

Summary
Acute lung injury (ALI) is a heterogeneous disease with the hallmarks of alveolar capillary membrane injury, increased pulmonary oedema and pulmonary inflammation. The most common direct aetiological factor for ALI is usually parenchymal lung infection or haemorrhage. Reactive oxygen species (ROS) generated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX2) are thought to play an important role in the pathophysiology of ALI. Glucose‐6‐phosphate dehydrogenase (G6PD) plays an important role both in production of ROS as well as their removal through the supply of NADPH. However, how G6PD modulation affects NOX2‐mediated ROS in the airway epithelial cells (AECs) during acute lung injury has not been explored previously. Therefore, we investigated the effect of G6PD inhibitor, 6‐aminonicotinamide on G6PD activity, NOX2 expression, ROS production and enzymatic anti‐oxidants in AECs in a mouse model of ALI induced by lipopolysaccharide (LPS). ALI led to increased G6PD activity in the AECs with concomitant elevation of NOX2, ROS, SOD1 and nitrotyrosine. G6PD inhibitor led to reduction of LPS‐induced airway inflammation, bronchoalveolar lavage fluid protein concentration as well as NOX2‐derived ROS and subsequent oxidative stress. Conversely, ALI led to decreased glutathione reductase activity in AECs, which was normalized by G6PD inhibitor. These data show that activation of G6PD is associated with enhancement of oxidative inflammation in during ALI. Therefore, inhibition of G6PD might be a beneficial strategy during ALI to limit oxidative damage and ameliorate airway inflammation.
Keywords: acute lung injury, airway epithelial cells, G6PD inhibitor, NOX2, reactive oxygen species
Introduction
Acute lung injury (ALI) is a heterogeneous disease characterized by injury to alveolar capillary membrane, increased pulmonary oedema due to increased permeability and pulmonary inflammation that can occur directly or indirectly 1, 2, 3. The most common direct aetiologies of ALI are usually parenchymal lung infection (i.e. pneumonia) or haemorrhage, whereas indirect aetiologies of ALI usually result from systemic insults such as sepsis, or trauma or acute kidney injury. However, both direct pulmonary injury and sepsis‐induced systemic inflammation can damage the lung, resulting in a similar clinical picture of ALI 1, 4, 5.
Numerous mechanisms are involved in the development of ALI. Direct injury results in the production of cytokines and chemokines by alveolar macrophages, T cells and epithelial cells leading to increased pulmonary permeability, neutrophil infiltration and subsequent ALI. Injury is amplified by the production of proteases, reactive oxygen species (ROS) and chemokines, further aggravating the inflammatory status. The generation of ROS that results from activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) plays a central role in generation as well as the perpetuation of ALI through production of oxidative stress 1, 6, 7, 8.
Glucose‐6‐phosphate dehydrogenase (G6PD) is a key enzyme involved in the regulation of redox balance in the cell through reduction of NADP+ into NADPH 9. It not only provides reducing equivalents to NADPH oxidase, but also reduction of ROS. ROS scavenging is considered to be beneficial because they affect cell function adversely through oxidative modifications of proteins, nucleotides and lipids 7, 10. Therefore, G6PD activity modulation has been shown to produce both harmful as well as beneficial effects in different inflammatory conditions either through its inhibition or over‐expression 11, 12, 13, 14. As NADPH is required for the reduction of ROS, G6PD deficiency usually results in increased inflammation and cell damage 9, 15. In particular, tissues that have low G6PD expression are more prone to oxidant‐induced injury 9. Therefore, over‐expression of G6PD has been shown to be protective in different organs 12, 16. However, under certain inflammatory conditions such as atherosclerosis, heart failure and obesity, increased availability of NADPH due to G6PD has been shown to promote NADPH oxidase‐derived ROS, which may lead to increased inflammation 17, 18, 19. In a previous study, G6PD activity has also been shown to be elevated in a rabbit model of ALI 20. In this scenario, pharmacological inhibition of G6PD may probably be beneficial due to the limited supply of NADPH.
Among the enzymatic systems employed by the cells to produce ROS, different isoforms of NOXs are at the forefront. NOX family consists of several isoforms, such as NOX1, 2, 3, 4 and 5, and dual oxidases 1 and 2. The most studied member of this family is, of course, NOX2, which consists of specialized cytosolic proteins, p47phox and p67phox, and the small regulatory proteins, all of which are translocated to the membrane upon cell stimulation for complete assembly after interaction with the membrane subunit. This leads to production of superoxide anions for anti‐microbial effects 10, 21, 22. NOX2 is found mainly in phagocytes such as neutrophils, but airway epithelial cells are also known to express it 7, 22. However, the role of G6PD in NOX2‐derived ROS in airway epithelial cells (AECs) during ALI has not been explored previously.
