

Abstract
Background & Aims
Fibrolamellar hepatocellular carcinoma (FL-HCC) is a primary liver cancer that predominantly affects young adults with no underlying liver disease. A somatic, 400 Kb deletion on chromosome 19 that fuses part of the DnaJ heat shock protein family (Hsp40) member B1 gene (DNAJB1) to the protein kinase cAMP-activated catalytic subunit alpha gene (PRKACA) has been repeatedly identified in patients with FL-HCC. However, the DNAJB1–PRKACA gene fusion has not been shown to induce liver tumorigenesis. We used the CRISPR/Cas9 technique to delete in mice the syntenic region on chromosome 8 to create a Dnajb1–Prkaca fusion and monitored the mice for liver tumor development.
Methods
We delivered CRISPR/Cas9 vectors designed to juxtapose exon 1 of Dnajb1 with exon 2 of Prkaca to create the Dnajb1–Prkaca gene fusion associated with FL-HCC, or control Cas9 vector, via hydrodynamic tail vein injection to livers of 8 week-old female FVB/N mice. These mice did not have any other engineered genetic alterations and were not exposed to liver toxins or carcinogens. Liver tissues were collected 14 months after delivery; genomic DNA was analyzed by PCR to detect the Dnajb1–Prkaca fusion, and tissues were characterized by histology, immunohistochemistry, RNA sequencing, and whole-exome sequencing.
Results
Livers from 12 of the 15 mice given the vectors to induce the Dnajb1–Prkaca gene fusion, but none of the 11 mice given the control vector, developed neoplasms. The tumors contained the Dnajb1–Prkaca gene fusion and had histologic and cytologic features of human FL-HCCs: large polygonal cells with granular, eosinophilic, and mitochondria-rich cytoplasm, prominent nucleoli, and markers of hepatocytes and cholangiocytes. In comparing expression levels of genes between the mouse tumor and non-tumor liver cells, we identified changes similar to those detected in human FL-HCC, which included genes that affect cell cycle and mitosis regulation. Genomic analysis of mouse neoplasms induced by the Dnajb1–Prkaca fusion revealed a lack of mutations in genes commonly associated with liver cancers, as observed in human FL-HCC.
Conclusions
Using CRISPR/Cas9 technology, we found generation of the Dnajb1–Prkaca fusion gene in wild-type mice to be sufficient to initiate formation of tumors that have many features of human FL-HCC. Strategies to block DNAJB1–PRKACA might be developed as therapeutics for this form of liver cancer.
Keywords: liver cancer, protein kinase A, genomic engineering, mouse model
Introduction
Fibrolamellar hepatocellular carcinoma (FL-HCC) is a rare form of liver cancer that typically arises in children and young adults with no history of cirrhosis or other liver diseases1. The cancer carries high mortality with 5-year survival below 45%2. Surgery is presently the only effective therapy if the cancer is diagnosed before the occurrence of metastases, and long-term survival is jeopardized by tumor recurrence2. FL-HCC has pathodiagnostic features distinct from the predominant liver cancers, classical hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA), which include large eosinophilic and mitochondria-rich polygonal cells with prominent nucleoli and lamellar bands of fibrosis3,4. FL-HCC expresses markers for hepatic progenitor, biliary and hepatocytic lineages; however, none of these are specific for this tumor5,6.
The molecular basis of FL-HCC has been enigmatic, as none of the major drivers of other liver cancers, as for instance CTNNB1, TP53 or KRAS, have been found mutated in FL-HCC. Recently, however, a ~400 kilobase (Kb) somatic deletion on chromosome 19 was identified in primary tumor samples from FL-HCC patients7,8. The deletion involves breakpoints that are positioned within intron one, or less frequently within exon 2, of DNAJB1, which encodes a heat shock 40 protein family member, and within intron one of PRKACA, which encodes the adenosine 3′,5′-monophosphate (cAMP)-dependent protein kinase A (PKA) catalytic subunit alpha. The deletion generates a fusion gene encompassing exon 1 (and in fewer cases a portion of exon 2) of DNAJB1 and exons 2-10 of PRKACA, producing a chimeric protein that retains PKA kinase activity7,8. Interestingly, the fusion gene is the only known recurrent genomic aberration in FL-HCC, where it has been identified in 80-100% of patients in various studies. By contrast, this aberration has not been reported in any other cancer7,9,10. Mutations in other genes have been detected in FL-HCC tumors along with the DNAJB1–PRKACA alteration8,11,12. However, at present, it remains to be established whether any of the genes found mutated in FL-HCC, most notably the DNAJB1–PRKACA fusion, may have a causative role.
Determining if DNAJB1–PRKACA is capable of initiating tumor formation is an outstanding question regarding the molecular basis of FL-HCC, as this would establish the principal genetic origin of this cancer and identify the fusion protein as a candidate for new targeted therapies. To address this issue, we engineered the first FL-HCC mouse model by using the CRISPR/Cas9 technique to elicit in mice the ~400 Kb chromosomal deletion that creates the DNAJB1–PRKACA fusion in patients. In addition to modeling the exact genomic alteration, we took several other measures to better mimic human FL-HCC. First, because DNAJB1–PRKACA mutation is somatic, we engineered this alteration in the liver of young adult mice through hydrodynamic tail vein delivery of the CRISPR/Cas9 reagents. Second, as we anticipated a low efficiency of the complex engineering task, we mirrored the stochastic and isolated mutational events that occur naturally during tumor formation. Third, because FL-HCC arises in healthy liver, we did not challenge the mice with any liver toxin, as for example CCl4, which is otherwise frequently used to enhance liver cancer by damaging the liver and mimicking liver disease13. Finally, to best test DNAJB1–PRKACA as a potentially sole genetic driver, we engineered the aberration in a wild-type mouse genetic background, nor did we treat the mice with any mutagen like N-nitrosodiethylamine (DEN) that is often employed to increase tumor susceptibility and produce a cancer phenotype, when testing a new, candidate cancer gene in genetically engineered mouse models13.
Strikingly, the Dnajb1–Prkaca engineered mice developed tumors with high frequency that replicated human FL-HCC, as scored on several cytological and histological parameters. Furthermore, similar to human FL-HCC, we found no evidence of other plausible co-driver mutations in Dnajb1–Prkaca elicited tumors.
Our findings demonstrate that the DNAJB1–PRKACA genomic alteration is sufficient to initiate and progress oncogenic transformation in FL-HCC. Together with the presence of the gene fusion in nearly all FL-HCC patients, our data strongly supports DNAJB1–PRKACA as the driver and specific diagnostic biomarker in FL-HCC. Furthermore, the fusion may constitute a promising and possibly sole candidate for targeted therapies in this cancer.
