

Abstract
The endogenous neurotransmitter, noradrenaline, exerts anti-inflammatory and neuroprotective effects in vivo and in vitro. Reduced noradrenaline levels results in increased inflammation and neuronal damage. The primary source of noradrenaline in the central nervous system is tyrosine hydroxylase (TH)-positive neurons, located in the locus coeruleus (LC). TH is the rate-limiting enzyme for noradrenaline synthesis; therefore, regulation of TH protein expression and intrinsic enzyme activity represents the central means for controlling the synthesis of noradrenaline. Catalpol is an iridoid glycoside purified from Rehmannia glutinosa Libosch, which exerts a neuroprotective effect in multiple sclerosis (MS). The present study used an experimental mouse model of autoimmune encephalomyelitis to verify the neuroprotective effects of catalpol. Significant improvements in the clinical scores were observed in catalpol-treated mice. Furthermore, catalpol increased TH expression and increased noradrenaline levels in the spinal cord. In primary cultures, catalpol exerted a neuroprotective effect in rat LC neurons by increasing the noradrenaline output. These results suggested that drugs targeting LC survival and function, including catalpol, may be able to benefit patients with MS.
Keywords: catalpol, experimental autoimmune encephalomyelitis, locus coeruleus, noradrenaline, tyrosine hydroxylase
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS), which is characterized by widespread regions of focal demyelination, loss of oligodendrocytes and axonal degeneration (1). It affects 0.1% people in the western world and results in chronic disability, primarily in young adults (2). The etiology and pathogenesis of the disease, however, remain unclear. Currently, MS is believed to be a complex multifactorial disease, involving genetic and environmental factors affecting the autoimmune responses that damage the myelin (3). Increasing evidence suggests that abnormalities of the locus coeruleus noradrenaline (LC-NA) system may be a contributing factor for myelin damage. A magnetic resonance study demonstrated an impairment of selective attention in MS, which was increased with axonal damage at the right LC (4). In mice immunized with myelin oligodendrocyte glycoprotein peptide MOG35–55, cortical and spinal cord levels of noradrenaline were significantly reduced compared with control mice (5). The immunized mice additionally exhibited increased astrocyte activation and neuronal damage in the LC (5). The LC-selective neurotoxin, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4), (6) significantly increases the clinical severity of experimental autoimmune encephalomyelitis (EAE) in mice (7), which suggests that the functional status of LC influences EAE disease. Numerous studies have demonstrated that noradrenaline levels are altered in the CNS of patients with multiple sclerosis, and that noradrenaline levels are reduced in the brainstem and spinal cord of rats with EAE (5,8,9).
The endogenous neurotransmitter, noradrenaline, exerts anti-inflammatory and neuroprotective effects in vivo and in vitro (10,11). Noradrenaline acting at β2-adrenergic receptors reduces the expression of numerous genes involved in inflammation, including those encoding adhesion molecules, chemokines, major histocompatibility class II molecules, inducible nitric oxide synthase (iNOS)2, interleukin (IL)-1β and tumor necrosis factor (TNF)-α via the activation of cyclic AMP-signaling pathways in astrocytes (12–16). Another important target of noradrenaline signaling in the CNS are microglia, which are often considered to be the resident immune cells of the CNS. Noradrenaline regulates the three key microglia actions: Migration, proliferation and phagocytosis, via activation of the β2-adrenergic receptor (17–19). As in astrocytes, noradrenaline reduces the expression of proinflammatory cytokines in microglia (20). In addition to suppressing the production of proinflammatory cytokines, noradrenaline increases neurotrophin expression in glia cells, including brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor and fibroblast growth factor-2 (21–23). Noradrenaline induces the production of the IL-1 receptor antagonist and IL-R2, which leads to an overall decrease in IL-1 signaling and IL-10 levels in the cortex and hippocampus. Noradrenaline additionally has beneficial effects on the maturation of oligodendrocyte progenitor cells, which may stimulate the myelination of axons and promote the recovery of MS (24). Therefore, a diminished noradrenaline level or perturbation of the noradrenaline-signaling system exacerbates neuro-inflammation in MS (25).
Increased levels of noradrenaline reduces neurotoxicity due to inflammatory or excitotoxic stimuli, or incubation with amyloid β. For example, using an α2-adrenergic antagonist reduces neuronal NOS2 expression due to aggregated amyloid β (26). Selective noradrenaline reuptake inhibitors reduce CNS cytokine, chemokine and adhesion molecule expression following systemic endotoxin injection and increased anti-inflammatory cytokines (27,28); and a synthetic noradrenaline precursor reduces astrocyte activation in EAE (7).
The primary source of noradrenaline in the CNS is tyrosine hydroxylase (TH)-positive neurons, which are located in the LC (29). The LC is located at the lower corners of the fourth cerebral ventricle, and releases noradrenaline over almost the entire CNS via nonjunctional varicosities (30). Degeneration or damage of the LC decreases the levels of noradrenaline in its projection areas (31). As reduced noradrenaline levels may lead to increased inflammation and neuronal damage, and as the LC is the primary source of brain noradrenaline and the sole source of noradrenaline fibers to the hippocampus and neocortex (32), methods to raise noradrenaline levels or improve LC function may benefit patients with MS (5). However, a better understanding of the interactions between the LC-NA and immune systems is required to develop novel therapeutic approaches for the treatment of MS.
Catalpol is an important iridoid glycoside, which is purified from the roots of Rehmannia glutinosa Libosch. It has been demonstrated to exert anti-inflammatory, antioxidant, anti-apoptotic and other neuroprotective properties, and serves a role in neuroprotection against ischemic injuries and neurodegenerative diseases (33–36). It is additionally found to exert neuroprotective effects age-associated neuroplasticity loss (37). It was recently reported that catalpol may improve memory defects in animal neurodegenerative models (38,39). However, the effects and underlying mechanisms of catalpol in EAE were not investigated extensively. The present study investigated the effects of catalpol as a putative TH inducer by measuring TH protein concentration and activities in the LC.
Materials and methods
Animals
Female C57BL/6 mice of specific pathogen-free grade (age, 4–6 weeks; weight, 14–16 g; n=80) and timed-pregnant Sprague-Dawley rats bearing E15 pups (age, 8–10 weeks; weight, 250–300 g; n=30) were purchased from Vital River Laboratories [Beijing, China; certification no. SCXK (JING) 2012–0001]. All animals had unrestricted access to food and water and were kept in a controlled 12 h light/dark environment. The animal facility was maintained at 21±2°C and 50±10% relative humidity. All animal experiments were approved by the Animal Experiments and Experimental Animals Management Committee and Animal Experiments and Experimental Animal Welfare Committee of Capital Medical University (Beijing, China; AEEI-2014-018).