As ROS play a major role in airway inflammation during ALI, we postulated that G6PD activity might regulate the flux of ROS through NOX2 in AECs. Our results show that G6PD expression/activity is up‐regulated during lipopolysaccharide (LPS)‐induced ALI which leads to increase in NOX2‐derived ROS in AECs and is associated with pulmonary inflammation/permeability. Our study shows further that pharmacological inhibition of G6PD might be beneficial during ALI through reduction in NOX2‐derived ROS and subsequent oxidative stress.
Materials and methods
Animals
Male BALB/c mice (10–12 weeks of age; 25–30 g) maintained under specific pathogen‐free conditions were used in the experiments. Animal care was according to the regulations set by the Experimental Animal Care Center, College of Pharmacy, King Saud University. The experimental protocols for the utilization of animals were approved by the Animal Care and Research Committee of College of Pharmacy, King Saud University.
LPS‐induced acute lung injury in mice
Mice were anaesthetized lightly with isoflurane and exposed to a single dose of LPS (50 µg/50 µl/mouse) intranasally (i.n.), as described earlier 23, 24. Control mice received saline i.n. in a similar volume.
Drug treatments
The effect of G6PD inhibition on LPS‐induced pulmonary inflammation in mice was assessed by administration of 6‐aminonicotinamide at 200 µg/mouse i.n. 1 h before and 12 h after administration of intranasal LPS or saline. Vehicle was also administered i.n. 1 h before and 12 h after administration of LPS or saline.
Mice were divided into the following groups – control group (CON): mice received only saline i.n.; LPS‐administered group (LPS): mice received LPS i.n. using the protocol described above; G6PD inhibitor, 6‐aminonicotinamide; and LPS‐treated group (G6PD‐inhibition + LPS): mice received 6‐aminonicotinamide i.n. before and intranasal LPS administration using the protocol described above. G6PD inhibitor, 6‐aminonicotinamide‐treated control group (G6PD‐inhib + control): mice received vehicle i.n. before and intranasal saline administration using the protocol described above.
Bronchoalveolar lavage (BAL) and histopathological analysis
Mice were anaesthetized with isoflurane, and BAL procedure was performed 1 day after LPS challenge by lavaging the lungs for collection of BAL fluid (BALF). Recovered cells were cytocentrifuged onto the slide for differential count (neutrophils, macrophages, lymphocytes) according to the standard morphology, as described previously 24, 25, 26. Cell number was expressed as mean ± standard error of the mean (s.e.m.) per ml for each group. Cell‐free BALF protein as an indicator of alveolar–capillary injury was assessed by measurement of total protein concentration using a commercial kit from Bio‐Rad (Hercules, CA, USA). For histological analysis, lungs were harvested followed by fixation with 10% formalin. Paraffin‐embedded tissue was cut into 5‐μm sections and stained with haematoxylin and eosin (H&E). Stained slides were analysed by light microscopy.
Myeloperoxidase (MPO) activity
MPO activity in lung samples was measured as described previously 27. Data were expressed in absorbance after normalization by the protein content.
G6PD activity
G6PD activity was measured in tracheal epithelial cells using a kit from BioVision (Milpitas, CA, USA), according to the manufacturer's instructions. Values were normalized to protein content in each sample and expressed as pmol/min/mg protein.
Glucose reductase (GR) activity
GR activity in tracheal epithelial cells was measured by the method of Carlberg and Mannervik 28, following the NADPH‐dependent reduction of oxidized glutathione through a decrease in absorbance at 340 nm. Values were normalized to protein content in each sample and expressed as nmol/min/mg protein.
Real‐time polymerase chain reaction (PCR)
Murine tracheal epithelial cells were scraped gently from the lumen of the trachea with a plastic polyethylene brush, as described previously 25. Total RNA from tracheal epithelial cells was extracted using the RNeasy microkit with DNase treatment (Qiagen, Hilden, Germany). cDNA was synthesized from 0·5 μg of total RNA using reverse transcription reagents (high‐capacity cDNA archive kit; Applied Biosystems, Foster City, CA, USA), as per the manufacturers’ protocols 24, 25, 26. Real‐time PCR was performed using Taqman Universal Mastermix (Applied Biosystems), cDNA and carboxyfluorescein (FAM)‐labelled gene‐specific primers from Applied Biosystems for the genes encoding NOX2, SOD1, SOD2, glutathione peroxidase 1 (GPx1), G6PD and 18S rRNA (ribosomal RNA). Real‐time PCR was carried out on a 7500 real‐time PCR system (Applied Biosystems). Data for each target gene were normalized to endogenous control gene and expressed as fold change using the comparative CT method, as described earlier 29.
