

Abstract
Background
Genome-wide studies identified pan-cancer genes and shared biological networks affected by epigenetic dysregulation among diverse tumor entities. Here, we systematically screened for hypermethylation of DNA damage repair (DDR) genes in a comprehensive candidate-approach and exemplarily identify and validate candidate DDR genes as targets of epigenetic inactivation unique to bladder cancer (BLCA), which may serve as non-invasive biomarkers.
Methods
Genome-wide DNA methylation datasets (2755 CpG probes of n = 7819 tumor and n = 659 normal samples) of the TCGA network covering 32 tumor entities were analyzed in silico for 177 DDR genes. Genes of interest were defined as differentially methylated between normal and cancerous tissues proximal to transcription start sites. The lead candidate gene was validated by methylation-specific PCR (MSP) and/or bisulfite-pyrosequencing in different human cell lines (n = 36), in primary BLCA tissues (n = 43), and in voided urine samples (n = 74) of BLCA patients. Urines from healthy donors and patients with urological benign and malignant diseases were included as controls (n = 78). mRNA expression was determined using qRT-PCR in vitro before (n = 5) and after decitabine treatment (n = 2). Protein expression was assessed by immunohistochemistry (n = 42). R 3.2.0. was used for statistical data acquisition and SPSS 21.0 for statistical analysis.
Results
Overall, 39 DDR genes were hypermethylated in human cancers. Most exclusively and frequently methylated (37%) in primary BLCA was RBBP8, encoding endonuclease CtIP. RBBP8 hypermethylation predicted longer overall survival (OS) and was found in 2/4 bladder cancer cell lines but not in any of 33 cancer cell lines from entities with another origin like prostate. RBBP8 methylation was inversely correlated with RBBP8 mRNA and nuclear protein expression while RBBP8 was re-expressed after in vitro demethylation. RBBP8 methylation was associated with histological grade in primary BLCA and urine samples. RBBP8 methylation was detectable in urine samples of bladder cancer patients achieving a sensitivity of 52%, at 91% specificity.
Conclusions
RBBP8 was identified as almost exclusively hypermethylated in BLCA. RBBP8/CtIP has a proven role in homologous recombination-mediated DNA double-strand break repair known to sensitize cancer cells for PARP1 inhibitors. Since RBBP8 methylation was detectable in urines, it may be a complementary marker of high specificity in urine for BLCA detection.
Electronic supplementary material
The online version of this article (10.1186/s13148-018-0447-6) contains supplementary material, which is available to authorized users.
Keywords: RBBP8/CtIP, DNA repair, Bladder cancer/BLCA, Epigenetics, Infinium HumanMethylation450 BeadChip, Urine biomarker
Background
Molecular characterization of cancer entities based on cohorts of tumor samples from all major organs created a wealth of data, allowing researchers to identify mutational landscapes across different cancer types. These studies provided novel insides into genomic signatures independent of tissue of origin [1], highlighting driver mutations potentially suitable for targeted therapies [2–4]. Next to druggable mutations, the cancer epigenome, known to regulate gene expression, holds clues to identify novel biomarkers and therapeutic approaches improving patient stratification. Genome-wide studies have recently identified pan-cancer DNA methylation (DNAm) patterns among diverse tumor entities, e.g., affected by genetic alterations in epigenetic regulators like DNMT3 (reviewed in Witte et al. [5]). Still, the co-existence of unique DNAm patterns indicates that also entity-specific and subtype-specific targets of epigenetic deregulation could lead to the development of distinct methylation phenotypes contributing to tumorigenesis. These specific epigenetic aberrations, also referred to as epimutations, may uncover novel targets to improve disease management in many respects.
So far, DNA methylation is proposed as a molecular biomarker for cancer detection [6] but also as a biomarker for prediction and stratification of patients with risk of distinct clinical outcome and response to therapies [7]. Owing to this, methylation of DNA repair genes in general seems to be a good pool for prediction [8–12] of how patients respond to treatment with conventional chemotherapies as well as novel classes of targets such as poly (ADP-ribose) polymerase (PARP) inhibitors. Examples of predictive methylated genes are MGMT in glioma (temozolomide) [13, 14], BRCA1 in breast cancer (PARP1 inhibitors, cisplatin, and chemotherapy) [15–18], and PRKCDBP in colon cancer (oxaliplatin) [19], among others.
In the presented study, we were, therefore, interested to reveal whether differential DNAm patterns of DNA repair genes of the DNA damage response (DDR) network were common epimutations across cancer entities, especially in those known to be impaired in DNA repair function, such as bladder cancer [20] which showed an essentially stagnant disease management since decades [21]. Since there is, to our knowledge, no systematic screen for hypermethylation of DNA repair genes, we performed a comprehensive candidate approach comprising 177 DDR genes [22] as targets of epigenetic deregulation in 32 tumor entities. Subsequently, we exemplarily aimed at the validation of the identified lead candidate gene, RBBP8, to assess potential biomarker performance based on a non-invasive detection approach.
Accumulating studies propose RBBP8, known to encode the endonuclease CtIP [23], as a novel susceptibility gene [24] whose functional loss increases sensitivity towards PARP1 inhibition [25, 26] similar to BRCA1 inactivation as, for instance, recently demonstrated in a mice xenograft model of breast cancer [27]. Mechanistically, RBBP8, a nuclear located protein that is conserved among vertebrates, interacts with tumor suppressors such as BRCA1 and the pRb family members through binding sites that are frequently mutated in human cancers [24]. As part of the DDR network, RBBP8/CtIP has a proven role as key factor in regulating DNA-end resection and double-strand break (DSB) repair mechanisms [28, 29] by supporting homologous recombination (HR) [30], classical non-homologous end-joining (c-NHEJ) [31, 32], and alternative non-homologous end-joining (alt-NHEJ) [33]. Thus, RBBP8/CtIP is thought to be involved in the maintenance of genome integrity in a cell cycle- and DNA damage-dependent manner [34, 35]. Here, we demonstrate that RBBP8 whose unique hypermethylation pattern in human bladder cancer was associated with its gene silencing might serve as a biomarker that can be accessed via urine tests.
Results
DNAm pattern in promoter regions of DDR genes in 32 different human cancer types
Our first aim was to identify novel DNA repair genes as targets of epigenetic inactivation unique to human cancer types, which may finally be used as a non-invasive methylation biomarker. The study design is illustrated in Additional file 1. Based on the Infinium HumanMethylation450 dataset of the publically available The Cancer Genome Atlas (TCGA) platform, genome-wide DNA methylation data of 7819 primary tumor samples and 659 normal samples comprising 32 tumor entities (Table 1) was assessed. Overall, we performed an integrated analysis by defining CpG probe groups including initially 2755 CpG probes (sites) located between 2000 bp downstream and 500 bp upstream of the reported transcription start sites (TSS) of 177 DDR genes. Subsequently, we focused on those genes whose CpG probe set with a putative regulatory impact met the following criteria: A probe set specific and (healthy-) normalized β value cutoff of ~ 0.25 (β value ~ 0.25 (90% CI 0.20–0.35)) after transformation to M values (see the “Methods” section). By that, 39 DDR genes were found to show methylation in a fraction of ≥ 5% of samples in one or more entities (see heatmap in Fig. 1). This includes known epigenetic silenced DDR genes with a therapeutic impact like BRCA1 [17, 18], MGMT [13, 14], and ERCC1 [36, 37] (Additional file 2). In addition (to these already known DDR genes), we were also able to identify novel potential targets like SLX1A which has not yet been associated with DNA methylation in tumors so far. The identified DDR genes can be classified into all essential subnetworks of DNA repair mechanisms [22], i.e., mismatch repair (MMR, e.g., MLH1), homologous repair (HR, e.g., RBBP8/CtiP), dissolution of joint DNA molecules (JMs, e.g., TOP3A), nucleotide excision repair (NER, e.g., ERCC1), and base excision repair (BER, e.g., PARP1).
Table 1.
TCGA studies and sample numbers used for DNA methylation analyses in this study
| Study | Study name/tumor entity | Tumors | Normals |
|---|---|---|---|
| ACC | Adrenocortical carcinoma | 79 | 0 |
| BLCA | Bladder urothelial carcinoma | 368 | 17 |
| BRCA | Breast invasive carcinoma | 772 | 96 |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma | 299 | 3 |
| CHOL | Cholangiocarcinoma | 35 | 9 |
| COAD | Colon adenocarcinoma | 265 | 30 |
| DLBC | Lymphoid Neoplasm Diffuse Large B-Cell Lymphoma | 47 | 0 |
| ESCA | Esophageal carcinoma | 182 | 13 |
| GBM | Glioblastoma multiforme | 129 | 2 |
| HNSC | Head and neck squamous cell carcinoma | 481 | 50 |
| KICH | Kidney chromophobe | 60 | 0 |
| KIRC | Kidney renal clear cell carcinoma | 271 | 141 |
| KIRP | Kidney renal papillary cell carcinoma | 243 | 34 |
| LGG | Brain lower grade glioma | 498 | 0 |
| LIHC | Liver hepatocellular carcinoma | 360 | 45 |
| LUAD | Lung adenocarcinoma | 399 | 22 |
| LUSC | Lung squamous cell carcinoma | 337 | 36 |
| MESO | Mesothelioma | 82 | 0 |
| OV | Ovarian adenocarcinomas | 10 | 0 |
| PAAD | Pancreatic adenocarcinoma | 155 | 7 |
| PCPG | Pheochromocytoma and paraganglioma | 152 | 3 |
| PRAD | Prostate adenocarcinoma | 476 | 40 |
| READ | Rectum adenocarcinoma | 91 | 7 |
| SARC | Sarcoma | 244 | 0 |
| SKCM | Skin cutaneous melanoma | 102 | 1 |
| STAD | Stomach adenocarcinoma | 388 | 2 |
| TGCT | Testicular germ cell tumors | 149 | 0 |
| THCA | Thyroid carcinoma | 484 | 54 |
| THYM | Thymoma | 122 | 2 |
| UCEC | Uterine Corpus Endometrial Carcinoma | 404 | 45 |
| UCS | Uterine carcinosarcoma | 56 | 0 |
| UVM | Uveal melanoma | 80 | 0 |
Fig. 1.
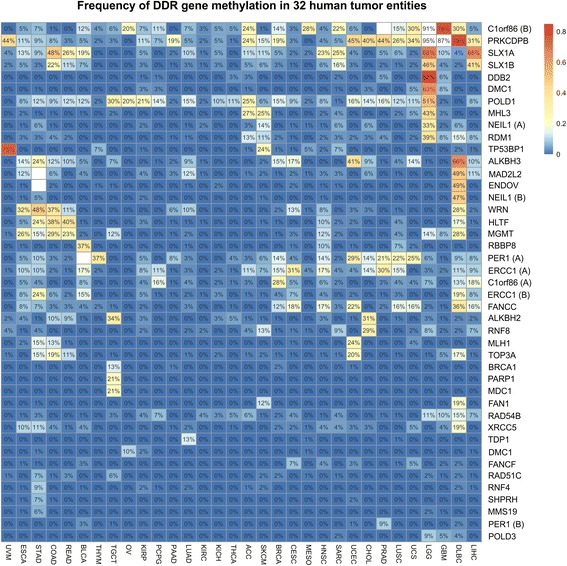
Discovery analysis of DNAm pattern in promoter regions of DNA damage repair genes across tumor entities. Promoter methylation of DDR genes was present in 32 different tumor entities. The heatmap summarizes the conducted analysis of the Infinium HumanMethylation450 BeadChip data available from the TCGA project (see the “Methods” section for details). Only those genes (CpG groups near TSS (s)) for which at least one tumor entity exhibited a hypermethylation of more than 5% of cases are shown. Fields for which corresponding normal tissues were hypermethylated in more than 15% are not shown (white). Note: The tumor entities ACC, DLBC, KICH, LGG, MESO, OV, SARC, TGCT, UCS, and UVM were included despite missing corresponding normal tissue samples. Fields were clustered hierarchically in both dimensions
We then reduced the selected list of candidates (i.e., CpG probe groups) by focusing on genes exhibiting hypermethylated in more than 15% of cases in at least one tumor entity with a minimum of ten analyzed normal tissue samples (Additional file 3). The list of genes with stringently tumor-specific hypermethylation includes, for instance, ERCC1, MGMT, POLD1, and RBBP8/CtiP. In all cases, hypermethylation was entity-specific as demonstrated by the conducted cluster analysis (see Fig. 1) and statistical testing applied to contingency tables of tumor samples (entity vs. relationship to the threshold, p < 10−6).