Induction of EAE and DSP-4-EAE
C57BL/6 female mice were injected subcutaneously with an emulsion of 300 µg MOG35–55 (catalog no. MEVGWYRSPFSRVVHLYRNGK; SciLight Biotechnology, Beijing, China) dissolved in 100 µl phosphate-buffered saline (PBS), mixed with 100 µl complete Freund's adjuvant containing 500 µg heat-inactivated Mycobacterium tuberculosis H37RA (Difco; BD Biosciences, Franklin Lakes, NJ, USA). Pertussis toxin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany; 200 ng/mouse) in 200 µl PBS was administered intraperitoneally (i.p.) on the day of immunization and repeated 2 days later. The mice received a booster MOG35–55 injection 1 week later (40) (Fig. 1). Subcutaneous injection of animals was conducted following anesthetizing the mice with isoflurane.
Figure 1.

Induction of EAE and DSP-4-EAE. EAE, experimental autoimmune encephalomyelitis; DSP-4, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine; MOG, myelin oligodendrocyte glycoprotein; PTX, pertussis toxin.
C57BL/6 female mice were treated i.p. with 50 mg/kg DSP-4 (Sigma-Aldrich; Merck KGaA) dissolved in PBS once a week for 2 weeks (41). The mice were subsequently immunized with MOG35–55 1 week later (14 days following the first DSP-4 injection) and again 1 week later (Fig. 1).
Mice were weighed and evaluated daily from the day of first immunization. Clinical signs were scored by a scale from 0–15 as follows: Tail function: 0, no symptoms; 1, partial paralysis; 2, full paralysis. Hind or forelimbs (assessed separately): 1, weak and abnormal gait with the particular limb; 2, paresis (dragging movement); 3, full paralysis. Scores were calculated as the total of the tail and all four limbs (42,43). Based on this scoring system, a fully paralyzed quadriplegic animal would attain a score of 14 and mortality would be given a score of 15. Scoring was conducted by a blinded investigator.
Treatments
A total of 80 mice were randomly assigned to four groups: Control (n=15), DSP-4-EAE mice (n=25), vehicle-treated EAE mice (n=20) and catalpol-treated EAE mice (n=20). Catalpol (110808–201210; purity, >99%; assayed by high-performance liquid chromatography analysis) was purchased from the Chinese Institute for Drug and Biological Product Control (Beijing, China). Catalpol (10 mg/kg) was administered i.p. daily from the day prior to MOG35–55 immunization and continued for 40 days (44). Catalpol was prepared by dissolving 2 mg catalpol in 1.0 ml PBS. The control mice, DSP-4-EAE mice and vehicle-treated EAE mice received PBS (i.p.) for 40 days.
LC cell culture and treatments
Culture plates (Corning Incorporated, Corning, NY, USA) were precoated with 100 µg/ml poly-L-lysine (Sigma-Aldrich; Merck KGaA) in sterile calcium-magnesium-free PBS (pH 7.2) and stored at 37°C overnight in an air-humidified, high efficiency particulate arrestance-filtered incubator. The plates were rinsed twice with sterile double-distilled water, following which 500 µl culture media was added and the plates were incubated at 37°C in a 5% CO2/95% air humidified incubator until further use.
Pregnant Sprague-Dawley rats were sacrificed at E15 by exposure to CO2. The uterine horns were removed under sterile conditions and placed in a Petri dish containing cold Dulbecco's Modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) on ice. The embryos were removed and further dissection was performed in the same medium. The area dissected corresponded to the proximal rhombencephalic ring between the distal part of the mesencephalic flexure and the proximal part of the pontine flexure (45). The tissues were completely dissociated by gentle trituration with fire-polished glass Pasteur pipettes. Cells were plated on previously prepared 24-well plates at a density of 2×105 cells/well. Cells were cultured in culture medium consisting of DMEM and Ham's F-12 nutrient mixture [1:1 (v/v); Gibco; Thermo Fisher Scientific, Inc], supplemented with 6 mg/ml glucose, 2 mmol/l glutamine, 5% horse serum (HyClone, GE Healthcare Life Sciences; Logan, UT, USA), 10% fetal bovine serum (Hyclone, GE Healthcare Life Sciences) and 50 U/ml penicillin and 50 µg/ml streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.). Cells were cultured at 37°C in a water-saturated 5% CO2 and 95% air incubator. At 6 h following plating, the original media was discarded and replenished with serum-free medium. The cells were maintained for 7 days in serum-free medium consisting of neurobasal medium with 1/50 B27 and bovine serum albumin (all purchased from Gibco; Thermo Fisher Scientific, Inc.) and penicillin-streptomycin, which yielded a relatively neuron-pure culture. The culture medium was replaced every other day.
Immunofluorescence staining
TH immunoreactivity was assessed in the LC as a marker of noradrenaline neurons. At 40 days following PBS or catalpol administration, mice were anesthetized with 10% chloral hydrate (5 ml/kg; Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) and the thoracic cavity was opened, and mice were intra-cardially perfused with PBS (0.1 M, pH 7.4) at 4°C, followed by phosphate-buffered 4% paraformaldehyde solution. Directly following perfusion, the brains were removed, post fixed for further 48 h at 4°C and dehydrated in 30% sucrose solution for 4–6 days. Coronal sections (25-µm thick) were cut from the blocks containing the LC using a Leica freezing microtome (Leica Microsystems GmbH, Wetzlar, Germany).
TH immunofluorescence detection was used to analyze TH-positive noradrenaline neurons. Following fixing with 4% paraformaldehyde for 15 min at 4°C, the cells were washed three times with PBS and the permeability of the cell membrane was increased with 0.2% Triton X-100. Subsequently, cells were blocked for 1 h with 10% goat serum (GS; Gibco, Thermo Fisher Scientific, Inc.) at room temperature and incubated overnight at 4°C with a rabbit polyclonal TH antibody (cat. no. IMG5070; Novus Biologicals, LLC, Littleton, CO, USA) diluted 1:400 in 10% GS. Subsequent incubations were performed at room temperature for 1 h with an Alexa Fluor 568 F (ab')2 fragment of a goat anti-rabbit immunoglobulin (Ig)G secondary antibody (cat. no. A-11079; Invitrogen; Thermo Fisher Scientific, Inc.). Neurons were identified with mouse anti-β III tubulin primary antibody diluted 1:1,000 (cat. no. ab195879; Abcam, Cambridge, MA, USA) followed by incubation with an Alexa Fluor 488 F (ab')2 fragment of a goat anti-mouse IgG secondary antibody (cat. no. A-11034; Invitrogen; Thermo Fisher Life Sciences, Inc.). Cells were imaged with the Leica Application Suite V4 software from the Leica Microsystems Framework using an inverted fluorescent microscope (DMI4000; Leica Microsystems GmbH).
Cell viability assay
Survival of LC neurons was determined by measuring their metabolic activity using a Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). Cells were cultured at a density of 1×105 in triplicate in a 96-well plate. A total of 7 days following plating, the culture media was replaced with fresh media (controls) or media containing 1, 5, 10 or 20 µM catalpol, followed by incubation for 24 h at 37°C. Oxidative damage was initiated by a water-soluble initiator 2,2′-azobis (2-amidinopropane) hydrocholoride (AAPH; 20 µM) (46). Following incubation for 3 h at 37°C, cell viability was analyzed by CCK-8. The resultant orange formazan product indicative of metabolic activity of the cells was solubilized and quantified at a wavelength of 450 nm using the SpectraMax M5 multifunctional microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA). Mean values were obtained from three replicates (n=3).