Flow cytometry
Whole tracheas were cut open longitudinally and cells from the lumen were scraped gently using a mini plastic spatula in RPMI‐1640. Scrapped epithelial cell clumps were passed through an 18G needle several times to prepare single‐cell suspensions for flow cytometry experiments. Thereafter, single‐cell suspensions were labelled with fluorescein/allophycocyanin/phycoerythrin (FITC/APC/PE)‐tagged antibodies against CD326 (Ep‐CAM; BioLegend, San Diego, CA, USA), NOX2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), nitrotyrosine (Santa Cruz Biotechnology) or superoxide dismutase 1 (SOD1) (Santa Cruz Biotechnology) for surface/intracellular labelling using immunostaining methods described earlier 26, 27. The stained cells were acquired on a flow cytometer (Beckman Coulter, Brea, CA, USA) and analysed for the expression of protein of interest using Cytomics FC 500 software, as described earlier 26, 27.
ROS measurement
The intracellular ROS generation in epithelial cells was measured for 30 min using a fluorescent dye 2′,7′‐dichlorofluorescein diacetate (DCFH‐DA; 100 μM) with a multi‐mode microplate spectrofluorometer (FLUOstar Omega; BMG LabTech, Cary, NC, USA) by the method of Wang and Joseph 30, as described earlier 24, 25. Data were expressed as fold mean fluorescence intensity (MFI) over control.
Chemicals
Highest‐grade chemicals/reagents were purchased from Sigma Chemicals (St Louis, MO, USA) unless stated otherwise.
Statistical analysis
The data were expressed as mean ± s.e.m. Comparisons among different groups were analyzed by one‐way analysis of variance (anova) followed by Tukey's multiple comparison tests. A P‐value of less than 0·05 was considered significant for all statistical tests. All the statistical analyses were performed using the GraphPad Prism statistical package.
Results
Effect of G6PD inhibitor on LPS‐induced ALI
Figure 1a–e shows that LPS effectively enhances airways inflammation (total leucocytes/neutrophils, MPO activity and histopathology) and permeability (BALF total protein concentration, an indicator of alveolar‐capillary leak; Fig. 1e). However, the G6PD inhibitor, 6‐aminonicotinamide, led to amelioration of LPS‐induced airway inflammation and permeability, as shown in Fig 1a–e. Moreover, there was no statistical difference between G6PD inhibitor‐treated and ‐untreated control mice in any of the above‐stated parameters; therefore, this group was not included in further analyses. These data show that inhibition of G6PD attenuates LPS‐induced airway inflammation.
Figure 1.

Effect of glucose‐6‐phosphate dehydrogenase (G6PD) inhibitor, 6‐aminonicotinamide on lipopolysaccharide (LPS)‐induced airway inflammation and related parameters in mice. (a) Total bronchoalveolar lavage (BAL) leucocytes, (b) total BAL neutrophils, (c) lung myeloperoxidase (MPO) activity, (d) bronchoalveolar lavage fluid (BALF) protein concentration and (e) histopathological analysis by haematoxylin and eosin (H&E) staining of lung tissue sections (upper horizontal panel; ×100) and BAL differential leucocyte count (lower horizontal panel; ×100). Values are expressed as mean ± standard error (s.e.), n = 6–8/group. *P < 0·05 versus lipopolysaccharide (LPS) group. [Colour figure can be viewed at wileyonlinelibrary.com]
Effect of LPS on G6PD expression/activity in AECs and its modulation by G6PD inhibitor
As oxidative inflammation (ROS generation as well as their scavenging) is dependent upon G6PD activity/expression, we therefore evaluated its expression and activity in AECs during ALI. Figure 2a,b shows that G6PD was elevated significantly during LPS‐induced ALI. However, G6PD inhibitor, 6‐aminonicotinamide, normalized the activity of G6PD without affecting its expression (Fig. 2a,b). These data show that the decrease in LPS‐induced airway inflammation/permeability as noted above after treatment with 6‐aminonicotinamide was a consequence of a decrease in G6PD activity in AECs.
Figure 2.
