

Calcitonin gene-related peptide mediates K+-evoked delayed and prolonged activation of cranial meningeal afferents but does not contribute to their enhanced responsiveness following cortical spreading depression.
Keywords: Headache, Migraine, Meninges, Trigeminal, Sensory afferents, CGRP
Abstract
Introduction:
Cortical spreading depression (CSD) is believed to promote migraine headache by enhancing the activity and mechanosensitivity of trigeminal intracranial meningeal afferents. One putative mechanism underlying this afferent response involves an acute excitation of meningeal afferents by cortical efflux of K+ and the ensuing antidromic release of proinflammatory sensory neuropeptides, such as calcitonin gene-related peptide (CGRP).
Objectives:
We sought to investigate whether (1) a brief meningeal K+ stimulus leads to CGRP-dependent enhancement of meningeal afferent responses and (2) CSD-induced meningeal afferent activation and sensitization involve CGRP receptor signaling.
Methods:
Extracellular single-unit recording were used to record the activity of meningeal afferents in anesthetized male rats. Stimulations included a brief meningeal application of K+ or induction of CSD in the frontal cortex using pinprick. Cortical spreading depression was documented by recording changes in cerebral blood flow using laser Doppler flowmetery. Calcitonin gene-related peptide receptor activity was inhibited with BIBN4096 (333 μM, i.v.).
Results:
Meningeal K+ stimulation acutely activated 86% of the afferents tested and also promoted in ∼65% of the afferents a 3-fold increase in ongoing activity, which was delayed by 23.3 ± 4.1 minutes and lasted for 22.2 ± 5.6 minutes. K+ stimulation did not promote mechanical sensitization. Pretreatment with BIBN4096 suppressed the K+-induced delayed afferent activation, reduced CSD-evoked cortical hyperemia, but had no effect on the enhanced activation or mechanical sensitization of meningeal afferents following CSD.
Conclusion:
While CGRP-mediated activation of meningeal afferents evoked by cortical efflux of K+ could promote headache, acute activation of CGRP receptors may not play a key role in mediating CSD-evoked headache.
1. Introduction
Migraine is the third most prevalent and seventh most disabling disease in the world, affecting about 15% of the adult population worldwide.61,63 A key migraine theory links the genesis of the headache to a cascade of inflammatory-driven nociceptive events, namely the prolonged activation and increased mechanosensitivity of pain sensitive afferents that innervate the intracranial meninges and their related large blood vessels.37,43,48,49
The origin of such meningeal inflammatory response in migraine is not completely understood, although a key hypothesis implicates the action of proinflammatory sensory neuropeptides that are released from peripheral nerve endings of activated primary afferents through an “axon reflex” process.30,47 According to this neurogenic inflammatory hypothesis, the initial stimulation of trigeminal meningeal afferents leads to an antidromic, calcium-mediated vesicular release of sensory neuropeptides, which act upon the meningeal vasculature and immune cells to promote sterile inflammation that subsequently leads to the prolonged increase in meningeal afferent responsiveness.7,9,37,53 While this hypothesis was initially focused on the neuropeptide substance P as the key mediator of meningeal inflammation and migraine pain,53 the failure of substance P receptor antagonists to abort migraine attacks10 has led to diminished support for the role of neurogenic inflammation in migraine headache origin.
Another key neuropeptide that is released from activated trigeminal afferents is calcitonin gene-related peptide (CGRP). Calcitonin gene-related peptide is a potent vasodilator of cerebral42 and dural arteries34,44 and may also contribute to dural plasma extravasation.29 Trigeminal CGRP is considered a major player in migraine pain: a notion that was based primarily on a study which detected elevated CGRP levels in the jugular vein during a migraine attack and its normalization by the antimigraine drug sumatriptan.26 It should be noted that a later study could not replicate these findings67; the validity of its methodology was questioned subsequently, however.16
A link between CGRP, meningeal axonal reflex, and migraine headache has been further argued based on studies showing that agents that block CGRP action and inhibit neurogenic meningeal vasodilatation52,66,71 can abort migraine headache.1,32 The notion that meningeal elaboration of CGRP can directly enhance the responsiveness of meningeal afferents, however, has been questioned by us previously based on data from experiments that tested the effects of exogenously applied CGRP.38 Nonetheless, the notion that antimigraine treatments that block CGRP action, including drugs from the gepant class of CGRP receptor antagonists and monoclonal antibodies, act peripherally15 suggests that neurogenic release of CGRP does, somehow, play a role in mediating migraine pain.
One key unanswered question is what process leads to the initial trigeminal peripheral release of CGRP in migraine. One such potential event might be cortical spreading depression (CSD), a putative trigger of migraine with aura.35 Cortical spreading depression is associated with a robust increase in cerebral blood flow,50 which is mediated in part by cerebrovascular release of CGRP.8,56,69 A more direct link between CSD and the origin of migraine headache has been suggested by our previous studies in a rat model, which documented in a substantial number of meningeal afferents a short-lasting increase in ongoing activity, during the passage of the CSD wave under the afferents' receptive field (RF) that is followed, after some delay, by a prolonged activation and mechanosensitization of the afferents.73,76,77 The mechanism underlying the short-lasting activation of meningeal afferents during the CSD event remains unknown but may involve the nociceptive action of diffusible excitatory molecules, including potassium ions64 that are briefly released in the cortex during the CSD wave.4,21,41,68 Based on the neurogenic inflammatory hypothesis of migraine in the wake of CSD, the initial activation of meningeal afferents by K+ or potentially other nociceptive factors indirectly leads to the prolonged and enhanced afferent responses through a mechanism that involves a trigeminal axon reflex and meningeal release of CGRP or other proinflammatory neuropeptides.53
In this study, we used single-unit recording of meningeal afferents in anesthetized rats to initially address the question, whether a brief excitation of meningeal afferents in response to short-term elaboration of K+, as would be expected during CSD, leads to prolonged change in the afferents' response properties. Having identified such K+-driven afferent response, and given that acute exposure of meningeal afferents to K+ likely promotes meningeal CGRP release,11,12,44 we further examined whether acute CGRP receptor signaling mediates this prolonged afferent response. Finally, given that CSD is associated with a CGRP-related meningeal vascular response, we investigated whether CGRP receptor signaling contributes to the prolonged meningeal afferent response following CSD.
2. Materials and methods
2.1. Animals and anesthesia
The current study employed male Sprague-Dawley rats (250–350 g). Animals were handled in compliance with the experimental protocol approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center. Animals were deeply anesthetized with urethane (1.5 g/kg, ip) and mounted on a stereotaxic frame (Kopf Instruments, Tujunga, CA). Core temperature was kept at 37.5 to 38°C using a homoeothermic control system. Animals breathed room air enriched with 100% O2 spontaneously. Physiological parameters were collected throughout the experiments using PhysioSuite (Kent Scientific,Torrington, CT) and CapStar-100 (CWE, Ardmore, PA). Data used in this report were obtained only from animals exhibiting physiological levels of oxygen saturation (>95%), heart rate (350–450 bpm), and end-tidal CO2 (3.5%–4.5%).