Enzyme-linked immunosorbent assay (ELISA)
Cell lysates were prepared from samples of the cortex and spinal cord of control, vehicle-treated EAE and catalpol-treated EAE mice. Tissues (100 mg) were rinsed with PBS, homogenized in 1 ml PBS and stored overnight at −20°C. Following performing two freeze-thaw cycles to break the cell membranes, the homogenates were centrifuged at 5,000 × g for 5 min at 4°C. The supernatant was collected and assayed immediately. ELISA for noradrenaline was performed using a Mouse Noradrenaline ELISA kit (cat. no. MBS701514; MyBioSource, Inc., San Diego, CA, USA) at a detection range of 7.5–300 pg/ml, according to the manufacturer's protocol.
Following drug treatment, the conditioned medium was collected and assayed immediately. Noradrenaline levels were assessed using a rat noradrenaline ELISA kit (cat. no. MBS701791; MyBioSource, Inc.) at a range of 0.49–2,000 pg/ml, according to the manufacturer's protocol.
Western blotting
Pons sections containing the LC of differently treated mice were homogenized in cell lysis buffer (cat. no. C3228; Sigma-Aldrich; Merck KGaA) supplemented with a protease inhibitor cocktail (cat. no. P3840; Sigma-Aldrich; Merck KGaA). The homogenate was subsequently centrifuged at 8,000 × g for 15 min at 4°C. Similar to the LC of animals, the homogenates prepared from the LC neurons were centrifuged at 8,000 × g for 15 min at 4°C. Protein concentration of the supernatants was quantified by the bicinchoninic acid assay kit (Thermo Fisher Scientific, Inc.). Proteins (40 µg) from each sample were separated by 10% SDS-PAGE and subsequently transferred onto polyvinylidene difluoride membranes (Merck KGaA) by a semi-dry electrophoretic transfer system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membranes were blocked with 5% skimmed milk (BD Biosciences) in TBS with Tween-20 (0.1%, TBST) for 2 h at room temperature and subsequently incubated with a rabbit polyclonal TH or mouse monoclonal anti-β-actin (cat. no. AC-15; Sigma-Aldrich; Merck KGaA) antibody at 4°C overnight. The antibodies were used at a dilution of 1:1,000. The primary antibody was removed and washed three times with TBST, following which the membranes were further incubated with horseradish perioxidase-conjugated anti-mouse or anti-rabbit IgG secondary antibodies (1:2,000; Abmart Inc., Shanghai, China) for 1 h at room temperature. Following three washes with TBS with Tween-20, the bands were visualized using Luminol reagent (Santa Cruz Biotechnology Inc., Dallas, TX, USA) by FluorChem FC2 (ProteinSimple, San Jose, CA, USA), and were quantified using Quantity One image analysis software (Version, 4.62; Bio-Rad Laboratories, Inc.).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (version, 5.0; GraphPad Software, La Jolla, CA, USA). Data are presented as the mean ± standard error. Repeated measures two-way analysis of variance (ANOVA) was performed to compare EAE score and body weight in treatment groups. Pair-wise comparison between the groups was performed by one-way ANOVA followed by Tukey's post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
DSP-4 administration exacerbates EAE
DSP-4 is widely used to reduce brain noradrenaline levels to study the functions of the central noradrenergic system. Numerous studies have demonstrated that DSP-4 may produce long-lasting reduction of endogenous noradrenaline in the brain of mice and rats (47–49). Administration of 50 mg/kg DSP-4 has previously resulted in a significant reduction in noradrenaline levels in EAE animals (a reduction of 73% vs. vehicle) (50). In the present study, 2 weeks following the first DSP-4 injection, mice were immunized with a MOG35–55 peptide. The results indicated that in the EAE group, 16/20 (80%) mice developed clinical symptoms, and the mean day of onset was 13.5±0.6 days post initial immunization. In the DSP-4-treated group, the incidence was 25/25 (100%), and the mean time of onset was at 11.0±0.08 days post initial immunization, which was markedly different from the EAE group (Fig. 2A). In addition, the mean daily clinical scores were significantly increased in DSP4-EAE mice compared with the EAE group at the start and end time points, particularly at days 11, 12 and 13 (P<0.001) and days 33, 34 and 35 (P<0.01; Fig. 2B). These results suggested that selectively reducing noradrenaline levels in the CNS exacerbates EAE. Noradrenaline depletion significantly exacerbated the magnitude of EAE-induced clinical signs, and incidence was higher in DSP-4-treated group compared with the control group.
Figure 2.

DSP-4 treatment exacerbates EAE. (A) Mean daily clinical scores and (B) mean daily incidence of disease for vehicle (n=20) and DSP-4 treated (n=25) mice. In the two groups, scores were significantly different from baseline values by day 11 post-immunization, and there were significant differences in the mean day of onset (13.5±0.6 and 11.0±0.08 for EAE and DSP-4-EAE groups, respectively). Additionally, there was a statistically significant difference between development of clinical scores in DSP-4 vs. vehicle groups (F[33,1]=2.73; P<0.001) Data are presented as the mean ± standard error from two independent experiments. *P<0.05; **P<0.01; ***P<0.001 vs. EAE group. EAE, experimental autoimmune encephalomyelitis; DSP-4, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine.
Catalpol treatment ameliorates EAE
The weight and disability scores of each mouse were obtained daily over the course of the 40-day experiment. The efficacy of catalpol on the neurological behaviors of EAE mice was investigated. Treatment with catalpol or vehicle was initiated the day prior to MOG35–55 immunization and continued for 5 weeks. In the vehicle group, there was a significant increase in clinical scores vs. baseline at day 12, and the maximum mean clinical score (6.4±0.10) was reached on day 20, which remained unaltered thereafter (Fig. 3A). From day 10 onwards, 3/20 mice in the vehicle group exhibited clinical improvement. In the catalpol group, clinical scores were significantly increased from baseline scores at day 13, and the maximum mean clinical score (2.5±0.06) was reached on day 18, after which it began to decrease, reaching 0.5±0.02 at the end of the study. From day 10 onwards, 10/20 catalpol-treated mice exhibited clinical improvement. Mean clinical scores of catalpol-treated EAE mice were significantly reduced compared with vehicle-treated EAE mice (P<0.05; Fig. 3A). Cumulative incidence scores of catalpol-treated EAE mice were also reduced compared with vehicle-treated EAE mice (Fig. 3B). The body weights of animals were monitored daily to confirm the results of the clinical scores. As presented in Fig. 3C, mice demonstrated persistent loss of body weight with the progression of EAE, and catalpol treatment prevented body weight loss of EAE mice. No obvious adverse effects of catalpol were observed at a dose of 10 mg/kg/day in EAE mice 35 days following injection. The overall disease burden for each mouse is represented as the sum of the daily scores over the course of the 40-day experiment (42). There was a decrease in disease burden in catalpol-treated animals (P<0.05; Fig. 4). These results indicated that catalpol suppressed the severity of EAE.
