

Abstract
Epithelial-mesenchymal transition (EMT) of retinal pigment epithelial (RPE) cells is inevitable change of age-related macular degeneration (AMD). Smoking is a major risk factor for the development of EMT in several diseases, including lung cancer. Cigarette smoke-induced stress promotes the production of epidermal growth factor (EGF) in RPE cells. However, the underlying signaling pathways induced by aberrant EGF receptor (EGFR) expression in cigarette smoke-exposed RPE cells remain largely unknown. In the present study, the morphological transformation and production of EMT-associated cytokines were investigated to analyze the effect of smoking on the retina. Furthermore, EGF-treated or cigarette smoke-exposed RPE cells, as well as the downstream targets of EGFR, were investigated to identify the key molecules involved in EMT of cigarette smoke-stimulated RPE cells via immunoblotting. Exposure of RPE cells to cigarette smoke extract (CSE) induced secretion of VEGF and TGF-β1, and increased the expression of EMT markers. CSE-mediated focal adhesion kinase (FAK) activation resulted in the phosphorylation and activation of spleen associated tyrosine kinase (Syk)/Src proto-oncogene, non-receptor tyrosine kinase (Src), leading to migration and invasion of RPE cells. Knockdown of FAK or pharmacological inhibition of Syk/Src abrogated CSE-mediated VEGF and TGF-β1 production and blocked the phosphorylation of Smad2/3 in CSE-stimulated RPE cells. Erlotinib (an EGFR inhibitor) suppressed EGF and CSE-mediated switch from an epithelial to mesenchymal phenotype. Baicalein, an inhibitor of 12/15-lipooxygenase, also efficiently suppressed CSE-induced EMT processes by inhibiting EGFR-associated downstream signaling transduction. The results identified a novel signaling pathway mediated by EGFR in CSE-activated RPE cells, and suggest baicalein as a potential new therapeutic drug for CSE-associated retinopathy.
Keywords: retinal pigment epithelial cell, cigarette smoke, focal adhesion kinase, epidermal growth factor receptor, epithelial mesenchymal transition, 12/15-lipooxygenase
Introduction
Proliferative vitreoretinopathy (PVR) is one of the most important causes of blindness following surgery for retinal detachment and in diabetic retinopathy (1). The formation of scar-like tissue causes subsequent traction retinal detachment resulting in serious vision complications (2). Retinal pigment epithelial (RPE) cells have a central role in the early stages of PVR induction and are modulated by a wide range of growth factors, including transforming growth factor-β (TGF-β) and epidermal growth factor (EGF) (3). EGF and TGF-β signaling has been shown to be one of the most potent inducers of epithelial-mesenchymal transition (EMT) in various types of cancer and is associated with both invasion and nodal metastasis (4,5). In addition, stimulation with EGF and TGF-β triggers a variety of PVR-associated phenotypical changes and proliferation of RPE cells (6,7).
Cigarette smoke (CS) is the single most important environmental risk factor for age-related macular degeneration, which is the major cause of legal blindness in developed countries (8,9). Exposure to CS results in oxidative stress and damage or modification to RPE cells (10). Oxidative stress can alter RPE cells to express growth factors, including EGF, proteins of the EGF-related growth factor family, and vascular endothelial growth factor (VEGF) (11–13). Stimulation of lung cancer cells with CS or hydrogen peroxide (H2O2) induces aberrant phosphorylation of EGF receptor (EGFR) and activates downstream signaling pathways (14). EGFR is expressed in various different types of cancer including lung cancer and has a critical role in aberrant cell proliferation and development of cancer (15,16). The combination of EGF and TGF-β1 induces dramatic fibroblast-like morphologies and characteristics of EMT in intestinal epithelium (17). CS-induced TGF-β1 release induces downstream signaling via the Sma- and Mad-related family 2/3 (Smad2/3) cascade to promote EMT processes in human bronchial epithelial cells (18,19). In addition, stimulation of human lung cancer cells with TGF-β1 induces migration and invasion through Smad2/3 and focal adhesion kinase (FAK)/Src proto-oncogene, non-receptor tyrosine kinase (Src) signaling (20). However, it remains unclear whether CS-induced abnormal EGFR activation has a critical role in the migration of RPE cells and how TGF-β1, which is released by CS-induced EGFR activation, transduces the signal needed to trigger the EMT process in RPE cells.
Erlotinib, an EGFR tyrosine kinase inhibitor (TKI), inhibits EGFR signaling, thereby impeding tumor proliferation (21). EGFR TKI treatment also inhibits TGF-β-induced cell motility (22). In addition, treatment with erlotinib inhibits pathological neovascularization of the retina and prevents posterior capsule opacification after cataract surgery (23,24). Stimulation with various growth factors including EGF, hepatocyte growth factor and keratinocyte growth factor induces 12-lipoxygenase (12-LOX), leading to a marked increase in 12(S)-hydroxyeicosatetraenoic acid (HETE). On the other hand, baicalein, an inhibitor of 12-LOX, blocks EGF-dependent cell growth and the production of 12(S)-HETE in rabbit corneal epithelial cells (25,26). 12-LOX and its product HETE also has an important role in retinal neovascularization by upregulating production of VEGF (27). These findings suggest that erlotinib and baicalein may have a beneficial effect on preventing and treating abnormal EGFR-associated retinal diseases by downregulating TGF-β1-mediated signaling in RPE cells. Furthermore, the cellular mechanisms by which cigarette smoke causes EMT in PVR through the regulation VEGF or TGF-β1 production remain unclear.
In this study, the signaling pathways downstream of CS extract (CSE)-induced abnormal EGFR activation that are involved in promoting EMT were investigated using ARPE-19 human RPE cells. Additionally, whether the EGF-like role of CSE induces EMT by modulating VEGF or TGF-β production was examined. Finally, the ability of erlotinib or baicalein to inhibit EMT processes was investigated in CSE-stimulated RPE cells.
Materials and methods
Cell culture and reagents
Primary human RPE (HRPEpi) cells were purchased from ScienCell Research Laboratories, Inc. (San Diego, CA, USA). ARPE-19, a human RPE cell line, was purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM)/F12 supplemented with 10% fetal bovine serum (FBS) (both from (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) and antibiotics under a humidified atmosphere with 5% CO2. Baicalein (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was dissolved in sterile dimethyl sulfoxide. PP1 (Src inhibitor) and Bay-61-3606 [spleen associated tyrosine kinase (Syk) inhibitor] were purchased from Calbiochem (Merck KGaA, Darmstadt, Germany). Erlotinib (EGFR TKI) was obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). Recombinant EGF (rEGF) and recombinant VEGF (rVEGF) were purchased from Tocris Biosciences (Bristol, UK). LY2109761 (dual TGF-β receptor I and II kinase inhibitor) was purchased from Selleck Chemicals (Houston, TX, USA).