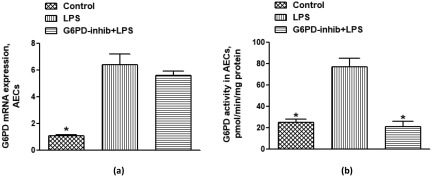
Effect of lipopolysaccharide (LPS) on glucose‐6‐phosphate dehydrogenase (G6PD) expression/activity and its modulation by G6PD inhibitor, 6‐aminonicotinamide in airway epithelial cells (AECs) of mice. (a) G6PD mRNA expression and (b) G6PD activity. Values are expressed as mean ± standard error (s.e.), n = 6–8/group. *P < 0·05 versus LPS group.
Effect of LPS on NOX2 expression/ROS generation in AECs and their modulation by G6PD inhibitor
NOX2‐derived ROS generation has been implicated in different models of ALI, and several immune/non‐immune cells have been shown to contribute to its pathogenesis. In this study we attempted to explore the relationship of G6PD and ROS generation in AECs with ALI in a mouse model. We hypothesized that G6PD may regulate NOX2‐mediated ROS due to availability of NADPH in AECs during ALI. Our data show that LPS induces NOX2 in AECs as shown by real‐time PCR and flow cytometry (Fig. 3a,b,d). As a further confirmation, there is a significant increase in ROS production in AECs after LPS administration (Fig. 3c). G6PD inhibition did not change expression of NOX2; however, it led to a decrease in ROS production, due probably to the limitation in supply of NADPH (Fig. 3a–d). These data show that NOX2‐derived ROS are elevated in AECs, during ALI which may be controlled by G6PD activity.
Figure 3.
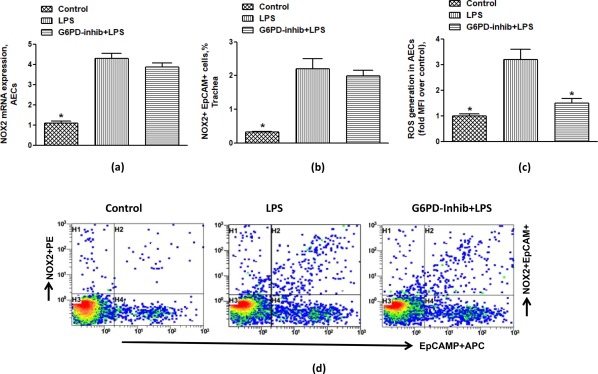
Effect of glucose‐6‐phosphate dehydrogenase (G6PD) inhibitor, 6‐aminonicotinamide on lipopolysaccharide (LPS)‐induced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX2) expression and reactive oxygen species (ROS) generation in airway epithelial cells (AECs) of mice. (a) NOX2 mRNA expression, (b) % NOX2 + epithelial cell adhesion molecule (EpCAM)+ cells, (c) ROS generation, and (d) representative dot‐plot for NOX2 immunostaining in EpCAM+ cells. Dot‐plots show events in tracheal epithelial cells taken from every group. Values are expressed as mean ± standard error (s.e.), n = 6–8/group. *P < 0·05 versus LPS group. [Colour figure can be viewed at wileyonlinelibrary.com]
Effect of LPS on anti‐oxidant enzymes in AECs and their modulation by G6PD inhibitor
Our next aim was to assess the enzymes responsible for detoxification of NOX2‐mediated ROS, i.e. SOD1/2 and GPx1. Our data show an increase in mRNA expression of SOD1 (Fig. 4a) but not SOD2/GPx1 (data not shown) after LPS administration in AECs, suggesting that ROS mainly induce SOD1 in AECs. Increased expression of SOD1 in AECs during ALI was also confirmed by flow cytometry, as is evident in Fig. 4a,b,e. Up‐regulated SOD1 expression was concomitant with increased nitrotyrosine expression, a marker of peroxynitrite‐mediated oxidative damage in LPS‐treated mice (Fig. 4d,f). Treatment of mice having ALI with G6PD inhibitor led to normalization of SOD1 and nitrotyrosine expression in AECs (Fig. 4a,b,d–f). GR activity was also measured to confirm the effect of ALI and G6PD inhibitor in AECs. LPS‐induced ALI led to a decrease in GR activity in AECs; however, it was normalized by G6PD inhibitor (Fig. 4c). These data show that up‐regulated SOD1 and decreased GR activity may lead to oxidative damage and inflammation during ALI. These data suggest that inhibition of G6PD activity normalizes LPS‐induced oxidant–anti‐oxidant imbalance in AECs. Overall, these data depict that inhibition of G6PD may be a good strategy to dampen airway inflammation/permeability associated with ALI.