Figure 3.

Catalpol treatment ameliorates EAE. (A) Mean daily clinical scores, (B) mean daily incidence of disease and (C) body weight alterations in vehicle- (n=20) and catalpol-treated (n=20) mice. There was a significant effect of catalpol on clinical scores from day 18 onwards (F[33,1]=3.17, P<0.001). Data are presented as the mean ± standard error from two independent experiments. *P<0.05; **P<0.01; ***P<0.001 vs. EAE group. EAE, experimental autoimmune encephalomyelitis; DSP-4, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine.
Figure 4.

Catalpol treatment decreases disease burden in animals. The overall disease burden for vehicle-(n=20), catalpol-(n=25) and DSP-4-treated mice are represented as the sum of scores, which is the sum of the disability scores obtained daily over the course of the 40-day experiment. Data are presented as the mean ± standard error. Catalpol vs. EAE, *P<0.05; DSP-4-EAE vs. EAE, *P<0.05. EAE, experimental autoimmune encephalomyelitis; DSP-4, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine.
Catalpol treatment effects on TH immunoreactivity in LC
Serial sections prepared from the LC of control, vehicle-treated and catalpol-treated mice were immunostained for TH. In the LC, where TH-immunoreactivity reflects noradrenergic neurons, examination of immunofluorescence in sections from control mice revealed numerous large spherical TH immunoreactivity neurons (Fig. 5A). In EAE mice (Fig. 5B), TH expression was greatly reduced compared with the control-group mice, whereas catalpol treatment (Fig. 5C) recovered this effect.
Figure 5.

Sagittal sections through (A) control, (B) vehicle-treated and (C) catalpol-treated mice were stained for the presence of TH+ neurons. Images are representative of three mice in each group, and demonstrate an increase in TH + staining following catalpol treatment. TH, tyrosine hydroxylase; EAE, experimental autoimmune encephalomyelitis; DSP-4, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine; Cont, control.
Effect of catalpol on the viability of LC cells
As assessed by CCK-8 assay, vehicle-treated LC cells demonstrated markedly decreased viability 3 h after treatment with DSP-4. Catalpol (10 and 20 µM) significantly inhibited this effect (Fig. 6).
Figure 6.
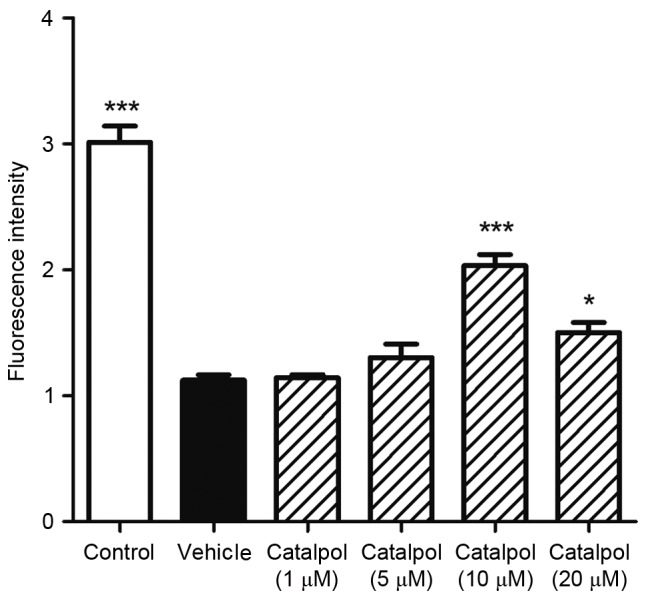
Catalpol protects against 2,2′-azobis (2-amidinopropane) hydrocholoride-induced oxidative damage in locus coeruleus neurons. Cells were treated with 1, 5 10 or 20 µM catalpol and incubated for 24 h. Oxidative damage was initiated by 20 µM DSP-4 treatment for 3 h. Cell viability was analyzed using a Cell Counting Kit-8. Cell viability was significantly decreased in the vehicle-treated group 3 h following DSP-4 treatment. Catalpol (10 and 20 µM) inhibited this effect. Data are presented as the mean ± standard error (n=8). *P<0.05; ***P<0.001 vs. vehicle. DSP-4, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine; Cont, control.
Catalpol increases noradrenaline levels in vivo and in vitro
Noradrenaline levels were determined by specific ELISA analyses in homogenates prepared from the cortex and spinal cords of control, vehicle-treated EAE and catalpol-treated EAE mice 20 and 40 days following immunization. In control mice, noradrenaline levels were increased in the cortex compared with the spinal cord. A significant decrease in noradrenaline levels was observed in the cortex (~38% reduction) and spinal cord (~62% reduction) of EAE mice compared with controls. Additionally, noradrenaline levels were significantly increased in catalpol-treated EAE mice (Fig. 7A).
Figure 7.

Catalpol increases noradrenaline levels in vivo and in vitro. (A) Noradrenaline levels were significantly decreased in the frontal cortex and spinal cord of EAE mice compared with controls. There were statistically significant differences between catalpol-treated and vehicle groups. Data are presented as the mean ± standard error. ###P<0.001 vs. EAE-d40; ***P<0.001 vs. EAE-d20. (B) Catalpol treatment markedly increased the amount of noradrenaline released into the medium. Data are presented as the mean ± standard error (n=6). **P<0.01; ***P<0.001 vs. damage. Damage, vehicle-treated neurons; NA, noradrenaline; CTX, cortex; SC, spinal cord; EAE, experimental autoimmune encephalomyelitis; d20, day 20 following immunization; d40; day 40 following immunization.
Treatment with 10 µM catalpol for 24 h markedly enhanced the amount of noradrenaline released into the medium compared with the control after DSP-4-induced damage (20 µM AAPH) for 3 h (Fig. 7B).
Catalpol treatment effects on TH expression in vivo and in vitro
Mice were injected with PBS or catalpol for 40 days. The LC was dissected from the mouse brains, and the protein expression levels of TH protein were determined. Protein expression levels of TH were increased when the animals were chronically treated with 10 mg/kg catalpol for 40 days (P<0.01; Fig. 8A). However, no significant differences were observed in TH protein expression levels between catalpol- and vehicle-treated LC neurons (P>0.05; Fig. 8B).
Figure 8.

Representative western blotting images and quantification of TH protein expression levels in (A) C57BL/6 mice and (B) Sprague-Dawley rat embryos. β-actin served as an internal control. Data are presented as the mean ± standard error (n=3). ***P<0.001 vs. control; ###P<0.01 vs. EAEd40. Con, control; TH, tyrosine hydroxylase; EAE, experimental autoimmune encephalomyelitis; DSP-4, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine; d20, day 20 following immunization; d40; day 40 following immunization.