Materials and Methods
Generation of gRNA constructs for engineering the Dnajb1–Prkaca fusion
Three gRNAs were designed to target intron 1 of either mouse Dnajb1 or mouse Prkaca and introduced into pX330-U6-chimeric_BB-CBh-hSpCas9 vector that co-expresses gRNA driven by U6 promoter and Cas9 driven by chicken β-actin hybrid promoter14 (Addgene plasmid #42230). Neuro-2a cells were transfected with the individual pX330 gRNA/Cas9 constructs along with empty pSpCas9(BB)-2A-GFP vector15 at a 6:1 molecular ratio to mark transfected cells with GFP, using X-tremeGENE HP transfection reagent according to the manufacturer’s instructions (Roche). Two days post-transfection, we analyzed the efficiency of the gRNA designs using our protocol for genome editing using FACS enrichment of nuclease expressing cells and indel detection by amplicon analysis (IDAA)16. Briefly, the top-10% most GFP fluorescent cells were isolated by FACS and lysed to 2000 cells/μl in QuickExtract DNA extraction solution (Epicentre). One μl cell extract was used as template in a tri-primer genomic PCR with locus-specific IDAA Fwd and Rev primers that amplified the gRNA target site and a common FamFwd primer identical to an overhang on the Fwd primer, which rendered the amplicons fluorescent (sequences of primers used in this study are provided in Supplementary Table 1). Subsequent analysis of amplicons in a 3500 Genetic Analyzer (Thermo Fisher) revealed the size and frequency of insertion and deletion (indel) mutations elicited by the gRNA designs.
To test the ability of the gRNA constructs to generate the Dnajb1–Prkaca fusion, Neuro-2a cells were co-transfected with combinations of pX330 pairs targeting intron 1 of Dnajb1 and Prkaca, using X-tremeGENE HP transfection reagent. Three days post-transfection, total RNA was extracted using QIAshredder spin columns (Qiagen) and RNeasy Mini Kits (Qiagen). Total RNA was reverse transcribed using TaqMan Reverse Transcription Reagents (Thermo Fisher Scientific) and analyzed by PCR using the primers Dnajb1–RT-Fwd and Prkaca-RT-Rev that amplify the junction of the Dnajb1–Prkaca fusion cDNA. In addition, genomic DNA was extracted and PCR amplified with the primers Dnajb1–Fwd1 and Prkaca–Rev1 that amplify the Dnajb1–Prkaca junction and resulting PCR products were cloned and Sanger sequenced to determine the sequence of the CRISPR/Cas9-induced genomic breakpoint. Amplicons were further analyzed by IDAA using the primers Dnajb1–intron1-IDAA-Fwd and Prkaca–intron1-IDAA-Rev to determine the number and frequency of different fusion alleles present in the population.
CRISPR/Cas9 engineering of Dnajb1–Prkaca in the adult mice liver
The pair of pX330 gRNAs that appeared most optimal with respect to generating the Dnajb1–Prkaca fusion in vitro was delivered to the liver of adult mice via hydrodynamic tail vein injection, which was performed essentially as previously described17,18. Briefly, 8 week-old female FVB/N mice (FVB/NRj, Janvier Labs) were anesthetized with isoflurane (Abbott) and hydrodynamically injected within 5-8 seconds with 50 μg of each pX330 gRNA plasmid suspended in a volume of Ringer’s solution corresponding to approximately 8% (vol/wt) of their body weight. The control cohort was injected with 100 μg of empty pX330 vector expressing Cas9 only. Animal housing and approvals are described in Supplementary Materials and Methods.
Histology
Livers were fixed in 10% neutral buffered formalin (Lillies fixative) for 24 h at 4 °C, submersed in 70% ethanol for 24-48 h, dehydrate d in ethanol and xylene in a tissue processor, and then embedded in paraffin. The livers were cut into 3-5 μM thick sections that were mounted onto glass slides and deparaffinized and rehydrated by standard procedures. Sections were then either stained with hematoxylin and eosin (H&E), with PicroSirius red, with periodic-acid Schiff reagent with or without pretreatment with diastase or subjected to immunohistochemistry using the following antibodies and conditions: rabbit monoclonal anti-cytokeratin 19 (1:200, ab52625, Abcam) and rabbit anti-Ki67 (1:100, ab16667, Abcam) used with antigen retrieval in citrate buffer, pH 6.0 for 20 min at 98 °C. Rabbit monoclonal anti-cytokeratin 7 (1:100, ab181598, Abcam), rabbit polyclonal anti-carcino embryonic antigen (1:30, ab33562, Abcam), rabbit polyclonal anti-carbamoyl phosphate synthetase-1 (HepPar-1) (1:50, ab3682, Abcam), rabbit polyclonal anti-glutamine synthetase (1:1000, ab73593, Abcam) used with antigen retrieval in Tris EGTA buffer, pH 9.0, for 15-20 min at 98°. A Shandon Sequenza slide rack system (Thermo Fisher Scientific) was used to incubate sections with primary antibodies overnight at 4°C and for 45 min at room temperature with EnVision+ System Labeled Polymer–horseradish peroxidase (HRP) anti-rabbit (#K4003, Dako), according to manufacturer’s instructions. Chromogen staining was performed using NovaRED HRP substrate kit (VWR International). All antibodies were diluted in Antibody Diluent with Background Reducing Components (Dako). Counterstaining was performed using Harris haematoxylin (Histolab).
Genomic PCR of the Dnajb1–Prkaca fusion on laser capture microdissected tumors
Parrafin-embedded livers were sectioned at 5 μM thickness, mounted on 1.0 PEN MembraneSlides (Zeiss) and stained with H&E. Desired tissue areas were microdissected onto CapSure HS LCM Caps (Thermo Fisher Scientific) in an Arcturus PixCell II laser capture microdissection system. Genomic DNA was extracted from the captured tissue through incubation of the tissue for 6 h at 65 °C in 10 μl proteinase K-containing extraction buffer followed by 10 min at 95 °C using the Arcturus PicoPure DNA Extraction Kit (Thermo Fisher Scientific). One μl of the extract was used as template in a multiplex PCR that simultaneously amplified the genomic Dnajb1–Prkaca breakpoint to test for the presence of the fusion as well as a nearby region in Prkaca that was not affected by the editing, thereby serving as an internal PCR control. PCR conditions are provided in Supplementary Materials and Methods.
Additional Methods can be found in Supplementary Materials and Methods
Results
CRISPR/Cas9-mediated engineering of Dnajb1–Prkaca in vitro and in vivo
In the mouse genome, Dnajb1 and Prkaca are located on chromosome 8 in a region syntenic to human chromosome 19. To engineer in mice the complex chromosomal rearrangement that generates DNAJB1–PRKACA in human FL-HCC, we made use of CRISPR/Cas9 technology. In our approach, guide (g)RNAs were designed to target Cas9 to intron 1 of murine Dnajb1 and intron 1 of murine Prkaca and thereby introduce DNA double-strand cuts in the regions, where chromosomal breaks occur in the vast majority of human FL-HCC analyzed (Figure 1A)7. When co-introduced in cells, we expected the CRISPR/Cas9 reagents to excise the desired ~400 Kb genomic DNA fragment, and the cellular repair machinery to join the DNA ends and create the Dnajb1–Prkaca fusion, as occurring in human FL-HCC.
Figure 1. Generation of CRISPR/Cas9 reagents that engineer the Dnajb1–Prkaca fusion in vitro and in adult mouse liver.
