

Abstract
Chemoresistance in gastric cancer is the leading cause of tumor recurrence and poses a substantial therapeutic challenge. The stem cell biomarker CD133 has been implicated in drug resistance of tumor-initiating cells in a number of cancers including gastric cancer. Therefore, we investigated the molecular mechanism of CD133-associated multidrug resistance in gastric cancer cells. Using CD133 overexpressing and knockdown gastric cancer cell lines, we demonstrated that loss of CD133 significantly increased the growth inhibition of chemotherapeutic agents; whereas, overexpression significantly reduced growth inhibition. Furthermore, CD133 knockdown significantly reduced the enzymatic activity of phosphatidylinositol-3 kinase (PI3K) and the expression of P-glycoprotein (P-gp), B-cell lymphoma 2 (BCL2), and phosphorylated-protein kinase B (p-AKT), but elevated the expression of BCL2 associated X (BAX). Conversely, overexpression of CD133 significantly increased PI3K enzymatic activity, expression of P-gp, BCL2, and p-AKT, and decreased BAX expression. The PI3K/AKT inhibitor LY294002 mirrored the effects of loss of CD133; whereas, the PI3K/AKT activator epidermal growth factor reproduced the effects of CD133 overexpression. To identify the interaction between CD133 and PI3K, we used site-directed mutagenesis to mutate individual tyrosine residues of CD133. We found that binding between CD133 and p85, the regulatory subunit of PI3K, was significantly reduced when tyrosine 852 was mutated. In summary, we have demonstrated that CD133 activates the PI3K/AKT signal transduction pathway through direct interaction with PI3K-p85, resulting in multidrug resistance of gastric cancer cells. These results suggest that the interaction between CD133 and PI3K-p85 may offer a novel therapeutic target in multidrug resistant gastric cancer.
Keywords: Gastric cancer, chemoresistance, CD133, PI3K-p85, phosphorylation
Introduction
Gastric cancer ranks second in terms of cancer-related deaths worldwide. The most effective treatment option is a combination of radical resection and chemotherapy. However, relapse and metastasis are common, and the 5-year survival rate is only 47.0% to 60.1% [1-5]. Tumor-initiating cells show strong self-renewal, proliferation, metastasis, and drug resistance. Innate or acquired drug resistance of tumor cells is the major cause of tumor recurrence, and poses a substantial challenge for traditional chemotherapy and the development of new biological agents [6]. Therefore, understanding the mechanism of multidrug resistance in gastric cancer is essential.
Currently, multidrug resistance involves multiple factors and mechanisms, including members of the B-cell lymphoma 2 (BCL2) family and the resistance-related protein P-glycoprotein (P-gp). Although the multidrug resistance mechanism is widely studied, the key factors underlying the phenomenon remain unclear [7]. Drug resistance and tumor-initiating cells have been associated with the cell surface marker CD133 [8-10]. In addition, CD133 may be associated with resistance to 5-Fluorouracil (5-FU) in gastric cancer cells [11].
CD133 has been shown to promote colon cancer progression by activating the protein kinase B (AKT) signal transduction pathway [14]. AKT is a cell survival signal that promotes drug resistance in many tumor cells [12]. AKT activation can occur through phosphatidylinositol 3-kinases (PI3Ks) that regulate many cellular processes, including apoptosis, via receptor tyrosine kinases [13]. The mechanism by which CD133 activates AKT in tumor-initiating cells remains unclear. CD133 has been shown to be activated by Src tyrosine kinases at intracellular tyrosine residues [15]. Additionally, in glioma stem cells CD133 activates the AKT pathway and promotes tumor formation via interaction with p85, the regulatory subunit of PI3K [16]. Therefore, we investigated the relationship between the phosphorylation of CD133 at inner tyrosine residues and resistance to chemotherapy in gastric cancer. We have identified an active site that offers a potential target to develop therapies against multidrug resistant gastric cancer.
Materials and methods
Chemicals
LY294002, epidermal growth factor (EGF), 5-FU, and cisplatin (DDP) were purchased from Sigma (St. Louis, MO, USA).
Cell lines and cultures
Human gastric cancer cell lines MKN45 and KATO-III were provided by the Shanghai Institute of Cell Biology (Shanghai, China). Cells were maintained in RPMI 1640 culture medium supplemented with 100 g/mL streptomycin, 100 U/mL penicillin, and 10% fetal bovine serum (both from HyClone, Utah, USA) at 37°C in a humidified atmosphere containing 5% carbon dioxide.
Plasmids
To knock down the endogenous CD133 expression, CD133 shRNA lentivirus vectors were generated by ligation of lentivirus vector pGreenPuro-H1 with the oligonucleotide (5’-GATCCGTGTACAGTAAACGGTGTATACTCGAGTATACACCGTTTACTGTACACTTTTTTG-3’).
Ectopic expression of CD133 was determined by designing a pGreenPuro-H1-CD133 plasmid containing full-length human CD133 cDNA spliced into the pGreenPuro-H1 lentivirus vector between BamHI and XhoI sites. CD133 phosphorylation mutants (Y818F, Y819F, Y828F, Y846F, and Y852F) were created using the Vazyme Mut Express® II Fast Mutagenesis kit from Vazyme Biotech (Nanjing, China). Mutant constructs were sequenced, and the correct plasmids were selected for further experiments.
The prokaryotic expression plasmid glutathione S-transferase (GST)-p85 was constructed by inserting the cDNA sequence encoding PI3K-p85 into pGEX-6P-1 prokaryotic expression vector between BamHI and XhoI sites.
Lentivirus construction and transfection
Briefly, using Lipofectamine 2000 reagent (Invitrogen, CA, USA), 293T cells were co-transfected with lentiviral vectors and the packaging vectors pCMV-dR8.2 and Pmd 2.G. Two days later, the supernatant was collected, filtered, concentrated, and used for experiments or frozen at -80°C. Cells were transduced using lentivirus with polybrene (Sigma). Puromycin was used to select for successfully infected cells.