Preparation of cigarette smoke extracts
Research-grade cigarettes (3R4F) were obtained from the Kentucky Tobacco Research Council (University of Kentucky, Lexington, KY, USA). Each cigarette contained 0.726 mg nicotine, 9.4 mg tar, and 10.9 mg total particulate matter. CSE consisted of an extract of mainstream CS. Briefly, smoke was bubbled through 25 ml DMEM/F12 medium without FBS for 2 min and the resulting solution was used as a stock (100%) and diluted further. The pH was adjusted to 7.2, the obtained CSE was filtered through a 0.22-µm pore filter (EMD Millipore, Billerica, MA, USA) for sterilization. CSE was standardized by measuring the absorbance at a wavelength of 320 nm and was used within 30 min after preparation (19). ARPE-19 cells were seeded into a 6-well plate at a density of 2×105 cells/2 ml in DMEM/F12 containing 10% FBS. Following overnight incubation at 37°C, the cells were either treated with rEGF (100 ng/ml) or exposed to 5% CSE for 4 h at 37°C, and then incubated with complete medium for an additional 24 h at 37°C. To analyze the effect of VEGF, ARPE-19 cells were treated with either 50 ng/ml rVEGF or 5% CSE alone, or co-treated (50 ng/ml rVEGF + 5% CSE) for 4 h at 37°C, subsequently washed out, and then incubated with complete medium for an additional 24 h at 37°C. To analyze the effect of EGFR and TGF-β blocking, ARPE19 cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h at 37°C, subsequently washed out, and then incubated with complete medium or 50 nM erlotinib (EGFK tyrosin kinase inhibitor) or 100 nM LY2109761 (TGF-β receptor I and II kinase inhibitor) for an additional 24 h at 37°C. To investigate the activation of the Syk/Src pathway, ARPE19 cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h at 37°C, subsequently washed out, and then incubated with complete medium, 200 nM PP1 (Src inhibitor) or 200 nM Bay 61–3606 (Syk inhibitor) for an additional 24 h at 37°C. To investigate the regulatory effect of baicalein on EMT, ARPE19 cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h at 37°C, subsequently washed out, and then incubated with complete medium or 20 µM baicalein for an additional 24 h at 37°C.
Small interfering RNA (siRNA) transfection
Experimentally verified human FAK-siRNA duplex (5′-GGUUCAAGCUGGAUUAUUTT-3′) and negative control-siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′) were obtained from Bioneer Corporation (Daejeon, Korea). Cells were seeded at a concentration of 1×105/well in a T75 flask and cultured overnight. Cells in each T75 flask were then transfected with 200 nM siRNAs using Lipofectamine® RNAiMAX reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer's instructions. Cells were used for further experiments 48 h after transfection.
Immunoblotting
Cells were harvested and lysed in NP-40 buffer (Elpis Biotech, Inc., Daejeon, Korea) supplemented with a protease inhibitor cocktail (Sigma-Aldrich; Merck KGaA). To address phosphorylation events, an additional set of phosphatase inhibitors (Cocktail II; Sigma-Aldrich; Merck KGaA) was added to the NP-40 buffer. Protein concentration was determined using a bicinchoninic acid assay kit (Pierce; Thermo Fisher Scientific, Inc.). The same volume of 2X Laemmli sample buffer (Elpis Biotech, Inc.) was added to each lysate, and each protein sample (10 µg/sample) was immediately boiled for 5 min at 100°C. Insoluble material was centrifuged at 14,000 × g at 25°C. Total cell lysates (5×106 cells/sample) were subjected to SDS-PAGE (10 µg/well) on a gel containing 15% (w/v) acrylamide under reducing conditions. Separated proteins were transferred to nitrocellulose membranes (EMD Millipore). The membranes were blocked with 5% skim milk and western blot analysis was performed. Chemiluminescence was detected using an enhanced chemiluminescence kit (Advansta, Inc., Menlo Park, CA, USA) and an Amersham Imager 600 (GE Healthcare Life Sciences, Little Chalfont, UK). Primary antibodies against the following proteins were incubated with membranes overnight at 4°C: E-cadherin (cat. no. 3195; 1:1,000), vimentin (cat. no. 5741; 1:1,000), tight junction protein 1 (ZO-1; cat. no. 8193; 1:1,000), EGFR (cat. no. 2232; 1:1,000), phospho-Smad2/3 (Ser465/467/Ser423/425; cat. no. 8828; 1:1,000), Smad2/3 (cat. no. 5678; 1:1,000), phospho-FAK (Tyr397; cat. no. 3283; 1:1,000), phospho-FAK (Tyr925; cat. no. 3284; 1:1,000), FAK (cat. no. 3285; 1:1,000), phospho-Syk (Tyr323; cat. no. 2715; 1:1,000), phospho-Syk (Tyr525/526; cat. no. 2710; 1:1,000), Syk (cat. no. 13,198; 1:1,000), phospho-Src (Tyr416; cat. no. 6943; 1:1,000), Src (cat. no. 2123; 1:1,000), extracellular signal-regulated kinase (ERK; cat. no. 9102; 1:1,000), phospho-ERK (cat. no. 9101; 1:1,000), phospho-p38 (cat. no. 9211; 1:1,000), p38 (cat. no. 9212; 1:1,000), c-Jun N-terminal kinase (JNK; cat. no. 9258; 1:1,000) phospho-JNK (cat. no. 4671; 1:1,000), and β-actin (cat. no. 4967; 1:1,000; Cell Signaling Technology, Inc.); and VEGF receptor 1 (VEGF-R1; cat. no. sc-271789; 1:500), VEGF-R2 (cat. no. sc-393163; 1:500), 12-LOX (cat. no. sc-365194; 1:500) and 15-LOX (cat. no. sc-133085; 1:500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA); and α-smooth muscle actin (α-SMA; cat. no. bs-10196R; 1:500; BIOSS, Beijing, China) Following this, the membranes were incubated with the following secondary antibodies for 1 h at room temperature: Goat anti-mouse-horseradish peroxidase (HRP; cat. no. K0211589; 1:3,000) or goat anti-rabbit-HRP (cat. no. K0211708; 1:3,000) (both from Koma Biotech, Seoul, Korea).