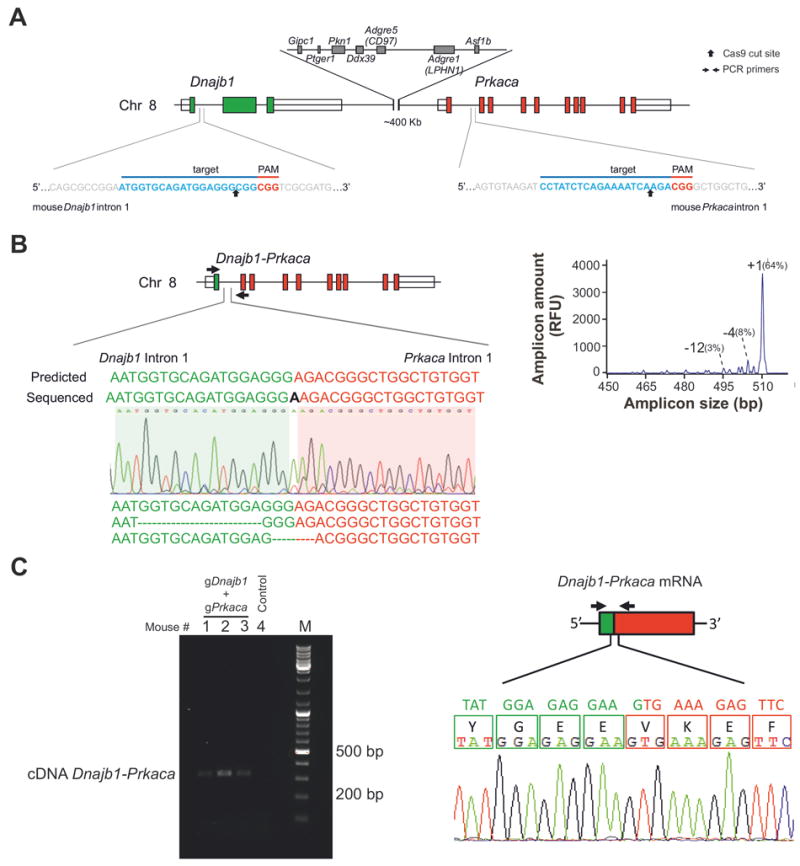
(A) In the mouse genome, Dnajb1 and Prkaca are located on chromosome 8 in a region syntenic to human chromosome19. The target sequences and locations of the gRNA pair used to engineer the Dnajb1–Prkaca fusion are shown. (B) Ability of the gRNA pair shown in (A) to engineer the Dnajb1–Prkaca fusion in transfected Neuro-2a cells. (Left) Schematic of the Dnajb1–Prkaca genomic fusion and location of primers (arrows) used to amplify the breakpoint. Sanger sequencing chromatogram and sequences for various fusion breakpoints are shown. “Predicted” sequence indicates gene fusion without any indel mutagenesis. (Right) IDAA profile showing the frequency of the various breakpoint amplicons. (C) Ability of the gRNA pair shown in (A) to engineer the Dnajb1–Prkaca fusion in the liver of hydrodynamically tail vein injected mice. (Left) Dnajb1–Prkaca specific PCR from cDNA derived from the liver of mice injected with the gRNA pair (Right) Schematic of the fusion transcript and location of primers (arrows) used to amplify the fusion breakpoint. The Sanger sequencing chromatogram demonstrates an in-frame fusion transcript.
Thus, we first engineered constructs expressing Cas9 and single-guide (sg)RNA in the pX330 vector14 targeting either Dnajb1 intron 1 or Prkaca intron 1 and assayed their efficiency in generating cuts at the respective target sites two days after their individual transfection into mouse Neuro-2a cells. To this end, we used our recent protocol16 to quantify the frequency of indel mutations generated by the cellular DNA repair machinery as a measure of the cutting efficiency of our CRISPR/Cas9 constructs (Supplementary Figure 1A). Next, we co-transfected the various construct pairs into Neuro-2a cells and assessed their ability to generate the Dnajb1–Prkaca fusion three days post-transfection. Analysis of genomic DNA and reverse-transcribed RNA by PCR and Sanger sequencing of the amplicons demonstrated the presence of the intended chromosomal rearrangement and the expression of the proper fusion transcript (Figure 1B, Supplementary Figure 1B).
Having confirmed the feasibility of our approach to engineer the desired chromosomal rearrangement in vitro, we combined our CRISPR/Cas9 reagents with hydrodynamic delivery in an attempt to create the Dnajb1–Prkaca aberration in the liver of young adult mice. Thus, we hydrodynamically tail vein injected 8 week-old mice with our most optimal CRISPR/Cas9 plasmid pair targeting Dnajb1 and Prkaca, or with empty pX330 control plasmid. Three days post-injection, the mice were sacrificed and their livers analyzed. Genomic DNA and mRNA analyses showed the presence of the Dnajb1–Prkaca fusion and detectable levels of fusion transcript in mice injected with the CRISPR/Cas9 plasmid pair (Figure 1C and Supplementary 1C). We estimated that the frequency of the Dnajb1–Prkaca genomic fusion was approximately 0.7-1.5 copy/100 cells in the CRISPR/Cas9 edited livers (data not shown).
The Dnajb1–Prkaca fusion induces FL-HCC in mice
A cohort of 8 week-old wild-type mice was hydrodynamically tail vein injected with the CRISPR/Cas9 plasmid pair for generation of the Dnajb1–Prkaca aberration or with empty pX330 control plasmid. Mice were sacrificed 14 months post-injection and their livers analyzed for the presence of alterations. Twelve out of 15 (80%) CRISPR/Cas9-injected mice and 0/11 control-injected mice presented oncogenic lesions in the liver parenchyma that varied in size (Figure 2, Supplementary Figure 3). Mouse FL-HCC driven by the fusion showed most of the specific cytological and histological features of human FL-HCC, as illustrated by a large brownish and fully surfaced tumor (10-12 mm) shown in Figure 2A. As in human, mouse FL-HCC appears as a relatively well circumscribed mass with pushing margin against the non-tumorous tissue, and composed of solid sheets and tightly packed cords and trabeculae of large polygonal hepatocyte-like cells separated by variably dilated sinusoids. Furthermore, tumor cells contained large nuclei with coarse chromatin and one or more prominent nucleoli (Figure 2B and 2C)19,20, and their abundant cytoplasm was characteristically granular and eosinophilic in analogy with the “oncocyte-like” aspects of the mitochondria-rich cytoplasm in cells of human FL-HCC21. Indeed, ultrastructural analysis obtained by transmission electron microscopy confirmed the presence of tightly packed mitochondria in cells from mouse FL-HCC and also evidenced the characteristic nuclear morphology of these oncocytic hepatocytes22,23 (Figure 2D and Supplementary Figure 2A). Additionally, tumor cells contained inclusions resembling the intracellular “pale bodies” as well as the hyaline globules often observed in human FL-HCC (Figure 2C, Supplementary Figure 2B), and accordingly, these globules contained material that was positive for the periodic acid-Schiff (PAS) stain and sensitive to diastase enzymatic digestion (Figure 2E, and data not shown)1. Importantly, the tumor was positive for the intended CRISPR/Cas9-induced Dnajb1–Prkaca fusion and expressed the chimeric in-frame transcript, as demonstrated by genomic DNA and reverse transcribed mRNA sequencing (Supplementary Figure 2C). No sign of off-target cutting elicited by the CRISPR/Cas9 tools was evident in the top ranking predicted off-target sites for both gRNAs24, as assessed by IDAA (Supplementary Figure 2D and data not shown).
Figure 2. The Dnajb1–Prkaca fusion induces FL-HCC in mice.
