

ABSTRACT
Herpes simplex virus 1 (HSV-1) establishes latent infection in neurons via a variety of epigenetic mechanisms that silence its genome. The cellular CCCTC-binding factor (CTCF) functions as a mediator of transcriptional control and chromatin organization and has binding sites in the HSV-1 genome. We constructed an HSV-1 deletion mutant that lacked a pair of CTCF-binding sites (CTRL2) within the latency-associated transcript (LAT) coding sequences and found that loss of these CTCF-binding sites did not alter lytic replication or levels of establishment of latent infection, but their deletion reduced the ability of the virus to reactivate from latent infection. We also observed increased heterochromatin modifications on viral chromatin over the LAT promoter and intron. We therefore propose that CTCF binding at the CTRL2 sites acts as a chromatin insulator to keep viral chromatin in a form that is poised for reactivation, a state which we call poised latency.
KEYWORDS: chromatin, epigenetics, herpes simplex virus, latent infection, regulation of gene expression
IMPORTANCE
Herpes simplex virus 1 (HSV-1) is a human pathogen that persists for the lifetime of the host as a result of its ability to establish latent infection within sensory neurons. The mechanism by which HSV-1 transitions from the lytic to latent infection program is largely unknown; however, HSV-1 is able to coopt cellular silencing mechanisms to facilitate the suppression of lytic gene expression. Here, we demonstrate that the cellular CCCTC-binding factor (CTCF)-binding site within the latency associated transcript (LAT) region is critical for the maintenance of a specific local chromatin structure. Additionally, loss of CTCF binding has detrimental effects on the ability to reactivate from latent infection. These results argue that CTCF plays a critical role in epigenetic regulation of viral gene expression to establish and/or maintain a form of latent infection that can reactivate efficiently.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) undergoes a lytic infection cycle at the primary mucosal site of infection, expresses approximately 80 lytic genes, and then spreads to sensory neurons, where it establishes a latent infection and expresses a minimal number of viral genes (1). Viral gene products recruit host epigenetic complexes to regulate the viral genome during lytic and latent infection (2, 3). HSV-1 persists as a latent infection in sensory ganglia, during which lytic genes are epigenetically silenced, and the only viral gene products expressed abundantly are a family of noncoding RNAs known as the latency-associated transcripts (LATs) and microRNAs (miRNAs) (4–8). The LAT gene is transcribed to yield a primary 8.3-kb transcript from which stable 1.5- and 2.0-kb introns and a number of miRNAs are processed (6–9). The LATs promote gene silencing and increased heterochromatin at lytic genes and have been associated with a reduction in lytic gene transcripts in both acute and latent infections of neurons (10–14). In addition, LAT-dependent gene repression during latent infection in a mouse model has been implicated in promoting neuronal survival and suppressing reactivation (15). No strong evidence for proteins encoded by the LAT genes has been found (16); however, long noncoding RNAs in other systems are known to mediate assembly of heterochromatin and maintenance through both direct and indirect mechanisms (16). In transfection assays, miRNAs originating from the LAT region can also repress expression of viral lytic proteins (6, 17–19).
During establishment of latency, lytic gene expression and viral replication are repressed and the viral genome progressively accumulates histones and heterochromatin on lytic genes (20, 21). Viral lytic genes are associated with histones that are hypoacetylated and enriched for markers of heterochromatin, such as histone H3 lysine 9 trimethyl (H3K9me3), which is a hallmark of constitutive heterochromatin, and H3 lysine 27 trimethyl (H3K27me3), which is a hallmark of facultative heterochromatin (9, 20–23). Elements of the LAT transcriptional unit, which include upstream regulatory sequences, a neuron-specific promoter, and a downstream enhancer, appear to be the exception to this chromatin phenotype (22–28). The LAT gene is the only viral region known to be enriched for acetylated histones and other markers of active euchromatin, while also maintaining association with markers of heterochromatin (22, 23). Upon reactivation in vitro, this pattern is reversed, with lytic genes associating increasingly with acetylated histones and markers of euchromatin and accumulating transcripts, while the LAT gene exhibits a corresponding decrease in euchromatin and transcript levels (29–32). Chromatin control of viral lytic gene expression is therefore thought to act as a regulator of the transitions between lytic infection, latent infection, and reactivation.
Transcribed from the strand antisense to the LAT gene in each of the long component repeats is the ICP0 gene (Fig. 1), which encodes an immediate-early (IE) protein that serves many functions to promote lytic infection, including the transactivation of viral genes and repression of the innate immune system and intrinsic resistance (33–39). ICP0 counters host-mediated chromatin silencing, intrinsic resistance, and innate immune responses through several mechanisms, including the degradation of promyelocytic leukemia (PML) protein in nuclear domain 10 (ND10) bodies (40), degradation of interferon-inducible protein 16 (IFI16) (36), and inhibition of the histone deacetylase (HDAC) RE1-silencing transcription factor (REST) corepressor to REST (CoREST)-HDAC repressor complex (41, 42). In latently infected neuronal populations, ICP0 is largely repressed despite its proximity to the LAT enhancer sequences and abundant expression from the adjacent LAT region (43); however, we recently found that ICP0 promotes LAT expression and latent infection (44).
FIG 1 .

Map of the LAT transcriptional unit of an HSV-1 viral mutant with a deletion of the CTCF-binding sites from the LAT intron sequences. (A) Schematic map of the HSV-1 genome with an expanded view showing the LAT coding region, with the LAT promoter (LAP), LAT enhancer (LTE), CTCF-binding sites (CTRL2), and ICP0 promoter (ICP0 P). Restriction endonuclease cleavage sites (H, HpaI; B, BamHI) used to generate the ΔCTRL2 virus are indicated. (B) The locations of qPCR primers are indicated as open arrows connected by dashed lines: I, LAP; II, LAT intron; III, ICP0 P. (C) Locations of the primary LAT, stable 2.0-kbp LAT intron, ICP0 transcript, and miRNAs.
The host factors regulating latent viral chromatin have not been well defined. One candidate, CCCTC-binding factor (CTCF), is an 11 zinc-finger DNA-binding protein that is essential, ubiquitously expressed, and highly conserved among metazoan species (45, 46). CTCF was initially classified as a transcriptional repressor with the ability to bind diverse DNA target sequences (45–48) and was later recognized as a transcriptional activator as well as an enhancer blocker and insulator (49–51). Consequently, CTCF emerged as an important chromatin regulator responsible for a diverse range of activities, including affecting pausing of RNA polymerase II (RNAPII) and RNA splicing, directing the specific positioning and phasing of nucleosomes (52, 53), and generating chromatin loops and mediating long-range chromatin interactions through manipulation of the chromatin three-dimensional (3D) architecture (54–56).