Figure 4.
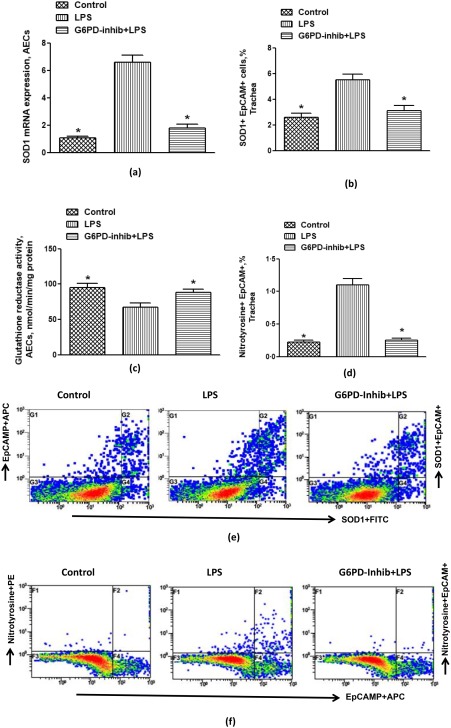
Effect of glucose‐6‐phosphate dehydrogenase (G6PD) inhibitor, 6‐aminonicotinamide on lipopolysaccharide (LPS)‐induced superoxide dismutase 1 (SOD1) and nitrotyrosine expression in airway epithelial cells (AECs) of mice. (a) SOD1 mRNA expression, (b) % SOD1 + epithelial cell adhesion molecule (EpCAM)+ cells, (c) glutathione reductase (GR) activity, (d) % nitrotyrosine + EpCAM+ cells, (e) representative dot‐plot for SOD1 immunostaining in EpCAM+ cells and (f) representative dot‐plot for nitrotyrosine immunostaining in EpCAM+ cells. Dot‐plots show events in tracheal epithelial cells taken from every group. Values are expressed as mean ± standard error (s.e.), n = 6–8/group. *P< 0·05 versus LPS group. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
NADPH‐producing enzyme, G6PD, plays key roles in the regulation of the redox balance as well as lipid/cholesterol synthesis. Under certain inflammatory conditions, it has been demonstrated that G6PD over‐expression leads to an increase in NOX2‐derived ROS due to the increased supply of NADPH 9, 31, 32. These observations suggest that up‐regulated G6PD might be one of the mediators for tissue inflammation. Therefore, to decipher the pathophysiological role of G6PD in LPS‐induced airway inflammation, we used the G6PD inhibitor. Our data show that G6PD inhibition leads to a decrease in airway inflammation, which is due probably to a decrease in NOX2‐derived ROS generation in AECs in the LPS model of ALI.
ROS generation is critically important in the pathogenesis of several lung diseases, such as acute respiratory distress syndrome, chronic obstructive pulmonary disease, asthma and cystic fibrosis. ROS may be released from conventional sources such as neutrophils. This usually involves activation of membrane‐bound NOX2, along with cytosolic members leading to the production of ROS 10, 21, 22. Over‐expression of NOX2 in the pulmonary system has been shown to lead to ROS generation. It has also been demonstrated that ROS generated by NOX2 mediates oxidative damage to pulmonary tissue in ALI 10, 22. Recently, AECs have also been shown to be a source of ROS production via NOX2 22, 33. However, the role of G6PD in modulation of NOX2‐mediated ROS in AECs during LPS‐induced ALI has not been explored previously.
The major biological importance of G6PD is to supply the cell with most abundant reducing co‐enzyme, NADPH. This co‐enzyme is produced by G6PD and utilized in several biosynthetic pathways, such as fatty acid and cholesterol biosynthesis. However, G6PD also plays an important role in the maintenance of the cellular redox pool, as well ROS production by NADPH to glutathione reductase and NOX2, respectively. Several studies have shown that G6PD may be proinflammatory as well as anti‐inflammatory, depending on its expression and organ involved. G6PD deficiency has been shown to be involved in oxidative stress and inflammation, as it supplies reducing equivalents for the reduction of ROS 11, 15, 34. This mechanism is responsible for the removal of ROS in cells under oxidative stress. However, our study shows increased G6PD activity along with NOX2‐mediated ROS generation in AECs, which could account for ALI‐associated pulmonary inflammation.