Discussion
Numerous studies have indicated that alterations in peripheral noradrenaline levels occur during MS and EAE (51–53). In contrast to its role in EAE and MS, the role of central noradrenaline remains to be fully elucidated. The results of the present study indicated that noradrenaline depletion significantly exacerbates clinical scores in EAE mice, and the incidence was increased in the DSP-4-treated group compared with the vehicle-treated EAE group. As DSP-4 has limited effects on peripheral noradrenaline levels (47), reduction in the noradrenaline levels in the CNS exacerbates the severity of EAE. These results were not completely consistent with previously published results, which reported that the incidence and average day of onset in the DSP-4-treated group were not significantly different from the control group (7).
The present study re-examined whether CNS noradrenaline level reduction occurs in EAE. Consistent with a previous study (5), it was observed that noradrenaline levels were decreased in the frontal cortex and spinal cord of EAE mice, and that the noradrenergic neurons in LC were additionally damaged, which confirmed the association between abnormalities of the LC-NA system with exacerbation of neuroinflammation and neural damage. However, decreased synthesis, increased metabolism or increased re-uptake may account for reduced noradrenaline levels, inflammation in the LC, and stress in TH-positive neurons. The present study demonstrated that noradrenaline metabolism in the cortex was increased in EAE mice compared with controls. Catecholamine-O-methyl transferase and monoamine oxidase are ubiquitous metabolic enzymes of noradrenaline; the two have been demonstrated to be increased in neurological diseases (54,55). Noradrenaline increases the expression of numerous neurotrophins, which are known to be critical for LC survival and maturation. These findings suggested that diminished LC function and reduced noradrenaline levels may contribute to damage by reducing neurotrophic support from glial cells and LC neurons.
Numerous tests to increase CNS noradrenaline levels have been demonstrated to provide benefit for MS and EAE. Noradrenaline levels may be increased by the use of selective α2-adrenoceptor antagonists, selective noradrenaline reuptake inhibitors (NARIs) and noradrenaline precursors. α2-adrenoceptors are inhibitory pre-synaptic neuronal autoreceptors and are primarily located in the noradrenergic neurons of LC. α2-antagonists increase noradrenaline release from LC noradrenergic neurons, reduce excitotoxicity (56), promote neuronal survival (57) and inhibit inflammatory responses (26) in vivo. However, their clinical use has been limited. Selective NARIs, including the non-tricyclic antidepressant atomoxetine, exhibit anti-inflammatory effects on microglial cells (58) and increase BDNF expression in the hippocampus (59). However, treatment with atomoxetine did not promote recovery in EAE mice (7), thereby suggesting that although extracellular noradrenaline levels may be increased by NARIs, the reduced endogenous noradrenaline levels are not sufficient to exert anti-inflammatory and neuroprotective effects. Noradrenaline levels may be directly increased by treatment with the synthetic noradrenaline precursor, L-threo-3,4-dihydroxyphenylserine (L-DOPS), which is converted to noradrenaline via decarboxylation by dopamine decarboxylase (60). In contrast, treatment with L-DOPS reduced clinical scores in EAE mice (7). Furthermore, treatment with L-DOPS and atomoxetine resulted in a synergistic effect on EAE mice (7). It was reported that co-treatment with L-phenylalanine and the NARI lofepramine improved clinical symptoms and reduced lesion volumes in MS patients (61,62).
TH catalyzes the rate-limiting and committed steps in the biosynthesis of noradrenaline (63). Therefore, regulation of TH protein expression levels and intrinsic enzyme activity may represent a key pathway for controlling the synthesis of noradrenaline. Chronic neuroinflammation leads to reduced TH enzyme levels in the LC and decreased noradrenaline levels in LC projections, including the cortex and hippocampus (64). Noradrenaline acts as an anti-inflammatory molecule, stimulating the production of BDNF, and supporting neurogenesis in the hippocampus. Therefore, cognitive decline may be associated with decreased noradrenaline levels in the cortex and hippocampus (65).
LC stress and damage are primary causes of noradrenaline dysfunction (66). In this regard, drugs that selectively increase LC neuronal viability or activity may be of benefit. A previous study reported that vindeburnol, a structural analog of the cerebral vasodilator vincamine, increased TH activity and protein expression in the LC (67). However, it was demonstrated that vindeburnol increases the expression of genes involved in LC survival and maturation, and increases central noradrenaline levels (66). Therefore, in relation to the present study, it may be hypothesized that binding of vindeburnol to a2-adrenergic receptors lead to a sustained increase in noradrenaline release and ultimately a requirement for de novo noradrenaline synthesis and increased TH expression.
Numerous approved first-line drugs, including interferon-β, glatiramer acetate, mitoxantrone and natalizumab, are either immunoregulators or immunosuppressants, and have significant adverse effects associated with long-term therapy, including infection, cardiotoxicity, anemia, nausea and depression (68). However, there are limited treatment options that reduce or inhibit the neurodegeneration, promote remyelination and improving neuron survival, which determines the outcome and prognosis of the disease.
Catalpol is widely used as a traditional Chinese herbal medicine for the treatment of various neurodegenerative diseases, including Alzheimer's, Parkinson's and ischemic diseases. Catalpol may additionally cross the blood-brain barrier (68). Furthermore, catalpol may enhance neuronal axon growth (69), implicating a potential role for the treatment of MS. Catalpol has been demonstrated to protect dopaminergic neurons from LPS-induced neurotoxicity (70). The present study used the most commonly used model for MS to confirm the neuroprotective effects of catalpol. In mice treated with catapol, a significant improvement in the clinical scores was observed in EAE. Catalpol exerts neuroprotective effects in cortical neurons (35); however, its role in exerting similar effects on LC cells, the primary source of noradrenaline in the CNS, remains unclear. The present study tested the effects of catalpol on LC neurons. In primary LC neuron cultures, catalpol exerted a neuroprotective effect and enhanced the generation of noradrenaline following DSP-4-induced neuronal damage. Furthermore, when the cultures were incubated with catalpol alone, there was no alteration in the production of noradrenaline, which may account for the fact that catalpol had fewer side effects at 10 µM. These results confirmed that catalpol serves as a potential therapeutic drug and may be useful for the treatment of MS.
In conclusion, these data suggested that catalpol treatment exerted positive effects on the synthesis of noradrenaline and LC physiology. However, as the study was restricted to the CNS, further investigation into whether catalpol is involved in regulation of peripheral lymphocytes and macrophage activation is required.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant nos. 81072765 and 81273742) and the Beijing Natural Science Foundation (grant no. 7142053).