Western blot
Protein lysates were resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA), and immunoblotted with mouse anti-human CD133/1 (1:100; Miltenyi Biotec, Krohne, Germany), P-gp (1:500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), rabbit anti-human p-AKT (Ser473, 1:1,000), AKT (1:1,000), BCL2 (1:1,000), BCL2 associated X (BAX) (1:1,000), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:2,000) (Cell Signaling Technology Inc., Boston, MA, USA). After immunoblotting, lysates were incubated with horseradish peroxidase-labeled goat or mouse anti-rabbit immunoglobulin G secondary antibody (1:2,000; Jackson, Mukilteo, WA, USA) at room temperature. Blots were visualized using enhanced chemiluminescence (Amersham Biosciences Inc., Piscataway, NJ, USA).
Quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from gastric cancer cells with TRIzol reagent (Invitrogen) following the manufacturer’s protocol. qPCR was performed using the isolated RNA with the following primer pairs for CD133: 5’-TTACGGCACTCTCACCT-3’ (forward) and 5’-TATTCCACAAGCAGCAAA-3’ (reverse); multidrug resistance protein 1 (MDR1): 5’-GCTTATGCGAAAGCTGGAGCAGTT-3’ (forward) and 5’-TGGCCGTGATGGCTTTCTTTATGC-3’ (reverse); BCL2: 5’-TTGGATCAGGGAGTT-3’ (forward) and 5’-TGTCCCTACCAACCAGAAGG-3’ (reverse); BAX: 5’-TGTCCCTACCAACCAGAAGG-3’ (forward) and 5’-AGCCACCCTGGTCTTG-3’ (reverse); and GAPDH as the internal control: 5’-ACGGATTTGGTCGTATTGGGCG-3’ (forward) and 5’-CTCCTGGAAGATGGTGATGG-3’ (reverse). Real-time qPCR was performed in triplicate with SYBR Green PCR master mix in Lightcycler 480 (Roche, Basel, Switzerland).
Cell proliferation and cytotoxicity assay
The Cell Counting Kit-8 (CCK-8) (Dojindo, Kyushu, Japan) assay was used to assess cell viability. Cells were seeded on 96-well plates at a concentration of 1 × 104/well and incubated overnight. Cells were exposed to 5-FU or DDP at various concentrations (0, 0.1, 1, 10, 100, and 1,000 μM; dissolved in dimethyl sulfoxide). After 10 μl of CCK-8 solution was added to each well, the plates were incubated for 1 h at 37°C. The absorbance of individual wells was read at 450 nm using a microplate reader (Bio-Rad Laboratories, CA, USA). The sensitivity of tumor cells to 5-FU or DDP was determined using IC50 values (doses that achieve 50% growth inhibition) from the respective dose-response curves.
Enzyme-linked immunosorbent assay (ELISA)
PI3K activity was determined by ELISA (Echelon Biosciences, Utah, USA) according to the manufacturer’s protocol. PI3K activity was estimated by the conversion of phosphatidylinositol 4,5-biphosphate [PI(4,5)P2] lipid to phosphatidylinositol 3,4,5-triphosphate [PI(3,4,5)P3] lipid. Briefly, PI3K-bound protein A Sepharose beads were incubated for 1 to 2 h with PI(4,5)P2 substrate at room temperature in 50 μL of buffer A [20 mM Tris-HCl (pH 7.4), 4 mM MgCl2, 10 mM NaCl, and 25 μM ATP]. The beads were removed by centrifugation, and the supernatant or known concentrations of PI(3,4,5)P3 were incubated for 1 h with 50 μL PI(3,4,5)P3 binding reagent and transferred to a plate coated with PI(3,4,5)P3. A peroxidase-linked secondary detection reagent was used to detect PI(3,4,5)P3 protein binding to the plate. The absorbance was measured at 450 nm, and the amount of PI(3,4,5)P3 generated was calculated from a standard curve using known concentrations of the lipid product.
GST pull-down assay
GST or GST-p85 proteins were purified from Escherichia coli BL21, bound to GST beads, and washed. The bound proteins were incubated with recombinant p85 (generated from E. coli BL21) in binding buffer at 4°C for 4 h. After washing with binding buffer, the pull-down products were subjected to SDS-PAGE and analyzed by Coomassie Brilliant Blue staining or western blot using the following antibodies: rabbit monoclonal anti-PI3K regulatory subunit p85 (Cell Signaling; 1:1,000) and mouse monoclonal anti-CD133 (1:100; Miltenyi Biotec).
Tumor formation assays
The tumor-initiating capacity of MKN45-vector, MKN45-CD133, and MKN45-CD133-Y852 cells was compared by intracranial injection of 50,000 cells into 6- to 8-week-old male nude mice. The tumor volume and weight were measured after 4 weeks when the mice were euthanized. The tumor volume was calculated using the formula: tumor volume = (length × width2)÷2.
Statistical analysis
Statistical analyses were performed using SPSS version 13.0 software (IBM, Chicago, IL, USA). The results were expressed as the mean ± the standard deviation (mean ± SD). Comparisons between groups were performed using one-way analysis of variance (ANOVA). P values less than 0.05 were considered to indicate a statistically significant difference.
