

Summary
Background
Hemophilia A results from the absence, deficiency or inhibition of factor VIII. Bleeding is treated with hemostatic agents (FVIII, recombinant activated FVII [rFVIIa], anti-inhibitor coagulation complex [FEIBA], or recombinant porcine FVIII [rpFVIII]). Despite treatment, some patients have prolonged bleeding. FXIII-A2B2 (FXIII) is a protransglutaminase. During clot contraction, thrombin-activated FXIII (FXIIIa) crosslinks fibrin and α2-antiplasmin, which promotes red blood cell retention and increases clot stability and weight. We hypothesized that FXIII cotreatment in hemophilia would accelerate FXIII activation, leading to increased fibrin crosslinking.
Methods
FVIII-deficient plasma and whole blood were clotted with or without hemostatic agents (FVIII, rFVIIa, FEIBA, or recombinant B-domain-deleted porcine FVIII [rpFVIII]) and/or FXIII. The effects on FXIII activation, thrombin generation, fibrin and α2-antiplasmin crosslinking, clot formation and clot weight were measured by western blotting, calibrated automated thrombography, thromboelastography, and clot contraction assays.
Results
As compared with FVIII-treated hemophilic plasma, FVIII + FXIII cotreatment accelerated FXIIIa formation without increasing thrombin generation. As compared with buffer-treated or FXIII-treated hemophilic plasma, FVIII treatment and FVIII + FXIII cotreatment increased the generation and amount of crosslinked fibrin, including α-chain-rich high molecular weight species and crosslinked α2-antiplasmin. In the presence of FVIII inhibitors, as compared with hemostatic treatments (rFVIIa, FEIBA, or rpFVIII) alone, FXIII cotreatment increased whole blood clot weight.
Conclusion
In hemophilia A plasma and whole blood, FXIII cotreatment with hemostatic agents accelerated FXIIIa formation, increased the generation and amount of fibrin α-chain crosslinked species, accelerated α2-antiplasmin crosslinking, and increased clot weight. FXIII cotreatment with hemostatic therapy may augment hemostasis through increased crosslinking of fibrin and α2-antiplasmin.
Keywords: α2-antiplasmin, factor XIII, fibrin, hemophilia, hemostasis
Introduction
Congenital hemophilia A and hemophilia B result from the absence or deficiency of factor VIII and FIX, respectively, and lead to delayed and reduced thrombin generation [1–4]. The initial hemophilic clot forms slowly, and has an abnormal fibrin structure with thick fibrin fibers [5–7]. As compared with normal clots, this abnormal fibrin structure leads to increased clot permeability and susceptibility to lysis [6,7]. Bleeding events are usually treated with replacement factors (recombinant or plasma-derived human FVIII or FIX) that restore thrombin generation and normalize clot structure and stability. However, approximately 25% of hemophilia A patients and 1–10% of hemophilia B patients develop inhibitors of replacement factors [8,9]. More rarely, non-hemophilic individuals develop hemophilia A because of acquired FVIII inhibitors [10]. Bleeding in hemophilic patients with inhibitors is treated with hemostatic agents, such as recombinant activated FVII (rFVIIa), plasma-derived anti-inhibitor coagulant complex (FEIBA), or recombinant porcine FVIII. These agents bypass or mimic the FVIII–FIX complex to increase thrombin generation and, ultimately, improve fibrin quality [2,4,6,7,11–14].
Unfortunately, despite hemostatic therapy, ~ 10–30% of hemophilic patients experience refractory bleeding [15]. In this setting, several treatment strategies may be used, including increased frequency or dose of the initial hemostatic therapy, sequential or concomitant use of hemostatic agents, or addition of antifibrinolytic agents [16–18]. However, use of these treatment strategies is limited by several factors, including insufficient laboratory monitoring capacity and concerns about thrombosis [19], leaving patients at increased risk of morbidity and mortality [20]. Consequently, better understanding of hemophilic clot formation and stability is needed to develop safer therapeutic options.
Hemostatic clot formation requires the thrombin-mediated cleavage of fibrinogen to fibrin, and activation of the plasma protransglutaminase FXIII-A2B2 (FXIII). FXIII activation involves the thrombin-catalyzed cleavage of activation peptide(s) from FXIII-A subunits to yield FXIII-A′, followed by the calcium-promoted dissociation of FXIII-B to yield activated FXIII (FXIIIa) [21]. FXIIIa introduces ε-N-(γ-glutamyl)-lysyl crosslinks between fibrin γ-chains and α-chains, and between fibrin and antifibrinolytic proteins (e.g. α2-antiplasmin), enhancing clot mechanical and biochemical stability [22–24]. Additionally, FXIIIa-mediated fibrin α-chain crosslinking promotes red blood cell (RBC) retention in clots, and consequently increases clot weight and determines clot composition [25,26].