Finally, an association between promoter methylation of defined CpG probe sets and mRNA expression of the corresponding gene across tumor types was assessed to narrow down potential candidate genes and entities for the subsequent biomarker discovery part of this study. To ensure valid statistics, only DDR genes exceeding a methylation frequency of > 5% in identified tumor entities were included. Based on that, 9 out of the identified 14 candidate genes (see Additional file 3) passed our criteria, i.e., showed a highly significant (i.e., p < 0.001) association with their gene expression in one or more tumor entities like PER1 and POLD1 (see Additional file 4). Among others (such as ALKBH2 and ALKBH3), MGMT promoter methylation, for instance, showed a strong inverse correlation in all analyzable tumor entities (i.e., COAD, DLBC, ESCA, GBM, HNSC, LGG, READ, STAD, TGCT).
RBBP8 is almost exclusively methylated in bladder cancer correlating with lower RBBP8 mRNA expression and a favorable prognosis
In bladder cancer, 3 out of these identified 39 DDR genes, i.e., SLX1A, ERCC1, and RBBP8, exhibited hypermethylation over 15%. Interestingly, RBBP8 was almost exclusively methylated in bladder cancer (Fig. 2a–c). Overall, 137 out of 368 (37%) analyzed bladder tumors exhibited a tumor-specific RBBP8 promoter methylation. The normal adjacent tissues showed methylation in 2 out of 17 cases. Nevertheless, the methylation was notably lower than that of the matching tumor tissues (β values 0.26 vs. 0.53 and 0.30 vs. 0.71) which might be due to an epigenetic field effect across the urothelium of bladders with cancer (e.g., [38]). Moreover, RBBP8 promoter methylation was only present at low frequency in head and neck squamous cell carcinoma (9.6%, n = 46/481), lung squamous carcinoma (7.1%, n = 24/337), uterine carcinosarcoma (1.8%, n = 1/56), cervical squamous cell carcinoma (1.3%, n = 4/299), esophageal carcinoma (1.1%, n = 2/182), and lung adenocarcinoma (1.0%, n = 4/399). In all other analyzed cancer types, RBBP8 methylation frequency was lower than 1.0%. A multiple t tests using the Holm correction for multiple comparison analysis confirmed a significantly increased methylation frequency of RBBP8 in bladder cancer compared to all other tested tumor entities (p < 0.019). In addition, a significantly inverse correlation between RBBP8 methylation (CpG probe set as defined) and RBBP8 mRNA expression (Spearman r − 0.32; p < 0.001) was only demonstrated for BLCA (Fig. 2d). No association was observed for HNSC and LUSC, and RBBP8 was thereof chosen as a lead candidate gene of this study for bladder cancer.
Fig. 2.
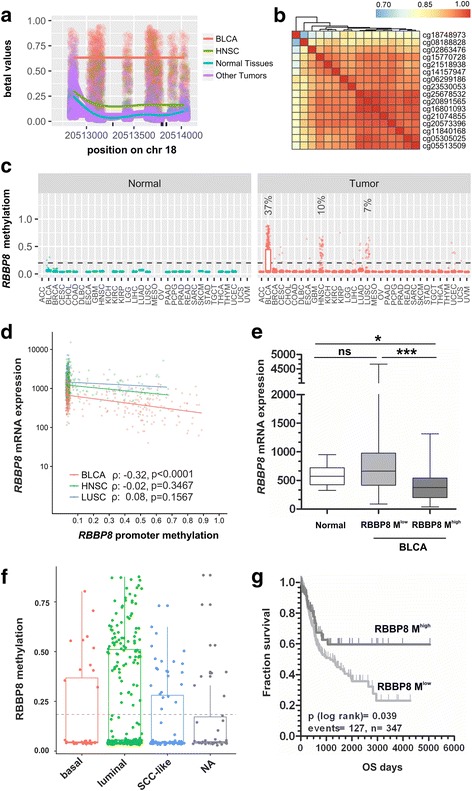
RBBP8 promoter methylation in bladder cancer of the TCGA data set. a Visualization of the promoter methylation of the RBBP8 gene as a scatterplot. The β values for each sample were jittered around the probe location and plotted as points. The per sample type 90% quantiles of methylation are shown as smoothed lines. The colors represent different sample groups (BLCA, bladder cancer; HNSC, head-neck squamous cell carcinoma). b The Pearson correlation coefficients (ρ) for probe pairs in the core region of the promoter are shown as heatmap demonstrating a high degree of correlation between probes (ρ > 0.7). c The beta values of all probes of the promoter region were summarized by their median value, stratified by the sample as well as tissue type, and visualized as a box plot. For RBBP8 gene loci, a frequent hypermethylation (β > 0.25 in > 5% of cases) was only observable in 37, 10, and 7% of bladder urothelial carcinoma (BLCA), head-neck squamous cell carcinoma (HNSC), and lung squamous cell carcinoma (LUSC), respectively. d Inverse correlation between RBBP8 methylation (defined CpG gene set) and RBBP8 mRNA expression in primary bladder cancer (BLCA), neck squamous cell carcinoma (HNSC), and lung squamous carcinoma (LUSC) samples of the TCGA data portal. Spearman correlation BLCA: − 0.32, p < 0.001; Spearman correlation HNSC: − 0.04, p = ns; Spearman correlation LUSC: 0.08, p = ns. e Box plot illustrates significant downregulation of RBBP8 methylation in primary tumors featuring increased RBBP8 promoter methylation (β value > 0.4). Horizontal lines — grouped medians. Boxes — 25 to 75% quartiles. Vertical lines — range, peak, and minimum. *p < 0.05, **p < 0.01, ns: not significant. f Box plot shows RBBP8 methylation in primary tumors classified by intrinsic subtypes. g Kaplan-Meier survival curves display overall survival (OS) of patients with high RBBP8 methylation (β value > 0.4, dark gray curve) compared to low RBBP8 methylation (β value ≤ 0.4, gray curve) based on TCGA datasets
Given entity-specific methylation of RBBP8 in BLCA, we aimed to provide a first insight on whether this epigenetic modification may provide a clinical impact in this entity. We divided the dataset (overall n = 405, for cohort characteristics see Additional file 5) into low methylated (RBBP8 β values (β ≤ 0.4)) and highly methylated (β > 0.4) tumor samples and found that a prevalent loss of RBBP8 mRNA expression was only present in tumors with high RBBP8 promoter methylation compared to normal bladder tissue (Fig. 2e). The close association between loss of RBBP8 mRNA expression and RBBP8 hypermethylation was confirmed by using a Fisher’s exact test (Table 2). Further associations of RBBP8 methylation with clinicopathological characteristics were evaluated as well (Table 2), which showed a significant association of RBBP8 methylation with higher histological tumor grade (p = 0.041). No further correlations between RBBP8 methylation and clinicopathological characteristics were found. Classifying tumor samples by subtype, i.e., “luminal,” “basal,” and “SCC-like,” [39] RBBP8 promoter methylation tended to be enriched in luminal-type bladder tumors (Fig. 2f).
Table 2.
Clinicopathological parameters in relation to RBBP8 promoter methylation of the BLCA TCGA dataset
| RBBP8 promoter methylationb | ||||
|---|---|---|---|---|
| n a | low | high | p valuec | |
| Parameter: | ||||
| Gender | ||||
| Male | 259 | 190 | 69 | 0.062 |
| Female | 94 | 78 | 16 | |
| Histological tumor grade | ||||
| Low grade | 20 | 19 | 1 | 0.041 |
| High grade | 330 | 247 | 83 | |
| Tumor stage | ||||
| pT1-pT2 | 109 | 86 | 23 | 0.49 |
| pT3-pT4 | 216 | 163 | 53 | |
| pN status | ||||
| Negative | 212 | 161 | 51 | 0.866 |
| Positive | 112 | 86 | 26 | |
| pM status | ||||
| Negative | 173 | 131 | 42 | 0.138 |
| Positive | 7 | 7 | 0 | |
| RBBP8 mRNAd | ||||
| Low | 200 | 124 | 76 | ≤ 0.001 |
| High | 200 | 175 | 25 | |
Significant p values are in italics
aOnly patients with primary bladder cancer without any neoadjuvant therapy were included
bBased on TCGA 450K DNA methylation analysis
cFisher’s exact test
dRBBP8 mRNA expression was dichotomized at the median expression level
As dysregulation of DDR repair is known to be associated with patients’ outcome, we examined overall survival (OS) as an indicator of potential clinical impact. By Kaplan-Meier analysis, we found that patients with high RBBP8 methylation have a longer overall survival (mean OS 3197.4 days ± 310.3; 95% CI 2589.3 to 3805.5 days) compared to low RBBP8 methylation (mean OS 1800.6 days ± 161.4; 95% CI 1484.1 to 2117.0 days) (Fig. 2g, Table 3). The multivariate hazard ratio of 0.650 (95% CI 0.312 to 1.354, p = 0.249) underlines a decreased risk for tumor death, although independency statistically failed in comparison to known prognostic parameters (Additional file 6).
Table 3.
Univariate analysis of clinicopathological parameters influencing OS
| Overall survival (OS) | |||
|---|---|---|---|
| n a | Events | p valueb | |
| Parameter: | |||
| RBBP8 methylationc | |||
| RBBP8 Mlow | 262 | 103 | 0.039 |
| RBBP8 Mhigh | 85 | 24 | |
| Gender | |||
| Male | 253 | 91 | 0.920 |
| Female | 94 | 36 | |
| Histological tumor graded | |||
| Low grade | 15 | 0 | 0.066 |
| High grade | 329 | 127 | |
| Tumor stage | |||
| pT1-pT2 | 105 | 23 | ≤ 0.001 |
| pT3-pT4 | 214 | 94 | |
| pN status | |||
| Negative | 207 | 53 | ≤ 0.001 |
| Nositive | 111 | 63 | |
| pM status | |||
| Negative | 168 | 52 | 0.004 |
| Positive | 7 | 5 | |
Significant p values are in italics
aOnly patients with primary bladder cancer without any neoadjuvant therapy were included
bLog-rank test at the two-sided significance level of 0.05
RBBP8 methylation is exclusively present in human bladder cancer cell lines and functionally associated with RBBP8 mRNA expression after DAC treatment
In silico analysis of the RBBP8 gene promoter sequence using genomic DNA information (ENSEMBL contig ENS00000101773) showed a CpG-rich island between genomic positions 20512942 and 20513943 (− 353 to + 648 bp relative to the expected transcription start site (TSS: transcript variante #1: position 20513295) on chromosome 18q which met the following criteria: DNA region ≥ 200 bp; Obs/Exp ≥ 0.6; and % GC ≥ 50. The DNA region including CpG sites that are closely associated to the TSS encodes a potential regulatory core promoter and ubiquitous elements for gene regulation as determined by in silico analysis using Genomatix [40]. In order to specify the TCGA data sets, we designed both a methylation-specific PCR (MSP) and bisulfite-pyrosequencing assay close to the TSS area to screen RBBP8 methylation status in a large set of normal and cancer cell lines from various cancer entities. The analyzed promoter region upstream of the transcription start is illustrated in Fig. 3a.
Fig. 3.