Results
CD133 promotes chemoresistance of gastric cancer cells
To determine the role of CD133 in multidrug resistance of gastric cancer, we constructed a CD133 silenced KATO-III cell line and a CD133 overexpressing MKN45 cell line using lentivirus. Under a fluorescence microscope, the majority of cells in each group expressed green fluorescent protein, which was indicative of high transfection efficiency (Figure S1A and S1B). Western blot and qPCR analyses showed that CD133 expression in the CD133 knockdown group was significantly lower than in the control group (P < 0.05). Additionally, CD133 expression in the CD133 overexpressing group was significantly higher than in the control group (P < 0.05) (Figure 1A), suggesting successful generation of CD133 silenced and overexpressing gastric cancer cell lines. Using these cell lines, we sought to determine the role of CD133 in drug resistance of gastric cancer cells. Resistance to increasing concentrations of 5-FU and DDP (0, 0.1, 1, 10, 100, 1000 µM) was assessed by cell viability following treatment. We found that loss of CD133 significantly increased the inhibitory capability of 5-FU and DDP in KATO-III cells with IC50 values of 54.03 ± 0.65 µM vs. 133.30 ± 4.92 µM and 2.31 ± 0.22 µM vs. 24.59 ± 2.16 µM, respectively (P < 0.05). In contrast, CD133 upregulation significantly reduced the inhibitory capability of 5-FU and DDP (P < 0.05) in MKN45 cells with IC50 values of 24.49 ± 2.13 µM vs. 11.62 ± 1.53 µM and 24.49 ± 2.13 µM vs. 11.62 ± 1.53 µM, respectively (Figure 1B and 1C). Interestingly, western blot analysis showed that loss of CD133 in KATO-III cells reduced the protein levels of P-gp and BCL2 and significantly enhanced BAX levels. CD133 overexpression in MKN45 cells significantly increased the expression of P-gp and BCL2 and reduced BAX levels (Figure 1D). MDR1, which encodes P-gp, and BCL2 levels in the CD133 knockdown group were significantly reduced, and the expression of BAX was downregulated (P < 0.05). CD133 overexpression significantly increased the expression of MDR1 and BCL2, and reduced the BAX level (P < 0.05) (Figure 1E).
Figure 1.
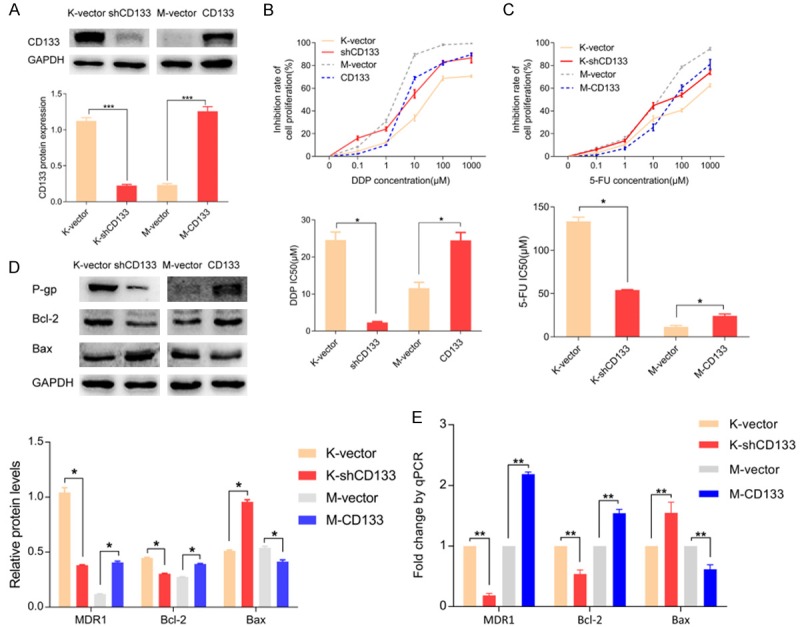
CD133 promotes chemoresistance of gastric cancer cells to 5-FU and DDP. A. CD133 knockdown and overexpressing gastric cancer cells were validated by western blot and qPCR. B, C. Cell viability after treatment with 5-FU or DDP was assessed by cell counting. CD133 knockdown reduced chemoresistance of gastric cancer cells, and CD133 overexpression increased chemoresistance to 5-FU and DDP. D, E. Protein and mRNA expression of P-gp (MDR1), BCL2, and BAX. By regulating the expression of apoptosis-related factors MDR1, BCL2, and BAX, CD133 could control the chemoresistance of gastric cancer cells. *P < 0.05, **P < 0.01, ***P < 0.001.
CD133 activates PI3K/AKT signaling
To study the effect of CD133 on the PI3K/AKT pathway, we assessed activation of the pathway in our CD133 knockdown and overexpressing cell lines. Western blot analysis revealed that p-AKT protein levels were reduced in the CD133 knockdown group and significantly increased in the CD133 overexpressing group compared to their respective controls (Figure 2A). Furthermore, ELISA results showed downregulation of PI3K enzymatic activity in KATO-III cells after CD133 knockdown, whereas CD133 overexpression significantly improved the enzymatic activity of PI3K in MKN45 cells (P < 0.05) (Figure 2B). To further confirm the interaction of CD133 and the PI3K/AKT pathway, we enhanced CD133 expression in KATO-III cells using 10 μmol/L of LY294002, a PI3K/AKT pathway inhibitor. We also induced low expression of CD133 in MKN45 cells with 5 ng/mL EGF, a PI3K/AKT pathway activator. To test the effects of LY294002 and EGF on PI3K enzyme activity, ELISA was performed after exposure to the compounds for 0, 1, 3, 6, 12, or 24 h. The activity of PI3K was reduced to a minimum 3 h after treatment with LY294002 and peaked 1 h after addition of EGF (Figure 2C). Cell viability analysis demonstrated that LY294002 increased the inhibitory response of KATO-III cells to 5-FU and DDP evidenced by IC50 values of 50.05 ± 3.65 µM vs. 143.30 ± 3.52 µM and 12.31 ± 1.23 µM vs. 24.02 ± 3.06 µM, respectively (P < 0.05). In contrast, EGF reduced the inhibitory response of 5-FU and DDP in MKN45 cells, with IC50 values of 34.49 ± 6.13 µM vs. 11.62 ± 1.53 µM and 25.01 ± 2.13 µM vs. 11.30 ± 0.93 µM, respectively (P < 0.05) (Figure 2D and 2E). Western blot showed similar downregulation of p-AKT levels by LY294002 and upregulation of p-AKT with EGF treatment, suggesting that PI3K/AKT signaling was inhibited and activated, respectively (Figure 3A and 3B). Additionally, western blot indicated that LY294002 and EGF regulated the expression of drug resistance and apoptosis-related factors including P-gp, BCL2, and BAX in a time-dependent manner. LY294002 reduced the protein levels of P-gp and BCL2, and increased the expression of BAX. Conversely, EGF addition enhanced the expression of P-gp and BCL2, and suppressed BAX (Figure 3A and 3B).
