

Abstract
The extracellular matrix (ECM) is readily enriched by decellularizing tissues with non-denaturing detergents to solubilize and deplete the vast majority of cellular components. This approach has been used extensively to generate ECM scaffolds for regenerative medicine technologies and in 3D cell culture to model how the ECM contributes to disease progression. A highly enriched ECM fraction can then be generated using a strong chaotrope buffer that is compatible with downstream bottom-up proteomic analysis or 3D cell culture experiments after extensive dialysis. With most tissues, an insoluble pellet remains after chaotrope extraction that is rich in structural ECM components. Previously we showed that this understudied fraction represented approximately 80 percent of total fibrillar collagen from the lung and other ECM fiber components that are known to be covalently cross-linked. Here we present a hydroxylamine digestion approach for chaotrope-insoluble ECM analysis with comparison to an established CNBr method for matrisome characterization. Because ECM characteristics vary widely among tissues, we chose five tissues that represent unique and diverse ECM abundances, composition and biomechanical properties. Hydroxylamine digestion is compatible with downstream proteomic workflows, yields high levels of ECM peptides from the insoluble ECM fraction and reduces analytical variability when compared to CNBr digestion.
Keywords: tissue extraction, extracellular matrix, matrisome, chemical digestion, proteomics, mass spectrometry, LC-SRM, collagen, insoluble matrix
Graphical Abstract
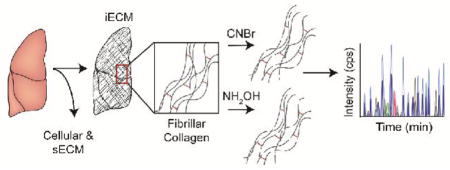
Introduction
The extracellular matrix (ECM) serves diverse functions and is a major component of the cellular microenvironment 1, 2. In addition to functioning as a physical scaffold for cells, the ECM initiates and maintains essential biochemical and biomechanical cues required for tissue development, differentiation and homeostasis through sequestration and release of growth factors and cryptic matrix sites, modulation of hydration levels, and pH of the local microenvironment, 3 to name a few. Although it is fundamentally composed of water, proteins and polysaccharides, the ECM exhibits an extraordinary level of tissue specificity as a result of reciprocal interactions with various cellular components that generate unique ECM compositions and architectures 1, 4. However, dysregulation can lead to the formation of fibrosis prevalent in many pathologies such as idiopathic pulmonary fibrosis, atherosclerosis and solid tumors 5–9. Despite these important functions and roles in disease, analytical methods to accurately quantify the abundance of ECM components in tissues are not standard. In particular, there are very few methods currently available to characterize insoluble ECM (iECM) components, which are rich in high molecular weight covalently cross-linked fiber proteins that undoubtedly play a dominant role in defining tissue architectures 10.
Isolation of insoluble tissue ECM requires enrichment through extensive decellularization steps to remove bulk cellular components that are solubilized by detergents. Post-decellularization, a chaotrope is often used to facilitate protein denaturation and the solubilization of a fraction of the remaining ECM components. After this extraction an insoluble pellet remains that contains a significant amount of protein based on strong acid hydrolysis and amino acid analysis 11. It was recently shown that greater than 80% of fibrillar collagen in murine liver and mammary gland resides in the iECM fraction 12 – a fraction excluded from traditional proteomic methods. Additionally, we have shown previously that the use of chemical digestion with cyanogen bromide (CNBr), prior to enzymatic cleavage with trypsin, is both necessary and sufficient to fully solubilize insoluble ECM components, such as fibrillar collagen from the liver and lungs 11, 12. This method was recently used to achieve higher levels of core matrisome protein sequence coverage compared to other ECM analysis strategies 13 and has been successfully applied to the characterization of ECM from mammary gland, liver, heart, lung, bone, and solid tumors, further supporting the general utility of the approach 8, 11, 12, 14–17.
Chemical digestion with cyanogen bromide (CNBr) is an obvious choice for ECM proteomics due to its extensive use in the biochemical analysis of collagens since its introduction as a cleavage agent in the early 1960s 18. Initially, in the presence of trifluoroacetic acid (TFA), a cyanosulfonium bromide derivative of protein methionine is formed. Under such acidic conditions, this intermediate leads to the formation of methylthiocyanate and homoserine iminolactone, which decompose spontaneously to (carboxy terminal) homoserine lactone and a new NH2-terminal fragment (Supplementary Figure S1-A) 18. Despite the ability of CNBr digestion to solubilize insoluble collagen matrices, there are several caveats associated with the use of CNBr. First, CNBr has a high degree of toxicity and requires careful handling during preparation, use and disposal 19. Second, the protocol for CNBr digestion is relatively laborious and involves a critical transfer step of the post-chaotrope iECM pellet into a glass vessel suitable for the strongly acidic conditions of digestion, introducing an opportunity for increased technical variability. Third, we have observed declining mass spectrometer performance during acquisition of large data sets of CNBr-digested iECM, necessitating additional sample clean-up steps and frequent instrument preventative maintenance measures.