Enzyme-linked immunosorbent assay (ELISA)
At 24 h, conditioned media was collected and the concentration of active TGF-β1 (cat. no. DY240) and VEGF (cat. no. DY293B) secreted by ARPE-19 or CSE-treated ARPE-19 cells was measured using the Single Cytokine ELISA Assay kit (R&D Systems, Inc., Minneapolis, MN, USA) according to the manufacturer's instructions. HETE concentrations in the culture supernatants were quantified using a single cytokine ELISA kit (Abcam, Cambridge, UK). Data are expressed as the mean value for biological replicates ± standard deviation (SD).
Wound healing assay
Wound healing assays were performed to measure the migration ability of ARPE-19 cells. CSE-treated or untreated ARPE-19 cells were plated in 6-well plates. As the cell layers reached confluence, a uniform wound consisting of a straight line was created in each well using a 200-µl micropipette tip and the wounded layers were washed with PBS to remove all cell debris. The cells were cultured in 5% CO2 at 37°C, and images were captured at 0 and 24 h after scratching using an inverted phase contrast microscope at ×100 magnification.
Statistical analysis
Data are expressed as the mean ± SD. Statistical analysis was conducted using one-way analysis of variance (ANOVA) using SigmaPlot software (version 10.0; Systat Software, Inc., San Jose, CA, USA). Bonferroni post hoc analysis was performed following the ANOVA for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
EGFR activation by CSE stimulates EMT processes in RPE cells by activating the TGF-β1-related pathway
It was initially analyzed whether CSE promotes the induction of EGFR expression and mesenchymal characteristics of RPE cells, and also whether exposure to CSE has an effect on the production of TGF-β1 and VEGF. Treatment with EGF or CSE increased the expression of EGFR, mesenchymal markers (vimentin and α-SMA), but reduced the levels of E-cadherin and ZO-1 compared with the control (Fig. 1A). Although CSE-exposed ARPE-19 cells acquired a fibroblast-like mesenchymal appearance (Fig. 1B), treatment with erlotinib restored epithelial characteristics and suppressed the cellular migration of CSE-stimulated ARPE-19 cells (Fig. 1C). Stimulation of RPE cells with a combination of EGF or CSE and erlotinib, an EGFR tyrosine kinase inhibitor (TKI), efficiently blocked the upregulation of mesenchymal markers and migration of CSE- or EGF-induced ARPE-19 cells (Fig. 1A and C). In addition, secretion of TGF-β1 and VEGF in CSE- or EGF-treated ARPE-19 cells was efficiently blocked by treatment with erlotinib (Fig. 1D). Stimulation with EGF or CSE had no effect on the activation of mitogen-activated protein kinases (MAPKs), including ERK, JNK and p38 MAPK (Fig. 2A). Additionally, ARPE-19 cells treated with EGF or CSE had increased expression of VEGFR and Smad2/3 phosphorylation, an intracellular signaling adaptor of TGF-β1 (Fig. 2A). Inhibition of the TGF-β1 pathway in EGF- or CSE-exposed ARPE cells using LY2109761, a small molecule inhibitor of the TGF-β receptor type I/type II kinases, disrupted Smad2/3 phosphorylation, blocked the suppression of E-cadherin and ZO-1 and decreased the expression of mesenchymal markers (Fig. 2A). Furthermore, considering that EGF or CSE treatment of ARPE-19 cells increased the production of VEGF (Fig. 1D), the present study also investigated whether co-treatment with CSE and VEGF enhanced the migration of ARPE-19 cells. Combined treatment of ARPE-19 cells with VEGF and CSE led to further enhancement of cell migration activity compared with either recombinant VEGF or CSE alone (Fig. 2B). These results suggested that CSE has an EGF-like effect on TGF-β1 and VEGF production through abnormal EGFR activation, and that TGF-β1-mediated signaling triggers EMT processes in cigarette smoke-exposed RPE cells.
Figure 1.

CSE-mediated EGFR activation induces EMT of RPE cells through secretion of VEGF and TGF-β1. ARPE-19 cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h, subsequently washed out, and then incubated with complete medium or 50 nM erlotinib for an additional 24 h. (A) Total cell lysates were immunoblotted with the indicated antibodies. β-actin served as an internal control. (B) CSE induces mesenchymal morphology in ARPE-19 cells. Morphology was observed with an inverted phase-contrast microscope. The scale bar represents 100 µm, and photographs were taken at ×100 magnification using a digital camera. (C) Cell motility was increased by CSE as measured by a wound-healing assay. Cells were wounded (0 h) and maintained for 24 h in complete medium. Dotted lines indicate the edges of the wounds. (D) Concentrations of active TGF-β1 and VEGF in the culture supernatants were quantified by ELISA assay. Data are presented as the mean ± standard deviation of three independent experiments. Results are representative of three independent experiments. *P<0.05 and **P<0.01. Ctrl, control; DMSO, dimethyl sulfoxide; rEGF, recombinant epidermal growth factor; CSE, cigarette smoke extract; EGFR, epidermal growth factor receptor; ZO-1, tight junction protein 1; α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor-β1; VEGF, vascular endothelial growth factor.
Figure 2.

CSE induces upregulation of VEGFR and activation of the transforming growth factor-β1-associated pathway. (A-C) ARPE-19 cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h, subsequently washed out, and then incubated with complete medium or 50 nM erlotinib or 100 nM LY2109761 for an additional 24 h. Total cell lysates were immunoblotted with the indicated antibodies. β-actin served as an internal control. (D) ARPE19 cells were treated with either 50 ng/ml rVEGF or 5% CSE alone, or co-treated (50 ng/ml rVEGF + 5% CSE) for 4 h, subsequently washed out, and then incubated with complete medium for an additional 24 h. Dotted lines indicate the edges of the wounds. Results are representative of three independent experiments. Ctrl, control; rEGF, recombinant epidermal growth factor; CSE, cigarette smoke extract; p, phospho; ERK, extracellular regulated kinase; JNK, c-Jun N-terminal kinase; DMSO, dimethyl sulfoxide; VEGF-R, vascular endothelial growth factor receptor; Smad, Sma- and Mad-related family; ZO-1, tight junction protein 1; α-SMA, α-smooth muscle actin; rVEGF, recombinant vascular endothelial growth factor.
CSE-mediated TGF-β1 activates the phosphorylation of FAK, Syk and Src to induce EMT processes in RPE cells
TGF-β induces EMT processes in various normal and cancer cells, and is also responsible for FAK-mediated scar formation in chronic fibrotic disease (28,29). The activation of the FAK/Src pathway in ARPE-19 cells was previously reported to be associated with collagen gel contraction in an in vitro model of PVR (30). It was subsequently investigated whether CSE-induced TGF-β1 activates the FAK signaling pathway in ARPE-19 cells. Stimulation with CSE induced FAK phosphorylation and stimulated the phosphorylation of Src and Syk kinase; however, pretreatment with erlotinib or LY2109761 efficiently blocked CSE-induced phosphorylation of FAK, Syk, and Src (Fig. 3A). In addition, treatment with LY2109761 significantly inhibited migratory activity, and TGF-β1 and VEGF production in CSE-stimulated ARPE cells (Fig. 3B and C). These results suggested that EGF-like CSE stimulation increases the production of TGF-β1 and activation of FAK-associated downstream signaling in ARPE-19 cells.