Figure 2.
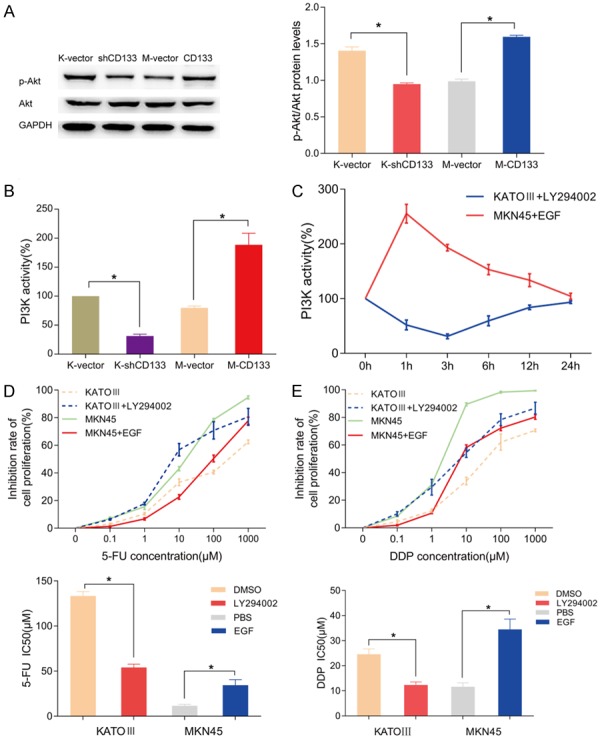
CD133 enhances the chemoresistance capacity of gastric cancer cells by activating the PI3K/AKT signaling pathway. A. Western blot analysis of p-AKT and AKT levels shows that loss of CD133 reduces p-AKT and overexpression of CD133 enhances p-AKT. B. PI3K enzymatic activity was assessed by ELISA. CD133 could significantly activate PI3K enzymatic activity, and this activation was lost with loss of CD133. C. ELISA results indicate that PI3K/AKT activator EGF and inhibitor LY294002 regulate PI3K activity in CD133 high and low expressing gastric cell lines. D, E. Cell viability after treatment with 5-FU or DDP demonstrated that LY294002 and EGF reduce and enhance the chemoresistance of gastric cancer cells in CD133 high and low expressing cells, respectively. *P < 0.05.
Figure 3.
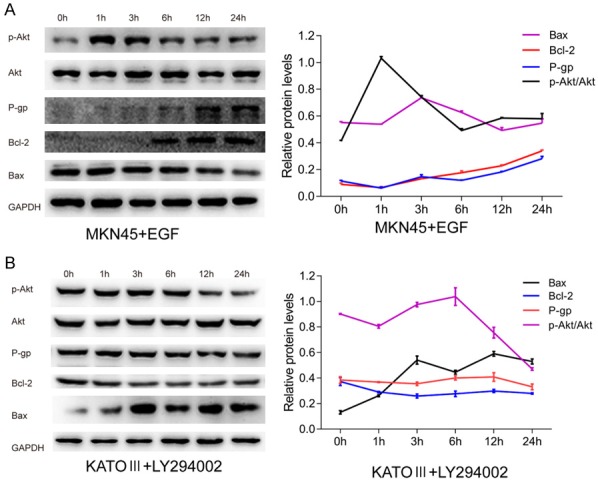
The molecular mechanisms of PI3K/AKT in the promotion of chemoresistance in gastric cancer cells. A, B. Western blot analysis of p-AKT, AKT, P-gp, BCL2, and BAX following treatment with EGF or LY294002. Manipulation of PI3K/AKT pathway activation regulates chemoresistant and apoptosis-related factors in a time-dependent manner.
Carboxyl tyrosine residues of CD133 and gastric cancer drug resistance
To further elucidate the interaction of CD133 with the downstream signaling pathway, we constructed five site-directed mutagenesis vectors of CD133 tyrosine residues 818, 819, 828, 846, and 852. After transfection into MKN45 cells, the IC50 values of 5-FU for the seven groups (vector, wide-type, 818, 819, 828, 846, 852) were assessed by cell viability and found to be 10.62 ± 1.534 μM, 24.49 ± 2.63 μM, 27.82 ± 3.11 μM, 23.11 ± 2.18 μM, 25.24 ± 3.72 μM, 28.82 ± 2.18 μM, and 12.72 ± 2.81 μM, respectively. The IC50 values for DDP were 6.62 ± 1.03 μM, 16.487 ± 2.137 μM, 15.734 ± 2.342 μM, 13.623 ± 1.872 μM, 14.734 ± 2.372 μM, 17.273 ± 2.321 μM, and 7.842 ± 1.734 μM, respectively. The IC50 values of wild-type, 818, 819, 828, and 846 groups were significantly higher than the vector group (all P < 0.05). However, the IC50 of the 852 group showed no significant difference from the vector group (Figure 4A). Using these tyrosine mutant cell lines, we examined PI3K/AKT pathway activation by western blot and ELISA. Results indicated that PI3K enzymatic activity in wild-type, 818, 819, 828, and 846 groups increased significantly (P < 0.05), while the activity of 852 group showed no significant difference compared with the vector group and was lower than other groups. The p-AKT protein level revealed a similar trend (Figure 4B and 4C). Altogether, this data suggests that the tyrosine residue 852 of CD133 may play an important role in the interaction with the downstream PI3K/AKT signaling pathway.
Figure 4.

The role of carboxyl terminal tyrosine residues of CD133 in drug-resistant gastric cancer. A. Cell viability after treatment with 5-FU or DDP in site-directed mutant tyrosine residue MKN45 cell lines. CD133 tyrosine mutants, except for tyrosine 852, significantly enhanced the chemoresistance of gastric cancer cells to 5-FU and DDP. B. Protein analysis of p-AKT and AKT reveals that only tyrosine 852 mutant cells fail to induce p-AKT expression. C. ELISA was performed on CD133 tyrosine mutant cell lines to assess PI3K enzymatic activity. CD133 tyrosine mutants, except for tyrosine 852, enhanced PI3K activity significantly. D. GST pull-down assay was used to determine the interaction of CD133 tyrosine residues with the PI3K regulatory subunit p85. Only the tyrosine 852 mutant impaired the binding of CD133 to PI3K-p85. E. CD133 overexpression could remarkably promote transplantation tumorigenesis in nude mouse, which was assessed by comparing tumor weight and volume. Mutation of CD133 tyrosine 852 significantly reduced both tumor weight and volume. F. A schematic diagram explaining the mechanisms of multidrug resistance in CD133-positive gastric cancer cells.