2.2. Surgery and electrophysiological recordings
A saline-cooled dental drill was used to perform craniotomy to expose the left transverse sinus as well as the adjacent cranial dura extending ∼2 mm rostral to the sinus. In animals tested for the effects of CSD, a small burr hole (0.5-mm diameter) was drilled to expose a small area of dura above the frontal cortex to induce CSD.77 The exposed dura was bathed with a modified synthetic interstitial fluid (SIF) containing 135 mm NaCl, 5 mm KCl, 1 mm MgCl2, 5 mm CaCl2, 10 mm glucose, and 10 mm HEPES, pH 7.2. The femoral vein was cannulated to allow for intravenous administration. Single-unit activity of meningeal afferents (1 afferent/rat) was recorded in the ipsilateral (left) trigeminal ganglion using a contralateral approach, as described recently75,76 (and see also Fig. 1). Briefly, a small craniotomy (2 × 2 mm) was made in the calvaria over the right hemisphere, 2 mm caudal, and 2 mm lateral to Bregma. A platinum-coated tungsten microelectrode (impedance 100 kΩ, FHC, Bowdoin, ME) was advanced through the right hemisphere into the left trigeminal ganglion using an angled (22.5°) trajectory. In earlier studies, we verified that inserting the recording electrode using this approach did not produce CSD in the left cortical hemisphere. Meningeal afferents were identified by their constant latency response to single shock stimulation applied to the dura above the ipsilateral transverse sinus (0.5-ms pulse, 5 mA, 0.5 Hz). The response latency was used to calculate conduction velocity (CV), based on a conduction distance to the trigeminal ganglion of 12.5 mm.64 Neurons were classified as either A-delta (1.5 < CV ≤ 5 m/s) or C-afferents (CV ≤ 1.5 m/s). Neural activity was digitized and a real-time waveform discriminator (Spike 2 software, CED) was used to create and store a template for the action potential evoked by electrical stimulation, which was used later to acquire and analyze the ongoing activity of the neurons and the activity evoked by mechanical stimulation and CSD.
Figure 1.
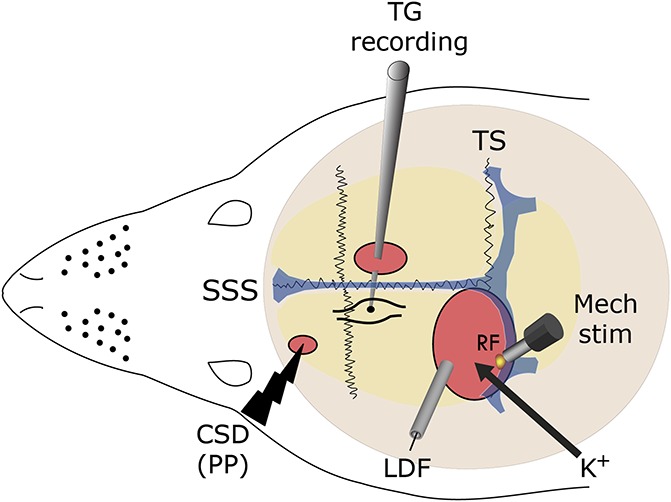
Experimental setup: 3 skull openings (red ovals) were made. A small burr hole was made over the left frontal cortex to elicit a single cortical spreading depression (CSD) event using a pinprick (PP) stimulation, by inserting a sharp glass microelectrode 2 mm into the cortex. Meningeal afferent activity was recorded in the left trigeminal ganglion (TG) using a tungsten microelectrode inserted through a craniotomy made over the contralateral hemisphere. An ipsilateral craniotomy was made to expose a part of the left transverse sinus (TS) and its vicinity to search for meningeal afferents with mechanical receptive field (RF). Quantitative mechanical stimuli were delivered to the afferents' RF using a feedback-controlled mechanical stimulator. Laser Doppler flowmetry (LDF) probe was placed over the cortex near the stimulated afferents' RF to validate the induction of the cortical spreading depression by testing related changes in cerebral blood flow. SSS, superior sagittal sinus. K+ stimulation was made by applying KCl to the exposed dural RF of the recorded afferent.
2.3. Mechanical stimulation and detection of sensitization
Mechanical RFs of meningeal afferents were first identified by probing the exposed dura with a 6.9 g von Frey filament (Stoelting). The site of the lowest mechanical threshold was further determined using monofilaments that exert lower forces (>0.03 g). For quantitative determination of mechanical responsiveness, we applied mechanical stimuli to the site with the lowest threshold, using a servo force-controlled mechanical stimulator (Series 300B Dual Mode Servo System; Aurora Scientific, Aurora, ON). The stimulus was delivered using a flat-ended cylindrical plastic probe that was attached to the tip of the stimulator arm. One of 3 probe diameters (0.5, 0.8 or 1.1 mm) was selected for each neuron, depending on the sensitivity of the neuron. Stimulus trials for testing mechanically sensitive were made using a custom-written script for Spike 2 and consisted of “ramp and hold” stimuli (rise time 100 ms, stimulus width 2 seconds, inter-stimulus interval 120 seconds). Each trial included a threshold stimulus (TH, which normally evoked 1-3-Hz responses) followed by a suprathreshold stimulus (STH, usually X2 of the threshold pressure; 8-10 Hz responses). To minimize response desensitization, stimulus trials were delivered every 15 minutes throughout the experiment.72,77 Ongoing afferent discharge rate was recorded continuously between the stimulation trials. Responses to mechanical stimulation were determined during at least 4 consecutive trials before the elicitation of CSD. Only afferents that exhibited consistent responses (variation of <0.5 Hz for TH responses and <1.5 Hz for STH responses) during baseline recordings were tested further.77
2.4. Induction and monitoring of cortical spreading depression
A single CSD episode was induced in the frontal cortex, using a pinprick stimulus with a fine glass micropipette (diameter 10 μm) at ∼2 mm depth for 2 seconds. Cortical spreading depression was induced in the frontal cortex to avoid potential damage to the meningeal tissue near the tested RF of the studied afferents, which could have led to their activation and/or sensitization.76 The occurrence of a CSD episode was determined noninvasively by recording changes in cerebral blood flow (CBF) using a laser Doppler flowmeter (LDF, Laserflo; Vasamedics) with the needle probe (0.8 mm diameter) positioned ∼1 mm above the exposed dura, 1 to 2 mm rostral to the RF of the recorded afferents (Fig. 1) in an area devoid of large blood vessels (>100 μm). Such an LDF system (using a near IR 785 nm probe and fiber separation of 0.25 mm) is expected to record CBF changes at a cortical depth of ∼1 mm.22 Laser Doppler flowmeter recordings were obtained with ambient lights turned off. Cerebral blood flow data were digitized and continuously sampled via an A/D interface (power 1401 and spike 2 software, CED, Cambridge, United Kingdom). Induction of CSD was considered successful when the typical hemodynamic signature characterized by a large transient (∼1–2 minutes) cerebral hyperemia followed by prolonged (>1 hour) post-CSD oligemia was observed.25,54,76 To verify that the K+ stimulation (see below) did not lead to CSD, in a few animals, we examined changes in CBF by positioning the LDF probe within a small craniotomy made ∼4 mm rostral to stimulation site.
2.5. Drugs
BIBN4096BS (Cat. No. 4561; Tocris, Minneapolis, MN) was dissolved in DMSO to make a 50-mM stock solution. For intravenous injection, a 400-μM solution was made by diluting with 0.9% saline. Potassium chloride (Cat. No. P9333; Sigma Aldrich) stock solution (1M) was made in distilled water and further diluted to 50 mM using SIF. For local meningeal K+ stimulation, we used a piece of cotton wool to create a circular pool to cover the exposed dural RF of the recorded afferent. The K+ stimulation protocol consisted of an initial application of 20 μL of a 50-mM KCl solution for 10 seconds, followed by 5 sequential applications of 20 μL of SIF (which contains 5 mM K+) every 10 seconds and a final wash using 500 μl of SIF.
2.6. Data analysis
Offline analyses for afferent responses were conducted using template matching in Spike 2. Statistical analyses were conducted using Prism 7 software. Average data are presented in the text as the mean ± SEM. Average data in figures are presented as the mean ± 95% confidence interval (CI). Criteria used to consider meningeal afferent activation and sensitization responses were based on our previous studies.76,77 In brief, an increase in the afferents ongoing activity was considered if the firing rate increased above the upper end point of the 95% CI calculated for the baseline mean. Acute afferent activation was considered when the ongoing activity level increased during the K+ stimulus or the arrival of the CSD wave (ie, the hyperemia stage) under the RF of the studied afferent and subsided within 2 minutes. Prolonged activation was considered when ongoing activity rate was increased for >10 minutes. Mechanical sensitization was considered only if the afferent responses changed according to the following criteria: TH and/or STH responses increased to a level greater than the upper end point of the 95% CI calculated for the baseline mean; increased responsiveness began during the first 60 minutes post-CSD; and sensitization lasted for at least 30 minutes. For analyses of CSD-related changes in the afferent responses, data from A-delta and C afferents were combined given their comparable values. Afferent responses to CSD in the presence of BIBN4096BS were compared to a pool of data from our earlier vehicle studies76,77 that was combined with additional data obtained from new vehicle experiments (total n = 36) conducted to increase sample size and add power. Group differences were analyzed using 2-tailed, Fisher exact test. Statistical differences were analyzed using 2-tailed unpaired t test or Mann–Whitney U test for data sets that failed normality test. Results were considered to be significant at P < 0.05.