Several previous studies have also reported increased G6PD activity with concomitant oxidative stress due to a greater supply of NADPH to NADPH oxidase 17, 18, 31. For example, the aberrant up‐regulation of G6PD in adipose tissue/macrophages has been shown to lead to oxidative stress and the expression of proinflammatory cytokines with concomitant dysregulation of lipid metabolism and insulin resistance in adipocytes 14, 35. Similarly, up‐regulation of G6PD in the heart, liver and pancreas of animals with different inflammatory diseases causes elevation in oxidative stress, thereby causing functional dysregulations in the respective tissues 18, 31, 36. LPS‐induced acute renal injury has also been shown to elevate G6PD activity and renal dysfunction 37. Inflammatory signals such as interleukin (IL)‐1β have also been shown to cause vascular damage associated with hyperglycaemia through the increased pentose phosphate pathway and NOX2‐derived ROS 19. Altogether, the previous findings, along with our study, suggest that aberrant G6PD up‐regulation in inflammatory conditions might channelize NADPH towards oxidant production rather than oxidant removal.
Suppression of ROS generation in AECs in response to pharmacological inhibition of G6PD without affecting NOX2 expression suggests that the diminished supply of NADPH might be responsible for the attenuation of ROS. An earlier study showed increased G6PD activity in a rabbit model of ALI; however, it did not investigate whether G6PD inhibition would be beneficial or harmful during ALI 20. Our study shows that G6PD inhibition leads to a decrease in ROS and airway inflammation. Vimercati et al. 38 also showed beneficial effects of the inhibition of G6PD in failing hearts due to a reduction in oxidative stress. Therefore, we propose that inhibition of G6PD and the resultant decrease in ROS could be a factor in attenuation of airway inflammation.
G6PD up‐regulation occurring under various pathological conditions could be either a protective or destructive mechanism, depending on whether ROS are produced or scavenged 11, 15, 17, 18, 31, 34. In the present scenario, instead of protecting the lung from LPS‐induced inflammation, G6PD up‐regulation turned into a detrimental response that contributed to ALI. Therefore, keeping NADPH production within physiological levels would limit pulmonary damage. The marked elevation of ROS and nitrotyrosine in AECs, which was prevented by 6‐aminonicotinamide, suggests strongly that elevated G6PD activity worsened pulmonary oxidative inflammation, due probably to excess ROS generated by NOX2.
Interestingly, up‐regulation of SOD1 seems to be an adaptive mechanism to remove increased ROS; however, it may have led to further elevation in oxidative inflammation, as depicted by increased nitrotyrosine formation in AECs. This could be due to unchanged GPx1 and decreased GR activity which would lead to a build‐up of hydrogen peroxide produced by SOD1. These observations suggest that although SOD1 may be up‐regulated as an adaptive anti‐oxidant mechanism, the overall balance shifts towards increased generation of ROS due to greater channelization of NADPH towards NOX2 during ALI. Decreased GR activity in our study is also an indication of increased utilization of NADPH towards NOX2‐derived ROS.
Oxidants such as peroxynitrite have been shown to cause reduction in GR activity 39. It was evident in our study by increased nitrotyrosine expression, which is a measure of peroxynitrite‐mediated damage 10, 39. In light of our observations, we propose that G6PD, NADPH oxidase and SOD1 become activated whereas, as GR activity falls, all these in combination lead to enhanced ROS production and oxidative inflammation observed during LPS‐induced ALI. Earlier studies have also shown alterations in different enzymatic anti‐oxidants such as SOD, GPx and GR during ALI 20, 40, 41.
ROS play an important role in increased epithelial/endothelial permeability as well as production of inflammation. The ROS produced by epithelial cells may promote endothelial dysfunction by oxidation of crucial cellular signalling proteins or disruption of junctional proteins 7, 10. For example, hydrogen peroxide may diffuse easily through cell membranes due to being an uncharged molecule. Hydrogen peroxide primarily attacks the endothelial cell membrane, resulting in endothelial cell damage and an early increase in vascular permeability. The effect of hydrogen peroxide may be amplified further in the presence of transition metals via the Fenton reaction leading to hydroxyl radical formation 7, 10. Our study shows a decrease in total BALF protein concentration with concomitant reduction in ROS generation. This suggests that a diminished ROS production due to inhibition of G6PD might be responsible for an overall decrease in pulmonary vascular permeability.
In summary we have demonstrated for the first time, to the best of our knowledge, that increase in NOX2‐derived ROS generation is dependent upon enhanced G6PD and decreased GR activity in the AECs of LPS‐treated mice. More importantly, suppression of G6PD activity by 6‐aminonicotinamide leads to a decrease in airway inflammation/permeability due probably to reduction in NOX2‐derived ROS and subsequent oxidative stress. Our results imply that G6PD might be a novel target for therapeutic intervention to reduce NOX2‐derived oxidative stress, and ameliorate pulmonary inflammation during ALI.