References
- 1.McQualter JL, Bernard CC. Multiple sclerosis: A battle between destruction and repair. J Neurochem. 2007;100:295–306. doi: 10.1111/j.1471-4159.2006.04232.x. [DOI] [PubMed] [Google Scholar]
- 2.Deckx N, Lee WP, Berneman ZN, Cools N. Neuroendocrine immunoregulation in multiple sclerosis. Clin Dev Immunol. 2013;2013:705232. doi: 10.1155/2013/705232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez JI, Cayrol R, Prat A. Disruption of central nervous system barriers in multiple sclerosis. Biochim Biophys Acta. 2011;1812:252–264. doi: 10.1016/j.bbadis.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 4.Gadea M, Martinez-Bisbal MC, Marti-Bonmati L, Espert R, Casanova B, Coret F, Celda B. Spectroscopic axonal damage of the right locus coeruleus relates to selective attention impairment in early stage relapsing-remitting multiple sclerosis. Brain. 2004;127:89–98. doi: 10.1093/brain/awh002. [DOI] [PubMed] [Google Scholar]
- 5.Polak PE, Kalinin S, Feinstein DL. Locus coeruleus damage and noradrenaline reductions in multiple sclerosis and experimental autoimmune encephalomyelitis. Brain. 2011;134:665–677. doi: 10.1093/brain/awq362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fritschy JM, Grzanna R. Experimentally-induced neuron loss in the locus coeruleus of adult rats. Exp Neurol. 1991;111:123–127. doi: 10.1016/0014-4886(91)90058-K. [DOI] [PubMed] [Google Scholar]
- 7.Simonini MV, Polak PE, Sharp A, McGuire S, Galea E, Feinstein DL. Increasing CNS noradrenaline reduces EAE severity. J Neuroimmune Pharmacol. 2010;5:252–259. doi: 10.1007/s11481-009-9182-2. [DOI] [PubMed] [Google Scholar]
- 8.White SR, Bhatnagar RK, Bardo MT. Norepinephrine depletion in the spinal cord gray matter of rats with experimental allergic encephalomyelitis. J Neurochem. 1983;40:1771–1773. doi: 10.1111/j.1471-4159.1983.tb08156.x. [DOI] [PubMed] [Google Scholar]
- 9.Krenger W, Honegger CG, Feurer C, Cammisuli S. Changes of neurotransmitter systems in chronic relapsing experimental allergic encephalomyelitis in rat brain and spinal cord. J Neurochem. 1986;47:1247–1254. doi: 10.1111/j.1471-4159.1986.tb00747.x. [DOI] [PubMed] [Google Scholar]
- 10.Marien MR, Colpaert FC, Rosenquist AC. Noradrenergic mechanisms in neurodegenerative diseases: A theory. Brain Res Brain Res Rev. 2004;45:38–78. doi: 10.1016/j.brainresrev.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 11.Madrigal JL, Kalinin S, Richardson JC, Feinstein DL. Neuroprotective actions of noradrenaline: Effects on glutathione synthesis and activation of peroxisome proliferator activated receptor delta. J Neurochem. 2007;103:2092–2101. doi: 10.1111/j.1471-4159.2007.04888.x. [DOI] [PubMed] [Google Scholar]
- 12.Frohman EM, Vayuvegula B, Gupta S, van den Noort S. Norepinephrine inhibits gamma-interferon-induced major histocompatibility class II (Ia) antigen expression on cultured astrocytes via beta-2-adrenergic signal transduction mechanisms; Proc Natl Acad Sci USA; 1988; pp. 1292–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benveniste EN, Huneycutt BS, Shrikant P, Ballestas ME. Second messenger systems in the regulation of cytokines and adhesion molecules in the central nervous system. Brain Behav Immun. 1995;9:304–314. doi: 10.1006/brbi.1995.1029. [DOI] [PubMed] [Google Scholar]
- 14.Feinstein DL. Suppression of astroglial nitric oxide synthase expression by norepinephrine results from decreased NOS-2 promoter activity. J Neurochem. 1998;70:1484–1496. doi: 10.1046/j.1471-4159.1998.70041484.x. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura A, Johns EJ, Imaizumi A, Abe T, Kohsaka T. Regulation of tumour necrosis factor and interleukin-6 gene transcription by beta2-adrenoceptor in the rat astrocytes. J Neuroimmunol. 1998;88:144–153. doi: 10.1016/S0165-5728(98)00109-X. [DOI] [PubMed] [Google Scholar]
- 16.Galea E, Heneka MT, Dello RC, Feinstein DL. Intrinsic regulation of brain inflammatory responses. Cell Mol Neurobiol. 2003;23:625–635. doi: 10.1023/A:1025084415833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujita H, Tanaka J, Maeda N, Sakanaka M. Adrenergic agonists suppress the proliferation of microglia through beta 2-adrenergic receptor. Neurosci Lett. 1998;242:37–40. doi: 10.1016/S0304-3940(98)00003-2. [DOI] [PubMed] [Google Scholar]
- 18.Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine; Proc Natl Acad Sci USA; 2010; pp. 6058–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong Y, Ruan L, Qian L, Liu X, Le Y. Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J Neurosci. 2010;30:11848–11857. doi: 10.1523/JNEUROSCI.2985-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell A. Inflammation, neurodegenerative diseases, and environmental exposures. Ann N Y Acad Sci. 2004;1035:117–132. doi: 10.1196/annals.1332.008. [DOI] [PubMed] [Google Scholar]
- 21.Zafra F, Lindholm D, Castrén E, Hartikka J, Thoenen H. Regulation of brain-derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J Neurosci. 1992;12:4793–4799. doi: 10.1523/JNEUROSCI.12-12-04793.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Remy S, Naveilhan P, Brachet P, Neveu I. Differential regulation of GDNF, neurturin, and their receptors in primary cultures of rat glial cells. J Neurosci Res. 2001;64:242–251. doi: 10.1002/jnr.1072. [DOI] [PubMed] [Google Scholar]
- 23.Kajitani N, Hisaoka-Nakashima K, Morioka N, Okada-Tsuchioka M, Kaneko M, Kasai M, Shibasaki C, Nakata Y, Takebayashi M. Antidepressant acts on astrocytes leading to an increase in the expression of neurotrophic/growth factors: Differential regulation of FGF-2 by noradrenaline. PLoS One. 2012;7:e51197. doi: 10.1371/journal.pone.0051197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghiani CA, Eisen AM, Yuan X, DePinho RA, McBain CJ, Gallo V. Neurotransmitter receptor activation triggers p27 (Kip1) and p21 (CIP1) accumulation and G1 cell cycle arrest in oligodendrocyte progenitors. Development. 1999;126:1077–1090. doi: 10.1242/dev.126.5.1077. [DOI] [PubMed] [Google Scholar]