Therefore, we sought to implement an orthogonal chemical digestion approach to circumvent these issues and improve sample throughput. Hydroxylamine (NH2OH) is a nucleophilic amine reported to cleave at Asn-Gly (NG) sites in proteins. In protein microenvironments at low pH, NG sites cyclize to an imide, which is then subjected to nucleophilic attack by NH2OH. Hydrolysis releases a C-terminal hydroxamate and N-terminal glycinyl residue (Supplementary Figure S1-B). In the past, this method has been used successfully to produce small soluble peptides, such as insulin-like growth factor 20. It has several advantages over CNBr including elimination of the transfer step after chaotrope extraction, safety, low cost, relatively low cross-reactivity, and has been reported to be selective for Asn-Gly linkages 21, 22. Therefore, we sought to address two major questions: 1) Compared to CNBr, will hydroxylamine digestion allow for comparable solubilization and quantification of ECM proteins while reducing technical variability? 2) Does tissue type influence the recovery of matrisome components across the chemical digestion approaches?
While traditional global proteomics approaches have improved our understanding of matrix composition, these approaches lack the ability to accurately quantify ECM protein abundance and stoichiometries in the microenvironment. Here we employ our previously described targeted LC-SRM (liquid chromatography-selected reaction monitoring) approach with ECM-specific stable isotope labeled (SIL) quantitative concatamers (QconCATs) to evaluate the two methods. Five previously described QconCAT proteins covering 170 peptides which represent 82 ECM, ECM-associated, and common cellular contaminant proteins of ECM preparations 8, 11, 12 are utilized here. We leveraged this approach to provide a quantitative comparison of insoluble ECM prepared by CNBr and NH2OH chemical digestion methods. In addition, we used a data-dependent LC-MS/MS approach to make qualitative assessments between methods. Our data reveal that NH2OH achieves equivalent or greater abundance of ECM components in all tissues analyzed. Overall, the results presented here provide an alternative ECM enrichment methodology that can be applied to a variety of tissues for identification and quantification of chaotrope-insoluble matrix proteins.
Materials and Methods
Reagents
Reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Sodium chloride was from Acros Organics (part of Thermo Fisher). Microcentrifuge tubes and other consumables were from Axygen Inc. (Union City, CA). Formic acid (FA), trifluoroacetic acid (TFA), and hydroxylamine hydrochloride were from Fluka (Buchs, Switzerland). Anhydrous potassium carbonate, guanidine hydrochloride, sodium hydroxide, and Optima acetonitrile were from Fisher Scientific (Pittsburgh, PA). Trypsin (sequencing grade, TPCK treated) was from Promega (Madison, WI).
QconCAT Design
Stable isotope-labeled QconCATs were designed as previously described 23. Six QconCATs were used to make 170 peptides covering 82 proteins in the Mus musculus proteome. Sequences are previously noted 12.
Sample Preparation
Three replicates of frozen bone, lung, liver, skin, and skeletal muscle harvested from a 6-week old female mouse (The Jackson Laboratory, Bar Harbor, ME) were powderized in liquid nitrogen using a ceramic mortar and pestle. Weighed tissue (approximately 5 mg of each) was homogenized in freshly prepared high-salt buffer (50 mM Tris-HCl, 3 M NaCl, 25 mM EDTA, 0.25% w/v CHAPS, pH 7.5) containing 1x protease inhibitor (Halt Protease Inhibitor, Thermo Scientific) at a concentration of 10 mg/mL. Homogenization took place in a bead beater (Bullet Blender Storm 24, Next Advance, 1 mm glass beads) for 3 min at 4 °C. Samples were then spun for 20 min 18,000 × g at 4 °C, and the supernatant removed and stored as Fraction 1. A fresh aliquot of high-salt buffer was added to the remaining pellet at 10 mg/mL of the starting weight, vortexed at 4 °C for 15 min, and spun for 15 min. The supernatant was removed and stored as Fraction 2. This high-salt extraction was repeated once more to generate Fraction 3, after which freshly prepared guanidine extraction buffer (6 M guanidinium chloride adjusted to pH 9.0 with NaOH) was added at 10 mg/mL and vortexed for 1 hour at room temperature. The samples were then spun for 15 min, the supernatant removed, and stored as Fraction 4. Fractions 1, 2, & 3 were combined and all fractions were stored at −20 °C until further analysis. The remaining pellets of each tissue representing insoluble ECM proteins were treated with either hydroxylamine or cyanogen bromide.
Hydroxylamine (NH2OH) Treatment
Following Fraction 4, pellets were treated with freshly prepared hydroxylamine buffer (1 M NH2OH-HCl, 4.5 M guanidine-HCl, 0.2 M K2CO3, pH adjusted to 9.0 with NaOH) at 10 mg/mL of the starting tissue weight 24. The samples were briefly vortexed, then incubated at 45 °C with end-over-end rotation for 17 hours. The tubes were fastened shut due to pressure build-up during incubation. Following incubation, the samples were spun for 15 min at 18,000 × g, the supernatant removed, and stored as Fraction 5 at −20 °C until further proteolytic digestion with trypsin. The final pellet was stored at −20 °C until further analysis.
