestinal stromal tumors after secondary imatinib resistance. Gastric Cancer. 2015;18:796–802. doi: 10.1007/s10120-014-0414-7. [DOI] [PubMed] [Google Scholar]
- 40.Hong JL, Li J, Li J, Shen L. [Secondary mutation of c-kit/PDGFRalpha genotypes after imatinib mesylate therapy and its relationship with efficacy of sunitinib] . Zhonghua Bing Li Xue Za Zhi. 2012;41:386–390. doi: 10.3760/cma.j.issn.0529-5807.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 41.Xu CW, Lin S, Wang WL, Gao WB, Lv JY, Gao JS, Zhang LY, Li Y, Wang L, Zhang YP, Tian YW. Analysis of mutation of the c-Kit gene and PDGFRA in gastrointestinal stromal tumors. Exp Ther Med. 2015;10:1045–1051. doi: 10.3892/etm.2015.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]