FXIII activation occurs early during coagulation, but is delayed in hemophilic whole blood [3]. Like FVIII-deficient or FIX-deficient patients, FXIII-deficient patients show intracranial and intramuscular bleeding [27], suggesting that hemophilic bleeding results, at least in part, from delayed or reduced FXIIIa-mediated crosslinking and fibrin stabilization. Several in vitro hemophilia studies have demonstrated that FXIII combined with either rFVIIa [28,29] or FVIII [30] accelerates FXIII activation, increases clot density, decreases clot permeability, and improves clot stability [28–30]. Clinically, Ng et al. [31] administered rFVIIa and FXIII sequentially to a severe hemophilia A patient with refractory bleeding; this approach attenuated bleeding and improved the patient’s clinical status. Collectively, these studies suggest that FXIII cotreatment enhances FXIIIa-mediated fibrin crosslinking; however, this mechanism has not been firmly established.
Herein, we demonstrate that, as compared with untreated and FVIII-treated hemophilia A plasma, FXIII cotreatment accelerates FXIII activation, and increases the rate and amount of fibrin α-chain and α2-antiplasmin crosslinking. Additionally, we used hemophilic whole blood with inhibitors to demonstrate that, as compared with rFVIIa-treated, FEIBA-treated or recombinant B-domain-deleted porcine FVIII (rpFVIII)-treated whole blood, FXIII cotreatment increases whole blood clot weight. Collectively, these findings provide a further mechanistic rationale for FXIII cotreatment in hemophilia.
Materials and methods
Proteins and materials
rpFVIII (Obizur, Shire, Lexington, MA, USA) was purchased from University of North Carolina Hospitals. Recombinant B-domain-deleted human FVIII (rhFVIII) (Xyntha; Pfizer, New York, NY, USA), rFVIIa (Novo Nordisk, Plainsboro, NJ, USA) and FEIBA (Shire, Lexington, MA, USA) were gifts from P. Monahan, University of North Carolina at Chapel Hill. Plasma-derived FXIII (Corifact; CSL Bering, King of Prussia, PA, USA) was a gift from the manufacturer. Factor concentrates were reconstituted according to the manufacturers’ directions, aliquoted, snap-frozen with liquid nitrogen, and stored at −80 °C. Bovine serum albumin was from Sigma-Aldrich (St Louis, MO, USA). Lipidated tissue factor (Innovin) was from Siemens (Newark, DE, USA). Polyclonal rabbit anti-human fibrinogen antibody was from Dako (Carpinteria, CA, USA). Polyclonal sheep anti-human α2-antiplasmin antibody was from Affinity Biologicals (Ancaster, Ontario, Canada). Human thrombin, FXIII and polyclonal sheep anti-human FXIII-A antibody were from Enzyme Research Laboratories (South Bend, IN, USA). Thrombin fluorogenic substrate (Z-glycine-arginine-AMC) and calibrator (α2-macroglobulin/thrombin) were from Diagnostica Stago (Parsippany, NJ, USA).
FXIII-A′ loading control was generated by reacting 20 μg mL−1 FXIII with 20 nM human thrombin in the presence of calcium for 120 min at 37 °C, followed by quenching with 50 mM dithiothreitol, 12.5 mM EDTA, and 8 M urea. The sample was diluted with 2.5% β-mercaptoethanol and Laemmli dye (Boston BioProducts, Boston, MA, USA), boiled at 95 °C, and stored at −20 °C.
Hemophilic patient plasma and whole blood samples
FVIII-deficient platelet-poor plasma (PPP) was from HRF (Raleigh, NC, USA). Whole blood samples were obtained from patients with congenital or acquired FVIII deficiency undergoing treatment at the University of North Carolina Hemophilia Treatment Center. Blood was taken from hemophilic patients if they had a documented FVIII inhibitor and had not received hemostatic therapy (FVIII, rFVIIa, FEIBA, or rpFVIII) for at least 24 h. Phlebotomy was performed on consenting patients in accordance with the Declaration of Helsinki and the University of North Carolina Institutional Review Board. Blood was collected by either antecubital venipuncture or central venous access, if present, into 0.105 M sodium citrate, pH 5.5 (10% v/v, final concentration). Clinical parameter assessment was performed at the University of North Carolina Hospital McLendon Clinical Laboratory (Table 1). Most patients had acquired hemophilia (n = 9, 75%; Table 1) with a median human FVIII inhibitor titer of 53 Bethesda units (range 2–554) and a median porcine inhibitor titer of 0.9 Bethesda units (range < 0.4–5; Table 1).