Importantly, CTCF is the only identified vertebrate insulator protein. As such, it acts as a boundary element to isolate adjacent domains of active and inactive chromatin by directing enhancer activity to prevent interaction with nearby but inappropriate promoters and by blocking the linear spread of heterochromatin (51, 57–59). Insulators are crucial in the spatial organization of complex genomes of both metazoan species and herpesviruses (60–63), throughout which essential active regions of transcription are frequently interspersed with silenced domains.
CTCF-binding sites have been identified in a number of herpesviruses and are associated with functions such as regulating latent gene expression in two gammaherpesviruses, Epstein-Barr virus (EBV) (60) and Kaposi’s sarcoma-associated herpesvirus (KSHV) (61, 62), as well as regulating expression of the major immediate-early gene in the betaherpesvirus human cytomegalovirus (HCMV) (63). A number of CTCF-binding sites have been identified in the HSV-1 genome, including the CTRL2 sites, which are located between the LAT and ICP0 promoter regions in the terminal repeats of the long component (Fig. 1) (64, 65). CTCF binds at the CTRL2 sites during latent infection but is lost during reactivation (66), and this site has been hypothesized to regulate distinct expression from the adjacent genetic elements (64–66). Additionally, in transfection assays, CTRL2 is capable of enhancer blocking, silencing, and prevention of heterochromatin spreading (64, 65). Indirect evidence also suggests that removal of CTCF-binding sites can alter gene expression from lytic promoters in cell culture latency models (67). Interestingly, in lytic infection, CTCF is not detected at CTRL2 or other latency-associated binding sites; however, CTCF does bind extensively to other regions of the viral genome and may promote viral transcription and prevent epigenetic silencing (68).
We were therefore interested in determining whether the presence of CTCF binding at the LAT intron is critical for establishing and/or maintaining the latent gene expression pattern and chromatin structure in the LAT region during latency. In this study, we eliminated the CTRL2 sites from HSV-1 to assess CTCF function in a mouse model system of latency. We found that the CTRL2 sites are essential for maintaining the chromatin at the LAT and ICP0 regions that promotes efficient reactivation.
RESULTS
Construction of an HSV-1 CTRL2 binding site deletion mutant virus.
Previous studies identified the CTRL2 DNA elements within the HSV-1 genome and demonstrated that CTRL2 was able to bind CTCF both in vitro and in vivo (64–66) and function as an insulator capable of blocking enhancer-promoter interactions (64) and the spread of heterochromatin in transfection assays (65). To test whether the CTRL2 region has specific effects on in vivo HSV-1 infection, we constructed a CTRL2 deletion mutant virus by removing a 370-bp fragment from within the 2.0-kbp LAT intron to generate the HSV-1 ΔCTRL2 mutant virus (Fig. 1). A control virus, called CTRL2R, was constructed in parallel by restoring an intact LAT-CTRL2 region in KOSΔLAT1.8eGFP virus, the parent of ΔCTRL2 mutant virus.
Growth kinetics in human foreskin fibroblast (HFF) and HeLa cells following infection at a low multiplicity of infection (0.1) confirmed that there were no differences in lytic replication between the ΔCTRL2 mutant and control CTRL2R viruses (Fig. 2).
FIG 2 .
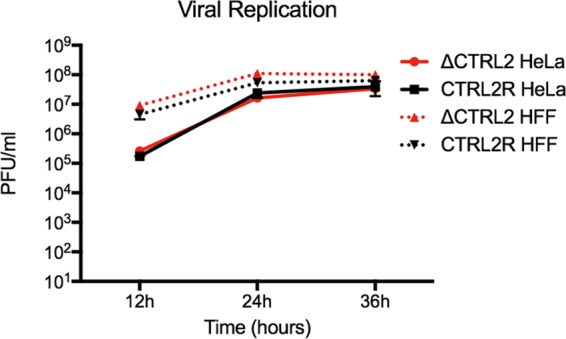
Analysis of ΔCTRL2 and CTRL2R lytic viral replication. HeLa or HFF cells were infected at 0.1 PFU/cell with ΔCTRL2 or CTRL2R virus. Progeny virus was collected at 12, 24, and 36 hours postinfection and titrated on Vero cells.
Analysis of CTCF binding to the ΔCTRL2 mutant and rescued viral genomes.
To confirm the loss of CTCF binding during in vivo infection due to deletion of the CTRL2 site and to validate the mutant virus, we infected mice via corneal scarification with 2 × 105 PFU/eye of ΔCTRL2 or CTRL2R virus and performed chromatin immunoprecipitation (ChIP) for CTCF. At 28 days postinfection (dpi), trigeminal ganglia (TGs) were removed and processed for ChIP analysis with a CTCF-specific antibody, and we quantified the immunoprecipitated DNA sequences by using quantitative PCR (qPCR) with primers specific to the LAT promoter, LAT intron, and ICP0 promoter (Fig. 3). ChIP analysis performed in parallel with a nonspecific rabbit IgG confirmed specificity. Statistical analyses were performed on results from 7 independent ChIP experiments from 4 independent infections, using Friedman’s test with Dunn’s post hoc test to account for multiple comparisons. Consistent with the absence of putative CTCF-binding sites, CTCF was not enriched at the LAT promoter (LAP) or at the cellular GAPDH pseudogene relative to the nonspecific antibody control upon infection with either ΔCTRL2 or CTRL2R virus (Fig. 3A and D). Comparison of the ΔCTRL2 mutant viral chromatin to control CTRL2R viral genome showed a significant decrease in CTCF binding at the LAT intron based on ChIP using primers specific to a region less than 500 bp downstream of the CTRL2 site (P ≤ 0.003) (Fig. 3B). Interestingly, ΔCTRL2 viral genomes also showed a significant reduction of CTCF binding within the ICP0 promoter region (P ≤ 0.05) (Fig. 3C). CTCF enrichment at the downstream ICP0 promoter site was consistent with a potential CTCF-binding site within the ICP0 promoter region or detection of CTCF binding to another CTCF-binding site, such as the CTa′m site, which is ~1.5 kbp away from CTRL2 (64). Collectively, these results demonstrated that deletion of the CTRL2 sequences in the ΔCTRL2 virus reduced CTCF association at the LAT intron sequences and at the ICP0 promoter region relative to that with the control CTRL2R virus.
FIG 3 .

Comparison of CTCF association at the LAT region during latent infection with ΔCTRL2 or CTRL2R virus in mice. Mice were infected with ΔCTRL2 or CTRL2R virus. At 28 dpi, the mice were sacrificed, and ChIP experiments were carried out on harvested TGs by using antibodies specific for CTCF. Three viral regions were queried: LAP (A), the LAT intron (B), and the ICP0 promoter (C). The cellular GAPDH sequences (D) were also analyzed. Percentages of immunoprecipitated DNA are shown as means and standard deviations from 7 ChIP experiments from 4 independent infections. Statistical significance was evaluated using Friedman’s test along with Dunn’s posttests for ΔCTRL2 versus CTRL2R (controlling for multiple comparisons); significant P values are shown on top of the brackets.