Cyanogen Bromide (CNBr) Treatment
Following Fraction 4, pellets were transferred to a glass vial and treated with 100 mM CNBr/86% TFA (v/v) solution at 10 mg/mL of the starting tissue weight. The samples were agitated in the dark at room temperature for 17 hours. Following incubation, the solvent was evaporated under N2, followed by 3 × 1 mL washes with 100 mM ammonium bicarbonate (pH 8) in hichw the samples were briefly vortexed and dried in a speedvac. The dried samples were stored as Fraction 5 at −20 °C until further proteolytic digestion with trypsin. Immediately prior to trypsin digestion, Fraction 5 was re-suspended in freshly prepared urea buffer (8 M urea in 100 mM ammonium bicarbonate) at 10 mg/mL, vortexed for 1 hour at room temperature, and the supernatant used for tryptic digestion.
Trypsin Digestion
200 μL of the Fraction 4 & 5 of all samples were subsequently subjected to enzymatic digestion with trypsin using a filter-aided sample preparation (FASP) approach and C18 cleanup as previously described 25. Prior to trypsin digestion, 200 fmols of each SIL peptide (170 peptides total) were spiked into 200 μL of sample to allow for two injections per sample (100 fmols eQ 1–6 per injection).
LC-SRM Analysis
Samples were analyzed by LC-SRM and LC-MS/MS as described 17. Equal volumes from each post-digestion sample were combined and injected every third run and used to monitor technical reproducibility. Skyline was used for method development and to extract the ratio of endogenous light peptides to heavy internal standards from LC-SRM data for protein quantification as described 26. LC-MS/MS data was processed as previously described 12. Limits of detection, quantification, and dynamic range were determined for each peptide as previously described 11. Final endogenous peptide values are expressed as fmol peptide/mg of starting wet tissue weight. Principal component analysis (PCA) and partial least squares-discriminant analysis (PLS-DA) were performed using the MetaboAnalyst 3.0 online platform with sum and range scaling normalizations 27. To control for any technical variability introduced prior to chemical digestion we normalized the CNBr and NH2OH treated samples to the measured protein abundance of the sECM fraction. Additionally, in order to ensure that the increased ratios (12C6/13C6) were a result of increased endogenous protein signal and not a result of decreased QconCAT digestion efficiency, we analyzed a subset of each tissue extract with pre-digested QconCAT and found no evidence of reduced QconCAT digestion efficiency. All data has been made publically available through the PRIDE database (PX-Submission #185523)
LC-MS/MS Analysis
Samples were analyzed on an LTQ Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific) coupled to an Eksigent nanoLC-2D system through a nanoelectrospray source. 8 μL of sample was injected onto a trapping column (Agilent Technologies ZORBAX 300SB-C18, dimensions 5 × 0.3 mm, 5 μm) and washed with 0.1% formic acid (FA) in water at a flow rate of 5 μL/min for 5 min. The analytical column (100 μm i.d. × 150 mm fused silica capillary packed in house with 4 μm 80 Å Synergi Hydro C18 resin (Phenomenex; Torrance, CA)) was then switched on-line at 600 nL/min for 10 min to load the sample. The flow rate was adjusted to 350 nL/min, and peptides were separated over a 120-min linear gradient of 2–40% ACN with 0.1% FA. Data acquisition was performed using the instrument supplied Xcalibur (version 2.1) software. The mass spectrometer was operated in positive ion mode. Full MS scans were acquired in the Orbitrap mass analyzer over the 300–1800 m/z range with 60,000 resolution at m/z 400. Automatic gain control (AGC) was set at 5.00E+05 and the ten most intense peaks from each full scan were fragmented via HCD with normalized collision energy of 35. MS/MS spectra were acquired in the Orbitrap mass analyzer with 15,000 resolution. All replicates of each tissue were run sequentially and pre-digested yeast alcohol dehydrogenase standard (nanoLCMS Solutions LLC, Rancho Cordova, CA) was run between tissue groups to monitor drift in analytical performance.
Database Searching and Protein Identification
MS/MS spectra were extracted from raw data files and converted into .mgf files using MS Convert (ProteoWizard, Ver. 3.0). Peptide spectral matching was performed with Mascot (Ver. 2.5) against the Uniprot mouse database (release 201701). Mass tolerances were +/− 10 ppm for parent ions, and +/− 0.01 Da for fragment ions. Trypsin + NH2OH or Trypsin + CNBr enzyme specificity was used, allowing for 1 missed cleavage. Met oxidation, Pro hydroxylation, protein N-terminal acetylation, and peptide N-terminal pyroglutamic acid formation were set as variable modifications (maximum number of 8) with Cys carbamidomethylation set as a fixed modification. Scaffold (version 4.4.6, Proteome Software, Portland, OR, USA) was used to validate MS/MS-based peptide and protein identifications. NH2OH cleavage at NG or NX sites was evaluated by merging Scaffold peptide reports for all tissues analyzed and reporting the total number of NG or NX cleavage observations in the entire dataset (Supplementary Table S4). Peptide identifications were accepted if they could be established at greater than 95.0% probability. Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least two identified unique peptides.
Error Tolerant Searches
Raw LC-MS/MS data was converted to peak lists using MS Convert (ProteoWizard, Ver. 3.0). Peptide spectral matching was performed with Mascot (Ver. 2.5) against the Uniprot mouse database (release 201701). Unbiased error tolerant searches were performed using Byonic 28 in order to identify unknown and unpredicted modifications induced by each chemical digestion method. PSMs (peptide spectral matches) were only used for analysis if their expectation value (analogous to p-value) was less than 0.05, and the total number of occurrences of each mass addition/subtraction was plotted using GraphPad Prism 6.0 (GraphPad Software Inc, La Jolla, CA).