3. Results
3.1. Brief meningeal stimulation with a cortical spreading depression–related K+ stimulus promotes prolonged activation of meningeal afferents
Earlier studies suggested that in response to CSD, the level of extracellular parenchymal K+ rises quickly within the superficial cortex to ∼50 mM followed by a rapid decrease within ∼1 minute to baseline levels.21,33,41,46,68 We therefore first asked whether a brief, local meningeal stimulation with a similar pattern of changes in K+ concentration (Fig. 2B), which did not lead to a CSD event (Fig. 3A), could promote long-term changes in the activity and/or mechanosensitivity of meningeal afferents, as observed in some afferents following CSD.73,76 The effect of K+ stimulation was tested on 14 meningeal afferents (5 A-delta, 9 C). Local administration of K+ gave rise, as expected,64 to a robust yet short-term (<70 seconds) increase in the ongoing activity rate (8.7 ± 2.4 fold) in 12 afferents (4/5 A-delta, 8/9 C). Following a wash with SIF (which contains 5 mM K+), 9/14 of the afferents (3/5 A-delta 6/9 C) also developed a delayed and prolonged increase in their ongoing activity rate. The responses of the A delta and C afferents were similar and therefore combined for further analyses. As Figure 3 depicts, the prolonged afferent activation following the K+ stimulus developed with an average delay of 23.3 ± 4.1 minutes (range 5–40 minutes) and lasted for 22.2 ± 5.6 minutes (range 10–55 minutes). Overall, the K+ stimulation evoked a 3.1 ± 0.4-fold (range 1.7- to 5.0-fold) increase in the afferent firing rate. When compared to the prolonged meningeal afferent activation evoked by CSD in animals treated with vehicle (9/19 A-delta, 9/17 C), K+ stimulation evoked afferent activation developed with a longer delay (compare to onset of 8.8 ± 2.6 minutes; range 0–35 minutes in the CSD group; P < 0.05, unpaired t test). The duration of the prolonged afferent activation following the K+ stimulation was also shorter (compared to duration of 32.8 ± 3.8 minutes; range 10–55 minutes in the CSD group; P < 0.05, t test). The magnitude of the prolonged response following K+ stimulation, however, was not different between the 2 groups (compare to 2.9 ± 0.5 fold; range 1.2- to 10.4-fold in the CSD group; P = 0.21, Mann–Whitney test). As Figure 4 illustrates, following K+ stimulation meningeal afferents did not become sensitized to mechanical stimulation. We detected a brief (30 minutes) increased in responsiveness in only 2/24 C afferent, which was not different from that observed in time–control experiments (0/6 A-delta and 1/6 C showed increased responsiveness for 30 minutes).
Figure 2.

Examples showing the development of delayed and prolonged activation of meningeal afferents following acute local stimulation with K+. (A) Local K+ stimulation-induced immediate and prolonged activation of a C meningeal afferent. Traces represent peristimulus time histograms (PSTH, 10-minutes epochs, 10-seconds bin-size). An overdrawn spike waveform of the afferent used for data analysis is shown on the left. Average ongoing activity rate (spikes/s) is denoted in parentheses. The last 5-minute recording period during the mechano-stimulation trials, conducted at the end of each 15-minute recording epoch, were omitted for clarity. (B) The protocol employed for meningeal application of K+. Note the acute increase in afferent activity during the stimulation. (C) Time course data depicting the ongoing activity level of the same afferent depicted in (A) during baseline and every 5 minutes after K+ stimulation. Red areas represent the acute and delayed phases of neuronal activation. (D) Acute and prolonged activation of an A-delta meningeal afferent following meningeal K+ stimulation. Traces represent PSTH as in (A). Time course data of the afferent response is depicted in (E).
Figure 3.
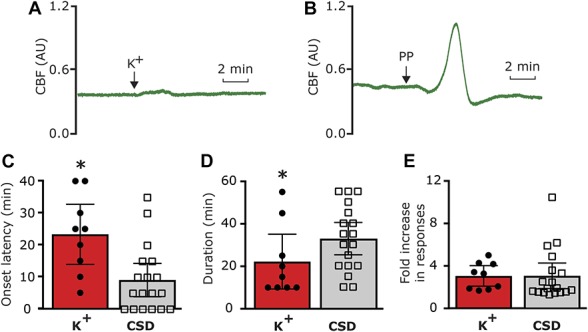
Characteristics of the delayed and prolonged meningeal afferent responses induced by local K+ stimulation compared to that induced by cortical spreading depression (CSD) in vehicle-treated animals. (A) Raw laser Doppler flowmeter (LDF) recording trace showing the lack of overt changes in cerebrovascular perfusion following meningeal K+ stimulation, indicating lack of CSD induction. AU, arbitrary units. (B) LDF recording trace demonstrating cortical pinprick (PP)—induced cortical hyperemia following by oligemia, indicative of a CSD event. (C) Onset latency of the prolonged afferent activation. (D) Duration of the afferent activation. (E) Magnitude of the increase in ongoing activity. *P < 0.05; unpaired t test.
Figure 4.
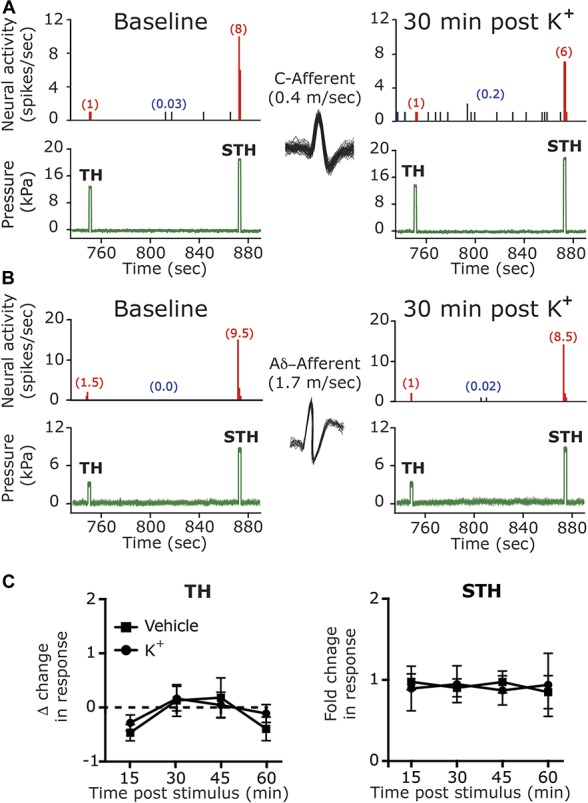
Meningeal K+ stimulus does not affect the mechanical responsiveness of meningeal afferents. Examples of experimental trials depicting the responses of 1 C afferent (A) and 1 A-delta afferent (B) (with matching overdrawn spike waveforms) to threshold (TH) and suprathreshold (STH) mechanical stimuli (green traces) applied to the their dural receptive field during baseline recording and then at 30 minutes following local K+ stimulation. Red numbers in parentheses denote mechanically evoked afferent responses (spikes/s). Note the lack of changes in TH or STH responses. Blue numbers in parentheses denote ongoing activity rate, note the K+ evoked activation. (C) Summary of time course data collected from meningeal afferents in time–control studies (vehicle) and following local K+ stimulation showing lack of changes in TH and STH responses.