Figure 3.

CSE-mediated TGF-β1 activates the phosphorylation of Syk, Src and FAK for EMT in retinal pigment epithelial cells. ARPE19 cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h, subsequently washed out, and then incubated with complete medium or 50 nM erlotinib or 100 nM LY2109761 for an additional 24 h. (A) Total cell lysates of each condition were harvested and immunoblotted with the indicated antibodies. β-actin served as an internal control. (B) CSE- or rEGF-induced cell migration was decreased by LY2109761 treatment as measured by a wound healing assay. Cells were wounded (0 h) and maintained for 24 h in complete medium. Dotted lines indicate the edges of the wounds. (C) Concentrations of active TGF-β1 and VEGF in the culture supernatants were quantified by ELISA. Data are presented as the mean ± standard deviation of three independent experiments. *P<0.05 and **P<0.01. Ctrl, control; rEGF, recombinant epidermal growth factor; CSE, cigarette smoke extract; p, phospho; FAK, focal adhesion kinase; Syk, spleen associated tyrosine kinase; Src, Src proto-oncogene non-receptor tyrosine kinase; TGF-β1, transforming growth factor-β1; VEGF, vascular endothelial growth factor.
FAK-mediated Syk/Src activation promotes the production of TGF-β1 and VEGF in CSE-stimulated RPE cells
It was also investigated whether CSE-mediated FAK activation has an effect on TGF-β1 production and cellular motility. The association between FAK and Syk or Src signaling in ARPE-19 cells stimulated with CSE was determined. Knockdown of FAK in ARPE-19 cells inhibited phosphorylation of Syk and Src kinase following CSE stimulation (Fig. 4A). Downregulation of FAK with siRNA significantly suppressed the phosphorylation of Smad2/3 and expression of mesenchymal markers (vimentin and α-SMA), and also restored the expression of epithelial markers (E-cadherin and ZO-1) in CSE-stimulated APRE-19 cells (Fig. 4A). Gene silencing of FAK by RNA interference resulted in significant reduction of CSE-activated VEGF and TGF-β1 release, and blocked cell migration activity (Fig. 4B and C). Pharmacological inhibitors of Src and Syk were also used to further analyze the role that these kinases have in TGF-β1-mediated FAK signaling in ARPE-19 cells exposed to CSE. Treatment with PP1 (Src inhibitor) and Bay 61–3606 (Syk inhibitor) effectively inhibited the activation of Src and Syk in CSE-treated ARPE cells, but did not block the phosphorylation of FAK (Fig. 5A). Levels of mesenchymal markers and expression of phosphorylated Smad2/3 in CSE-activated ARPE cells were reduced by the inhibitors (Fig. 5A). In addition, VEGF and TGF-β1 secretion was attenuated by addition of the inhibitors, as was the wound healing capacity of CSE-stimulated ARPE cells (Fig. 5B and C). Taken together, these results suggested that FAK-mediated Syk/Src signaling can activate EMT processes in CSE-treated ARPE-19 cells.
Figure 4.

FAK is an upstream signal of CSE-induced Syk/Src activation and secretion of VEGF and TGF-β1. ARPE19 cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h, subsequently washed out, and then incubated with complete medium for an additional 4 h. Cell were transfected with either si-FAK-siRNA (200 nM) or si-Ctrl for 36 h prior to experiments. (A) Following transfection, protein levels of cells were detected by western blotting with the indicated antibodies. β-actin was used as a loading control. (B) Concentrations of active TGF-β1 and VEGF in the culture supernatants were quantified by ELISA assay. Data are presented as the mean ± standard deviation of three independent experiments. (C) CSE- or rEGF-induced cell migration inhibited by FAK knockdown as measured by a wound healing assay. Cells were wounded (0 h) and maintained for 24 h in complete medium. Dotted lines indicate the edges of the wounds. Results are representative of three independent experiments. *P<0.05 and **P<0.01. Ctrl, control; rEGF, recombinant epidermal growth factor; si, small interfering RNA; FAK, focal adhesion kinase; CSE, cigarette smoke extract; p, phospho; Syk, spleen associated tyrosine kinase; Src, Src proto-oncogene non-receptor tyrosine kinase; EGFR, epidermal growth factor receptor; VEGF-R, vascular endothelial growth factor receptor; Smad, Sma- and Mad-related family; ZO-1, tight junction protein 1; α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor-β1; VEGF, vascular endothelial growth factor.
Figure 5.

Syk/Src activation regulates the secretion of VEGF and TGF-β1 in CSE-stimulated ARPE19 cells. ARPE19 cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h, subsequently washed out, and then incubated with complete medium, 200 nM PP1 (Src inhibitor) or 200 nM Bay 61–3606 (Syk inhibitor) for an additional 24 h. (A) Protein levels were detected by western blotting with the indicated antibodies. β-actin was used as a loading control. (B) Concentration of active TGF-β1 and VEGF in the culture supernatants were quantified by ELISA assay. Data are presented as the mean of three independent experiments, and error bars represent the standard deviation of the means. (C) PP1 or Bay 61–3606 treatment blocked CSE- or rEGF-induced cell migration as measured by wound healing assay. Cells were wounded (0 h) and maintained for 24 h in complete medium. Dotted lines indicate the edges of the wounds. Results are representative of three independent experiments. *P<0.05 and **P<0.01. Ctrl, control; DMSO, dimethyl sulfoxide; rEGF, recombinant epidermal growth factor; CSE, cigarette smoke extract; p, phospho; FAK, focal adhesion kinase; Syk, spleen associated tyrosine kinase; Src, Src proto-oncogene non-receptor tyrosine kinase; EGFR, epidermal growth factor receptor; VEGF-R, vascular endothelial growth factor receptor; Smad, Sma- and Mad-related family; ZO-1, tight junction protein 1; α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor-β1; VEGF, vascular endothelial growth factor.