Acute infection of mice with ΔCTRL2 and CTRL2R viruses.
We first examined acute infection in mice infected with the ΔCTRL2 and CTRL2R viruses by collecting shed virus with eye swabs for the initial 5 dpi and titrating the virus on Vero cells. We observed similar levels of viral shedding from mice infected with ΔCTRL2 and CTRL2R viruses at days 1 to 4; however, at day 5, virus levels collected from mice infected with ΔCTRL2 virus were slightly but significantly lower than in mice infected with CTRL2R (P ≤ 0.005) (Fig. 4A). To allow establishment of latent infection, we maintained infected mice for 28 days. For CTRL2R-infected mice, 90% of the mice survived to day 28 and generally did not succumb to infection before day 10. In contrast, for ΔCTRL2-infected mice, we observed slightly increased mortality beginning at day 7, with 15% fatality by day 10 and 19% fatality by day 28 (P ≤ 0.05 by log-rank test) (Fig. 4B). In general, acute viral replication and survival were largely similar for the mice infected with mutant or restored mutant viruses.
FIG 4 .

Eye swab titers and survival of mice infected with ΔCTRL2 or CTRL2R virus. Mice were infected with 2 × 105 PFU/eye of ΔCTRL2 or CTRL2R virus. (A) Viral replication in corneal epithelia. Eye swabs were collected daily at 1 to 5 dpi from 5 mice infected with each virus, from 4 independent infections. Collected virus was titrated on Vero cells, and the results are plotted as mean and standard errors for all infections. Statistical significance was evaluated using Student’s t test and is indicated with an asterisk (P < 0.05). (B) Infections were allowed to progress for 28 days, and survival curves are presented from 8 independent infections of groups of 5 to 20 mice per virus (infected in parallel in each infection group). Statistical significance was evaluated with the log-rank Mantel-Cox test.
Trigeminal ganglion infection with ΔCTRL2 and CTRL2R viruses.
To test whether the ΔCTRL2 mutation affected the viral genome load in the ganglia during acute or latent infection, we isolated total ganglion DNA and quantified viral genomes at 7 and 30 dpi from TGs of mice infected with ΔCTRL2 or CTRL2R virus. Viral DNA levels, when normalized to cellular DNA levels, showed no significant difference in genomes per TG for ganglia acutely infected with ΔCTRL2 or CTRL2R viruses at day 7 or for ganglia latently infected with ΔCTRL2 or CTRL2R viruses at day 30 (Fig. 5A), showing that acute infection of the trigeminal ganglia was similar with the two viruses and there were similar levels of latent infection with the ΔCTRL2 and CTRL2R viruses.
FIG 5 .

Viral DNA and transcript levels during acute and latent infection in mice infected with ΔCTRL2 or CTRL2R virus. TGs from infected mice were harvested at 7 dpi, 30 dpi, or following explant reactivation, and total RNA and DNA were isolated and quantified. (A) Viral genomes were measured by qPCR and normalized to the cellular DNA control. (B) Viral RNA transcript levels for LAT were measured at 7 and 30 dpi and following explant reactivation. (C to I) The miRNAs miR-H2 (C), miR-H4 (D), miR-H6 (E), and lytic transcripts ICP0 (F), ICP27 (G), tk (H), and gC (I) were measured by quantitative reverse transcription-PCR with specific primers and normalized to a cellular control and viral genome copy number. Statistical significance was evaluated with the Mann-Whitney test.
To measure latent viral gene expression with the two viruses, total RNA was also extracted from each sample and measured with primers specific for the viral LAT intron, the viral miRNAs, including miR-H2, miR-H4, and miR-H6, the lytic viral transcripts, including ICP0, ICP27 (IE), and the genes for thymidine kinase (tk; early kinetic class [E]), and glycoprotein C (gC; late kinetic class [L]), and also host GAPDH transcripts and let-7a miRNA. Viral transcripts were first normalized to cellular glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Total RNA levels were then normalized to total viral genome copy numbers for each mouse to calculate transcript levels per genome. Similarly, viral miRNAs were normalized to let-7a levels and then to the viral genome copy number for each mouse. In ganglia infected with the ΔCTRL2 virus, compared to those infected with the CTRL2R virus, we observed a small (~3-fold) but significant increase in levels of LATs at days 7 and 30 (Fig. 5B) (P ≤ 0.0001 by the Mann-Whitney test); however, the levels of LAT became indistinguishable during reactivation of ΔCTRL2 and CTRL2R viruses (Fig. 5B).
miR-H2 and miR-H4 are both derived from the primary LAT transcript (6, 17, 69); while miR-H6 is encoded upstream of the LAT transcription unit, its expression is dependent on a 200-bp sequence that includes the LAT promoter (6, 69). At day 7, miRNAs showed no significant difference; however, at day 30, we observed a small (~1.8-fold) increase in miR-H2 (Fig. 5C) in ΔCTRL2-infected ganglia, based on the Mann-Whitney test. Levels of miR-H4 (Fig. 5D) and miR-H6 (Fig. 5E) were not significantly different (~1.2-fold and ~1.4-fold higher, respectively). In addition, lytic transcript levels were not significantly different at day 7 (Fig. 5F to I). Because lytic transcript levels were too low for quantification at day 30, ganglia were scored as positive or negative for detectable transcripts. We did not find significant differences in the fraction of ganglia with detectable lytic viral transcripts between ΔCTRL2 and CTRL2R infections (Table 1). These results indicated that loss of CTCF binding at the CTRL2 site increased the accumulation of LATs slightly but did not affect the low levels of lytic gene expression during latent infection; however, LAT levels became indistinguishable upon explant reactivation.
TABLE 1 .
Expression of viral lytic transcripts in mouse TG at 30 days
| Virus | Expression of transcript (no. positive/total no.) |
|||
|---|---|---|---|---|
| ICP0 | ICP27 | tk | gC | |
| ΔCTRL2 | 8/34 | 8/34 | 7/34 | 7/34 |
| CTRL2R | 8/24 | 5/24 | 4/24 | 4/24 |
Increased H3K27me3 histone modification on the LAT region of ΔCTRL2 mutant virus.