Results
Chemical digestion of insoluble ECM components
Using previously reported conditions for hydroxylamine (NH2OH) 24 and our existing method for cyanogen bromide (CNBr) digestion 11 we evaluated NH2OH digestion of five tissues using several criteria including 1) quantitative yield using LC-SRM with stable isotope labeled standards 2) the number of unique proteins identified by data-dependent, global LC-MS/MS analysis and 3) potential side product generation (i.e. modifications) using error-tolerant database searches. In order to assess these criteria, we utilized an experimental workflow based on a modified version of our previously published protocol to characterize five murine organs: lung, liver, muscle, skin and bone (Figure 2A). These tissues were chosen as they represent organs or organ systems that vary greatly in the abundance of their ECM components relative to total protein content. For example, bone was chosen as a non-compliant and high ECM abundance tissue. Along these same lines, skin, muscle and lung were chosen as compliant tissues with high to medium ECM abundance, while liver is representative of tissues with low ECM abundance. Proteomic analysis of ECM components were compared across the two chemical digestion methods (CNBr vs. NH2OH) to determine the ability of each method to solubilize and, with the aid of trypsin, generate proteolytic peptides for ECM protein quantification.
Figure 2. Characterization of hydroxylamine cleavage specificity and fibrillar collagen abundance iECM fraction.

(A) Observed hydroxylamine cleavage specificity across all tissues (1534 peptides total) from data-dependent LC-MS/MS experiments (B) Fibrillar collagen abundance derived from LC-SRM data plotted as percentage of total quantified protein in the iECM fraction.
The tissue extraction/fractionation and digestion workflow used prior to LC-SRM data acquisition is shown as a schematic representation in Figure 1A. Our sample preparation protocol yields three distinct fractions: 1) cellular, 2) soluble ECM (sECM), and 3) insoluble ECM (iECM). Samples destined for CNBr or NH2OH digestion were prepared identically. The location of CNBr and NH2OH cleavage sites within the primary sequence of collagen alpha-1(I) are shown in Figure 1B. CNBr cleaves specifically at methionine residues, with collagen alpha-1(I) having 8 cleavage sites. In contrast, NH2OH is reported to cleave at Asn-Gly sites, with 6 sites for collagen alpha-1(I). However, we also observed cleavage at other Asn-X sites in our dataset, which accounts for an additional 7 potential cleavage sites in collagen alpha-1(I), although not all were not observed in our MS data (Figure 1B). Our hypothesis that NH2OH would be a good alternative to CNBr was based on early reports of hydroxylamine-sensitive bonds in collagen alpha-1(I) 29 – the most abundant iECM protein in the tissues analyzed.
Figure 1. Workflow diagram for comparative analysis of CNBr and NH2OH using quantitative QconCAT ECM proteomics.

(A) Tissues are sequentially extracted to obtain cellular, soluble ECM (sECM) fractions. The pellet remaining after chaotrope extraction is subjected to either CNBr or NH2OH digestion. QconCATs are spiked into CNBr or NH2OH iECM fractions and samples are then enzymatically digested prior to data acquisition using an LC-SRM approach. (B) Schematic representation of the mature fibrillar collagen fibers of collagen alpha-1(1). CNBr cleavage sites (8 for Col1a1) are denoted by a double black slash while NH2OH cleavage sites (13 for Col1a1) are denoted by a double red (NG) or double orange (NX) slash. Green hexagonal markings denote known sites of cross-linking in the telopeptide region fibrillar collagen. Transparent purple boxes mark sequence regions that match to stable isotope labeled (SIL) QconCAT peptides (Uniprot IDs: Col1a1: P11087 and Col1a2: Q01149). ‡ denotes cleavage sites that were detected by LC-MS.
Evaluation of hydroxylamine chemical cleavage specificity
NH2OH and CNBr cleavage were evaluated by performing database searches using dual cleavage specificity (i.e. trypsin + NH2OH or trypsin + CNBr). Although NH2OH has previously been reported to be specific for NG sites, we evaluated NX sites as well, where X is any other amino acid. Although we found that NG represented the combination with the highest number of cleavage events, all other NX combinations with the exception of cysteine were observed, with a preference toward small amino acids (Figure 2A). However, NG cleavage was observed, on average, 10-fold higher frequency compared to all other combinations (Supplementary Table S1). QG and QX sites were also investigated for potential cross-reactivity, but cleavage was not observed. As a negative control, these search conditions were used on a subset of the sECM fractions that had not been exposed to chemical digestion conditions. Cleavage C-terminal to Asn residues in this fraction was observed less than 1% of the time, indicating chemoselectivity of NH2OH treatment.
Fibrillar collagen abundance as a percentage of total protein in the hydroxylamine prepared iECM fraction was quantitated using LC-SRM data (Figure 2B). As expected, bone tissue showed the highest abundance of fibrillar collagen in the iECM fraction accounting for >90% of total quantified protein. In more compliant tissues, such as skin and lung, fibrillar collagen abundance represented 75% and 52% of total quantified protein in the iECM fraction, respectively. In the softest tissues analyzed with the lowest overall ECM content, muscle and liver, fibrillar collagen represented 43% and 25% of total quantified protein in the iECM fraction.