Baicalein treatment modulates CSE-mediated signaling pathways involved in EMT processes in ARPE cells
Baicalein, an inhibitor of 12-LOX, prevents the EGF-dependent signaling involved in cell growth in rabbit corneal epithelial cells (26). In the current study, it was also examined whether baicalein may alter the regulation of CSE-dependent TGF-β1 production, cell migration and associated signaling transduction in CSE-exposed ARPE-19 cells. Compared with ARPE-19 cells treated with EGF or CSE, treatment with baicalein reduced the expression of EGFR, VEGF-R1 and -R2, activation of Smad2/3 phosphorylation, and upregulation of mesenchymal markers in CSE-exposed ARPE-19 cells (Fig. 6A). Furthermore, baicalein efficiently blocked the phosphorylation of FAK, Src, and Syk in CSE-stimulated ARPE-19 cells (Fig. 6A). The activation of 12/15-LOX in CSE- or EGF-stimulated ARPE-19 cells was also markedly attenuated by baicalein treatment (Fig. 6A). FAK-knockdown or targeted inhibition of Syk/Src kinase attenuated the expression of 12/15-LOX in CSE-exposed ARPE-19 cells (Fig. 6B and C). In addition, pre-exposure to baicalein significantly suppressed the production of TGF-β1, VEGF and 12(S)-HETE in CSE-treated ARPE-19 cells (Fig. 6D and E). Finally, the effect of baicalein on migration activity and underlying signaling pathway in HRPEpi cells following treatment with EGF or CSE was determined. Levels of EGFR, VEGFR and phosphorylated Smad2/3 in CSE-treated HRPEpi cells were suppressed by treatment with baicalein or erlotinib (Fig. 7A). Treatment with baicalein of CSE-exposed HRPEpi resulted in restoration of epithelial markers (E-cadherin and ZO-1) and reduction of mesenchymal markers (vimentin and α-SMA; Fig. 7A). Furthermore, levels of phosphorylated FAK, Syk, and Src were reduced by baicalein treatment compared with erlotinib treatment (Fig. 7A). Cell migratory activity and secretion of EMT-associated cytokines, including TGF-β1 and VEGF, were blocked following treatment with baicalein or erlotinib in CSE-stimulated HRPEpi cells (Fig. 7B and C). Treatment of CSE-activated HRPEpi cells with baicalein or erlotinib markedly downregulated the expression of 12/15-LOX and the production of 12(S)-HETE compared with non-treated CSE-stimulated HRPEpi (Fig. 7D). Together, these results suggested that baicalein could suppress the activation of 12/15-LOX in RPE cells affected by cigarette smoking via the inhibition of FAK-related signaling pathway in retinal pathology.
Figure 6.

Baicalein treatment modulates the CSE-mediated signaling pathway for EMT processes in ARPE19 cells. (A) Cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h, subsequently washed out, and then incubated with complete medium or 20 µM baicalein for an additional 24 h. (B) Cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h, subsequently washed out, and then incubated with complete medium for an additional 4 h. Cell were transfected with either FAK-siRNA (200 nM) or control-siRNA for 36 h prior to experiments. (C) Cells were exposed to 5% CSE or 100 ng/ml rEGF for 4 h, subsequently washed out, and then incubated with complete medium, 200 nM PP1 (Src inhibitor), or 200 nM Bay 61–3606, (Syk inhibitor) for an additional 24 h. Total cell lysates of each condition were harvested and immunoblotted with the indicated antibodies. β-actin served as an internal control. (D) Concentrations of active TGF-β1 and VEGF in the culture supernatants were quantified by ELISA assay. Data are presented as the mean of three independent experiments, and error bars represent the standard deviation. (E) Concentrations of HETE in the culture supernatants were quantified by ELISA assay. Data are presented as the mean of three independent experiments, and error bars represent the standard deviation. *P<0.05 and **P<0.01; #P<0.005 and ##P<0.001. Results are representative of three independent experiments. Ctrl, control; DMSO, dimethyl sulfoxide; rEGF, recombinant epidermal growth factor; CSE, cigarette smoke extract; EGFR, epidermal growth factor receptor; VEGF-R, vascular endothelial growth factor receptor; Smad, Sma- and Mad-related family; ZO-1, tight junction protein 1; α-SMA, α-smooth muscle actin; p, phospho; FAK, focal adhesion kinase; Syk, spleen associated tyrosine kinase; Src, Src proto-oncogene non-receptor tyrosine kinase; LOX, lipoxygenase; TGF-β1, transforming growth factor-β1; VEGF, vascular endothelial growth factor; HETE, hydroxyeicosatetraenoic acid.
Figure 7.

Baicalein treatment modulates the CSE-mediated signaling pathway for EMT processes in HRPEpi. HRPEpi were exposed to 5% CSE or 100 ng/ml rEGF for 4 h, subsequently washed out, and then incubated with complete medium, 20 µM baicalein, or 50 nM erlotinib for an additional 24 h. (A) Total cell lysates of each condition were harvested and immunoblotted with the indicated antibodies. β-actin served as an internal control. (B) Baicalein treatment blocked CSE- or rEGF-induced cell migration as measured by wound healing assay. Cells were wounded (0 h) and maintained for 24 h in complete medium. Dotted lines indicate the edges of the wounds. Wound closure (measured after 24 h) was faster in cells treated with CSE or rEGF than in those treated with baicalein. (C) Concentrations of active TGF-β1 and VEGF in the culture supernatants were quantified by ELISA assay. Data are presented as the mean of three independent experiments, and error bars represent the standard deviation. (D) Upregulation of 12/15-LOX and production of HETE by CSE- or rEGF treatment was decreased by baicalein or erlotinib treatment. Total proteins were extracted from cell lysates and western blotting was performed for 12/15-LOX protein. Concentrations of HETE in the culture supernatants of ARPE19 cells were quantified by ELISA assay. Data are presented as the mean of three independent experiments, and error bars represent the standard deviation. Results are representative of three independent experiments. *P<0.05 and **P<0.01; #P<0.005 vs. rEGF/DMSO and ##P<0.001 vs. CSE/DMSO. HRPEpi, human primary retinal pigment epithelial cells; Ctrl, control; DMSO, dimethyl sulfoxide; rEGF, recombinant epidermal growth factor; CSE, cigarette smoke extract; EGFR, epidermal growth factor receptor; VEGF-R, vascular endothelial growth factor receptor; p, phospho; Smad, Sma- and Mad-related family; ZO-1, tight junction protein 1; α-SMA, α-smooth muscle actin; FAK, focal adhesion kinase; Syk, spleen associated tyrosine kinase; Src, Src proto-oncogene non-receptor tyrosine kinase; TGF-β1, transforming growth factor-β1; VEGF, vascular endothelial growth factor; LOX, lipoxygenase; HETE, hydroxyeicosatetraenoic acid.