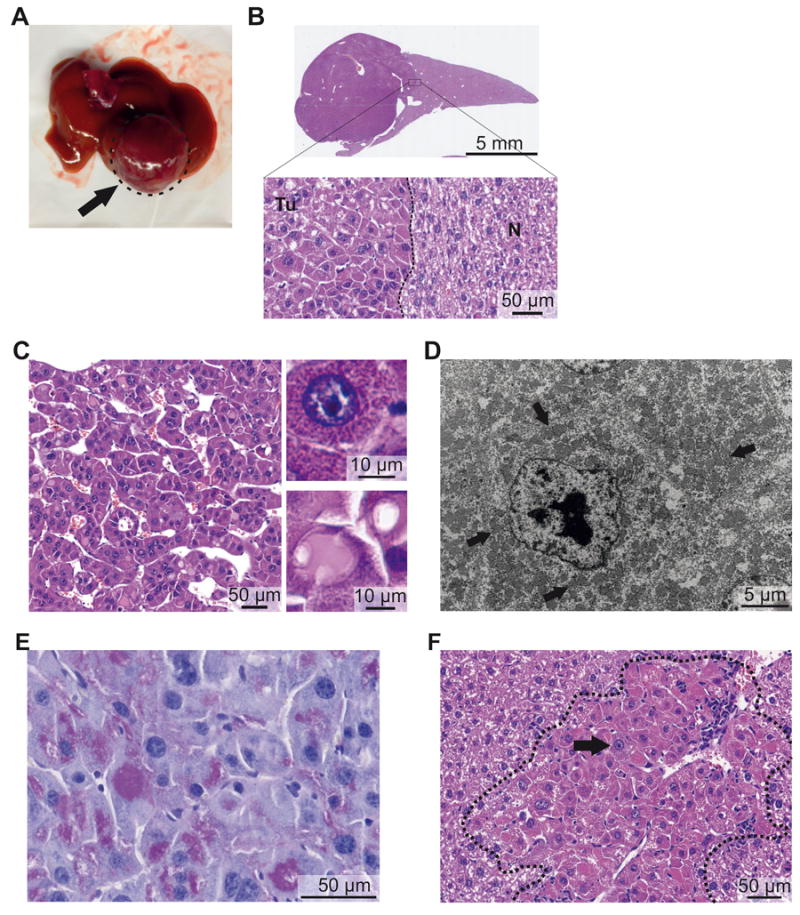
Representative macroscopic and microscopic images of mouse FL-HCC elicited by Dnajb1–Prkaca. (A) Macroscopic image of a tumor (arrow). (B) Whole-scan H&E image and magnification showing tumor (Tu)-non-tumor (N) border. (C) Microscopic H&E image of tumor area showing trabeculae of tumor cells separated by variably dilated sinusoids. (Upper inset) Detail of an “oncocytic” tumor cell with granular eosinophilic cytoplasm and large nucleus with prominent nucleolus. (Lower inset) Detail of “pale body” and hyaline globulus. (D) Transmission electron micrograph of tumor cell showing cytoplasm packed with mitochondria (arrows) and nucleus with prominent nucleolus and coarse chromatin. (E) Periodic acid-Schiff staining of hyaline globules. (F) Small neoplastic lesion with large oncocytic cells with granular eosinophilic cytoplasm and prominent nucleoli (black arrow) and leukocyte infiltration (white arrow).
The smaller neoplastic lesions (<1 mm) were also characterized by the distinctive “oncocytic” hepatocytes described in human FL-HCC. As observed for the large cells that constitute both human and murine FL-HCC, the “oncocytic” hepatocyte-like cells in these lesions were also large, also contained granular eosinophilic-orange cytoplasm, large nuclei and prominent nucleoli (Figure 2F and Supplementary Figure 3B). Such oncocytic cells were also present in transition areas between the large tumor shown in Figure 2A and the surrounding non-tumorous tissue, hence supporting the notion that these foci represent precursor lesions for FL-HCC. The small lesions often showed various degrees of leukocyte infiltration inside and around the foci (Figure 2F and Supplementary Figure 3B). All the large and small Dnajb1–Prkaca-elicited lesions showed the cytological characteristics of FL-HCC described above. Only one lesion, in fact, contained premalignant clear cells intermingled with oncocytic hepatocytes with prominent nucleoli and eosinophilic cytoplasm (Supplementary Figure 3C), in accordance with the observation that FL-HCC may occasionally appear as a mixed-FL-HCC, which has also been reported positive for the fusion25. Genomic DNA analysis of laser capture microdissected small lesions confirmed the presence of the oncogenic Dnajb1–Prkaca aberration (Supplementary Figure 3D).
PicroSirius red staining for collagen showed mild thickening of the collagen connective tissue network accompanying the tumor cells (Figure 3A), whereas the non-tumorous liver parenchyma was completely devoid of fibrotic change (Supplementary Figure 4A). The collagen bands in murine FL-HCC were not as thick as in human FL-HCC and no large, central collagen scar was observed, which however is consistent with the limited tendency of mouse liver to develop collagen fibrosis without chemical challenge26.
Figure 3. Histological characterization of Dnajb1–Prkaca elicited mouse FL-HCC.
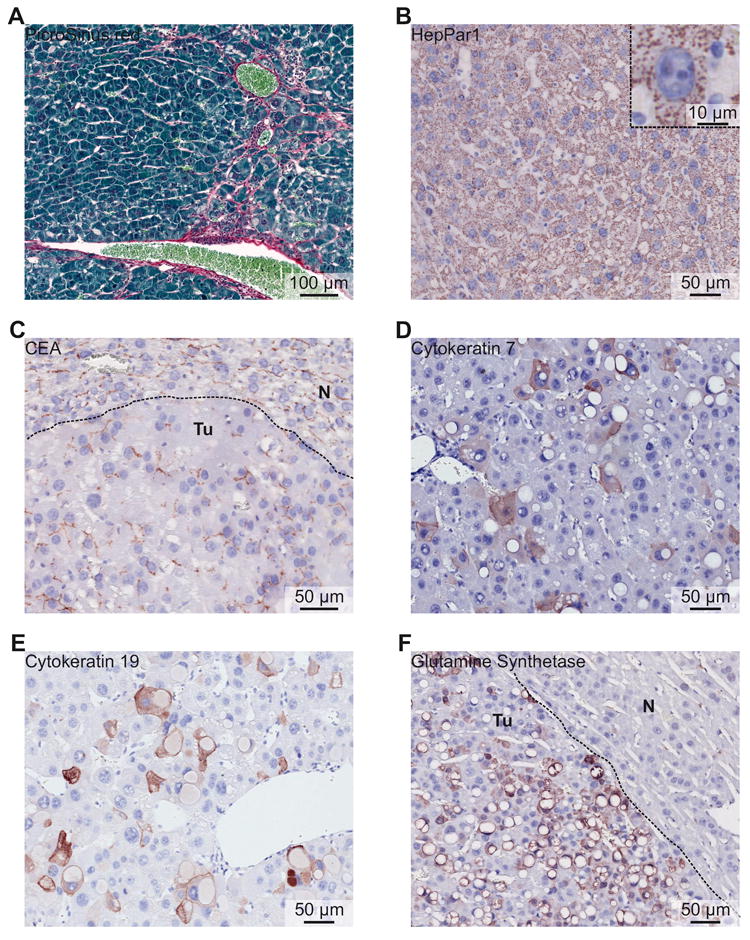
(A) PicroSirius red staining showing mild collagen fibrosis between tumor cells. (B) Tumor cells express HepPar1. (Inset) Detail of a tumor cell expressing HepPar1 with mitochondrial localization and illustrating the mitochondria-rich cytoplasm. (C) Carcinoembryonic antigen (CEA) staining showing canalicular distribution in tumor area (Tu), as well as in normal liver area (N). (D) Cytokeratin 7 expression in tumor area. (E) Cytokeratin 19 expression in tumor area. (F) Broad expression of glutamine synthetase by tumor cells (Tu), but not by hepatocytes of the normal liver area (N).