CNBr cleavage was specific to the C-termini of methionine residues as reported 12, 18. In the case of CNBr database searches, we also included two additional variable modifications, homoserine (Met to Hse) and homoserine lactone (Met to Hsl). The average observed missed cleavage percentage for trypsin across all CNBr and NH2OH digested tissues was found to be 8.5% and 8.3%, respectively. Semitryptic peptides with ragged N-termini represented 9.8% (CNBr) and 6.8% (NH2OH) of total peptides. Semitryptic peptides with ragged C-termini or non-tryptic peptides each represented less than 1% of total peptides identified.
Comparison of quantitative yield, protein and peptide identifications between CNBr and NH2OH digestion approaches
After characterization of NH2OH cleavage specificity, we sought to compare the quantitative yields of the CNBr and NH2OH digestion approaches to determine suitability of NH2OH digestion for analysis of the iECM. Total protein abundance as determined by LC-SRM data was used to make this quantitative comparison (Supplementary Table S2). We stratified proteins followed in our targeted assay into four major groups: cellular proteins, collagens, glycoproteins and proteoglycans. Total protein abundance in each group by CNBr or NH2OH digestion was plotted and compared for the five tissues tested (Figure 3A). Despite slight variations in yield within protein groups, the overall yields of CNBr and NH2OH were comparable with few notable differences. In general, NH2OH digestion followed by trypsin yielded a higher protein abundance than CNBr followed by trypsin. More specifically, cellular proteins quantified across the tissues tested were on average 2.5 fold higher in NH2OH preparations compared to CNBr. Along these same lines, collagens, glycoproteins and proteoglycans were found to be 1.6, 2.8 and 2.2-fold higher in NH2OH preparations, on average, for all tissues tested (Supplementary Table S3). While the general trend in the data suggests that NH2OH preparations are superior to CNBr preparations, there a few tissue specific phenomena revealed by the data. For example, collagens and proteoglycans quantified in bone tissue prepared with CNBr digestion and trypsin were found to be 1.4 and 1.3-fold higher abundance than those prepared with NH2OH and trypsin (Figure 3A and Supplementary Table S3). These differences are largely driven by variations in basement membrane (i.e. Col4a2, Lama2) and cytoskeletal (i.e. Act) component abundance between methods as discovered by performing principal component analysis (not shown) despite these proteins having fewer Asn-Gly motifs than Met residues. This indicates that trypsin access to these proteins is not dependent on release of the protein from crosslinked insoluble protein. Overall these proteins compose less than 3% of the total protein fraction in the insoluble bone ECM (Supplementary Table S2).
Figure 3. Comparison of quantitative yield, protein and peptide identifications between CNBr and NH2OH digestion approaches.

(A) Protein group abundance plots for cellular proteins, collagens, glycoproteins and proteoglycan protein groups derived from LC-SRM data for tissues prepared with cyanogen bromide (C) or hydroxylamine (H). (B) Venn diagrams of average shared and unique protein and peptide identifications from all tissues derived from data-dependent LC-MS/MS experiments.
We performed data-dependent LC-MS/MS experiments to perform qualitative assessments of iECM digestion with cyanogen bromide or hydroxylamine (Supplementary Table S4). Across all tissues tested, CNBr and NH2OH digestion methods shared 227 protein identifications with CNBr and NH2OH yielding 63 and 51 unique protein identifications, respectively (Figure 3B). However, comparison of the total peptide identifications averaged across all tissues tested was less congruent between methods owing to the stochastic sampling of the DDA method and nature of each chemical digestion mechanism and specificity. As such, CNBr and NH2OH were found to have 690 and 789 unique peptide identifications, respectively, and shared an average of 702 peptides across all tissues tested (Figure 3B and Supplementary Table S5).
Error tolerant searches
To investigate if observed differences between digestion methods were a result of side reactions generated during CNBr or NH2OH cleavage reactions, we performed error tolerant searches on data-dependent LC-MS/MS data (Supplementary Figure S2). Several expected abundant modifications were identified such as deamidation of Asp, oxidation of methionine and proline, and a +6 Da addition that results from the inclusion of our heavy labeled QconCAT SIL peptides. One of the most common modifications identified (+15.99 Da) is primarily derived from the endogenous fibrillar collagen hydroxy-proline residues (Supplementary Figure S2). Two modifications that are specific to the CNBr cleavage mechanism were also determined: 1. Formation of a homoserine lactone 18 and 2. The low frequency bromination of tyrosine, for example, both the brominated and non-brominated forms of the collagen alpha-2(I) peptide GYPGSIGPTGAAGAPGPHGSVGPAGK were identified (Figure 4).
Figure 4. CNBr selectively brominates tyrosine containing collagen peptides.

(A) Non-brominated form of collagen alpha-2(I) peptide GYPGSIGPTGAAGAPGPHGSVGPAGK. (B) Brominated form of collagen alpha-2(I) peptide GYPGSIGPTGAAGAPGPHGSVGPAGK at position Y2. (C) Isotopic distribution of non-brominated collagen alpha-2(I) peptide, theoretical pattern inset. (D) Isotopic distribution of brominated collagen alpha-2(I) peptide, theoretical pattern inset.