Table 1.
Clinical characteristics of hemophilic whole blood donors
| Reference range | Donors (n = 12) | |
|---|---|---|
| Age (years) | – | 61.8 ± 4.0 |
| Sex (% female) | – | 58.3 |
| White blood cells (× 109 L−1) | 4.5–11 | 8.8 ± 1.1 |
| Red blood cells (× 106 μL−1) | 4.2–5.4 | 3.9 ± 0.3 |
| Hemoglobin (g dL−1) | 13.5–17.5 | 10.7 ± 0.6 |
| Hematocrit (%) | 36–46 | 34.2 ± 1.8 |
| Platelets (× 109 L−1) | 150–400 | 295.9 ± 26.9 |
| Fibrinogen (mg dL−1) | 208–409 | 395.4 ± 35.5* |
| APTT (s) | 26.0–37.3 | 93.6 ± 7.9 |
| FVIII activity (%) | 54–161 | 3.6 ± 1.4 |
| FVIII inhibitor (Bethesda units) | ≤ 0.4 | 126.4 ± 48 |
| Porcine FVIII inhibitor (Bethesda units) | ≤ 0.4 | 1.5 ± 0.4† |
APTT, activated partial thromboplastin time. Mean ± standard error of the mean.
Value for n = 8 subjects.
Value for n = 11 subjects.
Plasma clot formation and detection of FXIII activation and crosslinked fibrin and α2-antiplasmin
FVIII-deficient PPP was incubated with buffer (HEPES-buffered saline [HBS]; 20 mM HEPES, pH 7.4, 150 mM NaCl), 2 IU mL−1 FXIII, 1 IU mL−1 rhFVIII or a combination of FXIII and rhFVIII (final concentrations of 2 IU mL−1 and 1 IU mL−1, respectively) for 2 min at 37 °C. Plasma clot formation was initiated by addition of CaCl2 (10 mM, final concentration) and tissue factor (0.5 pM, final concentration) at 37 °C. Clotting was terminated at the indicated time points by addition of quenching solution (50 mM dithiothreitol, 12.5 mM EDTA, 8 M urea); time zero had quenching solution present at the start. Samples were incubated at 60 °C for 1 h with occasional agitation. Samples were reduced, boiled, and separated on 7.5% or 10% Tris-glycine gels, and transferred to polyvinylidene difluoride membranes. Membranes were blocked with Odyssey Blocking Buffer (Licor, Lincoln, NE, USA), and probed with primary antibodies against human FXIII-A, fibrinogen, or α2-antiplasmin. After washing, membranes were incubated with fluorescence-labeled secondary antibodies, washed, and scanned with a GE Typhoon FLA-9000 Imager (GE Healthcare, Chicago, IL, USA). Band intensity (arbitrary units [AU]) was measured by densitometry with IMAGE J (version 1.5i). For FXIII, the FXIII-A′ band was normalized to an FXIII-A′ loading control. For fibrin crosslinking, bands were normalized to corresponding Bβ + β-chain. For α2-antiplasmin, crosslinked α2-antiplasmin bands were normalized to total α2-antiplasmin band at t = 0. Maximum rates of FXIII-A′, γ–γ dimer, high molecular weight (HMW) species and α2-antiplasmin formation were calculated by determining the slope of the points showing the maximum increase for each experiment. For this calculation, at least three points were used for all fits except for measurements of α2-antiplasmin crosslinking in the presence of FVIII + FXIII (two points).
Thrombin generation
FVIII-deficient PPP was spiked with buffer (HBS), 2 IU mL−1 FXIII, 1 IU mL−1 rhFVIII, or rhFVIII + FXIII (final concentrations of 2 IU mL−1 and 1 IU mL−1, respectively). Plasma samples (80 μL) were then added to 20 μL of PPP Reagent Low (final concentrations: 1 pM tissue factor and 4 μM lipid) or Calibration Reagent. After 10 min at 37 °C, reactions were initiated by the addition of 20 μL of fluorogenic substrate with calcium (final concentrations: 416 μM fluorogenic substrate and 16 mM CaCl2). Thrombin generation was detected with a Fluoroskan Ascent fluorometer (Thermo Labsystems, Waltham, MA, USA), and analyzed with Thrombinoscope software v5.0.0.742 (Stago, Parsippany, NJ, USA).