3.2. K+-induced prolonged activation of meningeal afferents is Calcitonin gene-related peptide dependent
Previous studies reported that a brief meningeal stimulation with high concentration of K+ similar to the current study evokes meningeal CGRP release.12,44,59 We therefore asked whether the K+-driven prolonged activation of meningeal afferents is CGRP dependent and thus potentially related to meningeal afferent axonal reflex. To examine the role of CGRP signaling, we administered the CGRP receptor antagonist BIBN4096 (333 μg/kg, i.v) 1 hour before testing the afferent response to meningeal K+ stimulation. The dose of BIBN4096 was adapted from previous studies showing efficacy in inhibiting neurogenically-evoked meningeal vasodilation27,52,66 and meningeal afferent volleys in the spinal trigeminal nucleus.23 We first tested the effect of BIBN4096 on the baseline response properties of meningeal afferents in 36 afferents (19 A-delta, 18 C). When compared to data obtained from animals administered only with saline (50 afferents; 25 A-delta, 25 C), BIBN4096 treatment neither affected the afferents' baseline ongoing activity rate (Table 1) nor influenced their baseline mechanosensitivity (Table 2). We next tested the effect of BIBN4096 treatment on the K+-evoked afferent responses in 6 A-delta and 7 C afferents. BIBN4096 did not inhibit the initial short excitatory response to K+ (10/13; 4/6 A-delta; 6/7 C afferents developed a 7.6 ± 3.7-fold increase in activity, P = 0.64, t test). However, as Figure 5 depicts, in the presence of BIBN4096, only 2/13 (2 C) of the afferents developed a prolonged increase in ongoing activity (compared with 9/14 units affected in vehicle control experiments, P < 0.05; 2-tailed, Fisher exact test). The onset latencies for the prolonged activation in these 2 afferents were 35 and 10 minutes with matching durations of 20 and 10 minutes. The magnitude of change in afferent activity in animals administered with BIBN4096 prior to K+ stimulation was also lower than that observed in units treated with vehicle prior to K+ stimulation (P < 0.05, t test).
Table 1.
Systemic BIBN4096 treatment does not affect baseline ongoing activity of meningeal afferents.
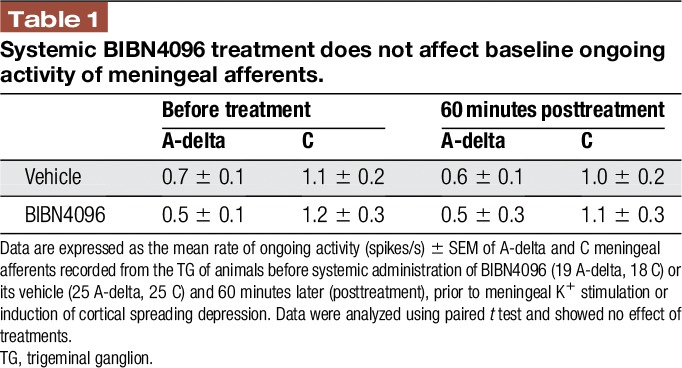
Table 2.
Systemic BIBN4096 treatment does not affect baseline mechanosensitivity of meningeal afferents.
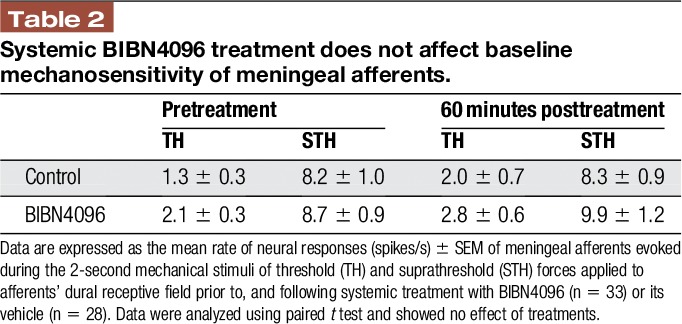
Figure 5.
Inhibition of K+-induced prolonged meningeal afferent activation by BIBN4096. In the presence of BIBN4096, only 2 afferents became activated (red symbols). (A) Onset latency. (B) Duration of the afferent activation. (C) Magnitude of change in ongoing activity rate. Open symbols denote changes in the activity of afferents not activated following K+ stimulation in BIBN4096 pretreated animals.
3.3. Enhanced meningeal afferent activation following cortical spreading depression is not mediated by acute calcitonin gene-related peptide receptor signaling
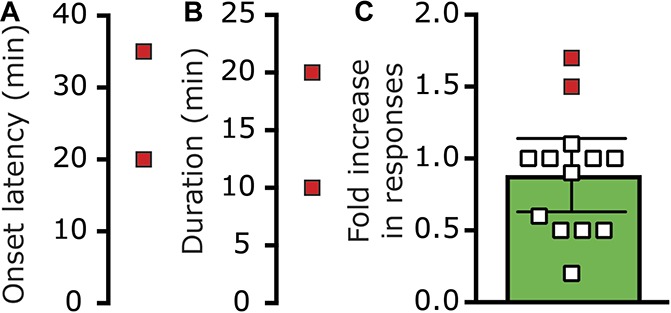
Given the ability of BIB4096 pretreatment to inhibit the K+-driven prolonged afferent activation, we next tested whether CGRP receptor activation also contributes to the prolonged meningeal afferents' responses in the wake of CSD. We initially verified that in the setting of CSD, BIBN4096 treatment inhibits a previously recognized CGRP-mediated meningeal response, namely the brief pial hyperperfusion8,69 (see also Fig. 3B). Systemic administration of BIBN4096 (n = 15) did not affect baseline CBF (Fig. 6A) but inhibited the cortical hyperemic response during CSD by ∼25% (Fig. 6B). Induction of CSD in BIBN4096 pretreated animals gave rise to a hyperemic response that was significantly lower than that observed in vehicle-treated animals (1.56 ± 0.1 fold vs 2.1 ± 0.1 fold; P < 0.01, Mann–Whitney test), confirming the efficacy of BIBN4096 as a CGRP receptor antagonist in this model. The effect of BIBN409 administration on CSD-evoked meningeal afferent activation was tested in 24 afferents (13 A-delta and 11 C). In the presence of BIBN4096, CSD resulted in the development of prolonged activation in 9/24 (37.5%) of the afferents tested (4/13 A-delta; 5/11 C). This response rate was not different from that observed in afferents recorded in animals treated only with vehicle (9/19 A-delta and 9/17 C; P = 0.80 overall; P = 0.47 for the A-delta afferent population; P = 1.00 for the C-afferent population, Fisher exact test). BIBN4096 treatment also did not influence the characteristics of the CSD-evoked prolonged afferent activation. The average onset latency for CSD-evoked afferent activation in the BIBN-treated group (3.9 ± 1.6 minutes; range 0–15 minutes; Fig. 7A) was not different from that observed in the vehicle-treated group (P = 0.29, t test, Fig. 3C). The duration of the post-CSD afferent discharge in the BIBN4096 treatment group (average 30.0 ± 5.3 minutes; range 10–55 minutes; Fig. 7B) was also not different from that observed in the vehicle control group (P = 0.15; t test, Fig. 3D). Finally, in the presence BIB4096, the magnitude of the increase in afferent discharge rate following CSD (average 2.9 ± 0.7 fold; range 1.5- to 8.3-fold; Fig. 7C) was also not different when compared to that observed in the vehicle treatment group (P = 0.41; t test, Fig. 3E).
Figure 6.
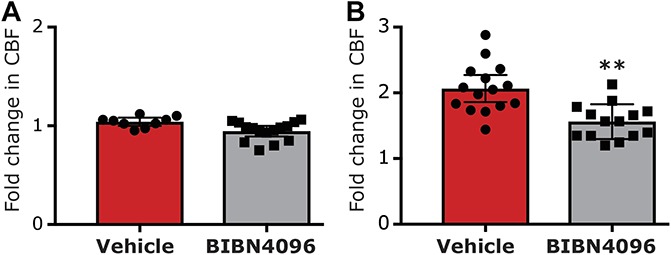
The effect of BIBN4096 on cortical spreading depression (CSD)-evoked cerebrovascular hyperemia. (A) Changes in baseline cerebrovascular perfusion. (B) Cerebrovascular dilation during CSD.