To examine the potential role of CTRL2 in regulating viral chromatin during latency and its potential function as an insulator, we collected TGs from mice latently infected with ΔCTRL2 or CTRL2R viruses and performed ChIP analysis with antibodies specific for total histone H3 or heterochromatin modifications H3K9me3 and H3K27me3. The relative fraction of HSV-1 DNA immunoprecipitated was measured by qPCR using primers specific for the viral LAT promoter (LAP), LAT intron, and ICP0 promoter normalized to the fraction of DNA immunoprecipitated by the cellular control, GAPDH.
Similar to previous results with wild-type (WT) KOS and 17syn+ HSV-1 strains (22, 23), the CTRL2R virus LAP sequences were associated with less silenced chromatin than the LAT intron or ICP0 promoter regions during latent infection (Fig. 6A and B). We observed ~3- to 5-fold less total H3, H3K9me3, and H3K27me3 associated at LAP relative to that at the ICP0 promoter (P ≤ 0.05, Wilcoxon matched-pairs signed-rank test). In contrast, during latent infection with the ΔCTRL2 mutant virus, the LAP sequences and ICP0 promoter region were associated with equivalent levels of H3 histones and H3-directed heterochromatin modifications. Specifically, we observed a mean fold enrichment of less than 1.5-fold at the ICP0 promoter relative to that of the LAPs for all antibodies tested, and this did not reach statistical significance. These results showed that CTCF binding at the CTRL2 region serves as a chromatin barrier to limit accumulation of H3K27me3 at the LAP and the LAT intron.
FIG 6 .

Deletion of CTRL2 increased H3K27me3 accumulation on LAT promoter and intron sequences. Mice were infected with 2 × 105 PFU/eye of ΔCTRL2 or CTRL2R virus. At 28 dpi, mice were sacrificed, TGs were harvested, and ChIP analysis was carried out with antibodies specific for total histone H3 (A), H3K9me3 (B), or H3K27me3 (C). Three viral regions, the LAT promoter (LAP), LAT intron, and ICP0 promoter (ICP0 P), were examined, and the results are expressed as means and standard deviations of the percent viral chromatin immunoprecipitated relative to immunoprecipitation of the cellular GAPDH region. Asterisks indicate significance (P < 0.05), which was evaluated using the Wilcoxon matched-pairs signed-rank test.
When we compared the chromatin of the ΔCTRL2 virus directly to that of the CTRL2R virus, we observed a significant increase of H3K27me3 heterochromatin marker accumulation at LAP and the LAT intron (P ≤ 0.05), but not at the ICP0 promoter (Fig. 6C). We also observed slight but not significant increases in total histone H3 or H3K9me3 accumulation. These results showed that CTCF binding to CTRL2 may prevent the spread of specific heterochromatin markers, such as H3K27me3, to the LAT region encompassing LAP and the LAT intron.
Explant reactivation from TGs infected with ΔCTRL2 virus is reduced.
To determine whether loss of CTCF binding at the CTRL2 sites affected the ability of the ΔCTRL2 mutant virus to reactivate, we harvested TGs latently infected with ΔCTRL2 or CTRL2R virus and explanted them onto a monolayer of Vero cells. We tested individual ganglia from two independent infections of 5 mice and 10 mice per virus, respectively. We assessed the emergence of infectious virus for 7 days postexplant by collecting overlay medium and, on day 7, also the underlying Vero cell monolayer and replating each on a fresh Vero monolayer. Infectious virus was first detected at 3 days from CTRL2R virus-infected ganglia but appeared first at 4 days from ΔCTRL2 virus-infected ganglia (Fig. 7). By 7 days postexplant, 77% of CTRL2R virus-infected ganglia produced infectious virus, while only 53% of ΔCTRL2 virus-infected ganglia produced detectable virus (P ≤ 0.03, by log-rank test). At 7 days, the underlying cells were also sampled, and ΔCTRL2 never reached the reactivation frequency of the CTRL2R virus, as evaluated by the log-rank Mantel-Cox test. These results indicated that despite similar numbers of latent viral genomes present at day 28, genomes from ΔCTRL2 virus infection showed significantly reduced explant reactivation.
FIG 7 .
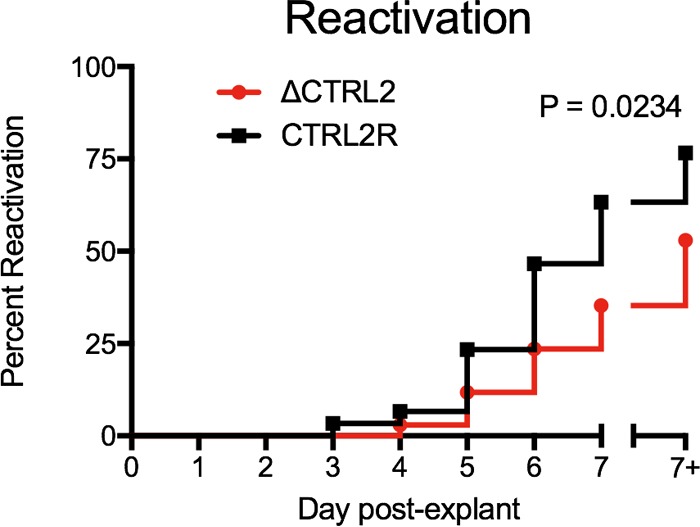
Explant reactivation was reduced by infection with ΔCTRL2 mutant virus. Mice were infected with 2 × 105 PFU/eye of ΔCTRL2 or CTRL2R virus. At 28 dpi, individual ganglia from each mouse, from 15 mice from two independent infections, were isolated and explanted onto Vero cell monolayers. Supernatant was collected daily from the overlay medium for 7 days. At 7 days, the underlying cells were also collected (7+). All samples were replated onto fresh Vero monolayers to detect infectious virus. Statistical significance was evaluated with the log-rank Mantel-Cox test.
DISCUSSION
The LAT and ICP0 transcriptional units are encoded on opposite strands of the HSV-1 genome (Fig. 1) but are regulated independently during latent infection, so that they maintain suppression of ICP0 gene transcription while allowing persistence of LAT transcription (4). The divergent expression patterns correlate with different chromatin modifications found at the LAT and ICP0 promoters that are maintained despite the relative proximity of these genetic elements (21–23, 70, 71). While ICP0 and other lytic gene promoters are associated with high levels of heterochromatin during latent infection, the LAT region also shows enrichment of euchromatin (23, 70). CTCF is a key mediator of the ability to maintain independently regulated but adjacent chromatin domains within mammalian and viral genomes (51, 72). Of the various sites on the HSV-1 genome that bind CTCF during latent infection, CTRL2 has been of particular interest due to its position between the transcriptionally active LAT promoter and the silenced ICP0 promoter (64–66).