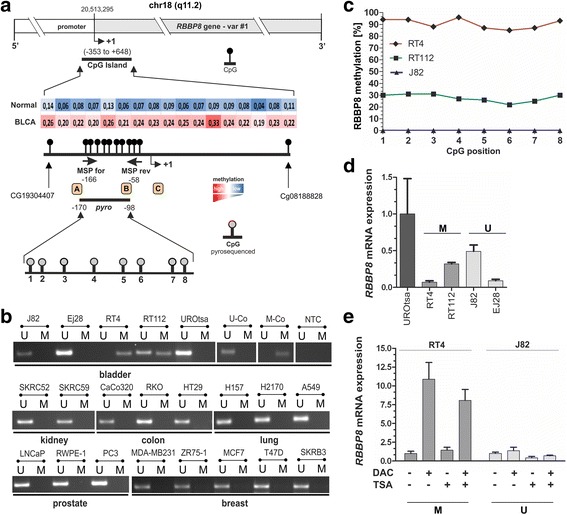
RBBP8 promoter methylation in human cancer cell lines. a Schematic map of the human RBBP8 gene including the relative positions and median β values of 17 CpG sites based on 450K methylation array profiling in bladder cancer (TCGA dataset) within a predicted CpG island (between base − 353 and + 648). Colored boxes present methylation level (mean ß-values for each CpG site) of the TCGA data set. Red, high methylation; blue, low methylation. + 1, RBBP8 transcription start site (TSS) of variant #1. The dots indicating the methylation sites closer to where they are depicted. CpG sites analyzed by MSP (black arrows) were indicated within the upstream promoter region close to the TSS. The relative position of the promoter area analyzed by bisulfite-pyrosequencing that comprises eight single CpG sites (gray dots) is shown as a black line. Orange boxes illustrate gene transcription-relevant regulatory core and ubiquitous elements statistically identified by using Genomatix [40] software (http://www.genomatix.de/). A — core promoter motif ten elements (− 200 to − 179); B — activator protein 2 (− 109 to − 94); C — activator-, mediator-, and TBP-dependent core promoter element for RNA polymerase II transcription from TATA-less promoters (+ 28 to + 39). b Representative MSP results of the RBBP8 promoter methylation status in cell lines of bladder, kidney, colon, lung, prostate, and breast cancer. Bands labeled with U and M reflect unmethylated and methylated DNA, respectively. Bisulfite-converted unmethylated, genomic (U-co), and polymethylated, genomic (M-co) DNA were used as positive controls. NTC, non-template control. c RBBP8 mean methylation values of analyzed CpG sites (1 to 8) using bisulfite-pyrosequencing of bladder cancer cell lines (RT4, RT112, and J82). d RBBP8 mRNA expression in normal urothelial cells (UROtsa) and bladder cancer cell lines (RT4, RT112, J82, and EJ28) arranged in relation to their RBBP8 promoter methylation status. U, unmethylated; M, methylated. Error bars: + s.e.m. e qPCR analysis for RBBP8 mRNA expression after in vitro demethylation analysis demonstrating a clear RBBP8 re-expression after treatment with both DAC (+) and TSA (+) only in RT4 with methylated RBBP8 promoter status (M) whereas in J82 cells without any RBBP8 methylation (U) RBBP8 expression was not further inducible. Non-treated cells served as controls and were set to 1. Error bars: + s.e.m.
Using MSP, a methylated RBBP8 promoter was detected in two (RT4 and RT112) out of four bladder cancer cell lines which derived from stage 2 and higher grade bladder tumors [41], while RBBP8 was unmethylated in normal urothelial cells (UROtsa) (Fig. 3b). The analyzed RBBP8 promoter region was also unmethylated in 33 further cancerous cell lines, i.e., cell lines from breast cancer, lung cancer, prostate cancer, colorectal cancer, and renal cancer (see the complete list in Additional file 7). Strong RBBP8 promoter methylation in RT4 (median methylation level 90.5%) and RT112 bladder cancer cells (median methylation level: 28.5%) was confirmed by using bisulfite-pyrosequencing covering the MSP product sequence (Fig. 3c).
In all bladder cancer cell lines (RT4, RT112, J82, and EJ28), RBBP8 mRNA was lower expressed than in normal UROtsa cells. Lowest mRNA expression was found in RT4 cells which featured the highest methylation level as well (Fig. 3d). Functionally, we confirmed this epigenetic modification as a molecular cause for RBBP8 gene regulation by in vitro demethylation experiments. Seventy-two hours after 5-aza-2′-deoxycytidine (decitabine (DAC)) and trichostatin A (TSA) treatment upregulation of RBBP8 mRNA expression (FC = 11) was demonstrated in RT4 tumor cells. DAC treatment without TSA supplementation already triggered an increase in RBBP8 mRNA expression in RT4 bladder cancer cells. Treatment with both DAC and TSA leads to a maximum of RBBP8 mRNA re-expression. In turn, RBBP8 mRNA was not further inducible by DAC/TSA in J82 bladder cancer cells (Fig. 3e) harboring an unmethylated RBBP8 promoter (see Fig. 3c). These findings indicate that epigenetic alterations of the RBBP8 gene may be caused by synergistic crosstalk between DNA methylation and histone modification.
Validation of DNAm pattern within the RBBP8 promoter close to the TSS in primary bladder tumors
Based on MSP and bisulfite-pyrosequencing, RBBP8 methylation was assessed in an independent cohort of primary bladder tumors. MSP analysis confirmed an unmethylated RBBP8 promoter in healthy urothelial tissues. RBBP8 methylation frequency was increased to 39.1% (9/23) in primary bladder tumors (Fig. 4a). None of the papillary low-grade tumors (n = 9) showed RBBP8 methylation, whereas 9 out of 14 invasive BLCAs were methylated for the RBBP8 locus. We subsequently analyzed the methylation status of eight CpG sites using pyrosequencing (Fig. 4b). The median RBBP8 methylation for each CpG is shown in Fig. 4b for both normal urothelium and bladder tumors (n = 20). Nine out of 20 tumor (45%) samples showed a median RBBP8 methylation level (including all eight CpGs) of > 5% (range 6 to 62.5%). Classifying the bladder tumors by papillary and invasive subtype, no clear differences in RBBP8 promoter methylation were detected (Fig. 4c). However, a significant increased RBBP8 promoter methylation was observed for high-grade bladder tumors, including pTa high-grade carcinomas, (mean methylation 20.9%; s.d. ± 4.4%) compared to low-grade BLCA (mean methylation 2.4%; s.d. ± 1.4%) (Fig. 4d). A close association between RBBP8 and higher histological tumor grade (p = 0.041) was also confirmed by Fisher’s exact test (Additional file 8).
Fig. 4.
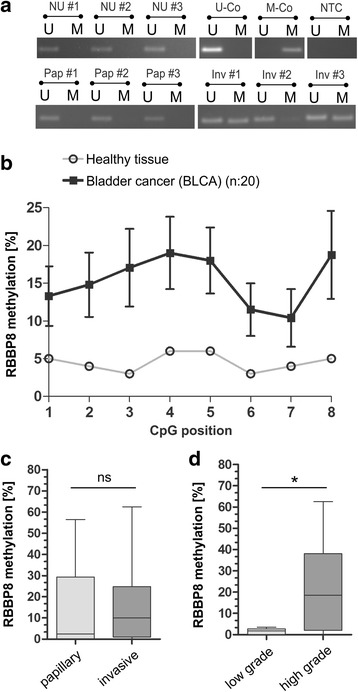
Validation of RBBP8 promoter methylation in primary bladder tumors. a Representative MSP analysis shows the RBBP8 promoter methylation status of normal urothelium (NU) and both papillary (Pap) and invasive (Inv) primary bladder cancer tissues. Band labels with U and M represent an unmethylated and methylated DNA locus, respectively bisulfite-converted unmethylated, genomic (U-co) and polymethylated, genomic (M-co) DNA were used as positive controls. NTC: non-template control. b RBBP8 mean methylation values of analyzed CpG sites (1 to 8) of healthy controls and bladder tumors demonstrating tumors-specific hypermethylation. c to d Box plot analysis of RBBP8 methylation in primary bladder tumors is based on mean values of pyrosequenced CpG sites 1–8. c RBBP8 methylation shows no significant differences between the two bladder cancer pathways (papillary and invasive tumors). d Significant enrichment of RBBP8 methylation is demonstrated in high-grade bladder tumors. Horizontal lines—grouped medians. Boxes—25 to 75% quartiles. Vertical lines—range, peak, and minimum; *p < 0.05. Horizontal lines—grouped medians. Boxes—25 to 75% quartiles. Vertical lines—range, peak, and minimum; ns, not significant, *p < 0.05
RBBP8 protein expression in bladder cancer
RBBP8 protein expression was characterized in normal urothelium and in bladder tumor tissues using immunohistochemistry. We found RBBP8 protein staining in the cytoplasm and frequently in the nuclei of the normal urothelium (Fig. 5a). Advanced bladder tumors showed a similar cytoplasmatic protein level, but only sporadically a nuclear protein localization was observed (Fig. 5c–e). Interestingly, low-grade non-invasive bladder tumors retained high levels of RBBP8 protein within the nucleus (Fig. 5f).
Fig. 5.
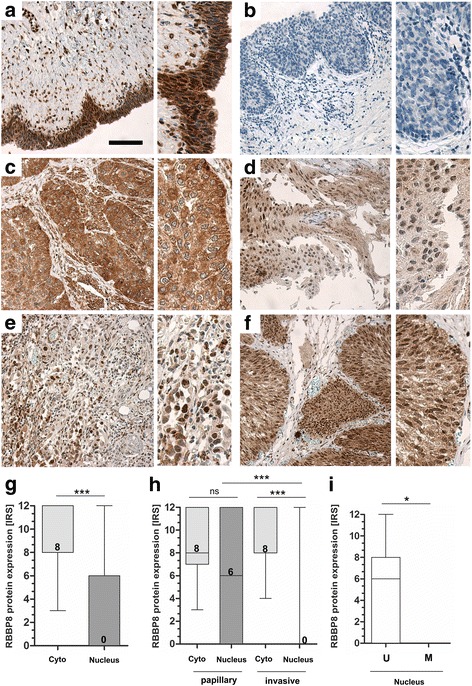
RBBP8 protein loss in nuclei of bladder tumors. Immunohistochemical RBBP8 protein staining of representative tissues are shown. a Strong RBBP8 immunoreactivity was detected in the cytoplasm and in the nuclei of a healthy urothelium, Scale bar: 100 μm. b Negative control of urothelial cell layers. The application of primary antibody was omitted. c Strong RBBP8 immunoreactivity in the cytoplasm of high grade, invasive tumor cells which completely lack nuclear staining. d Moderate cytoplasmatic and heterogeneously nuclear RBBP8 protein staining in invasive tumor cells. e Low RBBP8 protein expression in the cytoplasm of invasive bladder cancer showing strong RBBP8 staining in the nucleus. f Strong nuclear and cytoplasmic RBBP8 staining in non-invasive, papillary tumor cells. g Box plot demonstrating overall significant loss of RBBP8 protein only in the nucleus of bladder tumors. h Box plot graph illustrates the loss of RBBP8 protein within the nuclei of high-grade invasive bladder tumors. i Box plot shows a significant RBBP8 protein loss in tumors harboring RBBP8 promoter methylation. U, unmethylated; M, methylated. Horizontal lines — grouped medians. Boxes — 25 to 75% quartiles. Vertical lines — range, peak, and minimum; ns, not significant, *p < 0.05, ***p < 0.001
This observation was confirmed by quantification of RBBP8 protein staining according to an adapted immunoreactive score (IRS) developed by Remmele and Stegner [42] in both the cytoplasm and the nuclei. Overall, 42 bladder carcinomas were compared evaluating the average RBBP8 protein expression in a semi-quantitative manner (for cohort characteristics, see Additional file 9). Cytoplasmatic RBBP8 protein expression remained visible in all bladder tumors (median IRS, 8). In contrast, 71.4% (n = 30/42) of the analyzed bladder tumors exhibited a significant loss of RBBP8 protein expression in the nuclei with an IRS score below 3 (median IRS, 0) (Fig. 5g). Stratifying the tumor samples by substage, we revealed that the observed loss of nuclear RBBP8 protein significantly correlates with advanced invasive (pT1-pT4) bladder tumors, while pTa non-invasive tumors did not significantly differ in cytoplasmic and nuclear RBBP8 protein level (Fig. 5h).
We did not observe any association between cytoplasmatic RBBP8 staining and clinicopathological parameters. As RBBP8 is a known DNA repair protein that is functionally located in the nucleus (see for instance protein-staining pattern of breast cancer cells [43]), we further focused on nuclear RBBP8. Nuclear RBBP8 protein was significantly reduced in invasive tumors compared to papillary tumors (see Fig. 5h). A close association between loss of nuclear RBBP8 protein and both advanced tumor stages and high-grade BLCA was significantly illustrated by a Fisher’s exact test (Table 4). A significant downregulation of RBBP8 protein expression in nuclei was demonstrated for bladder tumors with an increased RBBP8 promoter methylation (> 5%) compared to those with a low RBBP8 methylation (Fig. 5i). In line, a statistically inverse correlation between nuclear RBBP8 protein level and RBBP8 promoter methylation was confirmed (Table 4), supporting a contribution of epigenetic RBBP8 alterations to its protein loss in primary bladder tumors.