Interaction of CD133 tyrosine residues and PI3K-p85
To confirm the interaction between CD133 tyrosine residues and PI3K-p85, we constructed a GST-PI3K-p85 prokaryotic expression vector and induced PI3K-p85 protein expression in vitro. After induction by isopropyl ß-D-1-thiogalactopyranoside (IPTG), the harvested bacteria were lysed and total protein was extracted. The expression of GST and GST-p85 was successfully identified by Coomassie Brilliant Blue staining and was enhanced compared to the control group without IPTG. Different concentrations of IPTG (0.2, 0.5, and 1.0 μM) were used for induction, and 0.5 μM was selected for follow-up (Figure S2A). Western blot also showed the successful expression of PI3K-p85 protein (Figure S2B). After purifying the prokaryotic expression protein, Coomassie Brilliant Blue staining indicated that the levels of GST and GST-p85 protein were dramatically increased (Figure S2C). Using these vectors we assessed the interaction between different mutant CD133 proteins and PI3K-p85 by GST pull-down. Results showed that the binding between CD133 protein and PI3K-p85 at tyrosine residue 852 was reduced compared to other groups, suggesting residue 852 may be important for binding efficiency (Figure 4D).
Tumor-initiating capacity of CD133 and CD133 tyrosine residue mutant
Four weeks after MKN45-vector, MKN45-CD133, and MKN45-CD133-Y852 cells were injected into nude mice, all the mice were euthanized. The length, width, and weight of the tumors were measured (Figure S3A and S3B). Results indicated that tumor volume and tumor weight in the protein with mutant residue 852 were significantly smaller than in the CD133 overexpressing group (P < 0.05) (Figure 4E).
Discussion
As one of the most important markers of tumor stem cells, CD133 is widely used in subgroup sorting and identification. Evidence suggests that CD133 regulates tumor biology including drug resistance. El-Khattouti et al. found that CD133-positive melanoma cells showed increased drug resistance to paclitaxel compared with CD133-negative cells [17]. Another study found that CD133 was capable of regulating multidrug resistance of glioma cells [18]. However, studies investigating the role of CD133 in multidrug resistance of gastric cancer are scarce. Our preliminary study showed that the resistance of CD133-positive gastric cells to 5-FU was stronger than that of CD133-negative cells [10]. Several studies have isolated specific subgroups based on CD133 expression. However, exclusion of factors that affect drug resistance in CD133-negative cells is difficult. Techniques such as genetic knockdown and overexpression are needed to exclude compounding factors.
To determine the role of CD133 in multidrug resistance of gastric cancer cells, we selected two gastric cancer cell lines, one with high CD133 expression and one with low CD133 expression, and knocked down or overexpressed CD133 using lentivirus-mediated techniques. CD133 knockdown significantly reduced the drug resistance of gastric cancer cells to 5-FU and DDP, whereas CD133 overexpression enhanced drug resistance. These results are consistent with previous studies involving gliomas and cancers of the liver, thyroid, colon, and other organs [19-22]. Although the chemoresistance of tumor cells is quite complex, factors that may be involved, such as drug resistance-related protein P-gp and apoptosis-related proteins BCL2 and BAX, have been identified. P-gp promotes cellular efflux of drugs, and BCL2 and BAX primarily regulate cellular apoptosis via cytochrome enzyme activity [18,23-26]. Our results indicated that CD133 knockdown significantly reduced the expression of P-gp and BCL2 and increased BAX expression. CD133 overexpression directly contrasted these results. Zhang et al. also showed that P-gp protein was involved in multidrug chemoresistance of gastric cancer cells [27]. Tsuchiya et al. studied the relationship between BCL2 expression and chemotherapy sensitivity in breast cancer, and established that BCL2 was involved in chemoresistance of breast cancer [28]. Other studies [29,30] showed that BAX was also involved in drug resistance and apoptosis of ovarian cancer and multiple myeloma. Altogether these results suggest that CD133-regulated multidrug resistance of gastric cancer cells may be mediated by P-gp, BCL2, and BAX.
PI3K/AKT signaling is widely involved in cell survival and activation, and its role in drug resistance of tumor cells is also well-established [12]. PI3K/AKT signaling is involved in proliferation, migration, and apoptosis of gastric cancer cells [31-33]. PI3Ks are divided into three groups. Class IA PI3Ks (hereafter referred to as PI3K) have been studied widely, and are specifically related to human tumors [34]. PI3K is mutated in many tumors, and is recognized as a diagnostic and therapeutic marker for tumors [35]. Our previous studies showed that Stromal cell-derived factor 1 (SDF1)/ C-X-C chemokine receptor type 4 (CXCR4) regulated the expression of CD133 via PI3K/AKT signaling, and the expression of AKT signaling in CD133-positive gastric cancer cells was greater than in CD133-negative cells [11,36]. To study the relationship between CD133 and the PI3K/AKT signaling pathway, we determined the expression of p-AKT and PI3K enzyme activity after CD133 knockdown and overexpression. Results indicated that after CD133 knockdown, the expression of p-AKT and PI3K activity in gastric cancer cells was significantly reduced, whereas CD133 overexpression yielded the opposite effect. The results are consistent with previous studies, which show that CD133 may regulate the activity of PI3K/AKT signaling pathways.