In this study, we constructed a mutant HSV-1 virus, ΔCTRL2, with deletions of the CTRL2 CTCF-binding sites to examine the role of CTCF binding to CTRL2 during in vivo latent infection in a mouse model system. The ΔCTRL2 mutant virus produced a relatively normal acute infection in the cornea and trigeminal ganglia and established latent infection to the same level as the CTRL2R virus. The ΔCTRL2 mutant virus showed slightly higher levels of LAT expression during latent infection, but this was indistinguishable during reactivation. Despite equal latent DNA genome loads per TG, the mutant virus showed reduced reactivation relative to that by the CTRL2R virus. Because the mutant virus showed higher levels of H3K27me3 heterochromatin on the LAT promoter and intron sequences, we hypothesize that CTCF binding at the CTRL2 site serves as a chromatin barrier to keep at least certain forms of heterochromatin off the LAT promoter/regulatory sequences.
CTCF is associated with the LAT intron sequences in a CTRL2-dependent manner.
We confirmed that deletion of the CTRL2 region was sufficient to reduce CTCF binding to the LAT intron to background levels. CTCF is known to mediate long-range, three-dimensional chromatin interactions through simultaneous association with distant binding sites; therefore, the loss of one binding site has the potential to alter CTCF binding to distant sites (54, 73–75). Our study also identified a reduction of CTCF binding at the ICP0 promoter region after deletion of the CTRL2 sites, suggesting that CTRL2 and a region near the ICP0 promoter may bind CTCF cooperatively. An interaction between these sites could form a closed chromatin loop surrounding the 5′ end of the ICP0 gene and separating the ICP0 promoter from the LAT promoter. However, there are CTCF-binding sites at many regions of the HSV-1 genome, and further work is required to characterize these sites, their role in the HSV-1 3D chromatin architecture, and their interaction with the LAT region.
Removal of CTCF binding to CTRL2 decreases viral reactivation in a mouse model.
Deletion of the CTCF binding site at the CTRL2 region did not affect viral replication in lytic infection of cultured HeLa or HFF cells or acute infection at the ocular epithelium for the first 4 days of infection. These results argue that lytic infection of nonneuronal cells is not affected by CTCF binding to CTRL2. This is consistent with the findings of a recent study that did not detect CTCF binding at the CTRL2 site during lytic infection (68). Although infection with the ΔCTRL2 virus showed a reduction in shed virus in the eyes of mice on one day, at 5 dpi, total viral genomes per TG were not significantly different between ΔCTRL2 and CTRL2R viruses at 7 or 30 dpi. However, the mutant virus showed reduced reactivation upon ganglionic explant, arguing that CTCF binding at CTRL2 promotes reactivation. Slightly higher levels of LAT were observed during latent infection with the mutant virus compared to the repaired virus, but during reactivation, the levels of LAT were equivalent. Therefore, the major effect of the CTRL2 sites on viral infection seems to be promotion of reactivation.
Results from related herpesviruses, EBV and KSHV, have also indicated that CTCF has a complex role in promoting latent infection. Removal of CTCF-binding sites from the intron of the LMP2A gene of EBV resulted in a higher viral genome copy number in latent infection, possibly as a result of partial lytic replication or aberrant latent replication (76). However, results with KSHV indicated that removal of CTCF-binding sites from the intron of its major latency-associated transcript reduced the latent genome copy number due to reduced viral episome maintenance (61). Given that CTCF binding has such diverse effects among related herpesviruses, it is possible that CTCF binding exhibits both positive and negative regulation during HSV-1 latent infection. To address these questions, future studies that address the progression of latency establishment before 28 days and the maintenance of long-term latent viral genomes are needed to differentiate between the initial viral dose entering the ganglia, viral replication within the neural tissue, and long-term viral genome maintenance.
Histone modifications at the LAT promoter sequences are affected by CTCF binding at the CTRL2 site.
The increased accumulation of H3K27me3-modified heterochromatin at LAP and the LAT enhancer regions of the ΔCTRL2 mutant viral genome is consistent with the hypothesis that CTCF binding at CTRL2 functions as an insulator to block the linear spread of heterochromatin from the lytic ICP0 region to the LAT transcriptional regulatory regions. Previous results demonstrated that the H3K27me3 modification accumulates gradually at viral lytic promoters from days 10 to 14 during the establishment of latent infection and is enhanced by LATs (20, 23). Therefore, increased LAT accumulation at 7 dpi with the ΔCTRL2 virus may also contribute to higher levels of H3K27me3 accumulation at the LAT sequences by day 28.
Furthermore, H3K27me3 can be found in bivalent chromatin domains (77) and poised chromatin domains (78) on developmentally regulated genes and genes in pluripotent stem cells. Our previous study found that increased levels of H3K27me3 at the ICP8 promoter relative to the viral UL48 promoter correlated with higher levels of ICP8 RNA than UL48 RNA (20). Therefore, H3K27me3 alone may be insufficient to silence transcription, or additional histone modifications, such as H3K4me3, may exert a dominant effect to maintain active transcription. Alternatively, a phospho switch proposed for reactivation (79) may work effectively on chromatin that has H3K27me3 modifications. Therefore, H3K27me3 may be important for maintaining repressed but poised genomes during latent infection.
Model for mechanism of action for CTCF binding at the CTRL2 site.
We have shown that binding of CTCF to the CTRL2 sites causes a statistically significant reduction in H3K27me3 and a trend toward reduction of H3K9me3 modification of chromatin on the LAT promoter and intron sequences. Furthermore, the CTRL2 sites promote the reactivation of latent virus upon explant in culture. We propose a model in which CTCF bound at CTRL2 dimerizes with a CTCF molecule bound at another site to form a 3D structure that promotes reactivation. The most likely targets, VP16 and ICP0, are viral genes whose products are involved in early stages of reactivation (1, 12, 80). We envision at least two ways in which CTCF promotes their expression during reactivation. First, we have shown that CTCF binding at the CTRL2 sites promotes CTCF binding in the ICP0 promoter, which could place the 5′ end of the ICP0 gene in a chromatin loop. Because CTCF-mediated chromatin loops are believed to often enclose inducible gene regulatory domains (81), enclosing the ICP0 gene promoter in a chromatin loop may promote its expression when reactivation is triggered. Second, CTCF may dimerize with a CTCF molecule bound near the VP16/UL48 gene promoter to keep the VP16 promoter in a chromatin state that can be readily induced when reactivation is triggered. Further studies are needed to define the 3D chromatin structure of viral chromatin that is dependent on CTCF binding at the CTRL2 sites and to test these hypotheses.
Establishing and maintaining a poised latent infection.