Figure 7.
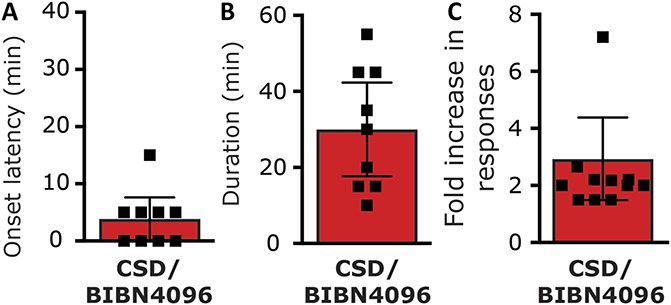
The effect of BIBN4096 on CSD-evoked prolonged activation of meningeal afferents. Onset latency (A), duration (B), and magnitude (C) of the prolonged increase in ongoing activity of meningeal afferents following cortical spreading depression (CSD) in animals pretreated with BIBN4096.
3.4. Acute calcitonin gene-related peptide receptor signaling does not mediate cortical spreading depression–evoked mechanical sensitization of meningeal afferents
To examine the effect of BIB4096 pretreatment on the CSD-evoked meningeal afferent mechanosensitization, we analyzed data collected from 21 afferents (11 A-delta and 10 C); all of which displayed consistent responses at baseline. In BIBN4096-treated animals, CSD-evoked mechanical sensitization in 11/21 afferents (5/11 A-delta and 6/10 C, see Fig. 8A, B). This rate of response was not different from that observed in animals treated with vehicle (7/10 A-delta and 9/13 C; P = 0.35 overall; P = 0.39 for the A-delta afferent population; P = 0.69 for the C afferent population, Fisher exact test). The magnitudes of the TH and STH sensitization responses in the BIBN-treated group were not different from those observed in the control group (TH, Δ1.6 ± 0.5 spikes/s; range 0.3–4.8 spikes/s vs Δ1.8 ± 0.4 spikes/s; range 0.3–4.8 spikes/s, P = 0.74 Mann Whitney test; STH, 1.6 ± 0.1 fold; range 1.2- to 2.3-fold vs 1.6 ± 0.1 fold; range 1.2- to 2.0-fold; P = 0.92; Mann Whitney test). BIBN4096 also did not affect the onset latency for the CSD-evoked sensitization. In the BIB4096-treated group, TH sensitization latency averaged 35.6 ± 6.3 minutes (range 15–60 minutes) and was not statistically different from that observed in the control group (21.0 ± 3.6 minutes, range 15–45 minutes; P = 0.09; Mann–Whitney test; Fig. 8C). The onset latency for the STH sensitization in the BIBN4096 group was 31.5 ± 5.7 minutes (range 15–60), which was also not statistically different from that observed in the control group (24.2 ± 3.2 minutes; range 15–60 minutes; P = 0.25; Mann–Whitney test; Fig. 8D). The duration of the TH sensitization response in the BIBN4096-treated group was 73.8 ± 9.9 minutes (range 30–120 minutes) and was also similar to that observed in the control group (71.3 ± 10.5 minutes; range 30–105 minutes, P = 0.89; Mann–Whitney test; Fig. 8E). Finally, the duration of the STH sensitization response in the BIBN4096 group was 64.5 ± 9.8 minutes (range 30–120 minutes) and was not statistically different from that observed in the control group (73.9 ± 8.7 minutes; range 30–120 minutes; P = 0.54; Mann–Whitney test; Fig. 8F).
Figure 8.
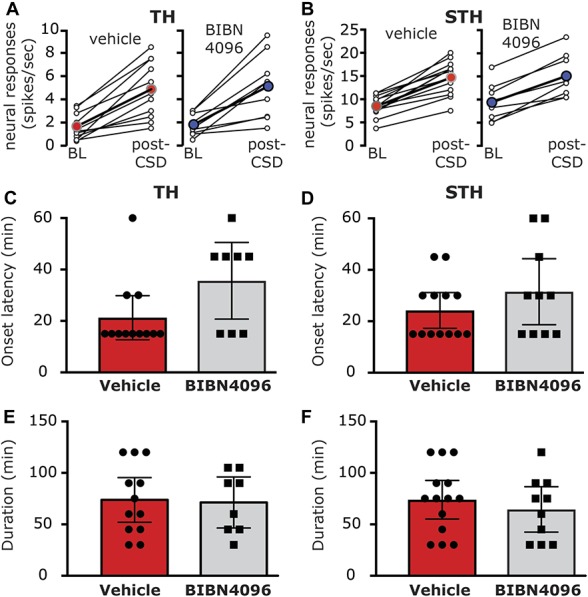
BIBN4096 does not inhibit cortical spreading depression (CSD)-induced mechanical sensitization of meningeal afferents. TH (A) and STH (B) responses in neurons that exhibited mechanical sensitization in response to CSD following treatment with vehicle or BIBN4096. Open circles depict the responses at baseline, before CSD, and during the time of peak response after CSD of individual afferents. Colored circles denote the mean response for the population. Note the similar magnitude of changes in the vehicle and BIBN4096-treated groups. BIBN4096 also had no effect on the onset latency of the prolonged TH (C) and STH (D) sensitization responses or the durations of the TH (E) and STH (F) sensitization responses following CSD.
4. Discussion
The main findings of the study suggest that (1) acute stimulation of meningeal afferents with high K+ concentration promotes a delayed and prolonged increase in their ongoing activity but does not increase their mechanosensitivity, (2) systemic pretreatment with BIBN4096, a CGRP receptor antagonist, blocks the K+-related prolonged activation of meningeal afferents, and (3) a similar BIBN4096 treatment does not inhibit the prolonged activation or mechanical sensitization of meningeal afferents in response to CSD.
Whether a trigeminovascular reflex, involving activity-dependent release of CGRP17,42 from meningeal afferent peripheral nerve endings, contributes to migraine pain remains unclear.58 Previous studies which examined the effect of acute stimulation of primary afferent neurons that innervate other tissues on the sensitivity of primary afferent nociceptive neurons have yielded conflicting data. For example, studies in monkeys45 and rats55 have shown that stimulation of cutaneous nociceptive afferents did not subsequently alter their ongoing activity, mechanosensitivity, or heat sensitivity. A study in rabbits, however, reported the development of heat sensitization of nociceptive afferents following antidromic afferent stimulation.24 Finally, a rat study which used local capsaicin stimulation to evoke acute excitation of cutaneous afferents documented a delayed and prolonged increase in the afferents' ongoing activity and mechanosensitivity that were suggested to involve CGRP signaling.40 Our present findings support a mechanism by which a strong, yet relatively brief (∼60 seconds), excitation of meningeal afferents at the level of their meningeal RF can give rise to a prolonged increase in their ongoing activity level.
The K+ stimulus paradigm used in the current study likely promoted meningeal CGRP release.12,44,59 Our finding that pretreatment with BIBN4096 abrogated the K+-induced prolonged activation of meningeal afferents suggests that meningeal release of CGRP and its action on nonneuronal meningeal receptors36 are likely to mediate the prolonged meningeal afferent response. However, given that BIBN4096 was administered systemically (to allow action also on meningeal tissue that remained unexposed in our preparation), we cannot exclude the possibility that inhibition of CGRP receptors localized to the cell body of meningeal afferents in the trigeminal ganglion, which is located outside the blood brain barrier,14,19,36 may have also contributed. Nonetheless, while dural CGRP release is an established phenomenon,12,44,59 evidence supporting CGRP release in the trigeminal ganglion (TG), as well as its action receptors localized to the TG cell bodies in vivo is currently lacking.
