

Introduction
Vigabatrin (VGB; γ–vinylGABA) is a unique antiepileptic directly elevating CNS GABA via inactivation of the GABA metabolic enzyme GABA-transaminase. VGB is effective in treating infantile spasms, a rare seizure disorder associated with significant morbidity. The potential for unexplained bilateral constriction of the visual field associated with VGB intervention can severely limit its temporal utility. Removal of this potential adverse effect with adjuvant intervention(s) would represent a significant advance in epilepsy therapeutics.
VGB is approved in many countries as adjuvant therapy for the treatment of resistant epilepsy, complex partial seizures (CPS), secondarily generalized seizures, and as monotherapy for infantile spasms (IS) (http://www.worldheritage.org/articles/Vigabatrin). Comprehensive clinical data have highlighted the utility of VGB as monotherapy for IS, as well as adjuvant in other disorders (secondarily generalized and complex partial seizures). The single alternative therapeutic for infantile spasms is adrenocorticotropic hormone (ACTH), with adverse effects including cardiovascular risks, onset of diabetes, and Cushing-like syndrome.
Despite VGB’s unique mechanism of action that directly elevates cerebral GABA (Fig. 1), its use may be compromised by the development of peripheral visual field defects (pVFD), the origins of which remain largely undefined. In a study of school age children who had received VGB as infants, pVFDs were detected in one-third, and the rate of VFDs increased from 9 to 63% as VGB treatment duration increased, although other studies have noted that bilateral visual field deficits are common in the treated epilepsy population. To track the retinal toxicity associated with VGB, pediatric neurologists variably rely on either serial funduscopic exams, electroretinogram (ERG), or optical coherence tomography (OCT) (Origlieri et al 2016). Nonetheless, there is no consensus as to what testing is sensitive enough to detect early onset pVFDs, since normal findings evolve with development, and the robustness of the findings in infants remains questionable. The potential occurrence of pVFD in patients receiving VGB requires clinicians to cautiously balance the potential risk of visual field disruption against the risk of catastrophic cognitive compromise associated with uncontrolled spasms.
Figure 1. Interrelationships of GABA metabolism.
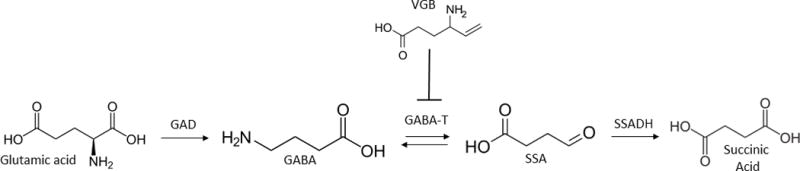
The site of action of vigabatrin (VGB) is shown at the level of aminobutyrate aminotransferase (GABA-transaminase). Abbreviations: GAD, glutamic acid decarboxylase; ABAT, aminobutyrate aminotransferase; SSADH, succinic semialdehyde dehydrogenase; VGB, vigabatrin.
There is not consensus regarding the pathomechanisms of pVFD associated with VGB intervention. Adults and infants treated with long-term VGB intervention manifest peripheral atrophy of the retinal nerve fiber layer. Rodents similarly treated manifest disorganization of the photoreceptor nuclear layer and damage to cone photoreceptors. Additional studies suggest that supraphysiologic GABA in the eye results in excitotoxicity associated with overstimulation of GABAergic receptors. A pathological role for amino acids with structural and biochemical properties similar to that of GABA (ornithine, taurine) has also been suggested in the etiology of retinal toxicity. Although there is not consensus, the preponderance of current data suggests that VGB may induce oxidative retinal damage, but the precise mechanisms by which such damage results in retinal cell loss and pVFDs remain unconfirmed. The fundamental unresolved question is whether VGB or elevated GABA (or both) are mechanistically linked to pVFDS.
Recent studies have begun to provide potential insights into the potential basis of pVFDs associated with VGB intervention. Lakhani et al (2014) unmasked a novel link between GABA metabolism, mTORC1 activity (Fig. 2), and mitophagy in mammals, and a more recent publication (Vogel et al 2015) indicates that this link may well extend to the capacity of VGB to elevate GABA. In yeast, elevated GABA inhibited mitophagy, resulting in elevated mitochondrial number and associated oxidant stress, all of which could be suppressed with rapamycin, a classical inhibitor of mTOR. These studies were extended to aldh5a1−/− mice, a phenocopy of the heritable Mendelian disorder succinic semialdehyde dehydrogenase deficiency (SSADHD) which also features accumulation of GABA and ablates the function of the second enzyme of GABA metabolism (Fig. 1). Elevated mitochondrial numbers in both brain and liver of aldh5a1−/− mice were observed, associated with increased markers of oxidative stress. As for yeast, rapamycin could override these effects in aldh5a1−/− mice. Immunoblotting studies of the ribosomal protein S6 in mice verified that mTORC1 was mechanistically in play in this pathology.