Immunohistochemical characterization showed that mouse FL-HCC elicited by Dnajb1–Prkaca expresses both hepatocyte and cholangiocyte markers, as reported for human FL-HCC5,11. Indeed, mouse FL-HCC was positive for the hepatocytic markers hepatocyte paraffin 1 (HepPar1) and carcinoembryonic antigen (CEA) with a canalicular distribution (Figure 3B and C), as frequently observed in human FL-HCC5,11. Furthermore, mouse FL-HCC showed scattered expression of cytokeratin 7 and cytokeratin 19 (Figure 3D and E), two markers that are frequently and occasionally expressed, respectively, by human FL-HCC5,11, whereas in normal liver these proteins are present only in the cholangiocytes of the bile ducts and in hepatic progenitor cells (Supplementary Figure 4B and C). Finally, mouse FL-HCC expressed glutamine synthetase (Figure 3F), as occurs in both HCC and CCA with similar frequency27, whereas in normal liver only hepatocytes adjacent to the central vein are positive (Supplementary Figure 4D).
Molecular analysis supports Dnajb1–Prkaca as the key driver of initiation and progression of FL-HCC
To gain further insight into the mechanisms underlying neoplastic transformation induced by Dnajb1–Prkaca, we performed transcriptomic and genomic analyses of the lesions. RNA-seq analysis of total RNA isolated from a tumor where freshly frozen tissue was available and from freshly frozen tissue from control injected mouse livers confirmed the expression of the proper Dnajb1–Prkaca fusion transcript in the tumor, with juxtaposition of exon 1 of Dnajb1 to exon 2 of Prkaca (Figure 4A and Supplementary Table 2). Gene set enrichment analysis (GSEA) of the differentially expressed genes between tumor and liver control against the Molecular Signatures Database (Broad Institute, C5.bp), revealed a robust enrichment of genes in processes associated with cell cycle control and mitosis regulation, and a down-regulation of genes involved in regulation of metabolic processes (GSEA: false discovery rate (FDR) p-value, <0.05) (Supplementary Table 3, 4 and 5). Further results for positively and negatively regulated enriched gene sets are presented in Supplementary Table 6 and 7.
Figure 4. Molecular analysis of Dnajb1–Prkaca elicited mouse FL-HCC.
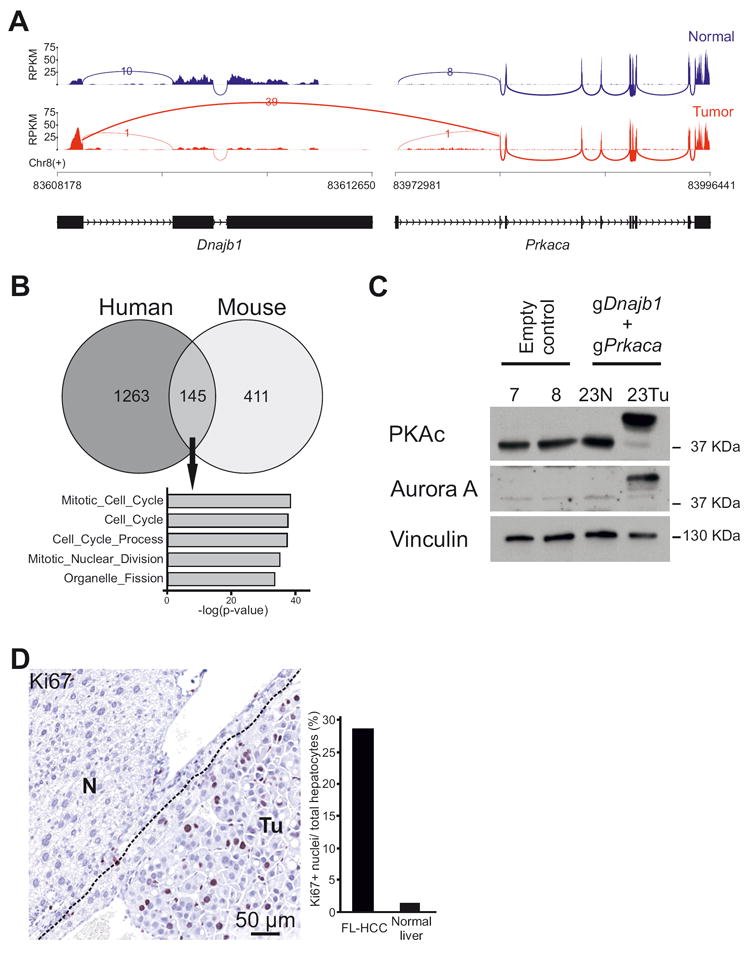
(A) Sashimi plot of RNA-seq read coverage for Dnajb1 and Prkaca in normal liver and mouse FL-HCC. Peaks depict reads per kilobase per million reads mapped (RPKM). Arcs depict reads spanning splice junctions. (B) Venn Diagram showing overlap of differentially expressed genes between human (Simon et al.29) and mouse FL-HCC. Differentially expressed genes for mouse FL-HCC were defined by a BaseMean value >25 and log2FC >1.5 over normal liver. Gene Ontology Biological Process (MSigDB, c5.bp) is shown for the 145 common differentially expressed genes, showing enrichment of mitosis and proliferation genes. (C) Immunoblotting of tissue lysates from mouse FL-HCC tumor (Tu), adjacent normal liver (N) or livers from control mice. PKAc: immunoblotting using an antibody raised against the catalytic subunit alpha of PKA. Vinculin is used as loading control. (D) Ki67-staining of tumor cells (Tu) and adjacent normal tissue (N). Bars represent quantification of Ki67 positive cells/number of hepatocytes.
Several studies have contributed to the characterization of the transcriptomic profile of human FL-HCC by comparing expression data from tumor specimen to that of normal liver or to that of other liver cancers10,11,28-30. We compared the differentially expressed genes between mouse FL-HCC and normal mouse liver to those from three different human FL-HCC studies8,11,29 and identified a strong enrichment (GSEA: familywise error rate (FWER) adjusted p-value, <0.001) of mouse FL-HCC genes in the downregulated and upregulated gene sets from these human studies, thus further confirming the resemblance of mouse tumor to human FL-HCC (Supplementary Figure 5A). In particular, common differentially expressed genes between RNA-seq data from human29 and mouse FL-HCC, include directionally concordant genes involved in cell cycle and mitosis regulation processes (Figure 4B) (List of genes in Supplementary Table 8 and 9). The mouse FL-HCC transcriptome also displayed a profile closest resembling that of mature hepatocytes as evidenced by a comparison between our dataset to mouse RNA-seq data available in the literature31 (Supplementary Figure 5B).
Notably, human FL-HCC was reported to express high levels of the cytokinesis and mitosis regulator Aurora kinase A29,30 and a clinical trial is ongoing to test the efficacy of its inhibition in FL-HCC patients32. Our RNA-seq data and immunoblotting of tissue lysate from mouse FL-HCC tumor showed high expression of Aurora kinase A, as compared to the non-tumorous area of the liver and control-injected livers (Supplementary Table 3 and Figure 4C) and, as expected, expression of the DNAJB1–PRKACA fusion protein, thus further corroborating the rationale of targeting Aurora kinase A in clinical trials. Moreover, tumors showed a high proliferation index revealed by Ki67 staining, as opposed to normal hepatocytes in the adjacent non-tumorous areas (Figure 4D, Supplementary Figure 3A).