We used a PI3K/AKT signaling inhibitor, LY294002, and activator, EGF, to culture cells, manipulate the PI3K/AKT pathway, monitor drug resistance, and detect the expression of apoptosis-related factors. We found that LY294002 significantly reduced the drug resistance of KATO-III cells to 5-FU and DDP. EGF significantly enhanced the chemoresistance of MKN45 cells to 5-FU and DDP. We also demonstrated the role of LY294002 and EGF in the inhibition and activation of PI3K/AKT signaling over time. Furthermore, we tested the expression of chemoresistance and apoptosis-related factors. We found that LY294002 significantly reduced the expression of chemoresistance-related factor P-gp and apoptosis inhibitor BCL2, and increased the expression of apoptotic factor BAX. EGF yielded contrasting results, which were consistent with the other studies [37,38]. These studies suggest that CD133 may regulate the expression of chemoresistance and apoptosis-related factors by activating the PI3K/AKT signaling pathway, and ultimately promote multidrug resistance in gastric cancer cells. However, the mechanism of CD133-mediated activation of downstream AKT signaling via interaction with PI3K needs to be elucidated.
A recent study showed that CD133 is a transmembrane protein [39]. The asparagine residue 548 at its extracellular N-terminal plays an important role in the proliferation of human hepatoma cells upon stimulation. Another study showed that phosphorylation of tyrosine residues 828 and 852 on the intracellular C-terminal of CD133 was involved in the activation of downstream signaling pathways [14-16]. In this study we constructed vectors with five mutant tyrosine residues located on the intracellular C-terminal of CD133 using site-directed mutagenesis. We found that the tyrosine 852 mutation of CD133 significantly reduced drug resistance in gastric cancer cells exposed to 5-FU and DDP compared to other groups. Furthermore, PI3K enzymatic activity and AKT phosphorylation were inhibited by the tyrosine 852 mutation of CD133. Thus, the phosphorylation of tyrosine 852 at the CD133 carboxyl terminal is a key molecular event that activates PI3K/AKT signaling and promotes chemoresistance of gastric cancer cells.
PI3K is composed of a regulatory subunit (p85) and a catalytic subunit (p110). The heterodimeric p85/p110 is recruited to receptor tyrosine residues on the cell membrane and is activated by either phosphorylation or binding of adaptor proteins to the SH2 domain of p85 [13,40-44]. As a key molecular target of PI3K, p85 plays an important role in the activation of the PI3K/AKT signaling pathway. Moreover, p85 is often mutated in solid tumors, and inhibition of p85 expression suppresses the proliferation and migration of tumor cells [45-48].
CD133 may activate the PI3K/AKT pathway via direct interaction with PI3K-p85. We constructed a GST-p85 prokaryotic expression plasmid, and used a GST pull-down method to elucidate the interaction between CD133 and PI3K-p85. GST pull-down showed that the binding between CD133 and PI3K-p85 might be mediated via the CD133 tyrosine 852 residue. The altered residue 852 also inhibited tumor formation in nude mice, which suggests that it may inhibit proliferation of gastric cancer cells. These results indicate that tyrosine 852 of CD133 may mediate chemoresistance in gastric cancer through activation of PI3K/AKT signaling. Our findings are similar to that of Wei et al. [15], although they involve different active sites and different tumors. Additional studies are needed to confirm the specific mechanism.
Overall, this study demonstrated that CD133 activated the PI3K/AKT signaling pathway via direct interaction with PI3K-p85 and regulated drug resistance factors, apoptosis-related factors, and multidrug resistance in gastric cancer. Importantly, the tyrosine 852 mutation of CD133 inhibited the development of drug resistance, and suggests a possible model for CD133-mediated multidrug resistance in gastric cancer (Figure 4F). Therefore, CD133 substructure and PI3K-p85 offer new possibilities for targeted therapy of cancer stem cells and resistant gastric cancer.
Acknowledgements
This work was sponsored by the Grants of the Shanghai Municipal Health Bureau Foundation of China (grant no. 201540202).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Yasui W, Sentani K, Sakamoto N, Anami K, Naito Y, Oue N. Molecular pathology of gastric cancer: research and practice. Pathol Res Pract. 2011;207:608–612. doi: 10.1016/j.prp.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Rocco A, Compare D, Nardone G. Cancer stem cell hypothesis and gastric carcinogenesis: experimental evidence and unsolved questions. World J Gastrointest Oncol. 2012;4:54–59. doi: 10.4251/wjgo.v4.i3.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao P, Li Y, Lu Y. Aberrant expression of CD133 protein correlates with Ki-67 expression and is a prognostic marker in gastric adenocarcinoma. BMC Cancer. 2010;10:218. doi: 10.1186/1471-2407-10-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samson PS, Escovidal LA, Yrastorza SG, Veneracion RG, Nerves MY. Re-study of gastric cancer: analysis of outcome. World J Surg. 2002;26:428–433. doi: 10.1007/s00268-001-0243-9. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Liu L, Zhang Y, Guan H, Wu J, Zhu X, Yuan J, Li M. MiR-503 targets PI3K p85 and IKK-beta and suppresses progression of non-small cell lung cancer. Int J Cancer. 2014;135:1531–1542. doi: 10.1002/ijc.28799. [DOI] [PubMed] [Google Scholar]