To establish a successful latent infection, HSV-1 must balance suppression of lytic gene expression with maintenance of a latent state that is poised for reactivation. The results in this study indicate that HSV-1 exploits the cellular protein CTCF to maintain its genome in a state that can be efficiently reactivated. Large domains of H3K27me3 with short domains of H3K4me3 histone modifications are known to define a bivalent chromatin structure that is silenced but poised to be reactivated at the appropriate time (77). We propose a model to explain how the CTRL2 sites can promote reactivation. In addition to the CTRL2 sites, HSV-1 gene products contribute to promoting H3K27me3 on HSV-1 lytic gene chromatin. HSV-1 LAT promotes H3K27me3 on lytic gene chromatin (23) as well as reactivation (12; P. Raja, J. S. Lee, and D. M. Knipe, unpublished results). Furthermore, ICP0 promotes LAT expression and H3K27me3 modifications on lytic gene chromatin (44) as well as reactivation (82, 83). Therefore, CTCF, LAT, and ICP0 may all be acting to promote the correct form of viral chromatin so that a poised latent infection is established and maintained and reactivation can take place at the appropriate time. Therefore, viral gene products play an active role in not only promoting epigenetic silencing of DNA viral genomes for latent infection (84) but also keeping them in a poised form that is capable of efficient reactivation, a state that we call poised latency.
MATERIALS AND METHODS
Cells and viruses.
Vero, HeLa, and HFF cells were obtained from ATCC. The ΔCTRL2 virus has a deletion of CTRL2 sites from within the LAT intron of both TRL repeats (HSV-1 KOS KT899744 bp 120136 to 120508 [85]).
The control CTRL2R virus was constructed in parallel by cotransfection of the WT DNA fragment. All viruses were propagated and titrated in parallel on Vero cells.
Mouse infections.
Mice were housed in accordance with institutional and National Institutes of Health guidelines on the care and use of animals in research. Ocular infection was carried out with 2 × 105 PFU/eye of virus (86). Eyes were swabbed on days 1 to 5 postinfection, and virus collected from tear films was titrated on Vero cells (87). Mice were monitored for survival for at least 28 dpi.
ChIP assays.
Immunoprecipitations were carried out on chromatin prepared from TGs (20) with 5 to 10 μg of anti-CTCF (catalog number 07-729; Millipore), 2.5 μg of anti-histone H3 (ab1791; Abcam, Inc.), 2.5 μg anti-H3K27me3 (39156; Active Motif, Inc.), 2.5 μg of anti-H3K9me3 (ab8580; Abcam, Inc.), or normal rabbit IgG (12-370; Millipore). Immunoprecipitated DNA was quantitated using the qPCR primers listed in Table 2.
TABLE 2 .
Primer sequences
| Target | Purpose |
Sequence (5′–3′) |
|
|---|---|---|---|
| Forward | Reverse | ||
| LAT promoter | qPCR | 5′-CCCGGCCCGCACGAT-3′ | 5′-CAACACCCCGCCGCTTT-3′ |
| LAT intron | qPCR | 5′-GGGTCATCCAGAGGCTGTTC-3′ | 5′-GTGGACCAGACGGGAAACAT-3′ |
| ICP0 promoter | qPCR | 5′-CGCCTTCCCGAAGAAACTCA-3′ | 5′-CGCTCAATGAACCCGCATT-3′ |
| ICP8 promoter | qPCR | 5′-GAGACCGGGGTTGGGGAATG AATC-3′ | 5′-CCCCGGGGGTTGTCTGTGAAG G-3′ |
| GAPDH pseudogene | qPCR | 5′-TTCGACAGTCAGCCGCATCTTCTT-3′ | 5′-CAGGCGCCCAATACGACCAA ATC-3′ |
Quantification of viral genomes and transcripts.
Nucleic acids were isolated using the Allprep RNA/DNA minikit (Qiagen), and RNA was reverse-transcribed with specific primers using the QuantiTect RT kit (Qiagen). Viral DNA and transcripts were quantified relative to host adipsin DNA and GAPDH mRNA (44, 88). For miRNA quantification, RNAs from TGs were purified using an RNeasyPlus minikit (Qiagen) and quantified using TaqMan miRNA assays (Applied Biosystems). Viral miRNA levels were normalized to cellular let-7a.
Reactivation.
TGs isolated from mice latently infected with HSV-1 ΔCTRL2 or CTRL2R viruses were bisected and explanted onto a confluent monolayer of Vero cells in Dulbecco’s modified Eagle’s medium supplemented with 10% (vol/vol) fetal bovine serum and 0.25 μg/ml amphotericin B. Culture medium was sampled daily for 7 days, and after 7 days (7+) the entire Vero monolayer and ganglia were collected, frozen, and replated onto a fresh Vero monolayer to score the number of ganglia that showed detectable infectious virus (see Text S1 for additional details on our methods).
Supplemental methods. Download TEXT S1, DOCX file, 0.1 MB (140.4KB, docx) .
Copyright © 2018 Lee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ACKNOWLEDGMENTS
We thank Jeho Shin for technical assistance and Patrick T. Waters for assistance with the manuscript.
This research was supported by NIH grant P01 AI098681 to D.M.C. and D.M.K.