Discussion
Here we have expanded upon existing ECM proteomics methods by developing an alternative method for insoluble protein characterization that avoids many of the drawbacks associated with the cyanogen bromide reagent. We utilized data-dependent LC-MS/MS experiments to qualitatively assess differences in protein and peptide identifications and a matrisome-targeted LC-SRM method to more accurately quantify ECM protein abundance and stoichiometries from five tissues. All samples presented in the study are technical replicates to provide the most direct comparison between the methods. The combination of our global LC-MS/MS and targeted LC-SRM approaches with ECM-specific stable isotope-labeled peptides revealed that an abundance of protein is present and available for analysis from tissues after chaotrope extraction.
Our previously described approach to solubilize iECM using CNBr was effective and capable of outperforming orthogonal methods for ECM composition analysis 13. Despite this success, we sought to develop an improved chemical digestion approach using NH2OH that minimizes sample loss (i.e. no pellet transfer step), avoids the reactive and toxic CNBr compound and strong acid used for digestion, and provides similar or improved quantification precision and reproducibility. We evaluated alternative proteases and chemical digestion methods and determined that NH2OH digestion was the top candidate for further development. Advantages of this approach include digestions being carried out in the same vessel as chaotrope extraction (no sample transfer to glass vial) in aqueous solution at pH 9 and a relatively modest chemical activity that is unlikely to generate extensive by-products that can hamper absolute quantification. In the case of CNBr, we suspect that liberated reactive species may negatively affect protein digestion with trypsin or account for the observed decreased MS instrument performance. Over time, a decrease in the resolution of low mass ions and in ion transfer efficiency (30–40%) of a calibration standard mix was observed after running samples prepared with CNBr. Generally, solid phase extraction (SPE) using C18 resin reduces, but does not eliminate, this effect.
From global LC-MS/MS analysis we determined that CNBr and NH2OH resulted in a similar number of identified ECM proteins irrespective of tissue type despite variations in the number of unique peptides generated between digestion approaches. These findings are supported by LC-SRM data that showed similarity between methods in the number of quantifiable peptides, however, the methods varied in terms of overall yield (i.e. total quantified protein). The NH2OH method yielded a higher concentration of ECM components and yielded more reproducible measures between replicates across all tissues except for bone. The specificity of the two chemical digestion approaches was evaluated by monitoring the occurrence of non-target cleavage events. While NG sites were the most preferred for cleavage, we found that NH2OH is relatively non-specific under the conditions used here. Although we did not observe NX cleavage on fibrillar collagen itself, it was observed in other proteins present in the iECM fraction. As a result, it is recommended that this be considered when performing database searches on data from NH2OH-treated samples. In addition, quantification of peptides that contain asparagine residues should be performed with caution.
A caveat of this work is that we did not perform deglycosylation which should increase sequence coverage, especially for proteoglycans 30–32. In our experience, the majority of proteoglycans and glycoproteins are extracted in the soluble ECM fraction prior to chemical digestion of the insoluble pellet 12. Further, we have intentionally avoided peptides with known glycosylation sites in our stable isotope-labeled standards used for quantification. However, deglycosylation of the prominent glycosaminoglycans (GAG) is required to maximize matrisome coverage and should be considered when this is an objective.
Not surprisingly, bone samples displayed the largest amount of structural ECM in the iECM fraction, the overwhelmingly majority being fibrillar collagen. Skin was second in total ECM abundance, followed by muscle, lung, and liver. Other major differences include the abundance of cytoskeletal components. As one might expect, muscle samples had a higher abundance of cytoskeletal components than all other tissues analyzed. In contrast, basement membrane appears to be most abundant in those organs with lower total ECM content and higher cellularity, such as lung, muscle, and liver. The most abundant basement membrane protein across all tissues was found to be collagen IV as has been previously reported in skeletal muscle 33. Not surprisingly, basement membrane components were found to be most abundant in lung tissue owing to large membranous surface area supporting the endothelial barrier as needed for gas exchange. Some of the largest differences between the two methods appeared to be a result of differential solubilization of basement membrane and cytoskeletal components such as Col4a2, Lamc2, Tubb, and Vim. Initially, we speculated that this was due to differences in the number of CNBr (methionine specific) and NH2OH (asparagine site specific) cleavage sites. Indeed, differences do exist in the number of cleavage sites, but the findings do not support the idea that more cleavage sites lead to more efficient digestion and resulting higher measured protein abundance in the iECM fraction of the tissues tested.
An alternative explanation is that a smaller peptide from chemical digestion is partially lost through the filter-aided sample preparation approach (FASP) approach. For example, one peptide that we utilized to follow Col1a1 abundance (GSEGPQGVR) resides in a CNBr cleavage product of 26.2 kDa, while NH2OH digestion results in a Col1a1 fragment of just 9.2 kDa. Thus, we postulated that our proportion of Col1a1 may be under-represented in the NH2OH samples resulting in a lower Col1a1/Col1a2 ratio due the smaller NH2OH fragment being partially lost through the 10 kDa MWCO filter employed in FASP digestion. However, Wisniewski et al. report efficient retention of small proteins (5 – 10 kDa) using a 10 kDa MWCO filter 34, 35.