- 6.Broxterman HJ, Gotink KJ, Verheul HM. Understanding the causes of multidrug resistance in cancer: a comparison of doxorubicin and sunitinib. Drug Resist Updat. 2009;12:114–126. doi: 10.1016/j.drup.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Baguley BC. Multiple drug resistance mechanisms in cancer. Mol Biotechnol. 2010;46:308–316. doi: 10.1007/s12033-010-9321-2. [DOI] [PubMed] [Google Scholar]
- 8.Lee HH, Seo KJ, An CH, Kim JS, Jeon HM. CD133 expression is correlated with chemoresistance and early recurrence of gastric cancer. J Surg Oncol. 2012;106:999–1004. doi: 10.1002/jso.23178. [DOI] [PubMed] [Google Scholar]
- 9.Kim KH, Yoo BC, Kim WK, Hong JP, Kim K, Song EY, Lee JY, Cho JY, Ku JL. CD133 and CD133-regulated nucleophosmin linked to 5-fluorouracil susceptibility in human colon cancer cell line SW620. Electrophoresis. 2014;35:522–532. doi: 10.1002/elps.201300364. [DOI] [PubMed] [Google Scholar]
- 10.Piao LS, Hur W, Kim TK, Hong SW, Kim SW, Choi JE, Sung PS, Song MJ, Lee BC, Hwang D, Yoon SK. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012;315:129–137. doi: 10.1016/j.canlet.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Yu J, Wang S, Lu R, Wu J, Jiang B. Overexpression of CD133 enhances chemoresistance to 5-fluorouracil by activating the PI3K/AKT/p70S6K pathway in gastric cancer cells. Oncol Rep. 2014;32:2437–2444. doi: 10.3892/or.2014.3488. [DOI] [PubMed] [Google Scholar]
- 12.Shimamura H, Terada Y, Okado T, Tanaka H, Inoshita S, Sasaki S. The PI3-kinase-AKT pathway promotes mesangial cell survival and inhibits apoptosis in vitro via NF-kappa B and Bad. J Am Soc Nephrol. 2003;14:1427–1434. doi: 10.1097/01.asn.0000066140.99610.32. [DOI] [PubMed] [Google Scholar]
- 13.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 14.Shimozato O, Waraya M, Nakashima K, Souda H, Takiguchi N, Yamamoto H, Takenobu H, Uehara H, Ikeda E, Matsushita S, Kubo N, Nakagawara A, Ozaki T, Kamijo T. Receptor-type protein tyrosine phosphatase kappa directly dephosphorylates CD133 and regulates downstream AKT activation. Oncogene. 2015;34:1949–60. doi: 10.1038/onc.2014.141. [DOI] [PubMed] [Google Scholar]
- 15.Boivin D, Labbe D, Fontaine N, Lamy S, Beaulieu E, Gingras D, Beliveau R. The stem cell marker CD133 (prominin-1) is phosphorylated on cytoplasmic tyrosine-828 and tyrosine-852 by Src and Fyn tyrosine kinases. Biochemistry. 2009;48:3998–4007. doi: 10.1021/bi900159d. [DOI] [PubMed] [Google Scholar]
- 16.Wei Y, Jiang Y, Zou F, Liu Y, Wang S, Xu N, Xu W, Cui C, Xing Y, Liu Y, Cao B, Liu C, Wu G, Ao H, Zhang X, Jiang J. Activation of PI3K/AKT pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad Sci U S A. 2013;110:6829–6834. doi: 10.1073/pnas.1217002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Khattouti A, Selimovic D, Haikel Y, Megahed M, Gomez CR, Hassan M. Identification and analysis of CD133(+) melanoma stem-like cells conferring resistance to taxol: an insight into the mechanisms of their resistance and response. Cancer Lett. 2014;343:123–133. doi: 10.1016/j.canlet.2013.09.024. [DOI] [PubMed] [Google Scholar]
- 18.Xi G, Hayes E, Lewis R, Ichi S, Mania-Farnell B, Shim K, Takao T, Allender E, Mayanil CS, Tomita T. CD133 and DNA-PK regulate MDR1 via the PI3K- or AKT-NF-kappaB pathway in multidrug-resistant glioblastoma cells in vitro. Oncogene. 2016;35:241–250. doi: 10.1038/onc.2015.78. [DOI] [PubMed] [Google Scholar]
- 19.Zhai B, Zhang X, Sun B, Cao L, Zhao L, Li J, Ge N, Chen L, Qian H, Yin Z. MK2206 overcomes the resistance of human liver cancer stem cells to sorafenib by inhibition of pAKT and upregulation of pERK. Tumour Biol. 2016;37:8047–55. doi: 10.1007/s13277-015-4707-1. [DOI] [PubMed] [Google Scholar]
- 20.Angelastro JM, Lame MW. Overexpression of CD133 promotes drug resistance in C6 glioma cells. Mol Cancer Res. 2010;8:1105–1115. doi: 10.1158/1541-7786.MCR-09-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kucerova L, Feketeova L, Kozovska Z, Poturnajova M, Matuskova M, Nencka R, Babal P. In vivo 5FU-exposed human medullary thyroid carcinoma cells contain a chemoresistant CD133+ tumor-initiating cell subset. Thyroid. 2014;24:520–532. doi: 10.1089/thy.2013.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G, Medema JP, Stassi G. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 23.Kapse-Mistry S, Govender T, Srivastava R, Yergeri M. Nanodrug delivery in reversing multidrug resistance in cancer cells. Front Pharmacol. 2014;5:159. doi: 10.3389/fphar.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moitra K. Overcoming multidrug resistance in cancer stem cells. Biomed Res Int. 2015;2015:635745. doi: 10.1155/2015/635745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marin JJ, Al-Abdulla R, Lozano E, Briz O, Bujanda L, Banales JM, Macias RI. Mechanisms of resistance to chemotherapy in gastric cancer. Anticancer Agents Med Chem. 2016;16:318–334. doi: 10.2174/1871520615666150803125121. [DOI] [PubMed] [Google Scholar]
- 26.Han Z, Hong L, Han Y, Wu K, Han S, Shen H, Li C, Yao L, Qiao T, Fan D. Phospho AKT mediates multidrug resistance of gastric cancer cells through regulation of P-gp, BCL2 and BAX. J Exp Clin Cancer Res. 2007;26:261–268. [PubMed] [Google Scholar]
- 27.Zhang D, Fan D. Multidrug resistance in gastric cancer: recent research advances and ongoing therapeutic challenges. Expert Rev Anticancer Ther. 2007;7:1369–1378. doi: 10.1586/14737140.7.10.1369. [DOI] [PubMed] [Google Scholar]
- 28.Tsuchiya M, Nakajima Y, Waku T, Hiyoshi H, Morishita T, Furumai R, Hayashi Y, Kishimoto H, Kimura K, Yanagisawa J. CHIP buffers heterogeneous BCL2 expression levels to prevent augmentation of anticancer drug-resistant cell population. Oncogene. 2015;34:4656–4663. doi: 10.1038/onc.2014.387. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Gao Y, Lu Y, Zhang J, Li L, Yin F. Oncogenes associated with drug resistance in ovarian cancer. J Cancer Res Clin Oncol. 2015;141:381–395. doi: 10.1007/s00432-014-1765-5. [DOI] [PubMed] [Google Scholar]