In addition to the evoked meningeal CGRP release, exogenous stimulation with K+ and perhaps CSD-evoked K+ release could have also stimulated adrenergic postganglionic autonomic fibers that innervate the meningeal vasculature,57 leading to local release and action of norepinephrine.11 Such local sympathetic response could have influenced the excitability of meningeal afferents indirectly by promoting the release of prostaglandins13 or other sensitizing factors from non-neuronal meningeal constituents, such as fibroblasts.70 However, the earlier finding that norepinephrine signaling inhibits CGRP release from trigeminal afferents3,28 suggest that activation of meningeal postganglionic sympathetic neurons does not play a role in mediating the prolonged afferent response following K+ stimulation.
Our previous finding that local or systemic application of vasodilating doses of CGRP does not activate or sensitize meningeal afferents38 seems to contradict the current data. The possibility that the levels of CGRP elaborated in response to the K+ stimulation in the present study exceeded the concentrations of CGRP used in our previous work may be entertained. However, previous data suggest that stimulation of meningeal afferents, either electrically or chemically with a mixture of inflammatory mediators or with 50 mM K+ leads to increased CGRP levels at the picomolar range12,20,59—much lower than the micromolar range that failed to influence meningeal afferent responsiveness in our previous study. Differences in the durations of CGRP action between the studies is also an unlikely explanation. In our previous study local CGRP action (measured using changes in local blood flow) lasted for at least 10 minutes when CGRP was applied locally and for ∼15 minutes following systemic application. Thus, the duration of CGRP action in our previous work was likely to be longer than that induced by the release of CGRP in response to the ∼1 minutes excitation of the afferents by the K+ stimulus in the present study. As a potential explanation for the seemingly discrepancy between the 2 studies, we propose that the process underlying the K+-evoked prolonged afferent activation, which requires CGRP, is multifactorial. The release of CGRP from activated meningeal afferents is likely to be accompanied by the release of other neuropeptides, and potentially by other factors such as glutamate. These mediators, in turn, could act upon meningeal immune, vascular, and Schwann cells, which produce the final algesic mediators that enhance the activity of meningeal afferents. We proposed that CGRP action plays a key part in this cascade of events, through a synergistic effect,5,6 by reinforcing or enhancing the release of other algesic mediators from meningeal nonneuronal cells which express CGRP receptors.36
In rodent models, CSD is associated with a brief cortical efflux of K+ that can reach ∼50 mM.46,51,68 Elevated K+ in the superficial cortical interstitial fluid could diffuse towards the meninges and is a likely major contributor to the acute activation of meningeal afferents observed in this model.53,64 Our current data suggest that the prolonged meningeal afferent response that emerges following a brief meningeal K+ stimulation occurs with a longer delay and has a shorter duration than that observed after a single CSD episode. These differences, as well as the lack of mechanical sensitization after K+ stimulation, suggest that local meningeal K+ action, or acute activation of meningeal afferents per se, cannot fully account for the prolonged enhancements in meningeal afferent responses that occur following CSD. The development of mechanical sensitization following CSD77 but not in response to K+ stimulation also points differences between the mechanisms that mediate the prolonged activation and mechanical sensitization of meningeal afferents. This notion is in agreement with our recent data that showed the lack of correlation between CSD-induced afferent activity and mechanosensitization77 as well as with previous findings from our laboratory, demonstrating the ability of nitric oxide and TNF-α signaling to elicit mechanical sensitization but no activation of meningeal afferents.39,72,74 It is worth noting nevertheless that CSD is associated with a wave of cortical K+ release that spreads throughout the hemicortex, a response that could lead to the acute activation a much larger population of meningeal afferents than following the local K+ stimulation used in the current study. Whether a spatial summation of such acute afferent responses plays a role in mediating the prolonged activation and sensitization of meningeal afferents following CSD remains to be determined.
Systemic administration of BIBN4096 is likely to target CGRP receptors primarily outside the central nervous system.14,18 Our data thus supports the view that acute activation of meningeal CGRP receptor signaling is unlikely to constitute a major contributing factor underlying the prolonged activation and sensitization of meningeal afferents following CSD. Our finding that BIBN4096 partially inhibited the CSD-evoked increase in CBF, a previously recognized CGRP-mediated event8,69 together with its ability to block neurogenic meningeal vasodilatation per se27,52 also support the claim that acute CGRP-related responses (including its pial vasodilatory effect) are also not major contributing factors that mediated the meningeal nociceptive effect of CSD. The discrepancy between the involvement of CGRP receptor signaling in mediating the K+-driven prolonged afferents activation data but not in response to CSD further calls into question the notion that the initial short-lasting cortical efflux of excitatory molecules during the CSD wave, such as K+, directly contributes to the prolonged meningeal nociceptive response via acute activation of peripheral CGRP receptors. While the current study suggests the acute CGRP receptor signaling may not mediate the meningeal nociceptive response to CSD, a recent finding suggested that several hours of systemic CGRP sequestering, using anti-CGRP monoclonal antibody (mAb), can inhibit the CSD-induced activation and sensitization of a subpopulation of trigeminal dorsal horn neurons that receive input from meningeal afferents. Such an effect, however, may be due to an action at the level of the central nerve ending in nucleus caudalis.23,62 Nonetheless, prolonged inhibition of basal CGRP action, unlike the acute effect of BIBN4096, could have also altered meningeal cellular processing, leading to decreased responsiveness of meningeal afferents to CSD in that model.
While CSD can be efficiently induced by a cortical pin-prick stimulus, another common induction method, which has been used in numerous in vivo studies of headache mechanisms, involves epidural application of K+ at high concentration (usually 1M). Our finding of acute and prolonged activation of meningeal afferents following epidural K+ stimulation at a much lower dose, that does not promote CSD, thus have important implications for the interpretation of previous studies that used K+ stimulation to study the effect of CSD on trigeminal/meningeal pain (eg, Refs. 2,60,65,73). Future studies that use the CSD paradigm to study related trigeminal responses and their underlying mechanisms thus should be conscientious about the CSD induction method used. The use of less invasive methods that induce CSD remotely, such as optogenetics,31 which may not influence meningeal afferents responsiveness per se may provide a better choice to study mechanisms of CSD-related meningeal nociception and its involvement in migraine headache.
Disclosures
The authors have no conflicts of interest to declare.
Supported by grants from the NIH/NINDS (NS086830, NS078263 to D.L.).
Parts of the manuscript have been presented previously only in an abstract form.