Disclosure
All authors declare no conflicts of interest.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group project no. RG‐1438‐019.
References
- 1. Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol 2005; 33:319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rubenfeld GD, Caldwell E, Peabody E et al Incidence and outcomes of acute lung injury. N Engl J Med 2005; 353:1685–93. [DOI] [PubMed] [Google Scholar]
- 3. Herold S, Gabrielli NM, Vadasz I. Novel concepts of acute lung injury and alveolar–capillary barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 2013; 305:L665–81. [DOI] [PubMed] [Google Scholar]
- 4. Faubel S, Edelstein CL. Mechanisms and mediators of lung injury after acute kidney injury. Nat Rev Nephrol 2016; 12:48–60. [DOI] [PubMed] [Google Scholar]
- 5. Singbartl K, Bishop JV, Wen X et al Differential effects of kidney–lung cross‐talk during acute kidney injury and bacterial pneumonia. Kidney Int 2011; 80:633–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal 2006; 8:1549–61. [DOI] [PubMed] [Google Scholar]
- 7. Mittal M, Siddiqui MR, Tran K et al Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 2014; 20:1126–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van der Vliet A. NADPH oxidases in lung biology and pathology: host defense enzymes, and more. Free Radic Biol Med 2008; 44:938–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hecker PA, Leopold JA, Gupte SA et al Impact of glucose‐6‐phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am J Physiol Heart Circ Physiol 2013; 304:H491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nadeem A, Siddiqui N, Alharbi NO, Alharbi MM. Airway and systemic oxidant–antioxidant dysregulation in asthma: a possible scenario of oxidants spill over from lung into blood. Pulm Pharmacol Ther 2014; 29:31–40. [DOI] [PubMed] [Google Scholar]
- 11. Sanna F, Bonatesta RR, Frongia B et al Production of inflammatory molecules in peripheral blood mononuclear cells from severely glucose‐6‐phosphate dehydrogenase‐deficient subjects. J Vasc Res 2007; 44:253–63. [DOI] [PubMed] [Google Scholar]
- 12. Salvemini F, Franzé A, Iervolino A, Filosa S, Salzano S, Ursini MV. Enhanced glutathione levels and oxidoresistance mediated by increased glucose‐6‐phosphate dehydrogenase expression. J Biol Chem 1999; 274:2750–7. [DOI] [PubMed] [Google Scholar]
- 13. Park J, Rho HK, Kim KH, Choe SS, Lee YS, Kim JB. Overexpression of glucose‐6‐phosphate dehydrogenase is associated with lipid dysregulation and insulin resistance in obesity. Mol Cell Biol 2005; 25:5146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ham M, Choe SS, Shin KC et al Glucose‐6‐phosphate dehydrogenase deficiency improves insulin resistance with reduced adipose tissue inflammation in obesity. Diabetes 2016; 65:2624–38. [DOI] [PubMed] [Google Scholar]
- 15. Xu Y, Zhang Z, Hu J et al Glucose‐6‐phosphate dehydrogenase‐deficient mice have increased renal oxidative stress and increased albuminuria. FASEB J 2010; 24:609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leopold JA, Zhang YY, Scribner AW, Stanton RC, Loscalzo J. Glucose‐6‐phosphate dehydrogenase overexpression decreases endothelial cell oxidant stress and increases bioavailable nitric oxide. Arterioscler Thromb Vasc Biol 2003; 23:411–7. [DOI] [PubMed] [Google Scholar]
- 17. Park J, Choe SS, Choi AH et al Increase in glucose‐6‐phosphate dehydrogenase in adipocytes stimulates oxidative stress and inflammatory signals. Diabetes 2006; 55:2939–49. [DOI] [PubMed] [Google Scholar]
- 18. Gupte RS, Floyd BC, Kozicky M et al Synergistic activation of glucose‐6‐phosphate dehydrogenase and NAD(P)H oxidase by Src kinase elevates superoxide in type 2 diabetic, Zucker fa/fa, rat liver. Free Radic Biol Med 2009; 47:219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peiró C, Romacho T, Azcutia V et al Inflammation, glucose, and vascular cell damage: the role of the pentose phosphate pathway. Cardiovasc Diabetol 2016; 15:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kozar RA, Weibel CJ, Cipolla J et al Antioxidant enzymes are induced during recovery from acute lung injury. Crit Care Med 2000; 28:2486–91. [DOI] [PubMed] [Google Scholar]