Figure 2. Mechanisms of molecular alterations contributing to potential VGB-induced visual field loss.
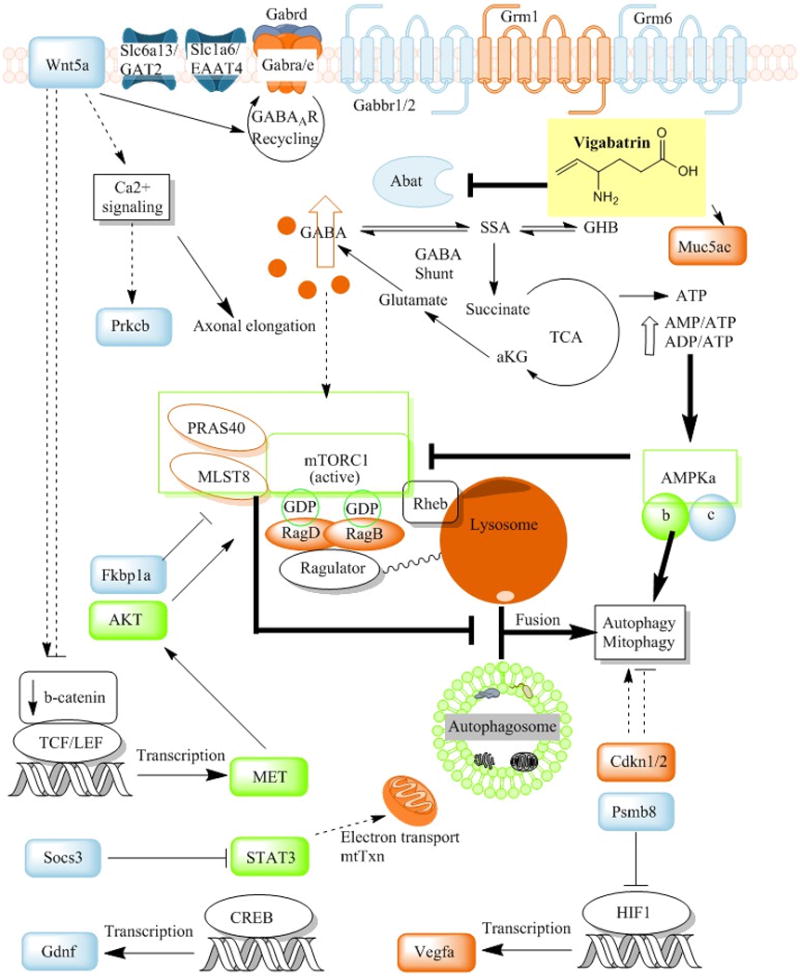
Vigabatrin (VGB) accumulates and increases GABA in the eye, resulting in molecular/cellular alterations in the mTOR pathway and GABAergic/glutamatergic receptor expression. VGB-treated mice manifest elevated mitochondrial number in the retinal pigment epithelia and elevated redox stress markers in whole eye extracts. Partial correction of these pathologies ensue with administration of the mTORC1/2 (mTOR complex) inhibitor Torin1, which correlate to involvement of hyper-p2448-mTOR activity (Vogel et al 2015). Gene expression studies demonstrated down-regulation of the γ subunit of the mTOR inhibitor AMPK (AMP-activated protein kinase), the latter sensitive to altered energy levels. AMPK activates autophagy indirectly via mTOR inhibition, while simultaneously activating autophagy through Ulk1 (Unc-51 like autophagy activating kinase 1) phosphorylation (e.g., during glucose starvation). mTOR responds to multiple cues (growth factor, tyrosine kinase, insulin, IGF (insulin growth factor) receptor signals), as well as amino acid levels, in its coordination of growth and catabolic processes. When amino acid levels are high, signaling to mTORC1 favors anabolic processes and inhibition of autophagy. Amino acids signal to mTOR through RAG (Ras-related GTP binding protein) GTPases and Ragulator at the lysosome. Elevation of RAG proteins and/or transcripts with conditions of elevated VGB/GABA suggests that GABA, a non-protein amino acid, may increase mTORC1 inhibition of autophagy, resulting in accumulation of mitochondria, and potentially other organelles or proteins. Impaired nutrient sensing in response to fasting conditions (metabolic changes), and accumulation of aged mitochondria, may further disrupt cellular homeostasis resulting in other pathological changes. Chronic elevation of GABA in eye downregulates GABA receptor expression (e.g., Gabra, Gabrd, Gabbr1/2) while simultaneously altering metabotropic glutamate receptors (Grm1/6), the GABA/taurine transporter GAT2 (Slc6a13), and the excitatory vesicular glutamate transporter EAAT4 (Slc1a6). These complex signaling processes (autophagic flux, mitochondrial accumulation, mTOR and AMPK signaling) and neurological mechanisms induced by elevated VGB/GABA may underlie the variable occurrence of ocular pathophysiology observed in some children, and less frequently in adults, utilizing VGB. Additional abbreviations: AKT, protein kinase B; HIF1, hypoxia inducible factor 1; Rheb, ras homolog enriched in brain; PRAS40, proline-rich Akt substrate (40 kDa); Wnt5a, Wnt family member 5A; Muc5ac, mucin 5AC protein; Prkcb, protein kinase c, beta subunit; aKG, a-ketoglutarate; MLST8, mTOR associated protein, LST8 homolog; Cdkn1/2, cyclin-dependent kinase inhibitors 1 and 2; Psmb8, proteasome subunit beta type-8; TCF/LEF, family of transcription factors; Fkbp1a, FK-506 binding protein 1a; MET, MET protooncogene; STAT3, Signal transducer and activator of transcription 3; Socs3, suppressor of cytokine signaling 3; Gdnf, glial cell-derived neurotrophic factor; CREB, cAMP response element binding protein; Vegfa, vascular endothelial growth factor A; mtTxn, mitochondrial transcription/translation. Arrows indicate metabolic sequences or activation, whereas indicates blockade.