Footnotes
Citation Lee JS, Raja P, Pan D, Pesola JM, Coen DM, Knipe DM. 2018. CCCTC-binding factor acts as a heterochromatin barrier on herpes simplex viral latent chromatin and contributes to poised latent infection. mBio 9:e02372-17. https://doi.org/10.1128/mBio.02372-17.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM (ed), Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 3.Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, Ott M, Margolis D, Nieto A, Nevels M, Parks RJ, Kristie TM. 2013. Snapshots: chromatin control of viral infection. Virology 435:141–156. doi: 10.1016/j.virol.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 5.Kramer MF, Coen DM. 1995. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J Virol 69:1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umbach JL, Nagel MA, Cohrs RJ, Gilden DH, Cullen BR. 2009. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J Virol 83:10677–10683. doi: 10.1128/JVI.01185-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, Coen DM. 2010. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J Virol 84:4659–4672. doi: 10.1128/JVI.02725-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deshmane SL, Fraser NW. 1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol 63:943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garber DA, Schaffer PA, Knipe DM. 1997. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J Virol 71:5885–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen SH, Kramer MF, Schaffer PA, Coen DM. 1997. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J Virol 71:5878–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Javier RT, Stevens JG, Dissette VB, Wagner EK. 1988. A herpes simplex virus transcript abundant in latently infected neurons is dispensable for establishment of the latent state. Virology 166:254–257. doi: 10.1016/0042-6822(88)90169-9. [DOI] [PubMed] [Google Scholar]
- 14.Steiner I, Spivack JG, Lirette RP, Brown SM, MacLean AR, Subak-Sharpe JH, Fraser NW. 1989. Herpes simplex virus type 1 latency-associated transcripts are evidently not essential for latent infection. EMBO J 8:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicoll MP, Hann W, Shivkumar M, Harman LE, Connor V, Coleman HM, Proença JT, Efstathiou S. 2016. The HSV-1 latency-associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PLoS Pathog 12:e1005539. doi: 10.1371/journal.ppat.1005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drolet BS, Perng GC, Cohen J, Slanina SM, Yukht A, Nesburn AB, Wechsler SL. 1998. The region of the herpes simplex virus type 1 LAT gene involved in spontaneous reactivation does not encode a functional protein. Virology 242:221–232. doi: 10.1006/viro.1997.9020. [DOI] [PubMed] [Google Scholar]
- 17.Tang S, Bertke AS, Patel A, Wang K, Cohen JI, Krause PR. 2008. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc Natl Acad Sci U S A 105:10931–10936. doi: 10.1073/pnas.0801845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flores O, Nakayama S, Whisnant AW, Javanbakht H, Cullen BR, Bloom DC. 2013. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J Virol 87:6589–6603. doi: 10.1128/JVI.00504-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang X, Chentoufi AA, Hsiang C, Carpenter D, Osorio N, BenMohamed L, Fraser NW, Jones C, Wechsler SL. 2011. The herpes simplex virus type 1 latency-associated transcript can protect neuron-derived C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-cell killing. J Virol 85:2325–2332. doi: 10.1128/JVI.01791-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cliffe AR, Coen DM, Knipe DM. 2013. Kinetics of facultative heterochromatin and Polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. mBio 4:e00590-12. doi: 10.1128/mBio.00590-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A 102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The Polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83:8173–8181. doi: 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobson AT, Sederati F, Devi-Rao G, Flanagan J, Farrell MJ, Stevens JG, Wagner EK, Feldman LT. 1989. Identification of the latency-associated transcript promotor by expression of rabbit beta-globin mRNA in mouse sensory nerve ganglia latently infected with a recombinant herpes simplex virus. J Virol 65:3844–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lokensgard JR, Bloom DC, Dobson AT, Feldman LT. 1994. Long-term promoter activity during herpes simplex virus latency. J Virol 68:7148–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lokensgard JR, Berthomme H, Feldman LT. 1997. The latency-associated promoter of herpes simplex virus type 1 requires a region downstream of the transcription start site for long-term expression during latency. J Virol 71:6714–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batchelor AH, O’Hare P. 1990. Regulation and cell-type-specific activity of a promoter located upstream of the latency-associated transcript of herpes simplex virus type 1. J Virol 64:3269–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zwaagstra JC, Ghiasi H, Slanina SM, Nesburn AB, Wheatley SC, Lillycrop K, Wood J, Latchman DS, Patel K, Wechsler SL. 1990. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J Virol 64:5019–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spivack JG, Fraser NW. 1988. Expression of herpes simplex virus type 1 latency-associated transcripts in the trigeminal ganglia of mice during acute infection and reactivation of latent infection. J Virol 62:1479–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amelio AL, Giordani NV, Kubat NJ, O’Neil JE, Bloom DC. 2006. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J Virol 80:2063–2068. doi: 10.1128/JVI.80.4.2063-2068.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kosz-Vnenchak M, Jacobson J, Coen DM, Knipe DM. 1993. Evidence for a novel regulatory pathway for herpes simplex virus gene expression in trigeminal ganglion neurons. J Virol 67:5383–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devi-Rao GB, Bloom DC, Stevens JG, Wagner EK. 1994. Herpes simplex virus type 1 DNA replication and gene expression during explant-induced reactivation of latently infected murine sensory ganglia. J Virol 68:1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J, Silverstein S. 1992. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J Virol 66:2916–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai W, Schaffer PA. 1992. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J Virol 66:2904–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orzalli MH, Conwell SE, Berrios C, Decaprio JA, Knipe DM. 2013. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci U S A 110:E4492–E4501. doi: 10.1073/pnas.1316194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chelbi-Alix MK, de Thé H. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941. doi: 10.1038/sj.onc.1202366. [DOI] [PubMed] [Google Scholar]
- 38.Everett RD, Murray J. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol 79:5078–5089. doi: 10.1128/JVI.79.8.5078-5089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jurak I, Silverstein LB, Sharma M, Coen DM. 2012. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J Virol 86:10093–10102. doi: 10.1128/JVI.00930-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol 80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gu H, Liang Y, Mandel G, Roizman B. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A 102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci U S A 104:17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Proença JT, Coleman HM, Connor V, Winton DJ, Efstathiou S. 2008. A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J Gen Virol 89:2965–2974. doi: 10.1099/vir.0.2008/005066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raja P, Lee JS, Pan D, Pesola JM, Coen DM, Knipe DM. 2016. A herpesviral lytic protein regulates the structure of latent viral chromatin. mBio 7:e00633-16. doi: 10.1128/mBio.00633-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lobanenkov VV, Nicolas RH, Adler VV, Paterson H, Klenova EM, Polotskaja AV, Goodwin GH. 1990. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 5:1743–1753. [PubMed] [Google Scholar]
- 46.Baniahmad A, Steiner C, Köhne AC, Renkawitz R. 1990. Modular structure of a chicken lysozyme silencer: involvement of an unusual thyroid hormone receptor binding site. Cell 61:505–514. doi: 10.1016/0092-8674(90)90532-J. [DOI] [PubMed] [Google Scholar]
- 47.Lobanenkov VV, Gudvin GG. 1989. CCCTC-binding protein: a new nuclear protein factor which interaction with 5′-flanking sequence of chicken c-myc oncogene correlates with repression of the gene. Dokl Akad Nauk SSSR 309:741–745. [PubMed] [Google Scholar]