Table 4.
Clinicopathological parameters in relation to nuclear RBBP8 protein expression
| RBBP8 IRSb nuclear | |||||
|---|---|---|---|---|---|
| n a | Low | High | p valuec | Spearman r | |
| Parameter: | |||||
| Age at diagnosis | |||||
| < 70 years | 23 | 14 | 9 | 0.213 | − 0.195 |
| ≥ 70 years | 19 | 15 | 4 | ||
| Gender | |||||
| Male | 33 | 25 | 8 | 0.075 | − 0.278 |
| Female | 9 | 4 | 5 | ||
| Tumor subtype | |||||
| Non-invasive papillary | 17 | 8 | 9 | 0.012 | − 0.392 |
| Invasive | 25 | 21 | 4 | ||
| Histological tumor graded | |||||
| Low grade | 12 | 4 | 8 | 0.002 | − 0.489 |
| High grade | 30 | 25 | 5 | ||
| Tumor staged | |||||
| pT1-pT2 | 13 | 9 | 4 | 0.040 | − 0.419 |
| pT3-pT4 | 12 | 12 | 0 | ||
| RBBP8 methylation | |||||
| Low | 11 | 5 | 6 | 0.020 | − 0.564 |
| High | 7 | 7 | 0 | ||
Significant p values are in italics
aOnly patients with primary bladder cancer were included
bScore (IRS) according to Remmele and Stegner [42]
cFisher’s exact test
dAccording to WHO 2004 classification
RBBP8 methylation is specifically detectable in urine samples derived from bladder cancer patients
As RBBP8 is highly methylated in bladder tumor tissue, RBBP8 methylated DNA might also be detectable in urine samples from bladder cancer patients. Therefore, urine sediments from bladder cancer patients (n = 22) and healthy controls (n = 10) were initially assessed for RBBP8 methylation by MSP (for cohort characteristics see Additional file 10). DNA methylation of RBBP8 was present in 11 out of 22 cancerous urine samples (50%) (Fig. 6a). In contrast, none of the control samples from healthy donors was tested positive for RBBP8 methylation.
Fig. 6.
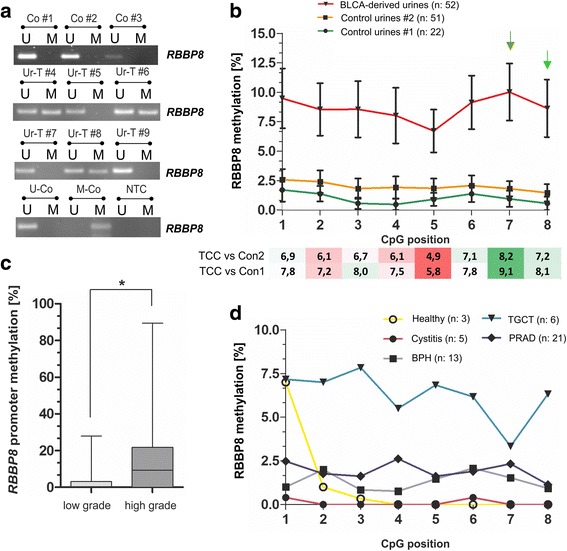
RBBP8 promoter methylation is detectable in urines from bladder cancer patients. a Representative MSP analysis shows the RBBP8 promoter methylation status of human urine samples derived from healthy controls (Co) and bladder tumors (Ur-T). Band labels with U and M represent an unmethylated and methylated DNA locus. Bisulfite-converted unmethylated, genomic (U-co), and polymethylated, genomic (M-co) DNA were used as positive controls. NTC, non-template control. b Upper graph: RBBP8 mean methylation values of analyzed CpG sites (1 to 8) in cancerous bladder diseases (BLCA-derived) and two control cohorts (benign: control urines #1 and malignant: control urines #2) using pyrosequencing. Lower heatmap: Differences of RBBP8 methylation between BLCA-derived urines and both control conditions (benign and malignant) highlighting GpG sites (#7 and #8) with the strongest impact for discrimination (see green arrows). c Box plot demonstrating a significant increase of RBBP8 methylation in high-grade bladder tumors. Horizontal lines — grouped medians. Boxes — 25 to 75% quartiles. Vertical lines — range, peak, and minimum; *p < 0.05. d RBBP8 mean methylation values of analyzed CpG sites (1 to 8) of controls classified by diseases. BPH, begin prostate hyperplasia; TGCT, testicular germ cell tumors; PRAD, prostate adenocarcinoma
Next, we assessed RBBP8 methylation using bisulfite-pyrosequencing in a collective (overall n = 103, see Table 5) comprising 52 BLCA-derived and 51 control urine samples. Control samples included inflammatory urological diseases and malignancies of the urological tract other than bladder cancer. Please note that the used control cohort which is enriched for urological malignancies does not reflect the epidemiological composition as found in populations, but served only to evaluate entity specificity of RBBP8. Quantification of RBBP8 methylation allows specification of cutoff levels, which are necessary for an objective assessment of biomarker performance. Summarized data of the mean RBBP8 methylation ratio for each CpG site of BLCA-derived and control urine set #1 (including healthy, inflammatory, and benign samples) and set #2 (including set #1 samples plus testicular germ cell tumors (TGCT) and prostate adenocarcinoma (PRAD) samples) is shown in Fig. 6b. RBBP8 methylation was clearly increased, up to 88%, in urines from bladder cancer patients. As previously observed in primary bladder cancer, RBBP8 promoter methylation detected in urines strongly correlates with high-grade tumors (Fig. 6c). This observation was statistically confirmed using a Fisher’s exact test (Table 6). Interestingly, RBBP8 mean methylation of control urines from patients diagnosed with cystitis and benign prostatic hyperplasia (BPH) was nearly similar to healthy controls. In turn, a few urines from young patients with a testicular neoplasm (mostly seminomas) showed an increased mean RBBP8 methylation (Fig. 6d).
Table 5.
Clinicopathological parameters of urine samples analyzed in this study by pyrosequencing
| Categorization | n | % analyzable |
|---|---|---|
| Controls: | 51 | |
| Age (median 69) | ||
| ≤ 69 years | 28 | 54.9 |
| > 69 years | 23 | 45.1 |
| Gender | ||
| Male | 48 | 94.1 |
| Female | 3 | 5.9 |
| Diagnosis | ||
| Disease-free | 3 | 5.9 |
| Cystitis | 5 | 9.8 |
| Benign prostatic hyperplasia | 13 | 25.5 |
| Prostate cancer | 21 | 41.2 |
| Testicular tumor | 6 | 11.8 |
| Other | 3 | 5.9 |
| BLCA asscociateda | 52 | |
| Age (median 71) | ||
| ≤ 71 years | 27 | 51.9 |
| > 71 years | 25 | 48.1 |
| Gender | ||
| Male | 42 | 80.8 |
| Female | 10 | 19.2 |
| Histological tumor gradeb | ||
| Low grade | 19 | 36.5 |
| High grade | 30 | 57.7 |
| NA | 3 | 5.8 |
| Tumor stageb | ||
| pTa | 21 | 40.4 |
| pTis | 3 | 5.8 |
| pT1 | 11 | 21.2 |
| pT2 | 8 | 15.4 |
| pT3 | 7 | 13.5 |
| pT4 | 2 | 3.8 |
aOnly urine samples of patients preoperatively diagnosed with primary bladder cancer (UC, without any other malignancy) were included
bAccording to WHO 2004 classification
Table 6.
Clinicopathological parameters in relation to RBBP8 promoter methylation in BLCA associated urine samples
| RBBP8 promoter methylationb | ||||
|---|---|---|---|---|
| n a | Low | High | p valuec | |
| Parameter: | ||||
| Gender | ||||
| Male | 42 | 31 | 11 | 0.687 |
| Female | 10 | 8 | 2 | |
| Histological tumor grade | ||||
| Low grade | 19 | 17 | 2 | 0.046 |
| High grade | 30 | 19 | 11 | |
| Tumor stage | ||||
| pTa | 21 | 19 | 2 | 0.021 |
| pT1-pT4 | 28 | 17 | 11 | |
| pN status | ||||
| Negative | 5 | 3 | 2 | 0.931 |
| Positive | 8 | 5 | 3 | |
Significant p values are in italics
aOnly patients with primary bladder cancer were included
bRBBP8 methylation was dichotomized at the Q75 (based on pyrosequencing)
cFisher’s exact test
In order to evaluate its diagnostic potential, we performed receiver operating characteristics (ROC) curve analysis to calculate the optimal cutoff value for RBBP8 as single marker, with a high specificity. ROC curve statistics were performed based on the two CpG sites with the strongest discrimination impact; the methylation ratio between cancerous BLCA urines and both the benign and malignant control set revealed strongest differences for CpG number #7 and #8 (see Fig. 6b). There was no significant difference between benign and malignant control urine samples, while RBBP8 methylation based on “best CpG sites” was highly significantly increased in BLCA-derived urines (Fig. 7a). Accordingly, RBBP8 methylation significantly discriminates between bladder cancer patients and non-malignant patients with a sensitivity of 51.9% and a specificity of 90.9% (AUC 0.730, 95% CI 0.616–0.844) (Fig. 7b). In case of 100% specificity, RBBP8 methylation as a single biomarker still showed a true positive rate of 25% (see Table 7). The control cohort consisted of patients with an inflammatory and benign diagnosis (benign control set). Additionally, the single RBBP8 marker was able to distinguish bladder cancer patients from patients with neoplasms of another urological origin (malignant control set) with high specificity (90.8%, AUC 0.686, 95% CI 0.583–0.789). Sensitivity was however reduced to 40.4% (Fig. 7c). Thus, beyond a possible prognostic or predictive relevance of RBBP8 loss in human cancers, its promoter methylation may be suitable for diagnostic or monitoring applications in bladder cancer. Since a RBBP8 methylation frequency of approximately 40% is found in bladder cancer, sensitivity is limited. Consequently, a combined application as part of a biomarker panel will be necessary to increase the true positive rate of detection.
Fig. 7.
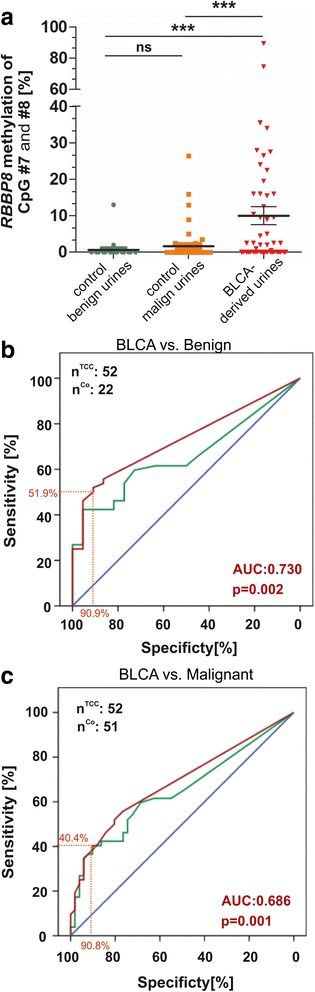
Biomarker performance of RBBP8 methylation based on a non-invasive approach. a RBBP8 methylation enables significant discrimination of cancerous bladder diseases from two control conditions (benign and malignant) using urine samples. The scatterplot shows the mean methylation values of the CpG sites #7 and #8; ns, not significant; ***p < 0.0001. b to c ROC curve analysis illustrating RBBP8 single-biomarker performance based on all analyzed CpG sites (green curve) and CpG site #7 and #8 (red curve). b ROC curve in benign disease controls. Red curve (CpG #7 and #8): the cutoff value of 1.25% methylation was defined for positive detection of disease; in that case, RBBP8 methylation achieved a specificity of 90.9% and a sensitivity of 51.9%. Area under the curve (AUC) 0.730 (95% CI, 0.616 to 0.844), p = 0.002. c ROC curve in malignant (prostate and testicular cancer) disease controls. Red curve (CpG #7 and #8): the cutoff value of 4.00% methylation was defined for positive detection of disease; in that case, RBBP8 methylation achieved a specificity of 90.2% and a sensitivity of 40.4%. Area under the curve (AUC) 0.686 (95% CI, 0.583 to 0.789), p = 0.001
Table 7.