Acknowledgments
Author Contributions: J. Zhao: performed research and analyzed data. D. Levy designed research, analyzed data and wrote the manuscript.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Bigal ME, Ferrari M, Silberstein SD, Lipton RB, Goadsby PJ. Migraine in the triptan era: lessons from epidemiology, pathophysiology, and clinical science. Headache 2009;49(suppl 1):S21–33. [DOI] [PubMed] [Google Scholar]
- [2].Bogdanov VB, Bogdanova OV, Koulchitsky SV, Chauvel V, Multon S, Makarchuk MY, Brennan KC, Renshaw PF, Schoenen J. Behavior in the open field predicts the number of KCl-induced cortical spreading depressions in rats. Behav Brain Res 2013;236:90–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bowles WR, Flores CM, Jackson DL, Hargreaves KM. Beta 2-Adrenoceptor regulation of CGRP release from capsaicin-sensitive neurons. J Dent Res 2003;82:308–11. [DOI] [PubMed] [Google Scholar]
- [4].Brinley FJ, Jr, Kandel ER, Marshall WH. Potassium outflux from rabbit cortex during spreading depression. J Neurophysiol 1960;23:246–56. [DOI] [PubMed] [Google Scholar]
- [5].Buckley TL, Brain SD, Collins PD, Williams TJ. Inflammatory edema induced by interactions between IL-1 and the neuropeptide calcitonin gene-related peptide. J Immunol 1991;146:3424–30. [PubMed] [Google Scholar]
- [6].Buckley TL, Brain SD, Rampart M, Williams TJ. Time-dependent synergistic interactions between the vasodilator neuropeptide, calcitonin gene-related peptide (CGRP) and mediators of inflammation. Br J Pharmacol 1991;103:1515–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain 2001;89:107–10. [DOI] [PubMed] [Google Scholar]
- [8].Colonna DM, Meng W, Deal DD, Busija DW. Calcitonin gene-related peptide promotes cerebrovascular dilation during cortical spreading depression in rabbits. Am J Physiol 1994;266:H1095–1102. [DOI] [PubMed] [Google Scholar]
- [9].Dalkara T, Zervas NT, Moskowitz MA. From spreading depression to the trigeminovascular system. Neurol Sci 2006;27(suppl 2):S86–90. [DOI] [PubMed] [Google Scholar]
- [10].Diener HC; RPR100893 Study Group. RPR100893, a substance-P antagonist, is not effective in the treatment of migraine attacks. Cephalalgia 2003;23:183–5. [DOI] [PubMed] [Google Scholar]
- [11].Dux M, Will C, Eberhardt M, Fischer MJ, Messlinger K. Stimulation of rat cranial dura mater with potassium chloride causes CGRP release into the cerebrospinal fluid and increases medullary blood flow. Neuropeptides 2017;64:61–8. [DOI] [PubMed] [Google Scholar]
- [12].Ebersberger A, Averbeck B, Messlinger K, Reeh PW. Release of substance P, calcitonin gene-related peptide and prostaglandin E2 from rat dura mater encephali following electrical and chemical stimulation in vitro. Neuroscience 1999;89:901–7. [DOI] [PubMed] [Google Scholar]
- [13].Ebersberger A, Takac H, Richter F, Schaible HG. Effect of sympathetic and parasympathetic mediators on the release of calcitonin gene-related peptide and prostaglandin E from rat dura mater, in vitro. Cephalalgia 2006;26:282–9. [DOI] [PubMed] [Google Scholar]
- [14].Edvinsson L. CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br J Clin Pharmacol 2015;80:193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Edvinsson L. The trigeminovascular pathway: role of CGRP and CGRP receptors in migraine. Headache 2017;57(suppl 2):47–55. [DOI] [PubMed] [Google Scholar]
- [16].Edvinsson L, Ekman R, Goadsby PJ. Measurement of vasoactive neuropeptides in biological materials: problems and pitfalls from 30 years of experience and novel future approaches. Cephalalgia 2010;30:761–6. [DOI] [PubMed] [Google Scholar]
- [17].Edvinsson L, Jansen Olesen I, Kingman TA, McCulloch J, Uddman R. Modification of vasoconstrictor responses in cerebral blood vessels by lesioning of the trigeminal nerve: possible involvement of CGRP. Cephalalgia 1995;15:373–83. [DOI] [PubMed] [Google Scholar]
- [18].Eftekhari S, Gaspar RC, Roberts R, Chen TB, Zeng Z, Villarreal S, Edvinsson L, Salvatore CA. Localization of CGRP receptor components and receptor binding sites in rhesus monkey brainstem: a detailed study using in situ hybridization, immunofluorescence, and autoradiography. J Comp Neurol 2016;524:90–118. [DOI] [PubMed] [Google Scholar]
- [19].Eftekhari S, Warfvinge K, Blixt FW, Edvinsson L. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain 2013;14:1289–303. [DOI] [PubMed] [Google Scholar]
- [20].Eltorp CT, Jansen-Olesen I, Hansen AJ. Release of calcitonin gene-related peptide (CGRP) from Guinea pig dura mater in vitro is inhibited by sumatriptan but unaffected by nitric oxide. Cephalalgia 2000;20:838–44. [DOI] [PubMed] [Google Scholar]
- [21].Enger R, Tang W, Vindedal GF, Jensen V, Johannes Helm P, Sprengel R, Looger LL, Nagelhus EA. Dynamics of ionic shifts in cortical spreading depression. Cereb Cortex 2015;25:4469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fabricius M, Akgoren N, Dirnagl U, Lauritzen M. Laminar analysis of cerebral blood flow in cortex of rats by laser-Doppler flowmetry: a pilot study. J Cereb Blood Flow Metab 1997;17:1326–36. [DOI] [PubMed] [Google Scholar]
- [23].Fischer MJ, Koulchitsky S, Messlinger K. The nonpeptide calcitonin gene-related peptide receptor antagonist BIBN4096BS lowers the activity of neurons with meningeal input in the rat spinal trigeminal nucleus. J Neurosci 2005;25:5877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fitzgerald M. The spread of sensitization of polymodal nociceptors in the rabbit from nearby injury and by antidromic nerve stimulation. J Physiol 1979;297:207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gariepy H, Zhao J, Levy D. Differential contribution of COX-1 and COX-2 derived prostanoids to cortical spreading depression-Evoked cerebral oligemia. J Cereb Blood Flow Metab 2017;37:1060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 1993;33:48–56. [DOI] [PubMed] [Google Scholar]
- [27].Gupta S, Akerman S, van den Maagdenberg AM, Saxena PR, Goadsby PJ, van den Brink AM. Intravital microscopy on a closed cranial window in mice: a model to study trigeminovascular mechanisms involved in migraine. Cephalalgia 2006;26:1294–303. [DOI] [PubMed] [Google Scholar]
- [28].Hargreaves KM, Jackson DL, Bowles WR. Adrenergic regulation of capsaicin-sensitive neurons in dental pulp. J Endod 2003;29:397–9. [DOI] [PubMed] [Google Scholar]
- [29].Hoehlig K, Johnson KW, Pryazhnikov E, Maasch C, Clemens-Smith A, Purschke WG, Vauleon S, Buchner K, Jarosch F, Khiroug L, Vater A, Klussmann S. A novel CGRP-neutralizing Spiegelmer attenuates neurogenic plasma protein extravasation. Br J Pharmacol 2015;172:3086–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Holzer P. Neurogenic vasodilatation and plasma leakage in the skin. Gen Pharmacol 1998;30:5–11. [DOI] [PubMed] [Google Scholar]
- [31].Houben T, Loonen IC, Baca SM, Schenke M, Meijer JH, Ferrari MD, Terwindt GM, Voskuyl RA, Charles A, van den Maagdenberg AM, Tolner EA. Optogenetic induction of cortical spreading depression in anesthetized and freely behaving mice. J Cereb Blood Flow Metab 2017;37:1641–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Karsan N, Goadsby PJ. Calcitonin gene-related peptide and migraine. Curr Opin Neurol 2015;28:250–4. [DOI] [PubMed] [Google Scholar]
- [33].Kraig RP, Nicholson C. Extracellular ionic variations during spreading depression. Neuroscience 1978;3:1045–59. [DOI] [PubMed] [Google Scholar]
- [34].Kurosawa M, Messlinger K, Pawlak M, Schmidt RF. Increase of meningeal blood flow after electrical stimulation of rat dura mater encephali: mediation by calcitonin gene-related peptide. Br J Pharmacol 1995;114:1397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metabol 2011;31:17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lennerz JK, Ruhle V, Ceppa EP, Neuhuber WL, Bunnett NW, Grady EF, Messlinger K. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. J Comp Neurol 2008;507:1277–99. [DOI] [PubMed] [Google Scholar]
- [37].Levy D. Endogenous mechanisms underlying the activation and sensitization of meningeal nociceptors: the role of immuno-vascular interactions and cortical spreading depression. Curr pain headache Rep 2012;16:270–7. [DOI] [PubMed] [Google Scholar]
- [38].Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol 2005;58:698–705. [DOI] [PubMed] [Google Scholar]