Human FL-HCC has been concordantly shown to have a relative stable genome with fewer mutations than other liver cancers and, with the exception of the DNAJB1–PRKACA fusion, no known liver cancer genes are recurrently altered8,11,12,33. These findings prompted us to analyze the mutational load of our CRISPR/Cas9 generated mouse FL-HCC. We therefore performed Whole-Exome Sequencing (WES) of paired normal liver and mouse FL-HCC (for one larger and one smaller tumor) to identify genomic alterations that could have contributed to tumorigenesis instigated by Dnajb1–Prkaca. A total of 80 single nucleotide variants (SNVs) and 1 deletion were identified in the larger tumor (Supplementary Figure 6A and Supplementary Table 10): of these, 35 were non-synonymous (Supplementary Figure 6B) that we scored for their potentially damaging effect using Polyphen234. Importantly, only one gene with a damaging SNV had a frequency of the mutant allele above 15%, i.e. Aldh2 which is involved in alcohol catabolism. Although an ALDH2 polymorphism (rs 671, missense mutation E457K) has been associated with increased susceptibility to alcohol-induced gastrointestinal cancer in East Asians35, it is perhaps not so likely that the Aldh2 SNV identified here contributes to Dnajb1–Prkaca elicited FL-HCC in the present mouse setting. We Sanger sequenced the mutated region of most of those genes expressed, as evidenced by the RNA-seq data, and harboring predicted deleterious mutations, and confirmed the presence of SNVs in 8 cases (Supplementary Table 10). We also confirmed by IDAA the 3 basepair intronic deletion identified in one gene (Supplementary Figure 6C). Importantly, none of the mutated genes were reported mutated in any of the human FL-HCC genomic studies, nor have these genes been found consistently mutated in human liver cancers or are obvious cancer drivers (Supplementary Table 10). The mutational load of the smaller tumor was characterized by fewer SNVs (9) than the larger tumor described above, and it confirmed the absence of recurrent mutations or mutations in known cancer genes in FL-HCC induced by Dnajb1–Prkaca, and supported the idea that the number of mutations in a tumor increases as a consequence of its evolution (Supplementary Table 11). Interestingly, only one indel mutation site with no resemblance to the gRNAs used was detected by the Whole-Exome Sequencing, demonstrating that CRISPR/Cas9 off-target mutagenesis had not occurred within the exome of the tumors. Lastly, of some of the most prevalently mutated genes in other liver cancers (CTNNB1, TP53 and KRAS)11 or in mouse models of liver cancers13 (Hras), none were found to be mutated by targeted analysis of the smaller lesions that we analyzed.
Discussion
The aim of this study was to determine if the DNAJB1–PRKACA fusion is oncogenic and by inference, is the causative mutation of fibrolamellar hepatocellular carcinoma. With the emerging recognition that DNAJB1–PRKACA occurs in most, if not all FL-HCC patients, the functional demonstration of DNAJB1–PRKACA as driver would establish the genetic origin of this cancer.
We provided this evidence by the stringent approach of engineering in adult mouse liver the same genomic Dnajb1–Prkaca alteration that occurs in human FL-HCC. To this end, we exploited the powerful CRISPR/Cas9 technology, which was recently used to engineer the complex genomic rearrangement that creates the well-established oncogenic EML4-ALK fusion in mouse models of human lung cancer36,37. Moreover, a key point of our study is that we engineered Dnajb1–Prkaca in wild-type mice and without administration of any mutagens, in order to conclude, upon tumor formation, that DNAJB1–PRKACA is the main driving mutation. Finally, our engineering of the aberration in a healthy mouse liver and, at a low frequency, are important additional features of our study that mimic the stochastic and rare events leading to tumor initiation in FL-HCC patients in absence of other liver disease. The development of neoplastic lesions in the majority of our CRISPR/Cas9 engineered mice demonstrated that Dnajb1–Prkaca is sufficient to induce FL-HCC and thus is the main genetic event that initiates this cancer. The tumor latency of >1 year in our FL-HCC model is not unusual for a single genetic alteration in mouse liver cancer models13, and is consistent with the notion that FL-HCC is considered to be a slowly progressing liver cancer in humans21.
Importantly, the lesions induced by Dnajb1–Prkaca in mice had a high resemblance with human FL-HCC. Gross-anatomically, the tumors were well circumscribed from adjacent normal liver tissue as seen for human FL-HCC. Perhaps most strikingly, the cytological characteristics of mouse FL-HCC were highly similar to those of human FL-HCC, which are used as major diagnostic features in the clinic (reviewed in Torbenson1, and in Ward and Waxman33). Indeed, mouse lesions were composed of unusually large, polygonal tumor cells, often presenting prominent macronucleoli and with an abundant granular eosinophilic cytoplasm characteristic of oncocytic hepatocytes. The cytological appearance of these oncocytic hepatocytes is due to the presence of densely packed mitochondria, as evidenced by ultrastructural examination of both human and mouse FL-HCC in electron microscopy studies. Mouse lesions also showed, as observed in nearly half of human FL-HCC, hyaline globules and round cytoplasmatic inclusions known as “pale bodies”1. It therefore appears that Dnajb1–Prkaca has the cell-autonomous capacity to elicit all the characteristics of the FL-HCC cell phenotype, both in mouse and human.
In one respect mouse FL-HCC did not fully recapitulate the human tumor. Although collagen fibrosis was observed, it was considerably less pronounced than in human FL-HCC, where it sometimes forms a large, central scar. The limited fibrosis may not be surprising, as mouse livers are known to display an attenuated fibrotic response, as observed in mouse models of cancer as well as liver fibrosis13. Little is known about how fibrosis is elicited in FL-HCC, nor is there any evidence that the collagen deposition has any role in the progression of FL-HCC. The present study suggests that FL-HCC arises and progress through a cell autonomous action of Dnajb1–Prkaca within the tumor cells, which does not depend on fibrosis in the tumor.
Mouse FL-HCC also resembles human FL-HCC by expressing both hepatocytic and cholangiocytic markers as evidenced by immunohistochemical characterization. Although widely used in clinical practice, none of the proposed markers are actually specific for fibrolamellar hepatocellular carcinoma and their diagnostic significance is therefore limited38. Mouse FL-HCC expressed high levels of hepatocytes markers (CEA and HepPar1) and displayed scattered positivity for the biliary epithelium marker cytokeratin 7 and a stronger staining for cytokeratin 19, a marker associated with both biliary differentiation and hepatic progenitors. Mouse FL-HCC failed to show staining for CD68 that has been shown to be expressed in human tumors (data not shown)39.
Transcriptomic analysis of mouse FL-HCC revealed similarities with human FL-HCC as well as differences. In fact, we did not identify in our dataset previously reported overexpressed genes associated with a neuroendocrine signature, as these genes were either expressed at a very low level or were not differentially expressed. An exception was carboxypeptidase which we found upregulated (Supplementary Table 3), in agreement with Xu et al8. However, although overexpression of some of these genes has been observed in several studies8,11,28,29, gene signatures have failed to consistently mark FL-HCC as a tumor of neuroendocrine origin29,33. Species differences in gene expression between human and mouse may explain the lack of select FL-HCC features, most notably the lamellar bands of fibrosis, in the murine tumor. However, a strong enrichment in directionally concordant differentially expressed genes in mouse and human tumor was evidenced by cross-species GSEA.