Since VGB enhances GABA production (Fig. 1), it was hypothesized that rodents undergoing VGB intervention would yield similar results to those found in aldh5a1−/− mice. In a report examining this hypothesis, quantification of total GABA in extracts of brain and eye derived from VGB- and sham-treated mice revealed dose-dependent increases of GABA. The dosages used approximated 1 and 7 mg VGB/day, equivalent to a human dose of 200 and 1400 mg/d, respectively. These dosages were consistent with those employed to treat epilepsy in rodents, and those prescribed to children and infants with infantile spasms (IS), and within the dosing range employed in patients with SSADHD. Subsequently, number and cross-sectional areas of mitochondria in VGB-treated mice were assessed with transmission electron microscopy in retina, hippocampus and liver. VGB (35 mg/kg for 7 days; i.p. administration) resulted in significant increases in mitochondrial number in mouse tissues, in addition to increased average cross-sectional areas of mitochondria in retina and hippocampus. Torin 1, a newer generation mTOR inhibitor, corrected mitochondrial proliferation in hippocampus, but not retina or liver. To seek a potential link between VGB-induced GABA elevation and mTOR signaling, the levels of caspase 3 (apoptosis marker) and phospho-mTOR (pSer2448; a marker of the activational status of mTOR) were significantly increased in VGB-treated mouse liver.
To further interrogate potential pathomechanism(s) of VGB intervention, and attempt to more accurately pinpoint the retinal cell-specific pathology, studies of VGB and various inhibitors of mTOR were performed in cultured human retinal pigmented epithelial cells (hRPE). These studies were designed to determine if potential cell-specific effects could be unmasked, although at this time there is not conclusive evidence that RPE cells contribute to the possible pathology of VGB (Vogel et al, in press). Using fluorescence microscopy and organelle-specific staining, VGB was observed to significantly enhanced peroxisome, lysosome and mitochondrial number, which could be normalized with the application of various mTOR inhibitors. Torin 2 was examined since it demonstrates superior bioavailability in comparison to rapamycin (Fpo=51 vs 5.5%) and improved metabolic stability (Cliv=19 vs. 23 ml/min/kg). Trehalose was evaluated as an mTOR-independent measure of autophagy. These studies appear to be the first cell-based studies indicating a systemic association between VGB and organelle proliferation, possibly pointing to the capacity of VGB to disrupt pexophagy, lysophagy and mitophagy, and suggesting photoreceptor degeneration as a component of pathology. In the retinal pigment epithelia (RPE), autophagy and mitophagy (and most likely pexophagy, lysophagy) are cytoprotective functions linked mechanistically to protection against all-trans-retinal- and light-induced damage and photoreceptor cell death.