Discussion
The presence of TGF-β1 and EGF in the subretinal fluid of patients with retinal detachment has been confirmed previously (2,3). Exposure of human airway epithelial cells to either CS or H2O2 results in an increase in EGFR activation and activation of metalloproteinases (31). APRE-19 cells stimulated with TGF-β1 and EGF have enhanced expression of α-SMA and vimentin, and upregulated metalloproteinases activity (32), indicating a role for these factors in the pathogenesis of PVR and EMT processes in RPE cells. Based on these results, blocking abnormal EGFR expression, and consequently its pathways, may inhibit cellular migration and invasion of RPE cells. Furthermore, the effects and underlying molecular mechanisms of smoking on EMT processes in PVR remain unclear. In the current study, ARPE cells exposed to CSE exhibited high levels of EGFR expression and activated downstream FAK-mediated Syk/Src signaling. CSE-mediated FAK activation also had an important effect on TGF-β1 and VEGF release in APRE-19 cells. Specifically, treatment with erlotinib or baicalein efficiently blocked CSE-dependent TGF-β1 release and downstream signal transduction, resulting in suppression of EMT processes (Fig. 8). These results suggest that suppression of EGFR-dependent FAK activation may be a key mechanism for preventing deterioration of PVR in chronic smokers.
Figure 8.
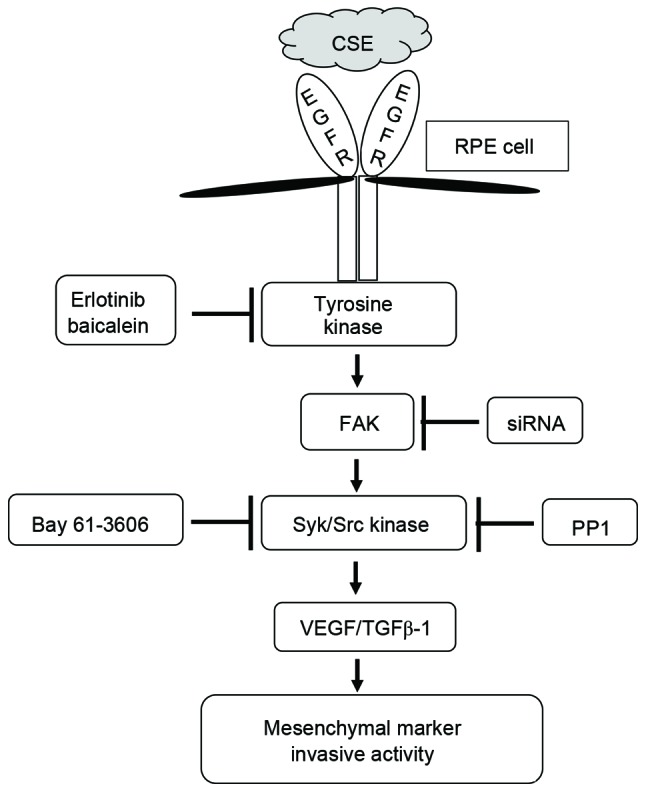
Schematic diagram of intracellular signaling mechanisms that induce epithelial mesenchymal transition during CSE-induced aberrant EGFR activation in human RPE cells. CSE induces the expression of EGFR and increases the activity of FAK, Syk, and Src in RPE cells. These signaling molecules promote the production of TGF-β1 and VEGF and induce expression of EMT markers. CSE, cigarette smoke extract; EGFR, epidermal growth factor receptor; RPE cell, retinal pigment epithelial cell; FAK, focal adhesion kinase; siRNA, small interfering RNA; Syk, spleen associated tyrosine kinase; Src, Src proto-oncogene non-receptor tyrosine kinase; VEGF, vascular endothelial growth factor; TGF-β1, transforming growth factor-β.
EGF synergistically stimulates TGF-β-mediated EMT processes (33), and FAK also acts as a critical mediator of EMT and metastasis stimulated by TGF-β in hepatocytes (34). Additionally, CSE-activated FAK signaling decreases cell motility and induces apoptosis in human bronchial cells (35,36), and CSE disrupts focal adhesion complexes and decreases FAK activity in cultured lung endothelial cells (37). These findings suggest that the role of FAK, particularly when induced by TGF-β1, is cell-type-dependent. In addition, it remains unclear whether FAK facilitates EGF-mediated TGF-β1-dependent signaling for enhancing EMT processes in retina-associated pathological conditions. In CSE-exposed RPE cells, the TGF-β1-mediated Smad2/3 signaling pathway was associated with FAK-induced migration activity. Activated FAK in CSE-stimulated APRE cells also promoted the production of TGF-β1 and VEGF. These results suggest that FAK activation is able to regulate the EMT processes of RPE cells in CSE-associated retinal diseases, and that FAK signaling may be a novel therapeutic target for regulating pathological conditions of the retina.
Src signaling promotes the transition of cells with an adhesive phenotype to a more motile phenotype in cancer (38). Unlike EGF-activated EGFR, CS-stimulated EGFR triggers aberrant Src phosphorylation in lung cancer cells (39). However, EGF-treated or CSE-exposed RPE cells enhanced the phosphorylation of Src in the present study. This result suggests that CSE stimulation of RPE cells may promote an EGFR-associated signaling pathway induced by EGF. The activated Src-induced FAK pathway facilitates TGF-β1-mediated EMT by diverse mechanisms required for migration and invasion in different cell types (38). TGF-β-induced phosphorylated FAK is also involved in EMT (40,41); however, direct evidence of the connection and role of Src and FAK in CSE-stimulated RPE cells has yet to been reported. Based on existing data, the link between Src phosphorylation and FAK activation in CSE-exposed ARPE19 cells was investigated in the current study. CSE stimulation induced the phosphorylation of FAK, Syk and Src. Although inhibitors of Syk or Src failed to block the induction of phosphorylated FAK, FAK knockdown in APRE-19 cells attenuated the activation of Syk/Src kinases following stimulation with CSE. Syk and Src kinase inhibitors efficiently suppressed the production of TGF-β1 and VEGF in CSE-treated APRE-19 cells, and the expression of mesenchymal markers and cell migration activity were reduced. These results suggest that FAK-dependent Syk/Src activation is a critical step for activating EMT processes in RPE cells following exposure to CSE.