RBBP8 biomarker performance
| Cutoff | Specificity (%) | Sensitivity (%) | AUC | p value | Control group |
|---|---|---|---|---|---|
| RBBP8 CpG #7 and #8 | |||||
| 14.25 | 100 | 25.0 | 0.730 | 0.002 | Benign |
| 1.25 | 90.9 | 51.9 | |||
| 14.25 | 96.1 | 25.0 | 0.686 | 0.001 | Malignant |
| 4.00 | 90.2 | 40.4 | |||
| RBBP8 CpG all | |||||
| 11.75 | 100 | 26.9 | 0.660 | 0.030 | Benign |
| 0.69 | 77.3 | 51.9 | |||
| 11.75 | 96.1 | 26.9 | 0.649 | 0.009 | Malignant |
| 3.94 | 90.2 | 38.5 | |||
Discussion
Dysfunction of DNA damage repair (DDR) genes in human cancer is common and, so far, mostly considered to be triggered by genetic alterations [44]. Yet, in 2015, Gao et al. proposed a notable clinical value of epigenetic alterations in distinct DDR genes in human cancer [12]. To our best knowledge, our study provides for the first time a systematic analysis of the epigenetic configuration of DNA repair genes involved in the DNA damage response across human cancer entities. As DDR genes are known to be important targets for novel therapeutic strategies [8–11] the genome-wide epigenetic configuration within promoter regions of DDR genes can be used to identify novel targets of epigenetic alterations. These targets may hold clues for non-invasive biomarker applications with predictive impact.
In the presented study, tumor-specific epigenetic deregulation of DDR genes was found to be a common event in human cancer. Statistical analysis of genome-wide 450K methylation array data identified 39 out of 177 DDR genes, whose promoter regions close to the TSS were hypermethylated in one or several tumor entities. Most tumor entities were characterized by hypermethylation of distinct DDR genes. Among others, polymerase delta (POLD1) methylation was enriched in 24 out of 32 different tumor types, implying an essential role in DNA repair or tumor suppression independent of tumor entity. This includes COAD and LIHC, i.e., entities which have been associated with increased somatic mutation rates [4]. Unrepaired DNA damage is a major source of potential mutagenic lesions driving tumorigenesis, and loss of DNA repair pathways is thought to accelerate the accumulation rate of additional mutations by 100 to 1000 times [45]. The POLD1 gene is highly conserved and encodes for the p125 subunit which provides the essential catalytic activity of polymerase δ (Polδ), mediating a key role in genome stability [46]. This raises the question whether or how methylation of the identified DDR genes may help to establish mutator phenotypes affecting the prevalence of somatic mutations in distinct tumor types. Weisenberger et al. previously revealed a close association between the CpG island methylator phenotype (CIMP), microsatellite instability, and increased BRAF mutation in COAD [47]. Moreover, sporadic cases of mismatch repair deficiency occur almost exclusively as a consequence of CIMP-associated MLH1 methylation, a DNA repair gene involved in MMR and microsatellite instability [48], whose hypermethylation close to the TSS was confirmed for COAD in the presented study.
Beyond, our findings may provide novel implications for therapies, as DNA damage repair dysregulation sensitizes cancer cells for DNA damaging agents [12]. Novel biomarkers with a clear prognostic or predictive impact are urgently required to identify those patients likely to respond favorably to such therapy, for instance, in BLCA. There is no significant progress in systemic therapy for muscle-invasive bladder cancer (MIBC) since decades [21, 49] with the exception of immunotherapeutic approaches like PDL-1 blocking antibodies [50]. Due to the lack of biomarkers [51], disease management of BLCA and hence the selection of an adequate therapy are difficult and often insufficient [52]. Here, we show that 2%, i.e., 6 out of 368 bladder patients, showed O6-methylguanine-DNA methylftransferase (MGMT) promoter hypermethylation which is in line with previous reports [53–55]. The MGMT promoter status has been shown to predict response to alkylating agents in glioblastoma patients [13, 14], and in times of personalized and precision medicine even such low case numbers could be of clinical significance. Furthermore, the excision repair cross-complementing group 1 (ERCC1) gene, encoding for a known key enzyme of the nucleotide excision repair (NER) pathway [56], was found frequently methylated in BLCA, up to 17%. Accumulating studies propose ERCC1 protein expression as a prognostic and predictive biomarker for platinum resistance in various tumor entities [37, 57] including bladder cancer [36, 58]. In BLCA, for instance, ERCC1-negative tumors could benefit from adjuvant chemotherapy combining gemcitabine and cisplatin [36], but its predictive impact remains controversial [59]. Future studies addressing the potential clinical impact of ERCC1 and the relation between its promoter methylation and gene silencing are important. Such a functional consequence would offer the development of predictive biomarkers based on a stable analyte (i.e., DNAm) which can be efficiently detected in bodily liquids like urine, in analogy to the here exemplarily identified and validated DDR lead candidate gene RBBP8/CtiP in BLCA.
RBBP8/CtiP has a proven role in homologous recombination-mediated DNA double-strand break repair (HR and NHEJ pathway) [30, 32, 33], impairment of which reduces DNA repair fidelity and may promote genome instability [34, 60] also in urothelial carcinomas. Already in 2006, genomic analysis described bladder cancer as significantly impaired in DNA repair [20]. Here, we demonstrate for the first time that RBBP8 is almost exclusively methylated in primary BLCA, suggesting the development of methylation phenotypes in a distinct molecular context. RBBP8 methylation was confirmed to be tumor-specific in up to 45% of analyzed BLCA patients. A close association between RBBP8 methylation and its gene expression in primary tumors as well as after demethylation treatment in vitro indicate that RBBP8 methylation could be responsible for its gene inactivation. Hence, it seems unlikely that entity-specific RBBP8 methylation is a silent passenger event without relevance, and we propose loss, or at least reduction, of RBBP8 mediated DNA repair function in BLCA. Interestingly, RBBP8 methylation correlates with a favorable prognosis in the BLCA TCGA dataset. Functionally, the RBBP8 protein is known to interact with BRCA1 [61], guiding HR by recruiting Dna2 to damage sites, thus ensuring a robust DSB resection necessary for efficient homologous recombination [30]. Previously, it has been shown that BRCA1/2 deficiency results in cellular sensitivity to chemotherapy [9, 62, 63]. For example, BRCA1 methylation has been revealed to predict significantly higher response rates to cisplatin treatment in breast and ovarian cancer patients [17], which is also traditionally used as the first-line agent in bladder cancer disease management [21]. Cisplatin-induced inter-strand adducts can lead to DNA lesions (double-strand breaks) which are regularly removed by the machinery of HR [64]. Unrepaired inter-strand crosslinks as consequence of reduced RBBP8 function may thereof sensitize tumor cells to chemotherapy treatment as well. Considering that a high RBBP8 methylation frequency correlates with increased survival of BLCA patients, RBBP8 methylation might be a potential biomarker for better therapy response in such subsets of patients; however, further investigation is necessary to validate this hypothetical notion. Nevertheless, accumulating studies already functionally demonstrated the involvement of RBBP8 on sensitizing breast [27] and ovarian cancer cells [26] for PARP inhibitor treatment in a similar way as BRCA1 mutations, thus unveiling novel therapeutic options for a significant subset of patients which may benefit from such approach.
The field of liquid biopsy is rapidly evolving [65] and shows that biomarkers can be detected in biological fluids like blood or urine, offering an easy and non-invasive application in diagnosis of and therapy prediction in cancer. Application of liquid biopsy tools are of importance in bladder cancer, as its clinical management remains challenging due to its high recurrence rate (up to 70%) [66] in the first 2 years after diagnosis, requiring a lifetime of surveillance, i.e., BLCA patients undergo multiple invasive procedures. One of these methods is the cystoscopy, representing the gold standard test for the detection of bladder cancer, achieving an operator-dependent sensitivity over 90%. Cystoscopy is however associated with significant discomfort, possible risk of infection, and high costs [67, 68]. Current guidelines recommend the application of non-invasive urine cytology testing complementary to cystoscopic assessment. Nevertheless, cytology is characterized by poor sensitivity, especially for low-grade and low-stage tumors like CIS [69]. In addition, previously developed non-invasive diagnostic tools, such as the “BladderCheck (point-of-care) Test” based on the protein marker NMP22, either lack sensitivity or specificity [69]. Urine-based tests assessing aberrant DNA methylation are emerging as a potential tool for cancer detection [6], and various studies described the identification of novel DNA methylation-based biomarkers. Nevertheless, although both multiple biomarker tests and single-biomarker candidates like Vimentin [70] were able to provide prognostic information, they did not show any predictive impact. In addition, specificity across tumor entities is disregarded in most studies. Here, we demonstrate that RBBP8 hypermethylation was accessible through a non-invasive urine test. Using RBBP8 as a methylation biomarker, BLCA could be detected with a 25% sensitivity (at maximal specificity of 100%) in urines from BLCA patients, i.e., enabling to distinguish early cancerous (CIS), high-grade urothelial, and even papillary subtype tumors, which are difficult to detect by urine cytology, from non-cancerous and benign ones. At 90.9% specificity, the sensitivity of RBBP8 was even increased up to 51.9%. Importantly, in comparison to a control urine cohort including urological malignancies like prostate and testicular cancer, we were still able to detect over 40% of primary bladder tumors with high specificity. Hence, we provide a novel biomarker candidate, i.e., RBBP8, which has previously been shown to sensitize tumor cells for PARP1 inhibitors and which may improve entity-specificity for monitoring, early detection, and clinical management of bladder cancer in a urine biomarker panel.
Conclusions
In the current study, we systemically screened for epigenetic configuration of DNA repair genes involved in the DNA damage response (DDR) across 32 human cancer entities and identified 39 methylated DDR genes which may be important targets for novel therapeutic strategies. This includes known epigenetic silenced repair genes like MGMT, MLH1, and ERCC1 but also novel targets of epigenetic dysregulation like RBBP8/CtiP, which has a proven role in homologous recombination-mediated DNA double-strand break repair and is further known to sensitize cancer cells for PARP1 inhibitors. RBBP8 was almost exclusively hypermethylated in bladder cancers and was detectable by a non-invasive approach in urines from bladder cancer patients. Hence, RBBP8 might serve as a complementary biomarker of high specificity that can be accessed through a urine test. Further investigation of mechanisms involving RBBP8 may deliver novel therapeutic options for bladder cancer patients, finally highlighting RBBP8 as a predictive biomarker as well.
Methods
Reagents
The DNA methyltransferase (DNMT) inhibitor 5-aza-2′-deoxycytidine (decitabine (DAC); 1 mM in phosphate-buffered saline (PBS)) and the histone deacetylase inhibitor trichostatin A (TSA; 1 mM in PBS) were obtained from Sigma-Aldrich. Drugs were stored in aliquots at − 80 °C.
Cell lines
Unless stated otherwise, cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and cultured as recommended by the vendor. ATCC provides molecular authentication in support of their collection through their genomics, immunology, and proteomic cores, as described, by using DNA barcoding and species identification, quantitative gene expression, and transcriptomic analyses. Cells were tested negative for mycoplasma infection before and after experiments. The 37 used cell lines were UROtsa (normal bladder urothelium), RT4, RT112, EJ28, J82 (bladder cancer), RWPE-1 (normal prostate epithelium), LNCaP, PC3, DU145 (prostate cancer), SKRC1, SKRC10, SKRC52, SKRC59 (clear cell renal cell carcinoma), HCT-116, RKO, CACO2, COLO205, HT29, SW480, CACO320 (colorectal cancer), MCF10A, MCF12A, MCF7, T-47D, ZR75-1, BT474, SKBR3, MDA-MB-231, MDA-MB-436, MDA-MB-468, UACC3199, HCC1937, BT20 (breast cancer), A549, H157, and H2170 (lung cancer).
Formalin-fixed paraffin-embedded tissue specimens
Tissues of primary bladder tumors and normal urothelium were obtained from the archives of the Institute of Pathology (RWTH Aachen University). The anonymized and retrospective study was approved by the local Ethics Committee (EK 122/04, 173/06, and 206/09). For cohort characteristics of analyzed samples, see Additional file 8. Only manually microdissected samples were used for RBBP8 methylation analysis. RBBP8 protein expression was evaluated using whole tissue sections or tissue microarrays with representative tissue cores of 1.5 mm diameter.