- 21. Diebold BA, Smith SM, Li Y, Lambeth JD. NOX2 as a target for drug development: indications, possible complications, and progress. Antioxid Redox Signal 2015; 23:375–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro‐inflammatory mediators‐induced airway and pulmonary diseases. Biochem Pharmacol 2012; 84:581–90. [DOI] [PubMed] [Google Scholar]
- 23. Matute‐Bello G, Winn RK, Martin TR, Liles WR. Sustained lipopolysaccharide‐induced lung inflammation in mice is attenuated by functional deficiency of the Fas/Fas ligand system. Clin Diagn Lab Immunol 2004; 11:358–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nadeem A, Siddiqui N, Al‐Harbi NO, Attia SM, AlSharari SD, Ahmad SF. Acute lung injury leads to depression‐like symptoms through upregulation of neutrophilic and neuronal NADPH oxidase signaling in a murine model. Int Immunopharmacol 2017; 47:218–26. [DOI] [PubMed] [Google Scholar]
- 25. Nadeem A, Alharbi NO, Vliagoftis H, Tyagi M, Ahmad SF, Sayed‐Ahmed MM. Proteinase activated receptor‐2‐mediated dual oxidase‐2 up‐regulation is involved in enhanced airway reactivity and inflammation in a mouse model of allergic asthma. Immunology 2015; 145:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nadeem A, Al‐Harbi NO, Ansari MA et al Psoriatic inflammation enhances allergic airway inflammation through IL‐23/STAT3 signaling in a murine model. Biochem Pharmacol 2017; 124:69–82. [DOI] [PubMed] [Google Scholar]
- 27. Nadeem A, Ahmad SF, Al‐Harbi NO, El‐Sherbeeny AM, Al‐Harbi MM, Almukhlafi TS. GPR43 activation enhances psoriasis‐like inflammation through epidermal upregulation of IL‐6 and dual oxidase 2 signaling in a murine model. Cell Signal 2017; 33:59–68. [DOI] [PubMed] [Google Scholar]
- 28. Carlberg I, Mannervik B. Glutathione reductase. Methods Enzymol 1985; 113:484–90. [DOI] [PubMed] [Google Scholar]
- 29. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐delta delta C(T)) method. Methods 2001; 25:402–8. [DOI] [PubMed] [Google Scholar]
- 30. Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med 1999; 27:612–6. [DOI] [PubMed] [Google Scholar]
- 31. Gupte RS, Vijay V, Marks B et al Upregulation of glucose‐6‐phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J Card Fail 2007; 13:497–506. [DOI] [PubMed] [Google Scholar]
- 32. Serpillon S, Floyd BC, Gupte RS et al Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose‐6‐phosphate dehydrogenase‐derived NADPH. Am J Physiol Heart Circ Physiol 2009; 297:H153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tickner J, Fan LM, Du J, Meijles D, Li JM. Nox2‐derived ROS in PPARγ signaling and cell‐cycle progression of lung alveolar epithelial cells. Free Radic Biol Med 2011; 51:763–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stanton RC. Glucose‐6‐phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012; 64:362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ham M, Lee JW, Choi AH et al Macrophage glucose‐6‐phosphate dehydrogenase stimulates proinflammatory responses with oxidative stress. Mol Cell Biol 2013; 33:2425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee JW, Choi AH, Ham M et al G6PD up‐regulation promotes pancreatic beta‐cell dysfunction. Endocrinology 2011; 152:793–803. [DOI] [PubMed] [Google Scholar]
- 37. Smith JA, Stallons LJ, Schnellmann RG. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin‐induced acute kidney injury. Am J Physiol Renal Physiol 2014; 307:F435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vimercati C, Qanud K, Mitacchione G et al Beneficial effects of acute inhibition of the oxidative pentose phosphate pathway in the failing heart. Am J Physiol Heart Circ Physiol 2014; 306:H709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 2007; 6:662–80. [DOI] [PubMed] [Google Scholar]
- 40. Ji L, Liu R, Zhang XD, Chen HL et al N‐acetylcysteine attenuates phosgene‐induced acute lung injury via up‐regulation of Nrf2 expression. Inhal Toxicol 2010; 22:535–42. [DOI] [PubMed] [Google Scholar]
- 41. Hochscheid R, Schuchmann U, Kotte E, Kranz S, Heinrichs S, Müller B. NO2‐induced acute and chronic lung injury cause imbalance of glutathione metabolism in type II pneumocytes. Med Sci Monit 2005;11:BR273–9. [PubMed] [Google Scholar]