Thromboelastography (TEG)
Citrated whole blood was pretreated for 30 min at 37 °C, with or without protein buffer (0.75% w/v bovine serum albumin in 0.9% sodium chloride solution), FXIII (2 IU mL−1, final concentration), rpFVIII (1 IU mL−1, final concentration), rFVIIa (25 nM, final concentration), or FEIBA (1 IU mL−1, final concentration), alone or with FXIII. Clot formation was initiated in 340 μL of whole blood by addition of 20 μL of tissue factor + CaCl2 (final concentrations of 0.4 pM and 10 mM, respectively), and measured with the TEG Hemostasis Analyzer System 5000 (Haemonetics, Braintree, MA, USA). TEG parameters were calculated with the manufacturer’s software: clot time (R, min), i.e. time between the start of the assay and the beginning of clot formation; clot formation (K, min), i.e. time from the beginning of clot formation until clot firmness reached 20 mm in amplitude; maximum amplitude (MA, mm), i.e. the maximum dynamic properties of fibrin and platelet bonding; and elasticity (G, dynes cm−2), i.e. clot strength at MA defined as G = 5000 × MA/100 − MA.
Whole blood clot contraction
Citrated whole blood was pretreated for 30 min at 37 °C, with or without protein buffer, FXIII (2 IU mL−1, final concentration), rpFVIII (1 IU mL−1, final concentration), rFVIIa (25 nM, final concentration), or FEIBA (1 IU mL−1, final concentration), alone or with FXIII. Clotting was triggered in recalcified (10 mM, final concentration) whole blood by addition of tissue factor (Innovin diluted 1 : 12 000; 1 pM, final concentration). Clot contraction proceeded at 37 °C for 120 min in siliconized multiwell plates. Contracted clots were removed and weighed.
Statistical methods
Descriptive statistics (mean, median, standard deviation, and standard error of the mean [SEM], normality) were calculated for each experiment. For densitometry values, two-way ANOVA was used to compare time and treatment. For other comparisons, repeated-measures ANOVA with Holm–Sidak post hoc tests for between-group analysis were used, with P < 0.05 being considered significant. Calculations were performed with GRAPHPAD v 7.02 (Synergy Software, Reading, PA, USA).
Results
As compared with FVIII treatment alone, FXIII cotreatment accelerates FXIII activation without increasing thrombin generation
Hemophilia is associated with delayed thrombin-dependent FXIII activation during plasma clot formation [3]. This observation suggests that mechanisms that enhance FXIII activation and/or activity may improve hemophilic clot quality. We tested this hypothesis by adding FVIII and FXIII, alone and in combination, to hemophilic plasma, and measuring the activation and level of FXIII-A′ by western blotting. As compared with no treatment or FVIII treatment (alone), FXIII, alone or in combination with FVIII, increased the baseline (t = 0) FXIII-A level ~ 1.8-fold (Fig. 1A). As compared with untreated hemophilic plasma, FVIII treatment (alone) increased the maximum rate of FXIII activation (0.01 ± 0.01 AU min−1 versus 0.20 ± 0.05 AU min−1, respectively, mean ± SEM; Fig. 1C) and increased the FXIII-A′ level (Fig. 1A, B). As compared with FVIII treatment (alone), FVIII + FXIII cotreatment significantly increased the maximum rate of FXIII activation (0.20 ± 0.05 AU min−1 versus 0.33 ± 0.05 AU min−1, respectively, P < 0.01; Fig. 1C), but did not increase the FXIII-A′ level (Fig. 1A, B). To determine whether accelerated FXIII activation was attributable to increased thrombin generation, we measured thrombin generation in hemophilic plasma treated with FVIII and FXIII, alone and in combination. As compared with untreated hemophilic plasma, FVIII treatment (alone) decreased the time to peak, and increased the rate (velocity index), thrombin peak, and endogenous thrombin potential (Table 2). However, addition of FXIII (alone or with FVIII) did not alter thrombin generation (Table 2). Collectively, these data indicate that cotreatment of hemophilic plasma with FXIII and FVIII accelerated FXIII activation without increasing thrombin generation.