Human urine samples
Voided urine samples from patients diagnosed with a primary bladder tumor (n = 52) were used to assess biomarker performance. Urine samples of patients with a known second malignancy such as prostate cancer were excluded from the study. Urines from healthy donors (n = 13) and samples derived from patients with 5 inflammatory (chronic cystitis), 13 benign (benign prostate hyperplasia), and 27 urological malignant diseases (testicular tumors and prostate cancer) served as controls. For cohort characteristics, see Table 5 and Additional file 10. All urine specimens were obtained from the RWTH centralized biomaterial bank (RWTH cBMB) and the Technical University of Munich. All patients gave written consent for retention and analysis of their samples according to local Institutional Review Board (IRB)-approved protocols of the Medical Faculty of RWTH Aachen University and the Technical University of Munich. For each sample, 20 ml of (morning) urines were centrifuged for 10 min at 2000×g, and sediments were stored at − 80 °C.
Genome-wide methylation analysis using TCGA data sets
Data retrieval
Infinium HumanMethylation450 BeadChip data (level 2) and RNASeqV2 data (level 3) of the tumor and normal tissue samples were obtained from the TCGA data portal [1–4]. Samples with any annotations reported via the TCGA annotation portal were excluded from the analyses. The β value of methylation was calculated according to Bibikova et al. [71] for all CpG probes with a detection p value ≤ 0.05. Probes with a detected p value above the threshold were classified as unmeasured. Multiple measurements per tumor were averaged after log-transformation of expression data and transformation of methylation data to M values [72]. In total, 7819 tumors and 659 normal samples were included in the differential CpG methylation analysis (Table 1). Five thousand one hundred ninety-one tumor samples were included in the correlation analysis of gene expression and CpG methylation. A summary of the TCGA samples that used both datasets can be found in Additional file 11.
Identification of CpG sites close to TSS of DDR genes
A list of 177 DNA damage repair genes was used for genome-wide analysis of which none was located on the sex chromosomes (Additional file 12). Information on gene location and transcription start sites (TSS) was obtained from the Ensembl database. For each gene, CpG probes (probe = CpG site) located 2000 bp downstream to 500 bp upstream of a reported TSS were determined.
Definition of probe sets near a TSS
An exploratory data analysis was performed to identify genomic regions with tumor-specific methylation, i.e., showing unmethylated CpGs in available normal tissue samples. In brief, clusters of TSS were identified (hierarchical clustering with single linkage; cut at a height of 2000 bp). Available methylation data from normal tissues were then visualized (see Additional file 13) in the regions surrounding the TSS clusters. Probes of CpGs in continuously unmethylated (β < 0.25) regions were grouped together as a set. Based on both the entity-specific patterns of methylation in tumor tissues (see Additional file 13) and the correlation coefficients between CpG sites (see Additional file 13), probe sets were further classified to define CpG (probe) sets with a putative regulatory impact within promoter regions of DDR genes. This approach led to the identification of one to four CpG (probe) sets for most candidate genes (170 out of 177). The majority (140 out of 170) of these genes showed only one CpG set, while for some genes, two (20 out of 170), three (2 out of 170) or four (1 out of 170) sets were considered. The detailed definition of used CpG probe sets for DDR genes can be found in the supplementary data (Additional file 14).
Determination of entity-specific frequency of hypermethylation
Given the definition of CpG probe sets as promoter regions potentially involved in gene regulation of the corresponding DDR gene, frequency of hypermethylation in tumor tissues from different entities was calculated for each gene. With respect to known characteristics of used tumor samples (i.e., the contamination with stromal cells (e.g., fibroblasts) in up to 40% of TCGA specimens; the copy number variations or whole genome duplications (e.g., [73]); and lastly, the prevalence of subclones) which could affect tumor-associated allele frequencies, a stepwise prioritization strategy with adjusted β value thresholds was conducted.
A CpG set specific methylation value was determined by the calculation of the median methylation level for each sample. Secondly, as the M value is more statistically valid for the differential analysis of methylation levels [72], a probe set specific (∆βco) cutoff for the tumor samples was derived jointly from all available normal samples after transformation to M values as the arithmetic mean plus three standard deviations. To further ensure robust confirmation of these findings by alternative methods such as pyrosequencing of sodium bisulfite converted DNA (e.g., [74, 75]), we increased this cutoff by a β value of 0.1 (see Additional file 14), hence defining minimum β value differences (∆βco) of 0.1–0.2 between normal and tumor samples which is in line with previous studies (e.g., [76]). In practice, a set specific β value above approximately 0.25 (90% CI 0.20–0.35) was considered to indicate hypermethylation of TSS regions in tumor samples. The fraction of samples with a hypermethylation event was calculated for each tumor entity separately.
In vitro genomic DNA demethylating treatment
For gene re-expression experiments, cells were seeded at 2.5 × 104 cells/cm2 in 6-well plates. After overnight attachment, the DNMT inhibitor DAC was added and renewed every 24 h. For a combined treatment with a histone deacetylase inhibitor, TSA was added at 300 nM to cells 16 h prior to RNA extraction. After 72 h, cells were harvested, and RNA was extracted.
Nucleic acid extraction from cell lines, FFPE tissues, and urines
Sections of archival formalin-fixed paraffin-embedded (FFPE) tissue samples were microdissected, when necessary, deparaffinized in xylene, and re-hydrated in a decreasing alcohol series prior to nucleic acid extraction. Genomic DNA of bladder samples was isolated using the QIAamp® DNA Mini kit (Qiagen, Hilden, Germany). Total RNA from cell lines was extracted using the TRIzol® (Life Technologies) method according to the manufacturers’ instructions.
Sediments of urine samples stored at − 80 °C were subjected to DNA extraction using the ZR Urine DNA Isolation Kit (ZR, Zymo Research, Orange, Ca, USA) following the manufacturer’s instructions. The DNA yield (ng/ml urine) and purity (A260/A280) were determined by using the NanoDrop (Thermo Fisher Scientific, Waltham, MA, USA). Only extractions from urines with a minimal amount of 100 ng genomic DNA and a ratio of > 1.5 were finally used for MSP and pyrosequencing analyses. Note that the extracted DNA derived from FFPE tissue samples are known to be more fragmented and altered compared, for instance, to DNA from urines. Therefore, results like methylation frequencies may slightly differ between both materials.
Bisulfite conversion and methylation-specific PCR
Five hundred nanogram of the genomic DNA was bisulfite-converted for 16 h using the EZ DNA Methylation™ kit (Zymo Research, Freiburg, Germany) according to the manufacturer’s instructions. Bisulfite-converted DNA was eluted in 20 μl of TRIS-EDTA buffer. MSP was performed as previously described [77]. In brief, 150 to 500 ng of converted DNA was amplified using MSP primers (designed by using MethyPrimer software [78]; for primers, see Additional file 15) that specifically met the following criteria: (1) In order to validate TCGA datasets, MSP primers recognizing either the unmethylated or methylated RBBP8 gene sequence, covered CpG sites also used in the TCGA data analysis. (2) To reveal whether the epigenetic alteration could be responsible for RBBP8 gene inactivation, which is a requirement for a causative impact on therapeutic responses, designed MSP primers annealed closely to the TSS (core promoter region). The EpiTect® PCR Control DNA Set (Qiagen) was used as positive controls for unmethylated and methylated DNA. Amplified products were visualized on 2% agarose gels (Biozym, Hessian Oldendorf, Germany) containing ethidium bromide (AppliChem, Darmstadt, Germany) and illuminated under UV light.
Bisulfite-pyrosequencing
In order to assess RBBP8 methylation in detail, pyrosequencing of eight CpG sites was performed that are located in the RBBP8 promoter area close to the transcription start site. The RBBP8 pyrosequencing assay was designed by using the Pyromark Assay Design Software (Qiagen), and all primers are listed in Additional file 16. Initially, a 136-bp fragment of the RBBP8 promoter region including CpG sites of both the complete MSP amplicon and the TCGA analysis was amplified by using the PyroMark PCR Kit (Qiagen). Degenerate PCR primers assured unbiased DNA amplification independently of the methylation status. A 73-pb region of the PCR amplicon, referred to as the sequence of interest, was subsequently sequenced covering eight CpG sites. Methylation ratio for each CpG of the RBBP8 promoter was quantified based on the PyroMark96 ID device and the PyroGoldSQA reagent Kit (Qiagen) as previously specified [79].
Reverse transcription and quantitative RT-PCR
One microgram of the total RNA was reverse transcribed using the Reverse Transcription System (Promega, Mannheim, Germany). qRT-PCR was accomplished on an iQ5 Real-Time PCR Detection System (Bio-Rad, München, Germany) together with the iQ™ SYBR® Green Supermix (Bio-Rad) including 5 μM of intron-spanning oligonucleotide primers (see Additional file 17) and 20 ng of cDNA in a 20 μl reaction volume. The cycling profile was 95 °C for 10 min, followed by 40 cycles of 95 °C for 30 s and 60 °C for 60 s. All reactions were run in triplicate, and post-amplification melting curve analyses assessed the product specificity. Gene expression relative to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was calculated by the ΔΔCT method [80].
RBBP8 immunohistochemistry
Immunohistochemical staining of RBBP8 protein was performed after heat-induced antigen retrieval (EnVision™ FLEX Target Retrieval Solution, Low pH, K8005, DAKO PT-Link, DAKO, Hamburg, Germany) according to the manufacturer’s protocols. Primary anti-RBBP8 antibody (dilution 1:200) (Atlas Antibodies, HPA039890, Sigma-Aldrich, Germany) was linked with DAKO EnVision™FLEX system and visualized with DAKO Liquid DAB Substrate Chromogen System in a DAKO Autostainer plus (K8024, K3468, DAKO). RBBP8 protein staining was quantified by a pathologist using an adapted immunoreactive scoring system (IRS) according to Remmele and Stegner [42].
Statistical analyses
Data analyses and statistical correlations of TCGA datasets were performed using R Statistical Software version 3.2.0 [81] (R Foundation for Statistical Computing, Vienna, Austria). The parameters of beta distributions were estimated by the maximum likelihood estimator implemented in the RPMM package [82]. Two-sided p values less than 0.05 were considered significant. In order to compare two groups, the non-parametric Mann-Whitney U test was implemented, whereas in case of more than two groups, the Dunn’s multiple comparison test was used. Statistical associations between clinicopathological and molecular factors were determined by Fisher’s exact test using SPSS software version 22.0 (SPSS Inc., Chicago, USA). Survival curves for overall survival (OS) were calculated using the Kaplan-Meier method with log-rank statistics. OS was measured from surgery until death and was censored for patients alive without evidence of death at the last follow-up date. Multivariate Cox regression analysis was performed to test for an independent prognostic value of RBBP8 methylation. Receiver operating characteristics (ROC) curves were calculated to assess biomarker performance of RBBP8 methylation in urine samples [83].
Additional files
Schematic map of the underlying study design comprising the discovery and the validation step. (DOCX 146 kb)
Detailed frequency of DNAm of BRCA1, MGMT, and ERCC1 across cancer entities and corresponding normal tissues. (DOCX 296 kb)
Heatmap of tumor-specific DNAm of DDR genes across cancer entities. (DOCX 345 kb)
Graphs illustrating the correlation between DDR gene hypermethylation and their gene expression tumor entities. (DOCX 358 kb)
This table illustrates the clinicopathological parameters of 405 bladder cancer specimens of the TCGA network analyzed in this study. (DOC 48 kb)
This table summarizes the results from the multivariate Cox regression analysis including all factors influencing overall survival. (DOC 34 kb)
This table lists the RBBP8 methylation in human cell lines analyzed by MSP. (DOC 52 kb)
This table shows the results of bivariate correlation statistics (Fisher exact test) between clinicopathological parameters and RBBP8 methylation. (DOC 49 kb)
A table is shown characterizing 42 bladder cancer specimens analyzed in this study by IHC. (DOC 46 kb)
This table summarizes the clinicopathological parameters of urine samples analyzed in this study by MSP. (DOC 48 kb)
List of 177 DDR genes analyzed in this study. (XLS 1076 kb)
This dataset illustrates a summary of the TCGA samples used in this study. (XLS 27 kb)
Illustrating the definition of probe sets near a TSS using three different genes (RBBP8, MGMT, and LIG4). (DOCX 1053 kb)
This dataset summarizes the beta values according to a defined cutoff of 0.1. (XLS 62 kb)
This table shows the sequences of all primers and conditions used in this study for MSP analysis. (DOC 33 kb)
This table lists the primer sequences for bisulfite-pyrosequencing of the RBBP8 promoter region. (DOC 32 kb)
Sequences of all primers and conditions for qPCR analysis are summarized in a table. (DOC 32 kb)
Acknowledgements
This paper is dedicated to the memory of our unforgotten colleague and head of the working group “Translational Pathology” Dr. Jürgen Veeck who designed and initiated this study but passed away after losing his short battle with cancer. We furthermore thank Timon Heide for his great support in analyzing the statistical datasets. The excellent technical assistance of Ursula Schneider is thankfully acknowledged. Genomic 450K methylation analysis is generated by using the TCGA Research Network datasets (http://cancergenome.nih.gov), and we would like to thank them for their efforts in making this data publicly available. We finally thank the following colleagues for kindly contributing cell lines: Sven Perner (University of Bonn; LNCaP and RWPE-1 cells) and Manon van Engeland (Maastricht University; HCT116, RKO, CACO2, COLO205, HT29, SW480, CACO320, SKRC1, SKRC10, SKRC52, and SKRC59 cells).