- 25.Kaneko YS, Mori K, Nakashima A, Sawada M, Nagatsu I, Ota A. Peripheral injection of lipopolysaccharide enhances expression of inflammatory cytokines in murine locus coeruleus: Possible role of increased norepinephrine turnover. J Neurochem. 2005;94:393–404. doi: 10.1111/j.1471-4159.2005.03209.x. [DOI] [PubMed] [Google Scholar]
- 26.Kalinin S, Polak PE, Madrigal JL, Gavrilyuk V, Sharp A, Chauhan N, Marien M, Colpaert F, Feinstein DL. Beta-amyloid-dependent expression of NOS2 in neurons: Prevention by an alpha2-adrenergic antagonist. Antioxid Redox Signal. 2006;8:873–883. doi: 10.1089/ars.2006.8.873. [DOI] [PubMed] [Google Scholar]
- 27.O'Sullivan JB, Ryan KM, Curtin NM, Harkin A, Connor TJ. Noradrenaline reuptake inhibitors limit neuroinflammation in rat cortex following a systemic inflammatory challenge: Implications for depression and neurodegeneration. Int J Neuropsychopharmacol. 2009;12:687–699. doi: 10.1017/S146114570800967X. [DOI] [PubMed] [Google Scholar]
- 28.O'Sullivan JB, Ryan KM, Harkin A, Connor TJ. Noradrenaline reuptake inhibitors inhibit expression of chemokines IP-10 and RANTES and cell adhesion molecules VCAM-1 and ICAM-1 in the CNS following a systemic inflammatory challenge. J Neuroimmunol. 2010;220:34–42. doi: 10.1016/j.jneuroim.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 29.Benarroch EE. The locus ceruleus norepinephrine system: Functional organization and potential clinical significance. Neurology. 2009;73:1699–1704. doi: 10.1212/WNL.0b013e3181c2937c. [DOI] [PubMed] [Google Scholar]
- 30.Szabadi E. Functional neuroanatomy of the central noradrenergic system. J Psychopharmacol. 2013;27:659–693. doi: 10.1177/0269881113490326. [DOI] [PubMed] [Google Scholar]
- 31.Iversen LL, Rossor MN, Reynolds GP, Hills R, Roth M, Mountjoy CQ, Foote SL, Morrison JH, Bloom FE. Loss of pigmented dopamine-beta-hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer's type. Neurosci Lett. 1983;39:95–100. doi: 10.1016/0304-3940(83)90171-4. [DOI] [PubMed] [Google Scholar]
- 32.Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003;42:33–84. doi: 10.1016/S0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- 33.Li DQ, Bao YM, Li Y, Wang CF, Liu Y, An LJ. Catalpol modulates the expressions of Bcl-2 and Bax and attenuates apoptosis in gerbils after ischemic injury. Brain Res. 2006;1115:179–185. doi: 10.1016/j.brainres.2006.07.063. [DOI] [PubMed] [Google Scholar]
- 34.Tian YY, Jiang B, An LJ, Bao YM. Neuroprotective effect of catalpol against MPP(+)-induced oxidative stress in mesencephalic neurons. Eur J Pharmacol. 2007;568:142–148. doi: 10.1016/j.ejphar.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 35.Jiang B, Du J, Liu JH, Bao YM, An LJ. Catalpol attenuates the neurotoxicity induced by beta-amyloid (1–42) in cortical neuron-glia cultures. Brain Res. 2008;1188:139–147. doi: 10.1016/j.brainres.2007.07.105. [DOI] [PubMed] [Google Scholar]
- 36.Liang JH, Du J, Xu LD, Jiang T, Hao S, Bi J, Jiang B. Catalpol protects primary cultured cortical neurons induced by Abeta(1–42) through a mitochondrial-dependent caspase pathway. Neurochem Int. 2009;55:741–746. doi: 10.1016/j.neuint.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, He QJ, Zou W, Wang HX, Bao YM, Liu YX, An LJ. Catalpol increases hippocampal neuroplasticity and up-regulates PKC and BDNF in the aged rats. Brain Res. 2006;1123:68–79. doi: 10.1016/j.brainres.2006.09.058. [DOI] [PubMed] [Google Scholar]
- 38.Zhang XL, An LJ, Bao YM, Wang JY, Jiang B. d-galactose administration induces memory loss and energy metabolism disturbance in mice: Protective effects of catalpol. Food Chem Toxicol. 2008;46:2888–2894. doi: 10.1016/j.fct.2008.05.032. [DOI] [PubMed] [Google Scholar]
- 39.Xia Z, Zhang R, Wu P, Xia Z, Hu Y. Memory defect induced by β-amyloid plus glutamate receptor agonist is alleviated by catalpol and donepezil through different mechanisms. Brain Res. 2012;1441:27–37. doi: 10.1016/j.brainres.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 40.Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L, Landreth GE, Pershadsingh HA, Weinberg G, Heneka MT. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol. 2002;51:694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- 41.Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O'Banion MK, Weinberg G, Klockgether T, Feinstein DL. Noradrenergic depletion potentiates beta-amyloid-induced cortical inflammation: Implications for Alzheimer's disease. J Neurosci. 2002;22:2434–2442. doi: 10.1523/JNEUROSCI.22-07-02434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giuliani F, Metz LM, Wilson T, Fan Y, Bar-Or A, Yong VW. Additive effect of the combination of glatiramer acetate and minocycline in a model of MS. J Neuroimmunol. 2005;158:213–221. doi: 10.1016/j.jneuroim.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 43.Weaver A, Goncalves da Silva A, Nuttall RK, Edwards DR, Shapiro SD, Rivest S, Yong VW. An elevated matrix metalloproteinase (MMP) in an animal model of multiple sclerosis is protective by affecting Th1/Th2 polarization. FASEB J. 2005;19:1668–1670. doi: 10.1096/fj.04-2030fje. [DOI] [PubMed] [Google Scholar]
- 44.Jiang B, Zhang H, Bi J, Zhang XL. Neuroprotective activities of catalpol on MPP+/MPTP-induced neurotoxicity. Neurol Res. 2008;30:639–644. doi: 10.1179/174313208X289543. [DOI] [PubMed] [Google Scholar]
- 45.Holm PC, Rodriguez FJ, Kresse A, Canals JM, Silos-Santiago I, Arenas E. Crucial role of TrkB ligands in the survival and phenotypic differentiation of developing locus coeruleus noradrenergic neurons. Development. 2003;130:3535–3545. doi: 10.1242/dev.00565. [DOI] [PubMed] [Google Scholar]
- 46.Flueraru M, So R, Willmore WG, Poulter MO, Durst T, Charron M, Wright JS. Cytotoxicity and cytoprotective activity of naphthalenediols in rat cortical neurons. Chem Res Toxicol. 2006;19:1221–1217. doi: 10.1021/tx060045z. [DOI] [PubMed] [Google Scholar]
- 47.Jonsson G, Hallman H, Ponzio F, Ross S. DSP4 (N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine)-a useful denervation tool for central and peripheral noradrenaline neurons. Eur J Pharmacol. 1981;72:173–188. doi: 10.1016/0014-2999(81)90272-7. [DOI] [PubMed] [Google Scholar]