Fig. 1.

As compared with FVIII treatment alone, FXIII cotreatment accelerates the maximum rate of FXIII activation. (A) Representative western blots for FXIII-A after tissue factor-initiated clotting in recalcified hemophilic plasma treated with buffer (HEPES-buffered saline), 2 IU mL−1 FXIII, 1 IU mL−1 FVIII, or FVIII + FXIII (final concentrations of 1 IU mL−1 and 2 IU mL−1, respectively). Cleavage of activation peptide(s) results in the formation of FXIII-A′ (lower band). (B) Quantification of FXIII activation (FXIII-A′) over time relative to FXIII-A′ loading control; n = 3–7 samples per time point, mean ± standard error of the mean (SEM), arbitrary units (AU). Two-way ANOVA was used to compare time and treatment. (C) Maximum rate of FXIII activation (mean ± SEM, AU min−1) for n = 7 samples. Treatments were compared by the use of repeated measures ANOVA with the Holm–Sidak multiple comparisons test.
Table 2.
Thrombin generation parameters
| Buffer (n = 7) | FXIII (n = 7) | FVIII (n = 7) | FVIII + FXIII (n = 7) | |
|---|---|---|---|---|
| Lag time (min) | 12.0 ± 2.9 | 10.2 ± 1.6 | 8.1 ± 0.6 | 8.6 ± 0.8 |
| Time to peak (min) | 28.4 ± 3.1 | 25.0 ± 1.8 | 13.6 ± 0.6* | 14.3 ± 1.1* |
| Velocity index (nM min−1) | 0.29 ± 0.1 | 0.44 ± 0.7 | 14.0 ± 2.5* | 13.1 ± 2.4* |
| Peak thrombin (nM) | 4.5 ± 0.6 | 6.1 ± 1.0 | 68.0 ± 7.0* | 68.7 ± 9.4* |
| ETP (nM min) | 95.5 ± 9.5 | 117.5 ± 16.0 | 638.1 ± 34.6* | 684.1 ± 70.5* |
ETP, endogenous thrombin potential. Mean ± standard error of the mean. P < 0.05 as compared with buffer-treated samples via repeated-measure ANOVA with Dunn’s or Holm–Sidak multiple comparisons for non-normal and normal distributions.
As compared with FVIII treatment alone, FXIII cotreatment increases fibrin crosslinking
Despite improving fibrin formation, increasing clot density, and decreasing clot permeability, rFVIIa treatment does not fully restore fibrin crosslinking rates in hemophilic plasma [2,6,7]. These results suggest that, despite a normalized fibrin structure, the FXIII-dependent effects on fibrin remain insufficient, and may be improved with FXIII cotreatment. To test this premise, we added FVIII and FXIII, alone and in combination, to hemophilic plasma, and measured fibrin crosslinking by western blotting. As compared with untreated hemophilic plasma, FXIII (alone) did not increase the formation rate or levels of crosslinked fibrin species (Fig. 2). However, as compared with untreated hemophilic plasma, FVIII treatment (alone) increased the maximum rate of γ–γ dimer formation (0.004 ± 0.001 AU min−1 versus 0.009 ± 0.003 AU min−1, mean ± SEM; Fig. 2A–C). Furthermore, as compared with FVIII alone, FVIII + FXIII cotreatment increased the maximum rate of γ–γ dimer formation (0.009 ± 0.003 AU min−1 versus 0.017 ± 0.004 AU min−1, respectively; Fig. 2A–C) and significantly increased overall γ–γ dimer level (0.48 ± 0.15 AU versus 0.98 ± 0.20 AU at 120 min, P < 0.01; Fig. 2A, B). As compared with untreated hemophilic plasma, FVIII (alone) increased the maximum rate of HMW species formation (0.002 ± 0.001 AU min−1 versus 0.010 ± 0.004 AU min−1, respectively; Fig. 2A, D–E). Importantly, as compared with FVIII alone, FVIII + FXIII cotreatment significantly increased the maximum rate of HMW species formation (0.01 ± 0.004 AU min−1 versus 0.03 ± 0.007 AU min−1, respectively, P < 0.02; Fig. 2E) and significantly increased overall HMW species levels (0.67 ± 0.20 AU versus 1.60 ± 0.30 AU at 120 min, respectively, P < 0.01; Fig. 2A, D). Combined, these data indicate that cotreatment of hemophilic plasma with FXIII and FVIII increases the rate of fibrin formation and the amount of fibrin crosslinking.