In mouse FL-HCC we confirmed the increased expression of Aurora kinase A, a mitotic serine/threonine kinase involved in cell cycle progression and implicated in tumorigenesis, which was recently reported upregulated in two human FL-HCC studies29,30. This finding may be important in light of the ongoing clinical trial aimed at testing Aurora kinase A inhibition in FL-HCC patients; also in that it identifies a common pathological event initiated by DNAJB1–PRKACA in both human and mouse FL-HCC, which represents an attractive therapeutic target40.
It remains unclear to what extent, if any, mutations other than the DNAJB1–PRKACA aberration contribute to FL-HCC. Prior to the discovery of DNAJB1–PRKACA, driver genes were searched for using comparative genomic amplification analyses or focused analyses of genes known to underlie other liver cancers, which, however, did not identify any plausible FL-HCC drivers33. After the discovery of DNAJB1–PRKACA, two studies performed whole-exome sequencing on a single tumor containing the fusion8,11, the latter extending the analysis of a few mutated genes to a larger FL-HCC tumor collection. The two studies identified relatively few coding mutations (11 and 90), which showed no overlap, were infrequently considered damaging, and did not affect known cancer genes, except for BRCA2, which was found mutated in 4.2% of FL-HCC samples in the extended analysis. Another study performed whole-genome sequencing of 10 FL-HCC patients and found no recurrent structural variants other than DNAJB1–PRKACA, few coding mutations per tumor (a median of 6) and some genes being recurrently mutated of which the significance was unclear12. The authors concluded that the lack of second-hit mutation in FL-HCC makes the DNAJB1–PRKACA fusion protein the best target for diagnostic and therapeutic advancement. Our study agrees with the human studies, as we found no relevant coding mutations in known driver genes for liver cancers or other malignancies.
Regardless of the possible involvement of other genes, our finding that DNAJB1–PRKACA is sufficient to elicit FL-HCC combined with its ubiquitous presence in FL-HCC patients firmly establish DNAJB1–PRKACA as the dominant genetic driver of this cancer. Indeed, testing of DNAJB1–PRKACA mRNA or protein as a drug target in FL-HCC is highly warranted38. DNAJB1–PRKACA could prove as excellent a drug target as the BCR-ABL1 fusion in chronic myeloid leukemia, where a single, oncogenic event elicits and drives the disease and predicts a very favorable response to its inhibition41. This scenario would require that once formed, FL-HCC tumors remain addicted to DNAJB1–PRKACA for growth and survival. FL-HCC mouse models generated as described here, along with a recently established FL-HCC transplantable tumor line42, might constitute preclinical settings where the DNAJB1–PRKACA fusion could be tested as a drug target in FL-HCC.
In summary, we report the first CRISPR/Cas9-engineered mouse model of liver cancer generated somatically by a single oncogenic modification in otherwise wild-type mice and without administration of toxin or carcinogen. Furthermore, we describe the first mouse model of FL-HCC, notably one that replicates to a high extent the genotype and phenotype associated with this human cancer. Importantly, our study demonstrates that the recently identified DNAJB1–PRKACA fusion is the cause of FL-HCC, and is responsible for oncogenic transformation and major pathodiagnostic features of this cancer. Our findings thereby provide strong support for DNAJB1–PRKACA as a target for new therapies in FL-HCC and our mouse model may be useful to study the initiation and progression of FL-HCC as well as pre-clinical testing of new treatments.
Supplementary Material
Acknowledgments
The authors would like to thank Britt Christoffersen, the HistoCore facility, Finsen Laboratory/BRIC for excellent support with histology; Olga Ostrup, Center for Genomic Medicine, Rigshospitalet, University of Copenhagen for help with whole-exome sequencing data; and Klaus Quortrup and Bente Stricker, Core Facility for Integrated Microscopy, University of Copenhagen for electron microscopy.
Grant Support: This work was supported by the Danish Cancer Society (R146-A9563-B3551 to FN and R124-A7632-15-S2 to MF), the Danish Council for Independent Research (DFF-Mobilex 4092-00235 to FN and 0602-02368B to MF), the Lundbeck Foundation (R165-2013-15743 to FN), the University of Copenhagen Excellence Programme for Interdisciplinary Research—CDO2016 (to MF) and NIH R01 CA205158, R21 CA184656 and an endowed investigatorship from the Roy and Lynne Frank Foundation (to SHH).
Abbreviations
- CRISPR/Cas9
clustered regularly interspaced short palindromic repeats/CRISPR-associated 9
- FL-HCC
fibrolamellar hepatocellular carcinoma
- IDAA
indel detection by amplicon analysis
- PAS
periodic acid-Schiff
- PKA
cAMP-dependent protein kinase A
- PKAc
cAMP-dependent protein kinase A catalytic subunit alpha
- gRNA
single-guide RNA
- WT
wild-type
Footnotes
Author Contributions: FN and MF: conception and design of the study; LHE, AR, DS, FD-H, JVJ, ES-R, SHH, FN and MF: generation, collection, assembly, analysis and interpretation of data; ES-R, FN and MF: drafting of the manuscript; all authors commented on the final version of the manuscript.