High levels of EGFR expression and paracrine EGF signaling are strongly associated with poor prognosis of patients with cancer, and with tumor cell invasion and dissemination (42). RPE cells also express various members of the EGF-related growth factor family. Heparin-binding-EGF-like growth factor produced by RPE cells stimulates proliferation, migration, and secretion of VEGF under conditions of oxidative stress (11). In addition, tissue inhibitor of matrix metalloproteinases-3 and erlotinib have emerged as attractive candidates for preventing and treating oxidative stress-induced proliferative retinopathies (23). Although erlotinib is an efficient and specific TKI for blocking EGFR activation and signaling, long-term exposure to CS causes alterations in EGFR conformation leading to smoking-associated TKI resistance (39). Based on these results, the development of novel therapeutic drugs is required to control cigarette smoke-associated retinal diseases. Baicalein, a 12-LOX inhibitor, may protect retinal cells from ischemia-induced apoptosis by downregulating hypoxia-inducible factor-1α, VEGF and matrix metalloproteinase-9 (43). In addition, treatment of rabbit corneal epithelial cells with baicalein efficiently blocks EGF-mediated cell growth (26). In the present study, treatment with baicalein also reduced the expression of TGF-β1 and suppressed the activation of FAK-associated signaling pathways in CSE-treated HRPEpi cells. These results suggest for the first time, to the best of our knowledge, that baicalein may be useful for treating PVR and associated retinal diseases. Furthermore, the use of human primary RPE cells supports the close association between CS and retinal diseases, rather than individual components of CSE.
In conclusion, the results of the present study suggest that treatment with erlotinib may prevent progression of PVR by suppressing the aberrant EGFR activation induced by CS, and also that baicalein is beneficial for inhibiting the development of smoke-associated retinal disease.
Acknowledgements
This study was supported by the Basic Science Research Program of Ministry of Education (grant no. NRF-2015R1D1A1A01056672) and Ministry of Science, ICT and Future Planning (grant no. NRF-2015R1C1A2A01053732) through the National Research Foundation of Republic of Korea. The sponsor had no role in the design or conduct of this research.
References
- 1.Ryan SJ. Traction retinal detachment. XLIX Edward Jackson Memorial Lecture. Am J Ophthalmol. 1993;115:1–20. doi: 10.1016/S0002-9394(14)73518-4. [DOI] [PubMed] [Google Scholar]
- 2.Charteris DG, Sethi CS, Lewis GP, Fisher SK. Proliferative vitreoretinopathy-developments in adjunctive treatment and retinal pathology. Eye (Lond) 2002;16:369–374. doi: 10.1038/sj.eye.6700194. [DOI] [PubMed] [Google Scholar]
- 3.Charteris DG. Proliferative vitreoretinopathy: Pathobiology, surgical management, and adjunctive treatment. Br J Ophthalmol. 1995;79:953–960. doi: 10.1136/bjo.79.10.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee MY, Chou CY, Tang MJ, Shen MR. Epithelial-mesenchymal transition in cervical cancer: Correlation with tumor progression, epidermal growth factor receptor overexpression and snail up-regulation. Clin Cancer Res. 2008;14:4743–4750. doi: 10.1158/1078-0432.CCR-08-0234. [DOI] [PubMed] [Google Scholar]
- 5.Ha GH, Kim JL, Breuer EK. TACC3 is essential for EGF-mediated EMT in cervical cancer. PLoS One. 2013;8:e70353. doi: 10.1371/journal.pone.0070353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Z, Chen CZ, Gong WR, Li JP, Xing YQ. Integrin-alpha5 mediates epidermal growth factor-induced retinal pigment epithelial cell proliferation and migration. Pathobiology. 2010;77:88–95. doi: 10.1159/000278290. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Wang H, Wang F, Gu Q, Xu X. Snail involves in the transforming growth factor β1-mediated epithelial-mesenchymal transition of retinal pigment epithelial cells. PLoS One. 2011;6:e23322. doi: 10.1371/journal.pone.0023322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakravarthy U, Wong TY, Fletcher A, Piault E, Evans C, Zlateva G, Buggage R, Pleil A, Mitchell P. Clinical risk factors for age-related macular degeneration: A systematic review and meta-analysis. BMC Ophthalmol. 2010;10:31. doi: 10.1186/1471-2415-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein R, Knudtson MD, Cruickshanks KJ, Klein BE. Further observations on the association between smoking and the long-term incidence and progression of age-related macular degeneration: The Beaver Dam Eye Study. Arch Ophthalmol. 2008;126:115–121. doi: 10.1001/archopht.126.6.840. [DOI] [PubMed] [Google Scholar]
- 10.Kunchithapautham K, Atkinson C, Rohrer B. Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J Biol Chem. 2014;289:14534–14546. doi: 10.1074/jbc.M114.564674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hollborn M, Iandiev I, Seifert M, Schnurrbusch UE, Wolf S, Wiedemann P, Bringmann A, Kohen L. Expression of HB-EGF by retinal pigment epithelial cells in vitreoretinal proliferative disease. Curr Eye Res. 2006;31:863–874. doi: 10.1080/02713680600888807. [DOI] [PubMed] [Google Scholar]
- 12.Higgins GT, Wang JH, Dockery P, Cleary PE, Redmond HP. Induction of angiogenic cytokine expression in cultured RPE by ingestion of oxidized photoreceptor outer segments. Invest Ophthalmol Vis Sci. 2003;44:1775–1782. doi: 10.1167/iovs.02-0742. [DOI] [PubMed] [Google Scholar]
- 13.Dong A, Xie B, Shen J, Yoshida T, Yokoi K, Hackett SF, Campochiaro PA. Oxidative stress promotes ocular neovascularization. J Cell Physiol. 2009;219:544–552. doi: 10.1002/jcp.21698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan EM, Lanir R, Danielson AR, Goldkorn T. Epidermal growth factor receptor exposed to cigarette smoke is aberrantly activated and undergoes perinuclear trafficking. FASEB J. 2008;22:910–917. doi: 10.1096/fj.06-7729com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 16.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–672. doi: 10.1016/S0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 17.Uttamsingh S, Bao X, Nguyen KT, Bhanot M, Gong J, Chan JL, Liu F, Chu TT, Wang LH. Synergistic effect between EGF and TGF-beta1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene. 2008;27:2626–2634. doi: 10.1038/sj.onc.1210915. [DOI] [PubMed] [Google Scholar]