Fig. 2.

As compared with FVIII treatment alone, FXIII cotreatment increases fibrin crosslinking. (A) Representative western blots for fibrin (ogen) after tissue factor-initiated clotting in recalcified hemophilic plasma treated as in Fig. 1. (B) Quantification of γ–γ dimer normalized to Bβ + β-chain. (C) Maximum rate of γ–γ formation (mean ± standard error of the mean [SEM], n = 7, arbitrary units [AU] min−1). (D) Quantification of high molecular weight (HMW) species normalized to Bβ + β-chain. (E) Maximum rate of HMW species formation (mean ± SEM, n = 7, AU min−1). For (B) and (D), two-way ANOVA with the Holm–Sidak multiple comparisons test was used to compare time and treatment. *P < 0.05 versus buffer-treated, FXIII-treated and FVIII-treated samples. For (C) and (E), treatments were compared by the use of repeated measures ANOVA with the Holm–Sidak multiple comparisons test.
As compared with FVIII treatment alone, FXIII cotreatment accelerates α2-antiplasmin crosslinking
Previous studies showed that coadministration of supraphysiologic concentrations of FXIII with rFVIIa or plasma-derived FVIII enhances hemophilic clot stability (area under the TEG curve) [32,33]. In addition to crosslinking fibrin, FXIIIa also crosslinks α2-antiplasmin to fibrin; this reaction is thought to retain a2-antiplasmin in clots during clot contraction and inhibit fibrinolysis. To test this premise, we added FVIII and FXIII, alone and in combination, to hemophilic plasma, and measured α2-antiplasmin crosslinking by western blotting. As compared with untreated hemophilic plasma, FXIII (alone) and FVIII (alone) increased the rate of α2-antiplasmin crosslinking (0.001 ± 0.0003 AU min−1 versus 0.002 ± 0.001 AU min−1 versus 0.004 ± 0.001 AU min−1, P < 0.02; Fig. 3A). As compared with FVIII-treated hemophilic plasma, FXIII cotreatment significantly increased the amount of fully crosslinked α2-antiplasmin (0.05 ± 0.01 AU versus 0.26 ± 0.07 AU at 30 min, P < 0.0001; Fig. 3B) and accelerated the rate of α2-antiplasmin crosslinking (0.013 ± 0.002 AU min−1, P < 0.04; Fig. 3C). Combined, these data indicate that FXIII cotreatment accelerates and increases the incorporation of antifibrinolytic α2-antiplasmin into the fibrin clot, and provides a rationale for the enhanced clot stability reported in previous studies [32,33].
Fig. 3.

As compared with FVIII treatment alone, FXIII cotreatment accelerates and increases α2-antiplasmin crosslinking. (A) Representative western blots for α2-antiplasmin crosslinking in recalcified hemophilic plasma treated as in Fig. 1. (B) Quantification of crosslinked α2-antiplasmin normalized to total α2-antiplasmin at t = 0 min (mean ± standard error of the mean [SEM], n = 7). Symbols are: buffer, closed circles; + FXIII, half-filled boxes; + FVIII, open triangles; + FVIII + FXIII, closed inverted triangles. *P < 0.05 versus buffer-treated, FXIII-treated and FVIII-treated samples. Two-way ANOVA with the Holm–Sidak multiple comparisons test was used to compare time and treatment. (C) Maximum rate of α2-antiplasmin crosslinking (mean ± SEM, n = 7, arbitrary units [AU] min−1). Treatments were compared by the use of repeated measures ANOVA with the Holm–Sidak multiple comparisons test.
As compared with standard hemostatic treatments alone, FXIII cotreatment increases clot weight in hemophilic whole blood with inhibitors
Finally, we investigated the effects of FXIII cotreatment on clotting parameters in whole blood from hemophilic patients with inhibitors. As compared with normal individuals, hemophilic patients have decreased plasma and whole blood clot weight [2,3], which has been attributed to decreased clot formation and fibrin mass [3]. Collectively, these results suggest that improving FXIIIa-mediated fibrin crosslinking may increase hemophilic clot formation and quality. To test this premise, we first added hemostatic agents (rFVIIa, FEIBA, or rpFVIII) alone or in combination with FXIII to hemophilic whole blood with inhibitors, and then measured clot formation by TEG. As compared with untreated hemophilic whole blood, treatment with hemostatic agents (rFVIIa, FEIBA, or rpFVIII), but not FXIII (alone), decreased R and K, and increased angle values (Table 3). However, as compared with the respective hemostatic agent(s) alone, FXIII cotreatment did not significantly change any clot formation parameters assessed by TEG (Table 3).