- 30.Zhao JJ, Chu ZB, Hu Y, Lin J, Wang Z, Jiang M, Chen M, Wang X, Kang Y, Zhou Y, Chonghaile TN, Johncilla ME, Tai YT, Cheng JQ, Letai A, Munshi NC, Anderson KC, Carrasco RD. Targeting the miR-221-222/PUMA/BAK/BAX pathway abrogates dexamethasone resistance in multiple myeloma. Cancer Res. 2015;75:4384–4397. doi: 10.1158/0008-5472.CAN-15-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu W, Chen GS, Shao Y, Li XL, Xu HC, Zhang H, Zhu GQ, Zhou YC, He XP, Sun WH. Gastrin acting on the cholecystokinin2 receptor induces cyclooxygenase-2 expression through JAK2/STAT3/PI3K/AKT pathway in human gastric cancer cells. Cancer Lett. 2013;332:11–18. doi: 10.1016/j.canlet.2012.12.030. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Zhang Y, Xu R, Du J, Hu Z, Yang L, Chen Y, Zhu Y, Gu L. PI3K/AKT-dependent phosphorylation of GSK3beta and activation of RhoA regulate Wnt5a-induced gastric cancer cell migration. Cell Signal. 2013;25:447–456. doi: 10.1016/j.cellsig.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Cao W, Yang W, Fan R, Li H, Jiang J, Geng M, Jin Y, Wu Y. miR-34a regulates cisplatininduce gastric cancer cell death by modulating PI3K/AKT/survivin pathway. Tumour Biol. 2014;35:1287–1295. doi: 10.1007/s13277-013-1171-7. [DOI] [PubMed] [Google Scholar]
- 34.Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci. 2005;30:194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Knapp S, Ahmed AA. The structural basis of PI3K cancer mutations: from mechanism to therapy. Cancer Res. 2014;74:641–646. doi: 10.1158/0008-5472.CAN-13-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang HG, Lu RQ, Wu JG, Zhou GC, Yu JW, Jiang BJ. Role of stromal cell derived factor/CXC chemokine receptor axis in regulation of expression of CD133 in gastric cancer cells via PI3K/AKT pathway in vitro. Chinese Journal of Experimental Surgery. 2012;29:378–380. [Google Scholar]
- 37.Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB. Inhibition of phosphatidylinositol 3’-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res. 2002;62:1087–1092. [PubMed] [Google Scholar]
- 38.Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010;28:1075–1083. doi: 10.1200/JCO.2009.25.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Ren S, Xie L, Cui C, Xing Y, Liu C, Cao B, Yang F, Li Y, Chen X, Wei Y, Lu H, Jiang J. Mutation of N-linked glycosylation at Asn548 in CD133 decreases its ability to promote hepatoma cell growth. Oncotarget. 2015;6:20650–20660. doi: 10.18632/oncotarget.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Zazzo E, Feola A, Zuchegna C, Romano A, Donini CF, Bartollino S, Costagliola C, Frunzio R, Laccetti P, Di Domenico M, Porcellini A. The p85 regulatory subunit of PI3K mediates cAMP-PKA and insulin biological effects on MCF-7 cell growth and motility. ScientificWorldJournal. 2014;2014:565839. doi: 10.1155/2014/565839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Breitkopf SB, Yang X, Begley MJ, Kulkarni M, Chiu YH, Turke AB, Lauriol J, Yuan M, Qi J, Engelman JA, Hong P, Kontaridis MI, Cantley LC, Perrimon N, Asara JM. A cross-species study of PI3K protein-protein interactions reveals the direct interaction of P85 and SHP2. Sci Rep. 2016;6:20471. doi: 10.1038/srep20471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JY, Chiu YH, Asara J, Cantley LC. Inhibition of PI3K binding to activators by serine phosphorylation of PI3K regulatory subunit p85alpha Src homology-2 domains. Proc Natl Acad Sci U S A. 2011;108:14157–14162. doi: 10.1073/pnas.1107747108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Comb WC, Hutti JE, Cogswell P, Cantley LC, Baldwin AS. p85alpha SH2 domain phosphorylation by IKK promotes feedback inhibition of PI3K and AKT in response to cellular starvation. Mol Cell. 2012;45:719–730. doi: 10.1016/j.molcel.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hofmann BT, Jucker M. Activation of PI3K/AKT signaling by n-terminal SH2 domain mutants of the p85alpha regulatory subunit of PI3K is enhanced by deletion of its c-terminal SH2 domain. Cell Signal. 2012;24:1950–1954. doi: 10.1016/j.cellsig.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring P, Dela Vega T, Kenski DM, Bowman KK, Lorenzo M, Li H, Wu J, Modrusan Z, Stinson J, Eby M, Yue P, Kaminker JS, de Sauvage FJ, Backer JM, Seshagiri S. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16:463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Folgiero V, Di Carlo SE, Bon G, Spugnini EP, Di Benedetto A, Germoni S, Pia Gentileschi M, Accardo A, Milella M, Morelli G, Bossi G, Mottolese M, Falcioni R. Inhibition of p85, the non-catalytic subunit of phosphatidylinositol 3-kinase, exerts potent antitumor activity in human breast cancer cells. Cell Death Dis. 2012;3:e440. doi: 10.1038/cddis.2012.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun M, Hillmann P, Hofmann BT, Hart JR, Vogt PK. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc Natl Acad Sci U S A. 2010;107:15547–15552. doi: 10.1073/pnas.1009652107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feola A, Cimini A, Migliucci F, Iorio R, Zuchegna C, Rothenberger R, Cito L, Porcellini A, Unteregger G, Tombolini V, Giordano A, Di Domenico M. The inhibition of p85alphaPI3KSer83 phosphorylation prevents cell proliferation and invasion in prostate cancer cells. J Cell Biochem. 2013;114:2114–2119. doi: 10.1002/jcb.24558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.