Funding
This work was supported by a grant from the Medical Faculty of the RWTH Aachen University (START program project 113/15).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- ATCC
American Type Culture Collection
- AUC
Area under curve
- BER
Base excision repair
- BLCA
Bladder cancer
- BPH
Benign prostatic hyperplasia
- BRCA1/2
Breast cancer 1/2, early onset
- cBMB
Centralized biomaterial bank
- cDNA
Copy number desoxyribonucleic acid
- CIMP
CpG island methylator phenotype
- CpG
5′-deoxycytidine-phosphate-deoxyguanosine-3′
- CtiP
CtBP-interacting protein
- DAC
Decitabine
- DDR
DNA damage repair
- DNA
Deoxyribonucleic acid
- DNAm
DNA methylation
- DNMT
DNA methyltransferase
- DSB
Double-strand break
- EK
Ethics committee
- ERCC1
Excision repair cross-complementing group 1
- FC
Fold change
- FFPE
Formalin-fixed paraffin-embedded
- GAPDH
Glyeradehyde 3-phosphate dehydrogenase
- HR
Homologous recombination
- IHC
Immunohistochemistry
- IRS
Immunoreactive score
- MGMT
O6-methylguanine-DNA methyltransferase
- MIBC
Muscle-invasive bladder cancer
- MLH1
MutL homolog 1
- MMR
Mismatch repair
- mRNA
Messenger ribonucleic acid
- MSP
Methylation-specific PCR
- n
Number
- NER
Nucleotide excision repair
- NHEJ
Non-homologous end-joining
- OS
Overall survival
- PARP
Poly (ADP-ribose) polymerase
- PBS
Phosphate-buffered saline
- PCR
Polymerase chain reaction
- POLD1
Polymerase delta 1
- qPCR
Quantitative PCR
- ROC
Receiver operating characteristic
- RWTH
Rheinisch-Westfälisch Technische Hochschule
- SCC
Squamous cell carcinoma
- s.d.
Standard deviation
- s.e.m.
Standard error of the margin
- TCGA
The Cancer Genome Atlas
- TSA
Trichostatin A
- TSS
Transcription start site
- USA
United States of America
Authors’ contributions
JM performed the methylation analysis and edited the manuscript. JV participated in the design of the study, drafted the manuscript, and was involved in bioinformatics. NTG, EB, TCR, and SG were involved in the experimental data acquisition and analysis. ED, DP, and SCS provided the human patients’ samples. RK participated in the design of the study. MR conceived and coordinated the study, was involved in data analysis, and wrote the manuscript. All authors read, critically revised, and approved the final manuscript.
Ethics approval and consent to participate
As stated in the “Methods” section, tissue and urine samples for the anonymized and retrospective study were obtained from the archives of the Institute of Pathology (RWTH Aachen University), the RWTH centralized biomaterial bank (RWTH cBMB), and the Technical University of Munich and approved by the local Institutional Review Board (IRB) of the Medical Faculty of RWTH Aachen University and the Technical University of Munich.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13148-018-0447-6) contains supplementary material, which is available to authorized users.
Contributor Information
Jolein Mijnes, Email: jmijnes@ukaachen.de.
Jürgen Veeck, Email: Juergen.Veeck@rwth-aachen.de.
Nadine T. Gaisa, Email: ngaisa@ukaachen.de
Eduard Burghardt, Email: Eduard.Burghardt@gmx.de.
Tim C. de Ruijter, Email: t.deruijter@maastrichtuniversity.nl
Sonja Gostek, Email: s.gostek@gmx.de.
Edgar Dahl, Email: edahl@ukaachen.de.
David Pfister, Email: david.pfister@uk-koeln.de.
Sebastian C. Schmid, Email: sebastian.schmid@tum.de
Ruth Knüchel, Email: rknuechel-clarke@ukaachen.de.
Michael Rose, Phone: +49-(0)241-80-89282, Email: mrose@ukaachen.de.
References
- 1.Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D, Zhu J. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci Rep. 2013;3:2652. doi: 10.1038/srep02652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell BR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witte T, Plass C, Gerhauser C. Pan-cancer patterns of DNA methylation. Genome Med. 2014;6:66. doi: 10.1186/s13073-014-0066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa-Pinheiro P, Montezuma D, Henrique R, Jerónimo C. Diagnostic and prognostic epigenetic biomarkers in cancer. Epigenomics. 2015;7:1003–1015. doi: 10.2217/epi.15.56. [DOI] [PubMed] [Google Scholar]
- 7.Miozzo M, Vaira V, Sirchia SM. Epigenetic alterations in cancer and personalized cancer treatment. Future Oncol. 2015;11:333–348. doi: 10.2217/fon.14.237. [DOI] [PubMed] [Google Scholar]
- 8.Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 9.Asakawa H, Koizumi H, Koike A, Takahashi M, Wu W, Iwase H, Fukuda M, Ohta T. Prediction of breast cancer sensitivity to neoadjuvant chemotherapy based on status of DNA damage repair proteins. Breast Cancer Res. 2010;12:R17. doi: 10.1186/bcr2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang J, D’Andrea AD, Kozono D. A DNA repair pathway-focused score for prediction of outcomes in ovarian cancer treated with platinum-based chemotherapy. J Natl Cancer Inst. 2012;104:670–681. doi: 10.1093/jnci/djs177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gkotzamanidou M, Sfikakis PP, Kyrtopoulos SA, Bamia C, Dimopoulos MA, Souliotis VL. Chromatin structure, transcriptional activity and DNA repair efficiency affect the outcome of chemotherapy in multiple myeloma. Br J Cancer. 2014;111:1293–1304. doi: 10.1038/bjc.2014.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao D, Herman JG, Guo M. The clinical value of aberrant epigenetic changes of DNA damage repair genes in human cancer. Oncotarget. 2016;7:37331–37346. doi: 10.18632/oncotarget.7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–1354. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 14.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 15.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 16.Veeck J, Ropero S, Setien F, Gonzalez-Suarez E, Osorio A, Benitez J, Herman JG, Esteller M. BRCA1 CpG island hypermethylation predicts sensitivity to poly (adenosine diphosphate)-ribose polymerase inhibitors. J Clin Oncol. 2010;28:563–564. doi: 10.1200/JCO.2010.30.1010. [DOI] [PubMed] [Google Scholar]
- 17.Stefansson OA, Villanueva A, Vidal A, Martí L, Esteller M. BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics. 2012;7:1225–1229. doi: 10.4161/epi.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drew Y, Mulligan EA, Vong WT, Thomas HD, Kahn S, Kyle S, Mukhopadhyay A, Los G, Hostomsky Z, Plummer ER, et al. Therapeutic potential of poly (ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst. 2011;103:334–346. doi: 10.1093/jnci/djq509. [DOI] [PubMed] [Google Scholar]
- 19.Moutinho C, Martinez-Cardús A, Santos C, Navarro-Pérez V, Martínez-Balibrea E, Musulen E, Carmona FJ, Sartore-Bianchi A, Cassingena A, Siena S, et al. Epigenetic inactivation of the BRCA1 interactor SRBC and resistance to oxaliplatin in colorectal cancer. J Natl Cancer Inst. 2014;106:djt322. doi: 10.1093/jnci/djt322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X, Gu J, Grossman HB, Amos CI, Etzel C, Huang M, Zhang Q, Millikan RE, Lerner S, Dinney CP, et al. Bladder cancer predisposition: a multigenic approach to DNA-repair and cell-cycle-control genes. Am J Hum Genet. 2006;78:464–479. doi: 10.1086/500848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knollman H, Godwin JL, Jain R, Wong YN, Plimack ER, Geynisman DM. Muscle-invasive urothelial bladder cancer: an update on systemic therapy. Ther Adv Urol. 2015;7:312–330. doi: 10.1177/1756287215607418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arcas A, Fernández-Capetillo O, Cases I, Rojas AM. Emergence and evolutionary analysis of the human DDR network: implications in comparative genomics and downstream analyses. Mol Biol Evol. 2014;31:940–961. doi: 10.1093/molbev/msu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu X, Baer R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J Biol Chem. 2000;275:18541–18549. doi: 10.1074/jbc.M909494199. [DOI] [PubMed] [Google Scholar]
- 24.Chinnadurai G. CtIP, a candidate tumor susceptibility gene is a team player with luminaries. Biochim Biophys Acta. 2006;1765:67–73. doi: 10.1016/j.bbcan.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Gaymes TJ, Mohamedali AM, Patterson M, Matto N, Smith A, Kulasekararaj A, Chelliah R, Curtin N, Farzaneh F, Shall S, et al. Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP-ribose) polymerase inhibitors in myeloid malignancies. Haematologica. 2013;98:1397–1406. doi: 10.3324/haematol.2012.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin ZP, Ratner ES, Whicker ME, Lee Y, Sartorelli AC. Triapine disrupts CtIP-mediated homologous recombination repair and sensitizes ovarian cancer cells to PARP and topoisomerase inhibitors. Mol Cancer Res. 2014;12:381–393. doi: 10.1158/1541-7786.MCR-13-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Ding Q, Fujimori H, Motegi A, Miki Y, Masutani M. Loss of CtIP disturbs homologous recombination repair and sensitizes breast cancer cells to PARP inhibitors. Oncotarget. 2016;7:7701–7714. doi: 10.18632/oncotarget.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruz-García A, López-Saavedra A, Huertas P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014;9:451–459. doi: 10.1016/j.celrep.2014.08.076. [DOI] [PubMed] [Google Scholar]
- 30.Hoa NN, Kobayashi J, Omura M, Hirakawa M, Yang SH, Komatsu K, Paull TT, Takeda S, Sasanuma H. BRCA1 and CtIP are both required to recruit Dna2 at double-strand breaks in homologous recombination. PLoS One. 2015;10:e0124495. doi: 10.1371/journal.pone.0124495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quennet V, Beucher A, Barton O, Takeda S, Löbrich N. CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic Acids Res. 2011;39:2144–2152. doi: 10.1093/nar/gkq1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makharashvili N, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, Deshpande RA, Lee JH, Lobrich M, Sleckman BP, et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol Cell. 2014;54:1022–1033. doi: 10.1016/j.molcel.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu B, Chen PL. Expression of PCNA-binding domain of CtIP, a motif required for CtIP localization at DNA replication foci, causes DNA damage and activation of DNA damage checkpoint. Cell Cycle. 2009;8:1409–1420. doi: 10.4161/cc.8.9.8322. [DOI] [PubMed] [Google Scholar]
- 35.Lafranchi L, de Boer HR, de Vries EG, Ong SE, Sartori AA, van Vugt MA. APC/C (Cdh1) controls CtIP stability during the cell cycle and in response to DNA damage. EMBO J. 2014;33:2860–2879. doi: 10.15252/embj.201489017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffmann AC, Wild P, Leicht C, Bertz S, Danenberg KG, Danenberg PV, Stöhr R, Stöckle M, Lehmann J, Schuler M, et al. MDR1 and ERCC1 expression predict outcome of patients with locally advanced bladder cancer receiving adjuvant chemotherapy. Neoplasia. 2010;12:628–636. doi: 10.1593/neo.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhlmann JD, Wimberger P, Bankfalvi A, Keller T, Schöler S, Aktas B, Buderath P, Hauch S, Otterbach F, Kimmig R, et al. ERCC1-positive circulating tumor cells in the blood of ovarian cancer patients as a predictive biomarker for platinum resistance. Clin Chem. 2014;60:1282–1289. doi: 10.1373/clinchem.2014.224808. [DOI] [PubMed] [Google Scholar]