Additional in vivo studies were undertaken to examine potential clinical correlates between visual evoked potentials (VEPs) and associated transcriptional changes in mTOR and GABA receptors associated with VGB intake. VEPs were employed as a global readout of visual function related to VGB intake. VGB-enhanced production of GABA was initially predicted to reduce the VEP amplitude, which would be consistent with recent electrophysiological measures using VEPs in VGB-treated children. In the latter, altered ocular processing employing VEP was observed in VGB-exposed children. In their animal study, Vogel and colleagues employed wild-type mice receiving continuous systemic VGB infusion via subcutaneous osmotic mini-pump. Results verified that VGB attenuated VEP amplitude and dark adaptation potentiated it, consistent with studies in VGB-treated patients. The data suggest that the VGB-induced deficits seen with VEP extend to the brain, which can be gauged with VEP, but may well be missed with electroretinography (ERG). On the other hand, ERG could provide specific information on retinal toxicity through the evaluation of detailed analysis of dark-adapted (rod) vs. light-adapted (cone) testing. Moreover, looking at the response amplitudes and latencies (time from stimulus flash to peak of response) holds the potential to elucidate whether potential retinal insult is at the photoreceptor vs. inner retinal cells. Vogel and colleagues extended their VEP studies to gene expression analysis in brain and eye, documenting differential expression of transcripts in the mTOR pathway, GABAA/B receptors, metabotropic glutamate receptors 1/6 and glutamate transporters in VGB-treated mice.
Of interest, recent studies have noted that a majority of earlier studies on the association of VGB with pVFDs had failed to evaluate baseline visual function before implementation of VGB intervention (Sergott et al 2016), suggesting that abnormalities of the afferent visual system represent comorbidities of refractory complex partial seizures (rCPS) or IS. Moreover, there remains considerable variability in visual field assessment for epileptic patients, regardless of VGB usage. Sergott and colleagues (2016) found that up to 1 year of adjuvant VGB treatment in patients with rCPSs did not significantly alter near visual fields, although limitations of that study were its focus solely on the adult population with rCPS, a single-arm open-label design, and limited vigabatrin exposure. Nonetheless, recent work described above has begun to define a novel link between VGB, mTOR and autophagy, which may lead to the consideration of an mTOR inhibitor as adjuvant intervention with VGB. Pharmaceuticals that eliminate the potential for pVFDs associated with VGB would provide a major advance in epilepsy treatment.
Acknowledgments
The authors acknowledge the support of NIH R21NS85369 for portions of this work.
References
- Lakhani R, Vogel KR, Till A, Liu J, Burnett SF, Gibson KM, Subramani S. Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition. EMBO Mol Med. 2014 Apr;6(4):551–66. doi: 10.1002/emmm.201303356. Epub 2014 Feb 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origlieri C, Geddie B, Karwoski B, Berl MM, Elling N, McClintock W, Alexander J, Bazemore M, de Beaufort H, Hutcheson K, Miller M, Taylormoore J, Jaafar MS, Madigan W. Optical coherence tomography to monitor vigabatrin toxicity in children. J AAPOS. 2016 Apr;20(2):136–40. doi: 10.1016/j.jaapos.2015.10.020. [DOI] [PubMed] [Google Scholar]
- Sergott RC, Johnson CA, Laxer KD, Wechsler RT, Cherny K, Whittle J, Feng G, Lee D, Isojarvi J. Retinal structure and function in vigabatrin-treated adult patients with refractory complex partial seizures. Epilepsia. 2016 Sep 1; doi: 10.1111/epi.13495. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, Jansen EE, Salomons GS, Gibson KM. Torin 1 partially corrects vigabatrin-induced mitochondrial increase in mouse. Ann Clin Transl Neurol. 2015 Jun;2(6):699–706. doi: 10.1002/acn3.200. Epub 2015 Apr 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KR, Ainslie GR, Schmidt MA, Wisor JP, Gibson KM. mTOR inhibition mitigates molecular and biochemical alterations of vigabatrin-induced visual field toxicity in mice. Pediatr Neurol. doi: 10.1016/j.pediatrneurol.2016.09.016. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]