Our results provide an updated ECM enrichment methodology for identification of chaotrope insoluble matrix proteins. Based on these findings, the NH2OH digestion approach for chaotrope-insoluble matrisome characterization is preferred to the CNBr-based chemical digestion method. In general, the NH2OH method for iECM solubilization yielded more reproducible results as demonstrated by smaller coefficients of variance (CVs) compared to CNBr iECM preparations. Eliminating the use of concentrated acid, along with the reactive and toxic compound CNBr reduces personal and environmental hazards. Overall, the semi-quantitative and quantitative results yielded more protein identifications and higher peptide levels for the new approach, suggesting equal or improved ability to solubilize the insoluble matrix. For bone analysis, we found the two approaches to be relatively similar in their overall yield suggesting either approach would be suitable for iECM characterization, however, it is likely that a standard demineralization step will be required to achieve equivalent or superior results for the mineralized matrix using this new method. For all other tissues tested, hydroxylamine performed the same or better than cyanogen bromide and is the recommended approach to characterize this challenging fraction of the proteome.
Supplementary Material
Supplementary Figure S1 – Cyanogen bromide and hydroxylamine reaction mechanisms
Supplementary Figure S2 – Error Tolerant Search Results from LC-MS/MS data
Supplementary Table S1 – Observed hydroxylamine cleavage results
Supplementary Table S2 – LC-SRM individual protein results
Supplementary Table S3 – Fold change analysis of ECM protein groups from LC-SRM data
Supplementary Table S4 – Data-dependent LC-MS/MS results
Supplementary Table S5 – Protein and peptide identifications from data-dependent LC-MS/MS experiments
Acknowledgments
The work included in this manuscript includes funding from NIH/CTSA 63007957 and NIH T32HL007171 (to ASB), NIH/NCI R33CA183685 (to KCH) and the Shared Instrument grant by the NIH S10OD021641 (to KCH).
Footnotes
Conflict of Interest disclosure
The authors declare no competing financial interest.
Author Information
ASB and KCH were responsible for hypothesis development, conceptual design and final data integrity and interpretation. MJW and JAR optimized hydroxylamine cleavage conditions. ASB, MJW, and RCH performed all proteomic analyses. ASB, KCH, AD, JAR, and MD contributed to the conceptual design of the manuscript and to data interpretation. ASB and KCH wrote the manuscript and all authors edited and approved the final manuscript submission.
References
- 1.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. Journal of cell science. 2010;123(24):4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nature Reviews Molecular Cell Biology. 2014;15(12):771–785. doi: 10.1038/nrm3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DuFort CC, Paszek MJ, Weaver VM. Balancing forces: architectural control of mechanotransduction. Nature reviews Molecular cell biology. 2011;12(5):308–319. doi: 10.1038/nrm3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson CM, Bissell MJ. Of extracellular matrix scaffolds and signaling: tissue architecture regulates development homeostasis and cancer. Annual review of cell and developmental biology. 2006;22:287. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO reports. 2014:e201439246. doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King TE, Pardo A, Selman M. Idiopathic pulmonary fibrosis. The Lancet. 2011;378(9807):1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 7.Schuppan D. Seminars in liver disease. 1990. Structure of the extracellular matrix in normal and fibrotic liver: collagens and glycoproteins; pp. 1–10. © 1990 by Thieme Medical Publishers, Inc.: 1990. [DOI] [PubMed] [Google Scholar]
- 8.Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nature medicine. 2016;22(5):497–505. doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heeneman S, Cleutjens JP, Faber BC, Creemers EE, van Suylen RJ, Lutgens E, Cleutjens KB, Daemen MJ. The dynamic extracellular matrix: intervention strategies during heart failure and atherosclerosis. The Journal of pathology. 2003;200(4):516–525. doi: 10.1002/path.1395. [DOI] [PubMed] [Google Scholar]
- 10.Eyre DR, Paz MA, Gallop PM. Cross-linking in collagen and elastin. Annual review of biochemistry. 1984;53(1):717–748. doi: 10.1146/annurev.bi.53.070184.003441. [DOI] [PubMed] [Google Scholar]
- 11.Hill RC, Calle EA, Dzieciatkowska M, Niklason LE, Hansen KC. Quantification of Extracellular Matrix Proteins from a Rat Lung Scaffold to Provide a Molecular Readout for Tissue Engineering. Molecular & Cellular Proteomics. 2015;14(4):961–973. doi: 10.1074/mcp.M114.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goddard ET, Hill RC, Barrett A, Betts C, Guo Q, Maller O, Borges VF, Hansen KC, Schedin P. Quantitative extracellular matrix proteomics to study mammary and liver tissue microenvironments. The International Journal of Biochemistry & Cell Biology. 2016;81:223–232. doi: 10.1016/j.biocel.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krasny L, Paul A, Wai P, Howard BA, Natrajan RC, Huang PH. Comparative proteomic assessment of matrisome enrichment methodologies. Biochemical Journal. 