- 38.Wolff EM, Chihara Y, Pan F, Weisenberger DJ, Siegmund KD, Sugano K, Kawashima K, Laird PW, Jones PA, Liang G. Unique DNA methylation patterns distinguish noninvasive and invasive urothelial cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res. 2010;70:8169–8178. doi: 10.1158/0008-5472.CAN-10-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 41.Masters JR, Hepburn PJ, Walker L, Highman WJ, Trejdosiewicz LK, Povey S, Parkar M, Hill BT, Riddle PR, Franks LM. Tissue culture model of transitional cell carcinoma: characterization of twenty-two human urothelial cell lines. Cancer Res. 1986;46:3630–3636. [PubMed] [Google Scholar]
- 42.Remmele W, Stegner HE. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe. 1987;8:138–140. [PubMed] [Google Scholar]
- 43.Soria-Bretones I, Sáez C, Ruíz-Borrego M, Japón MA, Huertas P. Prognostic value of CtIP/RBBP8 expression in breast cancer. Cancer Med. 2013;2:774–783. doi: 10.1002/cam4.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abbotts R, Thompson N, Madhusudan S. DNA repair in cancer: emerging targets for personalized therapy. Cancer Manag Res. 2014;6:77–92. doi: 10.2147/CMAR.S50497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tumini E, Barroso S, Pérez-Calero C, Aguilera A. Roles of human POLD1 and POLD3 in genome stability. Sci Rep. 2016;6:38873. doi: 10.1038/srep38873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weisenberger DJ, Siegmund KD, Campan M, Young D, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 48.Thibodeau SN, French AJ, Roche PC, Cunningham JM, Tester DJ, Lindor NM, Moslein G, Baker SM, Liskay RM, Burgart LJ, et al. Altered expression of hMSH2 and hMLH1 in tumors with microsatellite instability and genetic alterations in mismatch repair genes. Cancer Res. 1996;56:4836–4840. [PubMed] [Google Scholar]
- 49.Sonpavde G, Gordetsky JB, Lockhart ME, Nix JW. Chemotherapy for muscle-invasive bladder cancer: better late than never? J Clin Oncol. 2016;34:780–785. doi: 10.1200/JCO.2015.65.4442. [DOI] [PubMed] [Google Scholar]
- 50.Inman BA, Longo TA, Ramalingam S, Harrison MR. Atezolizumab: a PD-L1-blocking antibody for bladder cancer. Clin Cancer Res. 2017;23:1886–1890. doi: 10.1158/1078-0432.CCR-16-1417. [DOI] [PubMed] [Google Scholar]
- 51.Bertz S, Otto W, Denzinger S, Wieland WF, Burger M, Stohr R, Link S, Hofstadter F, Hartmann A. Combination of CK20 and Ki-67 immunostaining analysis predicts recurrence, progression, and cancerspecific survival in pT1 urothelial bladder cancer. Eur Urol. 2014;65:218-26. [DOI] [PubMed]
- 52.Otto W, Denzinger S, Fritsche HM, Burger M, Wieland WF, Hofstadter F, Hartmann A, Bertz S. The WHO classification of 1973 is more suitable than the WHO classification of 2004 for predicting survival in pT1 urothelial bladder cancer. BJU Int. 2011;107:404–408. doi: 10.1111/j.1464-410X.2010.09515.x. [DOI] [PubMed] [Google Scholar]
- 53.Maruyama R, Toyooka S, Toyooka KO, Harada K, Virmani AK, Zöchbauer-Müller S, Farinas AJ, Vakar-Lopez F, Minna JD, Sagalowsky A, et al. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res. 2001;61:8659–8663. [PubMed] [Google Scholar]
- 54.Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–3229. [PubMed] [Google Scholar]
- 55.Christmann M, Verbeeck B, Roos WP, Kaina B. O(6)-methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: enzyme activity, promoter methylation and immunohistochemistry. Biochim Biophys Acta. 2011;1816:179–90. [DOI] [PubMed]
- 56.Westerveld A, Hoeijmakers JH, van Duin M, de Wit J, Odijk H, Pastink A, Wood RD, Bootsma D. Molecular cloning of a human DNA repair gene. Nature. 1984;310:425–429. doi: 10.1038/310425a0. [DOI] [PubMed] [Google Scholar]
- 57.Usanova S, Piée-Staffa A, Sied U, Thomale J, Schneider A, Kaina B, Köberle B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol Cancer. 2010;9:248. doi: 10.1186/1476-4598-9-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun JM, Sung JY, Park SH, Kwon GY, Jeong BC, Seo SI, Jeon SS, Lee HM, Jo J, Choi HY, et al. ERCC1 as a biomarker for bladder cancer patients likely to benefit from adjuvant chemotherapy. BMC Cancer. 2012;12:187. doi: 10.1186/1471-2407-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klatte T, Seitz C, Rink M, Rouprêt M, Xylinas E, Karakiewicz P, Susani M, Shariat SF. ERCC1 as a prognostic and predictive biomarker for urothelial carcinoma of the bladder following radical cystectomy. J Urol. 2015;194:1456–1462. doi: 10.1016/j.juro.2015.06.099. [DOI] [PubMed] [Google Scholar]
- 60.Eid W, Steger M, El-Shemerly M, Ferretti LP, Peña-Diaz J, König C, Valtorta E, Sartori AA, Ferrari S. DNA end resection by CtIP and exonuclease 1 prevents genomic instability. EMBO Rep. 2010;11:962–968. doi: 10.1038/embor.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chaudhry P, Srinivasan R, Patel FD. Utility of gene promoter methylation in prediction of response to platinum-based chemotherapy in epithelial ovarian cancer (EOC) Cancer Investig. 2009;27:877–884. doi: 10.1080/07357900902849699. [DOI] [PubMed] [Google Scholar]
- 63.Zhang S, Yuan Y, Hao D. A genomic instability score in discriminating nonequivalent outcomes of BRCA1/2 mutations and in predicting outcomes of ovarian cancer treated with platinum-based chemotherapy. PLoS One. 2014;9:e113169. doi: 10.1371/journal.pone.0113169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 65.Wan JC, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R, Rosenfeld N. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 66.Kaufman DS, Shipley WU, Feldman AS. Bladder cancer. Lancet. 2009;374:239–249. doi: 10.1016/S0140-6736(09)60491-8. [DOI] [PubMed] [Google Scholar]
- 67.van der Aa MN, Steyerberg EW, Sen EF, Zwarthoff EC, Kirkels WJ, van der Kwast TH, Essink-Bot ML. Patients’ perceived burden of cystoscopic and urinary surveillance of bladder cancer: a randomized comparison. BJU Int. 2008;101:1106–1110. doi: 10.1111/j.1464-410X.2007.07224.x. [DOI] [PubMed] [Google Scholar]
- 68.Sievert KD, Amend B, Nagele U, Schilling D, Bedke J, Horstman M, Hennenlotter J, Kruck S, Stenzl A. Economic aspects of bladder cancer: what are the benefits and costs? World J Urol. 2009;27:295–300. doi: 10.1007/s00345-009-0395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ye F, Wang L, Castillo-Martin M, McBride R, Galsky MD, Zhu J, Boffetta P, Zhang DY, Cordon-Cardo C. Biomarkers for bladder cancer management: present and future. Am J Clin Exp Urol. 2014;2:1–14. doi: 10.11648/j.ajcem.20140201.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Monteiro-Reis S, Leça L, Almeida M, Antunes S, Monteiro P, Dias P, Morais A, Oliveira J, Henrique R, Jerónimo C. Accurate detection of upper tract urothelial carcinoma in tissue and urine by means of quantitative GDF15, TMEFF2 and VIM promoter methylation. Eur J Cancer. 2014;50:226–233. doi: 10.1016/j.ejca.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 71.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM DD, Zhang L, Schroth GB, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 72.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinf. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Van Loo P, Nordgard SH, Lingjærde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, Perou CM, Børresen-Dale AL, Kristensen VN. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A. 2010;107(39):16910–5. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wessely F, Emes RD. Identification of DNA methylation biomarkers from Infinium arrays. Front Genet. 2012;3 10.3389/fgene.2012.0016. [DOI] [PMC free article] [PubMed]
- 75.Emes RD, Farrell WE. Make way for the ‘next generation’: application and prospects for genome-wide, epigenome-specific technologies in endocrine research. J Mol Endocrinol. 2012;49:19–27. doi: 10.1530/JME-12-0045. [DOI] [PubMed] [Google Scholar]
- 76.Shinawi T, Hill VK, Krex D, Schackert G, Gentle D, Morris MR, Wei W, Cruickshank G, Maher ER, Latif F. DNA methylation profiles of long- and short-term glioblastoma survivors. Epigenetics. 2013;8:149–156. doi: 10.4161/epi.23398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Veeck J, Niederacher D, An H, Klopocki E, Wiesmann F, Betz B, Galm O, Camara O, Durst M, Kristiansen G, et al. Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis. Oncogene. 2006;25:3479–3488. doi: 10.1038/sj.onc.1209386. [DOI] [PubMed] [Google Scholar]
- 78.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 79.Rose M, Gaisa NT, Antony P, Fiedler D, Heidenreich A, Otto W, Denzinger S, Bertz S, Hartmann A, Karl A, et al. Epigenetic inactivation of ITIH5 promotes bladder cancer progression and predicts early relapse of pT1 high grade urothelial tumours. Carcinogenesis. 2013;35:727–736. doi: 10.1093/carcin/bgt375. [DOI] [PubMed] [Google Scholar]
- 80.Fink L, Seeger W, Ermert L, Hanze J, Stahl U, Grimminger F, Kummer W, Bohle RM. Real-time quantitative RT-PCR after laser-assisted cell picking. Nat Med. 1998;4:1329–1333. doi: 10.1038/3327. [DOI] [PubMed] [Google Scholar]
- 81.R Core Team . R: a language and environment for statistical computing. R foundation for statistical computing. 2016. [Google Scholar]
- 82.Koestler DC, Marsit CJ, Christinsen BC, Kelsey KT, Houseman EA. A recursively partitioned mixture model for clustering time-course gene expression data. Transl Cancer Res. 2014;3:217–232. doi: 10.3978/j.issn.2218-676X.2014.06.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zweig MH, Campbell B. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin Chem. 1993;39:561–577. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic map of the underlying study design comprising the discovery and the validation step. (DOCX 146 kb)
Detailed frequency of DNAm of BRCA1, MGMT, and ERCC1 across cancer entities and corresponding normal tissues. (DOCX 296 kb)
Heatmap of tumor-specific DNAm of DDR genes across cancer entities. (DOCX 345 kb)
Graphs illustrating the correlation between DDR gene hypermethylation and their gene expression tumor entities. (DOCX 358 kb)
This table illustrates the clinicopathological parameters of 405 bladder cancer specimens of the TCGA network analyzed in this study. (DOC 48 kb)
This table summarizes the results from the multivariate Cox regression analysis including all factors influencing overall survival. (DOC 34 kb)
This table lists the RBBP8 methylation in human cell lines analyzed by MSP. (DOC 52 kb)
This table shows the results of bivariate correlation statistics (Fisher exact test) between clinicopathological parameters and RBBP8 methylation. (DOC 49 kb)
A table is shown characterizing 42 bladder cancer specimens analyzed in this study by IHC. (DOC 46 kb)
This table summarizes the clinicopathological parameters of urine samples analyzed in this study by MSP. (DOC 48 kb)
List of 177 DDR genes analyzed in this study. (XLS 1076 kb)
This dataset illustrates a summary of the TCGA samples used in this study. (XLS 27 kb)
Illustrating the definition of probe sets near a TSS using three different genes (RBBP8, MGMT, and LIG4). (DOCX 1053 kb)
This dataset summarizes the beta values according to a defined cutoff of 0.1. (XLS 62 kb)
This table shows the sequences of all primers and conditions used in this study for MSP analysis. (DOC 33 kb)
This table lists the primer sequences for bisulfite-pyrosequencing of the RBBP8 promoter region. (DOC 32 kb)
Sequences of all primers and conditions for qPCR analysis are summarized in a table. (DOC 32 kb)
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.