- 48.Köhne AC, Baniahmad A, Renkawitz R. 1993. NeP1. A ubiquitous transcription factor synergizes with v-ERBA in transcriptional silencing. J Mol Biol 232:747–755. doi: 10.1006/jmbi.1993.1428. [DOI] [PubMed] [Google Scholar]
- 49.Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, Lobanenkov VV. 1993. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol 13:7612–7624. doi: 10.1128/MCB.13.12.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, Neiman PE, Collins SJ, Lobanenkov VV. 1996. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol 16:2802–2813. doi: 10.1128/MCB.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bell AC, West AG, Felsenfeld G. 1999. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98:387–396. doi: 10.1016/S0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 52.Fu Y, Sinha M, Peterson CL, Weng Z. 2008. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet 4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. 2011. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479:74–79. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, de Laat W. 2006. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev 20:2349–2354. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, Wong E, Sheng J, Zhang Y, Poh T, Chan CS, Kunarso G, Shahab A, Bourque G, Cacheux-Rataboul V, Sung WK, Ruan Y, Wei CL. 2011. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ong CT, Corces VG. 2014. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 58.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. 2007. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung JH, Whiteley M, Felsenfeld G. 1993. A 5′ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell 74:505–514. doi: 10.1016/0092-8674(93)80052-G. [DOI] [PubMed] [Google Scholar]
- 60.Tempera I, Wiedmer A, Dheekollu J, Lieberman PM. 2010. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog 6:e1001048. doi: 10.1371/journal.ppat.1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM. 2008. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J 27:654–666. doi: 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kang H, Wiedmer A, Yuan Y, Robertson E, Lieberman PM. 2011. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog 7:e1002140. doi: 10.1371/journal.ppat.1002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martínez FP, Cruz R, Lu F, Plasschaert R, Deng Z, Rivera-Molina YA, Bartolomei MS, Lieberman PM, Tang Q. 2014. CTCF binding to the first intron of the major immediate early (MIE) gene of human cytomegalovirus (HCMV) negatively regulates MIE gene expression and HCMV replication. J Virol 88:7389–7401. doi: 10.1128/JVI.00845-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amelio AL, McAnany PK, Bloom DC. 2006. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J Virol 80:2358–2368. doi: 10.1128/JVI.80.5.2358-2368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Q, Lin L, Smith S, Huang J, Berger SL, Zhou J. 2007. CTCF-dependent chromatin boundary element between the latency-associated transcript and ICP0 promoters in the herpes simplex virus type 1 genome. J Virol 81:5192–5201. doi: 10.1128/JVI.02447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ertel MK, Cammarata AL, Hron RJ, Neumann DM. 2012. CTCF occupation of the herpes simplex virus 1 genome is disrupted at early times postreactivation in a transcription-dependent manner. J Virol 86:12741–12759. doi: 10.1128/JVI.01655-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harkness JM, Kader M, DeLuca NA. 2014. Transcription of the herpes simplex virus 1 genome during productive and quiescent infection of neuronal and nonneuronal cells. J Virol 88:6847–6861. doi: 10.1128/JVI.00516-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lang F, Li X, Vladimirova O, Hu B, Chen G, Xiao Y, Singh V, Lu D, Li L, Han H, Wickramasinghe JM, Smith ST, Zheng C, Li Q, Lieberman PM, Fraser NW, Zhou J. 2017. CTCF interacts with the lytic HSV-1 genome to promote viral transcription. Sci Rep 7:39861. doi: 10.1038/srep39861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kramer MF, Jurak I, Pesola JM, Boissel S, Knipe DM, Coen DM. 2011. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 417:239–247. doi: 10.1016/j.virol.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kubat NJ, Amelio AL, Giordani NV, Bloom DC. 2004. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol 78:12508–12518. doi: 10.1128/JVI.78.22.12508-12518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kubat NJ, Tran RK, McAnany P, Bloom DC. 2004. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J Virol 78:1139–1149. doi: 10.1128/JVI.78.3.1139-1149.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Felsenfeld G, Burgess-Beusse B, Farrell C, Gaszner M, Ghirlando R, Huang S, Jin C, Litt M, Magdinier F, Mutskov V, Nakatani Y, Tagami H, West A, Yusufzai T. 2004. Chromatin boundaries and chromatin domains. Cold Spring Harb Symp Quant Biol 69:245–250. doi: 10.1101/sqb.2004.69.245. [DOI] [PubMed] [Google Scholar]
- 73.Majumder P, Boss JM. 2010. CTCF controls expression and chromatin architecture of the human major histocompatibility complex class II locus. Mol Cell Biol 30:4211–4223. doi: 10.1128/MCB.00327-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Phillips JE, Corces VG. 2009. CTCF: master weaver of the genome. Cell 137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohlsson R, Renkawitz R, Lobanenkov V. 2001. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet 17:520–527. doi: 10.1016/S0168-9525(01)02366-6. [DOI] [PubMed] [Google Scholar]
- 76.Chen HS, Martin KA, Lu F, Lupey LN, Mueller JM, Lieberman PM, Tempera I. 2014. Epigenetic deregulation of the LMP1/LMP2 locus of Epstein-Barr virus by mutation of a single CTCF-cohesin binding site. J Virol 88:1703–1713. doi: 10.1128/JVI.02209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 78.Lesch BJ, Page DC. 2014. Poised chromatin in the mammalian germ line. Development 141:3619–3626. doi: 10.1242/dev.113027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M. 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18:649–658. doi: 10.1016/j.chom.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sawtell NM, Thompson RL. 2016. De novo herpes simplex virus VP16 Expression gates a dynamic programmatic transition and sets the latent/lytic balance during acute infection in trigeminal ganglia. PLoS Pathog 12:e1005877. doi: 10.1371/journal.ppat.1005877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oti M, Falck J, Huynen MA, Zhou H. 2016. CTCF-mediated chromatin loops enclose inducible gene regulatory domains. BMC Genomics 17:252. doi: 10.1186/s12864-016-2516-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leib DA, Coen DM, Bogard CL, Hicks KA, Yager DR, Knipe DM, Tyler KL, Schaffer PA. 1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J Virol 63:759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Halford WP, Schaffer PA. 2000. Optimized viral dose and transient immunosuppression enable herpes simplex virus ICP0-null mutants to establish wild-type levels of latency in vivo. J Virol 74:5957–5967. doi: 10.1128/JVI.74.13.5957-5967.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Knipe DM, Raja P, Lee J. 2017. Viral gene products actively promote latent infection by epigenetic silencing mechanisms. Curr Opin Virol 23:68–74. doi: 10.1016/j.coviro.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Colgrove RC, Liu X, Griffiths A, Raja P, Deluca NA, Newman RM, Coen DM, Knipe DM. 2016. History and genomic sequence analysis of the herpes simplex virus 1 KOS and KOS1.1 sub-strains. Virology 487:215–221. doi: 10.1016/j.virol.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tenser RB, Dunstan ME. 1979. Herpes simplex virus thymidine kinase expression in infection of the trigeminal ganglion. Virology 99:417–422. doi: 10.1016/0042-6822(79)90021-7. [DOI] [PubMed] [Google Scholar]
- 87.Coen DM, Irmiere AF, Jacobson JG, Kerns KM. 1989. Low levels of herpes simplex virus thymidine-thymidylate kinase are not limiting for sensitivity to certain antiviral drugs or for latency in a mouse model. Virology 168:221–231. doi: 10.1016/0042-6822(89)90261-4. [DOI] [PubMed] [Google Scholar]
- 88.Pan D, Flores O, Umbach JL, Pesola JM, Bentley P, Rosato PC, Leib DA, Cullen BR, Coen DM. 2014. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe 15:446–456. doi: 10.1016/j.chom.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Download TEXT S1, DOCX file, 0.1 MB (140.4KB, docx) .
Copyright © 2018 Lee et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.