- [39].Levy D, Strassman AM. Modulation of dural nociceptor mechanosensitivity by the nitric oxide—cyclic GMP signaling cascade. J Neurophysiol 2004;92:766–72. [DOI] [PubMed] [Google Scholar]
- [40].Lin Q, Zou X, Willis WD. Adelta and C primary afferents convey dorsal root reflexes after intradermal injection of capsaicin in rats. J Neurophysiol 2000;84:2695–8. [DOI] [PubMed] [Google Scholar]
- [41].Mayevsky A, Zeuthen T, Chance B. Measurements of extracellular potassium, ECoG and pyridine nucleotide levels during cortical spreading depression in rats. Brain Res 1974;76:347–9. [DOI] [PubMed] [Google Scholar]
- [42].McCulloch J, Uddman R, Kingman TA, Edvinsson L. Calcitonin gene-related peptide: functional role in cerebrovascular regulation. Proc Natl Acad Sci U S A 1986;83:5731–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Messlinger K. Migraine: where and how does the pain originate? Exp Brain Res 2009;196:179–93. [DOI] [PubMed] [Google Scholar]
- [44].Messlinger K, Hanesch U, Kurosawa M, Pawlak M, Schmidt RF. Calcitonin gene related peptide released from dural nerve fibers mediates increase of meningeal blood flow in the rat. Can J Physiol Pharmacol 1995;73:1020–4. [DOI] [PubMed] [Google Scholar]
- [45].Meyer RA, Campbell JN, Raja SN. Antidromic nerve stimulation in monkey does not sensitize unmyelinated nociceptors to heat. Brain Res 1988;441:168–72. [DOI] [PubMed] [Google Scholar]
- [46].Moghaddam B, Schenk JO, Stewart WB, Hansen AJ. Temporal relationship between neurotransmitter release and ion flux during spreading depression and anoxia. Can J Physiol Pharmacol 1987;65:1105–10. [DOI] [PubMed] [Google Scholar]
- [47].Moskowitz MA, Reinhard JF, Jr, Romero J, Melamed E, Pettibone DJ. Neurotransmitters and the fifth cranial nerve: is there a relation to the headache phase of migraine? Lancet 1979;2:883–5. [DOI] [PubMed] [Google Scholar]
- [48].Noseda R, Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain 2013;154(suppl 1):S44–53. [DOI] [PubMed] [Google Scholar]
- [49].Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol 2009;8:679–90. [DOI] [PubMed] [Google Scholar]
- [50].Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed by spreading oligemia and impaired activation of rCBF in classic migraine. Ann Neurol 1981;9:344–52. [DOI] [PubMed] [Google Scholar]
- [51].Padmawar P, Yao X, Bloch O, Manley GT, Verkman AS. K+ waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nat Methods 2005;2:825–7. [DOI] [PubMed] [Google Scholar]
- [52].Petersen KA, Birk S, Doods H, Edvinsson L, Olesen J. Inhibitory effect of BIBN4096BS on cephalic vasodilatation induced by CGRP or transcranial electrical stimulation in the rat. Br J Pharmacol 2004;143:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol 2012;75:365–91. [DOI] [PubMed] [Google Scholar]
- [54].Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab 2009;29:1517–27. [DOI] [PubMed] [Google Scholar]
- [55].Reeh PW, Kocher L, Jung S. Does neurogenic inflammation alter the sensitivity of unmyelinated nociceptors in the rat? Brain Res 1986;384:42–50. [DOI] [PubMed] [Google Scholar]
- [56].Reuter U, Weber JR, Gold L, Arnold G, Wolf T, Dreier J, Lindauer U, Dirnagl U. Perivascular nerves contribute to cortical spreading depression-associated hyperemia in rats. Am J Physiol 1998;274:H1979–1987. [DOI] [PubMed] [Google Scholar]
- [57].Rice FL, Xie JY, Albrecht PJ, Acker E, Bourgeois J, Navratilova E, Dodick DW, Porreca F. Anatomy and immunochemical characterization of the non-arterial peptidergic diffuse dural innervation of the rat and Rhesus monkey: implications for functional regulation and treatment in migraine. Cephalalgia 2016. DOI: 10.1177/0333102416677051. [DOI] [PubMed] [Google Scholar]
- [58].Russo AF. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol 2015;55:533–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schueler M, Messlinger K, Dux M, Neuhuber WL, De Col R. Extracranial projections of meningeal afferents and their impact on meningeal nociception and headache. Pain 2013;154:1622–31. [DOI] [PubMed] [Google Scholar]
- [60].Shatillo A, Koroleva K, Giniatullina R, Naumenko N, Slastnikova AA, Aliev RR, Bart G, Atalay M, Gu C, Khazipov R, Davletov B, Grohn O, Giniatullin R. Cortical spreading depression induces oxidative stress in the trigeminal nociceptive system. Neuroscience 2013;253:341–9. [DOI] [PubMed] [Google Scholar]
- [61].Steiner TJ, Stovner LJ, Birbeck GL. Migraine: the seventh disabler. J Headache Pain 2013;14:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Storer RJ, Akerman S, Goadsby PJ. Calcitonin gene-related peptide (CGRP) modulates nociceptive trigeminovascular transmission in the cat. Br J Pharmacol 2004;142:1171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Stovner L, Hagen K, Jensen R, Katsarava Z, Lipton R, Scher A, Steiner T, Zwart JA. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia 2007;27:193–210. [DOI] [PubMed] [Google Scholar]
- [64].Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature 1996;384:560–4. [DOI] [PubMed] [Google Scholar]
- [65].Tepe N, Filiz A, Dilekoz E, Akcali D, Sara Y, Charles A, Bolay H. The thalamic reticular nucleus is activated by cortical spreading depression in freely moving rats: prevention by acute valproate administration. Eur J Neurosci 2015;41:120–8. [DOI] [PubMed] [Google Scholar]
- [66].Troltzsch M, Denekas T, Messlinger K. The calcitonin gene-related peptide (CGRP) receptor antagonist BIBN4096BS reduces neurogenic increases in dural blood flow. Eur J Pharmacol 2007;562:103–10. [DOI] [PubMed] [Google Scholar]
- [67].Tvedskov JF, Lipka K, Ashina M, Iversen HK, Schifter S, Olesen J. No increase of calcitonin gene-related peptide in jugular blood during migraine. Ann Neurol 2005;58:561–8. [DOI] [PubMed] [Google Scholar]
- [68].Vyskocil F, Kritz N, Bures J. Potassium-selective microelectrodes used for measuring the extracellular brain potassium during spreading depression and anoxic depolarization in rats. Brain Res 1972;39:255–9. [DOI] [PubMed] [Google Scholar]
- [69].Wahl M, Schilling L, Parsons AA, Kaumann A. Involvement of calcitonin gene-related peptide (CGRP) and nitric oxide (NO) in the pial artery dilatation elicited by cortical spreading depression. Brain Res 1994;637:204–10. [DOI] [PubMed] [Google Scholar]
- [70].Wei X, Yan J, Tillu D, Asiedu M, Weinstein N, Melemedjian O, Price T, Dussor G. Meningeal norepinephrine produces headache behaviors in rats via actions both on dural afferents and fibroblasts. Cephalalgia 2015;35:1054–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zeller J, Poulsen KT, Sutton JE, Abdiche YN, Collier S, Chopra R, Garcia CA, Pons J, Rosenthal A, Shelton DL. CGRP function-blocking antibodies inhibit neurogenic vasodilatation without affecting heart rate or arterial blood pressure in the rat. Br J Pharmacol 2008;155:1093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhang X, Kainz V, Zhao J, Strassman AM, Levy D. Vascular extracellular signal-regulated kinase mediates migraine-related sensitization of meningeal nociceptors. Ann Neurol 2013;73:741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R. Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 2010;30:8807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zhang XC, Kainz V, Burstein R, Levy D. Tumor necrosis factor-alpha induces sensitization of meningeal nociceptors mediated via local COX and p38 MAP kinase actions. Pain 2010;152:140–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhao J, Levy D. The sensory innervation of the calvarial periosteum is nociceptive and contributes to headache-like behavior. PAIN 2014;155:1392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zhao J, Levy D. Modulation of intracranial meningeal nociceptor activity by cortical spreading depression: a reassessment. J Neurophysiol 2015;113:2778–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zhao J, Levy D. Cortical spreading depression promotes persistent mechanical sensitization of intracranial meningeal afferents: implications for the intracranial mechanosensitivity of migraine. eNeuro 2016;22:3. pii ENEURO.0287-16.2016. 10.1523/ENEURO.0287-16.2016. eCollection 2016 Nov-Dec. [DOI] [PMC free article] [PubMed] [Google Scholar]