- 18.Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus VC, Sheppard D, et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest. 2007;117:3551–3562. doi: 10.1172/JCI32526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen HJ, Sun YH, Zhang SJ, Jiang JX, Dong XW, Jia YL, Shen J, Guan Y, Zhang LH, Li FF, et al. Cigarette smoke-induced alveolar epithelial-mesenchymal transition is mediated by Rac1 activation. Biochim Biophys Acta. 2014;1840:1838–1849. doi: 10.1016/j.bbagen.2014.01.033. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Hwan Kim S. CK2 inhibitor CX-4945 blocks TGF-β1-induced epithelial-to-mesenchymal transition in A549 human lung adenocarcinoma cells. PLoS One. 2013;8:e74342. doi: 10.1371/journal.pone.0074342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 22.Muraoka-Cook RS, Dumont N, Arteaga CL. Dual role of transforming growth factor beta in mammary tumorigenesis and metastatic progression. Clin Cancer Res. 2005;11:937s–943s. [PubMed] [Google Scholar]
- 23.Hewing NJ, Weskamp G, Vermaat J, Farage E, Glomski K, Swendeman S, Chan RV, Chiang MF, Khokha R, Anand-Apte B, Blobel CP. Intravitreal injection of TIMP3 or the EGFR inhibitor erlotinib offers protection from oxygen-induced retinopathy in mice. Invest Ophthalmol Vis Sci. 2013;54:864–870. doi: 10.1167/iovs.12-10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wertheimer C, Liegl R, Kernt M, Mayer W, Docheva D, Kampik A, Eibl-Lindner KH. EGF receptor inhibitor erlotinib as a potential pharmacological prophylaxis for posterior capsule opacification. Graefes Arch Clin Exp Ophthalmol. 2013;251:1529–1540. doi: 10.1007/s00417-013-2257-z. [DOI] [PubMed] [Google Scholar]
- 25.Seth RK, Haque MS, Zelenka PS. Regulation of c-fos induction in lens epithelial cells by 12(S)HETE-dependent activation of PKC. Invest Ophthalmol Vis Sci. 2001;42:3239–3246. [PubMed] [Google Scholar]
- 26.Ottino P, Taheri F, Bazan HE. Growth factor-induced proliferation in corneal epithelial cells is mediated by 12(S)-HETE. Exp Eye Res. 2003;76:613–622. doi: 10.1016/S0014-4835(03)00003-4. [DOI] [PubMed] [Google Scholar]
- 27.Al-Shabrawey M, Mussell R, Kahook K, Tawfik A, Eladl M, Sarthy V, Nussbaum J, El-Marakby A, Park SY, Gurel Z, et al. Increased expression and activity of 12-lipoxygenase in oxygen-induced ischemic retinopathy and proliferative diabetic retinopathy: Implications in retinal neovascularization. Diabetes. 2011;60:614–624. doi: 10.2337/db10-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- 29.Liu S, Xu SW, Kennedy L, Pala D, Chen Y, Eastwood M, Carter DE, Black CM, Abraham DJ, Leask A. FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: Implications for acquisition of a matrix-remodeling phenotype. Mol Biol Cell. 2007;18:2169–2178. doi: 10.1091/mbc.E06-12-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morales SA, Mareninov S, Wadehra M, Zhang L, Goodglick L, Braun J, Gordon LK. FAK activation and the role of epithelial membrane protein 2 (EMP2) in collagen gel contraction. Invest Ophthalmol Vis Sci. 2009;50:462–469. doi: 10.1167/iovs.08-3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Adiseshaiah P, Reddy SP. Matrix metalloproteinase/epidermal growth factor receptor/mitogen-activated protein kinase signaling regulate fra-1 induction by cigarette smoke in lung epithelial cells. Am J Respir Cell Mol Biol. 2005;32:72–81. doi: 10.1165/rcmb.2004-0198OC. [DOI] [PubMed] [Google Scholar]
- 32.Lee H, O'Meara SJ, O'Brien C, Kane R. The role of gremlin, a BMP antagonist, and epithelial-to-mesenchymal transition in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2007;48:4291–4299. doi: 10.1167/iovs.07-0086. [DOI] [PubMed] [Google Scholar]
- 33.Buck E, Eyzaguirre A, Barr S, Thompson S, Sennello R, Young D, Iwata KK, Gibson NW, Cagnoni P, Haley JD. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther. 2007;6:532–541. doi: 10.1158/1535-7163.MCT-06-0462. [DOI] [PubMed] [Google Scholar]
- 34.Cicchini C, Laudadio I, Citarella F, Corazzari M, Steindler C, Conigliaro A, Fantoni A, Amicone L, Tripodi M. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res. 2008;314:143–152. doi: 10.1016/j.yexcr.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 35.Carter CA. Multiplexed high content screening reveals that cigarette smoke condensate-altered cell signaling pathways are accentuated through FAK inhibition in human bronchial cells. Int J Toxicol. 2012;31:257–266. doi: 10.1177/1091581812440890. [DOI] [PubMed] [Google Scholar]
- 36.Carter CA, Hamm JT. Multiplexed quantitative high content screening reveals that cigarette smoke condensate induces changes in cell structure and function through alterations in cell signaling pathways in human bronchial cells. Toxicology. 2009;261:89–102. doi: 10.1016/j.tox.2009.04.039. [DOI] [PubMed] [Google Scholar]
- 37.Lu Q, Sakhatskyy P, Grinnell K, Newton J, Ortiz M, Wang Y, Sanchez-Esteban J, Harrington EO, Rounds S. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol. 2011;301:L847–L857. doi: 10.1152/ajplung.00178.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avizienyte E, Frame MC. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Opin Cell Biol. 2005;17:542–547. doi: 10.1016/j.ceb.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 39.Filosto S, Becker CR, Goldkorn T. Cigarette smoke induces aberrant EGF receptor activation that mediates lung cancer development and resistance to tyrosine kinase inhibitors. Mol Cancer Ther. 2012;11:795–804. doi: 10.1158/1535-7163.MCT-11-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka Y, Kobayashi H, Suzuki M, Kanayama N, Terao T. Transforming growth factor-beta1-dependent urokinase up-regulation and promotion of invasion are involved in Src-MAPK-dependent signaling in human ovarian cancer cells. J Biol Chem. 2004;279:8567–8576. doi: 10.1074/jbc.M309131200. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura K, Yano H, Schaefer E, Sabe H. Different modes and qualities of tyrosine phosphorylation of Fak and Pyk2 during epithelial-mesenchymal transdifferentiation and cell migration: Analysis of specific phosphorylation events using site-directed antibodies. Oncogene. 2001;20:2626–2635. doi: 10.1038/sj.onc.1204359. [DOI] [PubMed] [Google Scholar]
- 42.Ueno NT, Zhang D. Targeting EGFR in triple negative breast cancer. J Cancer. 2011;2:324–328. doi: 10.7150/jca.2.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chao HM, Chuang MJ, Liu JH, Liu XQ, Ho LK, Pan WH, Zhang XM, Liu CM, Tsai SK, Kong CW, et al. Baicalein protects against retinal ischemia by antioxidation, antiapoptosis, downregulation of HIF-1α, VEGF and MMP-9 and upregulation of HO-1. J Ocul Pharmacol Ther. 2013;29:539–549. doi: 10.1089/jop.2012.0179. [DOI] [PMC free article] [PubMed] [Google Scholar]