- 48.Hallman H, Jonsson G. Pharmacological modifications of the neurotoxic action of the noradrenaline neurotoxin DSP4 on central noradrenaline neurons. Eur J Pharmacol. 1984;103:269–278. doi: 10.1016/0014-2999(84)90487-4. [DOI] [PubMed] [Google Scholar]
- 49.Landa ME, Rubio MC, Jaim-Etcheverry G. The neurotoxic compound N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride (DSP4) depletes endogenous norepinephrine and enhances release of [3H]norepinephrine from rat cortical slices. J Pharmacol Exp Ther. 1984;231:131–136. [PubMed] [Google Scholar]
- 50.Cassano T, Gaetani S, Morgese MG, Macheda T, Laconca L, Dipasquale P, Taltavull J, Shippenberg TS, Cuomo V, Gobbi G. Monoaminergic changes in locus coeruleus and dorsal raphe nucleus following noradrenaline depletion. Neurochem Res. 2009;34:1417–1426. doi: 10.1007/s11064-009-9928-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dowdell KC, Gienapp IE, Stuckman S, Wardrop RM, Whitacre CC. Neuroendocrine modulation of chronic relapsing experimental autoimmune encephalomyelitis: A critical role for the hypothalamic-pituitary-adrenal axis. J Neuroimmunol. 1999;100:243–251. doi: 10.1016/S0165-5728(99)00211-8. [DOI] [PubMed] [Google Scholar]
- 52.Cosentino M, Zaffaroni M, Marino F, Bombelli R, Ferrari M, Rasini E, Lecchini S, Ghezzi A, Frigo G. Catecholamine production and tyrosine hydroxylase expression in peripheral blood mononuclear cells from multiple sclerosis patients: Effect of cell stimulation and possible relevance for activation-induced apoptosis. J Neuroimmunol. 2002;133:233–240. doi: 10.1016/S0165-5728(02)00372-7. [DOI] [PubMed] [Google Scholar]
- 53.Rajda C, Bencsik K, Vécsei LL, Bergquist J. Catecholamine levels in peripheral blood lymphocytes from multiple sclerosis patients. J Neuroimmunol. 2002;124:93–100. doi: 10.1016/S0165-5728(02)00002-4. [DOI] [PubMed] [Google Scholar]
- 54.Helkamaa T, Reenilä I, Tuominen RK, Soinila S, Väänänen A, Tilgmann C, Rauhala P. Increased catechol-O-methyltransferase activity and protein expression in OX-42-positive cells in the substantia nigra after lipopolysaccharide microinfusion. Neurochem Int. 2007;51:412–423. doi: 10.1016/j.neuint.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 55.Samantaray S, Knaryan VH, Butler JT, Ray SK, Banik NL. Spinal cord degeneration in C57BL/6N mice following induction of experimental parkinsonism with MPTP. J Neurochem. 2008;104:1309–1320. doi: 10.1111/j.1471-4159.2007.05091.x. [DOI] [PubMed] [Google Scholar]
- 56.Martel J, Chopin P, Colpaert F, Marien M. Neuroprotective effects of the alpha2-adrenoceptor antagonists, (+)-efaroxan and (+/-)-idazoxan, against quinolinic acid-induced lesions of the rat striatum. Exp Neurol. 1998;154:595–601. doi: 10.1006/exnr.1998.6942. [DOI] [PubMed] [Google Scholar]
- 57.Veyrac A, Didier A, Colpaert F, Jourdan F, Marien M. Activation of noradrenergic transmission by alpha2-adrenoceptor antagonists counteracts deafferentation-induced neuronal death and cell proliferation in the adult mouse olfactory bulb. Exp Neurol. 2005;194:444–456. doi: 10.1016/j.expneurol.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 58.Hashioka S, Klegeris A, Monji A, Kato T, Sawada M, McGeer PL, Kanba S. Antidepressants inhibit interferon-gamma-induced microglial production of IL-6 and nitric oxide. Exp Neurol. 2007;206:33–42. doi: 10.1016/j.expneurol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 59.Russo-Neustadt A, Beard RC, Cotman CW. Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology. 1999;21:679–682. doi: 10.1016/S0893-133X(99)00059-7. [DOI] [PubMed] [Google Scholar]
- 60.Goldstein DS. L-Dihydroxyphenylserine (L-DOPS): A norepinephrine prodrug. Cardiovasc Drug Rev. 2006;24:189–203. doi: 10.1111/j.1527-3466.2006.00189.x. [DOI] [PubMed] [Google Scholar]
- 61.Puri BK, Bydder GM, Chaudhuri KR, Al Saffar BY, Curati WL, White SJ, Mitchell L, Hajnal JV, Horrobin DF. MRI changes in multiple sclerosis following treatment with lofepramine and L-phenylalanine. Neuroreport. 2001;12:1821–1824. doi: 10.1097/00001756-200107030-00012. [DOI] [PubMed] [Google Scholar]
- 62.Loder C, Allawi J, Horrobin DF. Treatment of multiple sclerosis with lofepramine, L-phenylalanine and vitamin B (12): Mechanism of action and clinical importance: Roles of the locus coeruleus and central noradrenergic systems. Med Hypotheses. 2002;59:594–602. doi: 10.1016/S0306-9877(02)00261-X. [DOI] [PubMed] [Google Scholar]
- 63.Stjarne L, Lishajko F, Roth RH. Regulation of noradrenaline biosynthesis in nerve tissue. Nature. 1967;215:770–772. doi: 10.1038/215770a0. [DOI] [PubMed] [Google Scholar]
- 64.Bardou I, Kaercher RM, Brothers HM, Hopp SC, Royer S, Wenk GL. Age and duration of inflammatory environment differentially affect the neuroimmune response and catecholaminergic neurons in the midbrain and brainstem. Neurobiol Aging. 2014;35:1065–1073. doi: 10.1016/j.neurobiolaging.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging. 2007;28:327–335. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 66.Polak PE, Kalinin S, Braun D, Sharp A, Lin SX, Feinstein DL. The vincamine derivative vindeburnol provides benefit in a mouse model of multiple sclerosis: Effects on the Locus coeruleus. J Neurochem. 2012;121:206–216. doi: 10.1111/j.1471-4159.2012.07673.x. [DOI] [PubMed] [Google Scholar]
- 67.Weissmann D, Labatut R, Gillon J, Richard F, Pujol J. Measurement of brain tyrosine hydroxylase concentrations by quantitative autoradiography after transfer of soluble proteins to nitro-cellulose. C R Acad Sci III. 1988;306:457–460. (In French) [PubMed] [Google Scholar]
- 68.He Y, Zhu H, Li W, Chen G, Li Z, Xu X. HPLC determination of catalpol in cerebrospinal fluid of rats. Zhong Guo Zhong Yao Za Zhi. 2009;34:1717–1719. (In Chinese) [PubMed] [Google Scholar]
- 69.Wan D, Zhu HF, Luo Y, Xie P, Xu XY. Study of catalpol promoting axonal growth for cultured cortical neurons from rats. Zhongguo Zhong Yao Za Zhi. 2007;32:1771–1774. (In Chinese) [PubMed] [Google Scholar]
- 70.Tian YY, An LJ, Jiang L, Duan YL, Chen J, Jiang B. Catalpol protects dopaminergic neurons from LPS-induced neurotoxicity in mesencephalic neuron-glia cultures. Life Sci. 2006;80:193–199. doi: 10.1016/j.lfs.2006.09.010. [DOI] [PubMed] [Google Scholar]