2016;473(21):3979–3995. doi: 10.1042/BCJ20160686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calle EA, Hill RC, Leiby KL, Le AV, Gard AL, Madri JA, Hansen KC, Niklason LE. Targeted proteomics effectively quantifies differences between native lung and detergent-decellularized lung extracellular matrices. Acta Biomaterialia. 2016;46:91–100. doi: 10.1016/j.actbio.2016.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goddard ET, Hill RC, Nemkov T, D’Alessandro A, Hansen KC, Maller O, Mongoue-Tchokote S, Mori M, Partridge AH, Borges VF. The Rodent Liver Undergoes Weaning-Induced Involution and Supports Breast Cancer Metastasis. Cancer Discovery. 2016:CD-16-0822. doi: 10.1158/2159-8290.CD-16-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill RC, Wither MJ, Nemkov T, Barrett A, D’Alessandro A, Dzieciatkowska M, Hansen KC. Preserved proteins from extinct bison Latifrons identified by tandem mass spectrometry; hydroxylysine glycosides are a common feature of ancient collagen. Molecular & Cellular Proteomics. 2015;14(7):1946–1958. doi: 10.1074/mcp.M114.047787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson TD, Hill RC, Dzieciatkowska M, Nigam V, Behfar A, Christman KL, Hansen KC. Quantification of decellularized human myocardial matrix: a comparison of six patients. Proteomics-Clinical Applications. 2016;10(1):75–83. doi: 10.1002/prca.201500048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gross E. The cyanogen bromide reaction. Methods in enzymology. 1967;11:238–255. [Google Scholar]
- 19.ToxNet. HSDB: Cyanogen Bromide. US National Library of Medicine; 2014. p. 708. [Google Scholar]
- 20.Antorini M, Breme U, Caccia P, Grassi C, Lebrun S, Orsini G, Taylor G, Valsasina B, Marengo E, Todeschini R. Hydroxylamine-induced cleavage of the asparaginyl–glycine motif in the production of recombinant proteins: the case of insulin-like growth factor I. Protein expression and purification. 1997;11(1):135–147. doi: 10.1006/prep.1997.0771. [DOI] [PubMed] [Google Scholar]
- 21.Park HB, Pyo SH, Hong SS, Kim JH. Optimization of the hydroxylamine cleavage of an expressed fusion protein to produce a recombinant antimicrobial peptide. Biotechnology letters. 2001;23(8):637–641. [Google Scholar]
- 22.Bornstein P, Balian G. [14] Cleavage at Asn Gly bonds with hydroxylamine. Methods in enzymology. 1977;47:132–145. doi: 10.1016/0076-6879(77)47016-2. [DOI] [PubMed] [Google Scholar]
- 23.Dzieciatkowska M, D’Alessandro A, Hill RC, Hansen KC. Plasma QconCATs reveal a gender-specific proteomic signature in apheresis platelet plasma supernatants. Journal of Proteomics. 2015;120:1–6. doi: 10.1016/j.jprot.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simpson RJ. Cleavage of asn-gly bonds by hydroxylamine. CSH protocols. 2007;2007 doi: 10.1101/pdb.prot4697. [DOI] [PubMed] [Google Scholar]
- 25.Wither MJ, Hansen KC, Reisz JA. Mass Spectrometry-Based Bottom-Up Proteomics: Sample Preparation, LC-MS/MS Analysis, and Database Query Strategies. Current Protocols in Protein Science. 2016:16.4. 1–16.4. 20. doi: 10.1002/cpps.18. [DOI] [PubMed] [Google Scholar]
- 26.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic acids research. 2015;43(W1):W251–W257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bern M, Kil YJ, Becker C. Byonic: advanced peptide and protein identification software. Current Protocols in Bioinformatics. 2012:13.20. 1–13.20. 14. doi: 10.1002/0471250953.bi1320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bornstein P. The nature of a hydroxylamine-sensitive bond in collagen. Biochemical and Biophysical Research Communications. 1969;36(6):957–964. doi: 10.1016/0006-291x(69)90297-6. [DOI] [PubMed] [Google Scholar]
- 30.Leymarie N, Zaia J. ACS Publications. 2012. Effective use of mass spectrometry for glycan and glycopeptide structural analysis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Molecular & Cellular Proteomics. 2010;9(9):2048–2062. doi: 10.1074/mcp.M110.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson R, Norris EL, Brachvogel B, Angelucci C, Zivkovic S, Gordon L, Bernardo BC, Stermann J, Sekiguchi K, Gorman JJ. Changes in the chondrocyte and extracellular matrix proteome during post-natal mouse cartilage development. Molecular & Cellular Proteomics. 2012;11(1):M111.014159. doi: 10.1074/mcp.M111.014159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanes JR. The basement membrane/basal lamina of skeletal muscle. Journal of Biological Chemistry. 2003;278(15):12601–12604. doi: 10.1074/jbc.R200027200. [DOI] [PubMed] [Google Scholar]
- 34.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nature methods. 2009;6(5):359. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 35.Wiśniewski JR, Zougman A, Mann M Combination of FASP. StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. Journal of proteome research. 2009;8(12):5674–5678. doi: 10.1021/pr900748n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 – Cyanogen bromide and hydroxylamine reaction mechanisms
Supplementary Figure S2 – Error Tolerant Search Results from LC-MS/MS data
Supplementary Table S1 – Observed hydroxylamine cleavage results
Supplementary Table S2 – LC-SRM individual protein results
Supplementary Table S3 – Fold change analysis of ECM protein groups from LC-SRM data
Supplementary Table S4 – Data-dependent LC-MS/MS results
Supplementary Table S5 – Protein and peptide identifications from data-dependent LC-MS/MS experiments