Disclosure: The authors disclose no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Torbenson M. Fibrolamellar carcinoma: 2012 update. Scientifica (Cairo) 2012;2012:743–790. doi: 10.6064/2012/743790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kakar S, Burgart LJ, Batts KP, et al. Clinicopathologic features and survival in fibrolamellar carcinoma: comparison with conventional hepatocellular carcinoma with and without cirrhosis. Mod Pathol. 2005;18(11):1417–23. doi: 10.1038/modpathol.3800449. [DOI] [PubMed] [Google Scholar]
- 3.Craig JR, Peters RL, Edmondson HA, et al. Fibrolamellar carcinoma of the liver: a tumor of adolescents and young adults with distinctive clinico-pathologic features. Cancer. 1980;46(2):372–9. doi: 10.1002/1097-0142(19800715)46:2<372::aid-cncr2820460227>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 4.El-Serag HB, Davila JA. Is fibrolamellar carcinoma different from hepatocellular carcinoma? A US population-based study. Hepatology. 2004;39(3):798–803. doi: 10.1002/hep.20096. [DOI] [PubMed] [Google Scholar]
- 5.Ward SC, Huang J, Tickoo SK, et al. Fibrolamellar carcinoma of the liver exhibits immunohistochemical evidence of both hepatocyte and bile duct differentiation. Mod Pathol. 2010;23(9):1180–90. doi: 10.1038/modpathol.2010.105. [DOI] [PubMed] [Google Scholar]
- 6.Abdul-Al HM, Wang G, Makhlouf HR, et al. Fibrolamellar hepatocellular carcinoma: an immunohistochemical comparison with conventional hepatocellular carcinoma. Int J Surg Pathol. 2010;18(5):313–8. doi: 10.1177/1066896910364229. [DOI] [PubMed] [Google Scholar]
- 7.Honeyman JN, Simon EP, Robine N, et al. Detection of a recurrent DNAJB1–PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science. 2014;343(6174):1010–4. doi: 10.1126/science.1249484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu L, Hazard FK, Zmoos AF, et al. Genomic analysis of fibrolamellar hepatocellular carcinoma. Hum Mol Genet. 2015;24(1):50–63. doi: 10.1093/hmg/ddu418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham RP, Jin L, Knutson DL, et al. DNAJB1–PRKACA is specific for fibrolamellar carcinoma. Mod Pathol. 2015;28(6):822–9. doi: 10.1038/modpathol.2015.4. [DOI] [PubMed] [Google Scholar]
- 10.Dinh TA, Vitucci EC, Wauthier E, et al. Comprehensive analysis of The Cancer Genome Atlas reveals a unique gene and non-coding RNA signature of fibrolamellar carcinoma. Sci Rep. 2017;7:44653. doi: 10.1038/srep44653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cornella H, Alsinet C, Sayols S, et al. Unique genomic profile of fibrolamellar hepatocellular carcinoma. Gastroenterology. 2015;148(4):806–18.e10. doi: 10.1053/j.gastro.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darcy DG, Chiaroni-Clarke R, Murphy JM, et al. The genomic landscape of fibrolamellar hepatocellular carcinoma: whole genome sequencing of ten patients. Oncotarget. 2015;6(2):755–70. doi: 10.18632/oncotarget.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol. 2009;90(4):367–86. doi: 10.1111/j.1365-2613.2009.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lonowski LA, Narimatsu Y, Riaz A, Delay CE, et al. Genome editing using FACS enrichment of nuclease-expressing cells and indel detection by amplicon analysis. Nat Protoc. 2017;12(3):581–603. doi: 10.1038/nprot.2016.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;0(10):1735–7. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 18.Dagnaes-Hansen F, Holst HU, Søndergaard M, et al. Physiological effects of human growth hormone produced after hydrodynamic gene transfer of a plasmid vector containing the human ubiquitin promotor. J Mol Med (Berl) 2002;80(10):665–70. doi: 10.1007/s00109-002-0371-1. [DOI] [PubMed] [Google Scholar]
- 19.Torbenson M. Review of the clinicopathologic features of fibrolamellar carcinoma. Adv Anat Pathol. 2007;14(3):217–23. doi: 10.1097/PAP.0b013e3180504913. [DOI] [PubMed] [Google Scholar]
- 20.Berman MA, Burnham JA, Sheahan DG. Fibrolamellar carcinoma of the liver: an immunohistochemical study of nineteen cases and a review of the literature. Hum Pathol. 1988;19(7):784–94. doi: 10.1016/s0046-8177(88)80261-2. [DOI] [PubMed] [Google Scholar]
- 21.Hodgson HJ. Fibrolamellar cancer of the liver. J Hepatol. 1987;5(2):241–7. doi: 10.1016/s0168-8278(87)80580-9. [DOI] [PubMed] [Google Scholar]
- 22.Payne CM, Nagle RB, Paplanus SH, et al. Fibrolamellar carcinoma of liver: a primary malignant oncocytic carcinoid? Ultrastruct Pathol. 1986;10(6):539–52. doi: 10.3109/01913128609007211. [DOI] [PubMed] [Google Scholar]
- 23.Farhi DC, Shikes RH, Silverberg SG. Ultrastructure of fibrolamellar oncocytic hepatoma. Cancer. 1982;50(4):702–9. doi: 10.1002/1097-0142(19820815)50:4<702::aid-cncr2820500414>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 24.Haeussler M, Schönig K, Eckert H, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17(1):148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffith OL, Griffith M, Krysiak K, et al. A genomic case study of mixed fibrolamellar hepatocellular carcinoma. Ann Oncol. 2016;27(6):1148–54. doi: 10.1093/annonc/mdw135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujii T, Fuchs BC, Yamada S, et al. Mouse model of carbon tetrachloride induced liver fibrosis: Histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. 2010;10:79. doi: 10.1186/1471-230X-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lagana SM, Moreira RK, Remotti HE, et al. Glutamine synthetase, heat shock protein-70, and glypican-3 in intrahepatic cholangiocarcinoma and tumors metastatic to liver. Appl Immunohistochem Mol Morphol. 2013;21(3):254–7. doi: 10.1097/PAI.0b013e3182642c9c. [DOI] [PubMed] [Google Scholar]
- 28.Malouf GG, Job S, Paradis V, et al. Transcriptional profiling of pure fibrolamellar hepatocellular carcinoma reveals an endocrine signature. Hepatology. 2014;59(6):2228–37. doi: 10.1002/hep.27018. [DOI] [PubMed] [Google Scholar]
- 29.Simon EP, Freije CA, Farber BA, et al. Transcriptomic characterization of fibrolamellar hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2015;112(44):E5916–25. doi: 10.1073/pnas.1424894112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorenson EC, Khanin R, Bamboat ZM, et al. Genome and transcriptome profiling of fibrolamellar hepatocellular carcinoma demonstrates p53 and IGF2BP1 dysregulation. PLoS One. 2017;12(5):e0176562. doi: 10.1371/journal.pone.0176562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee DH, Park JO, Kim TS, et al. LATS-YAP/TAZ controls lineage specification by regulating TGFβ signaling and Hnf4α expression during liver development. Nat Commun. 2016;7:11961. doi: 10.1038/ncomms11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.https://clinicaltrials.gov/ct2/show/NCT02234986?term=fibrolamellar&rank=2
- 33.Ward SC, Waxman S. Fibrolamellar carcinoma: a review with focus on genetics and comparison to other malignant primary liver tumors. Semin Liver Dis. 2011;31(1):61–70. doi: 10.1055/s-0031-1272835. [DOI] [PubMed] [Google Scholar]
- 34.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui R, Kamatani Y, Takahashi A, et al. Functional variants in ADH1B and ALDH2 coupled with alcohol and smoking synergistically enhance esophageal cancer risk. Gastroenterology. 2009;137(5):1768–75. doi: 10.1053/j.gastro.2009.07.070. [DOI] [PubMed] [Google Scholar]
- 36.Maddalo D, Manchado E, Concepcion CP, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. 2014;516(7531):423–7. doi: 10.1038/nature13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blasco RB, Karaca E, Ambrogio C, et al. Simple and rapid in vivo generation of chromosomal rearrangements using CRISPR/Cas9 technology. Cell Rep. 2014;9(4):1219–27. doi: 10.1016/j.celrep.2014.10.051. [DOI] [PubMed] [Google Scholar]
- 38.Riggle KM, Turnham R, Scott JD, et al. Fibrolamellar Hepatocellular Carcinoma: Mechanistic Distinction From Adult Hepatocellular Carcinoma. Pediatr Blood Cancer. 2016;63(7):1163–7. doi: 10.1002/pbc.25970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ross HM, Daniel HD, Vivekanandan P, et al. Fibrolamellar carcinomas are positive for CD68. Mod Pathol. 2011;24(3):390–5. doi: 10.1038/modpathol.2010.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katsha A, Belkhiri A, Goff L, et al. Aurora kinase A in gastrointestinal cancers: time to target. Mol Cancer. 2015;14:106. doi: 10.1186/s12943-015-0375-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosti G, Castagnetti F, Gugliotta G, et al. Tyrosine kinase inhibitors in chronic myeloid leukaemia: which, when, for whom? Nat Rev Clin Oncol. 2017;14(3):141–154. doi: 10.1038/nrclinonc.2016.139. [DOI] [PubMed] [Google Scholar]
- 42.Oikawa T, Wauthier E, Dinh TA, et al. Model of fibrolamellar hepatocellular carcinomas reveals striking enrichment in cancer stem cells. Nat Commun. 2015;6:8070. doi: 10.1038/ncomms9070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.