Table 3.
Effect of hemostatic therapies with and without FXIII cotreatment on thromboelastography parameters
| Healthy controls (n = 4) | Buffer (n = 10) | FXIII (n = 10) | rFVIIa (n = 3) | rFVIIa + FXIII (n = 3) | FEIBA (n = 4) | FEIBA + FXIII (n = 4) | rpFVIII (n = 5) | rpFVIII + FXIII (n = 5) | |
|---|---|---|---|---|---|---|---|---|---|
| Clot time (R, min) | 13.1 ± 1.5 | 38.2 ± 9.1 | 38.3 ± 10.4 | 5.3 ± 1.1* | 5.6 ± 0.9* | 7.8 ± 2.5* | 7.7 ± 2.3* | 30.6 ± 6.8 | 22.8 ± 4.9 |
| Clot formation (K, min) | 5.5 ± 0.9 | 17.4 ± 5.1 | 19.7 ± 6.4 | 2.4 ± 0.4* | 2.6 ± 0.6* | 2.3 ± 0.5* | 1.85 ± 0.3* | 12.6 ± 4.1 | 10.1 ± 3.7 |
| Angle (°) | 35.9 ± 6.3 | 20.9 ± 4.3 | 20.4 ± 4.6 | 59.6 ± 4.0* | 58.3 ± 5.4* | 60.7 ± 5.2* | 65.2 ± 3.6* | 25.6 ± 6.6 | 29.1 ± 7.0 |
| Maximum amplitude (MA, mm) | 53.7 ± 5.9 | 56.9 ± 7.0 | 59.5 ± 6.9 | 69.8 ± 1.3 | 68.6 ± 0.9 | 63.8 ± 4.6 | 71.2 ± 3.6 | 59.9 ± 5.0 | 63.0 ± 543 |
| Elasticity (G, dynes cm−2) | 6482 ± 1837 | 8180 ± 1712 | 8788 ± 1571 | 11 644 ± 6913 | 10 945 ± 435.5 | 9146 ± 2781 | 13 424 ± 2909 | 8819 ± 1547 | 9983 ± 2629 |
FEIBA, plasma-derived anti-inhibitor coagulant complex; rFVIIa, recombinant activated FVII; rpFVIII, recombinant B-domain-deleted porcine FVIII. Mean ± standard error of the mean. P < 0.05 as compared with buffer-treated samples via ANOVA with Dunn’s or Holm–Sidak multiple comparisons for non-normal and normal distributions.
Recently, our laboratory determined that FXIIIa-mediated α-chain crosslinking promotes RBC retention and increases the weight of contracted whole blood clots [25,26]. Given the effect of FXIII cotreatment on fibrin α-chain crosslinking, we tested the effect of FXIII cotreatment on contracted clot weight. In these assays, as compared with untreated hemophilic whole blood, FXIII (alone) did not increase whole blood clot weight (mean clot weight of 42.7 ± 5.0 mg versus 50.5 ± 6.6 mg, respectively, n = 12). As compared with untreated hemophilic whole blood, rFVIIa (alone) increased clot weight (46.0 ± 6.3 mg versus 61.9 ± 5.3 mg, respectively, n = 8; Fig. 4A), but FEIBA or rpFVIII (alone) did not (FEIBA, 40.9 ± 7.2 mg versus 54.1 ± 2.4 mg, n = 5 [Fig. 4B]; rpFVIII, 35.3 ± 5.7 mg versus 42.4 ± 6.0 mg, n = 8 [Fig. 4C]). Interestingly, as compared with treatment with the respective hemostatic agent(s) alone, FXIII cotreatment with rFVIIa, FEIBA or rpFVIII significantly increased clot weight (to 73.1 ± 6.7 mg, 64.9 ± 2.5 mg and 57.6 ± 7.1 mg, respectively; Fig. 4A–C). Combined, these data suggest that FXIII cotreatment with rFVIIa, FEIBA or rpFVIII does not alter clot formation parameters assessed by TEG, but does increase contracted whole blood clot weight and can do so in the presence of platelets and inhibitors.
Fig. 4.
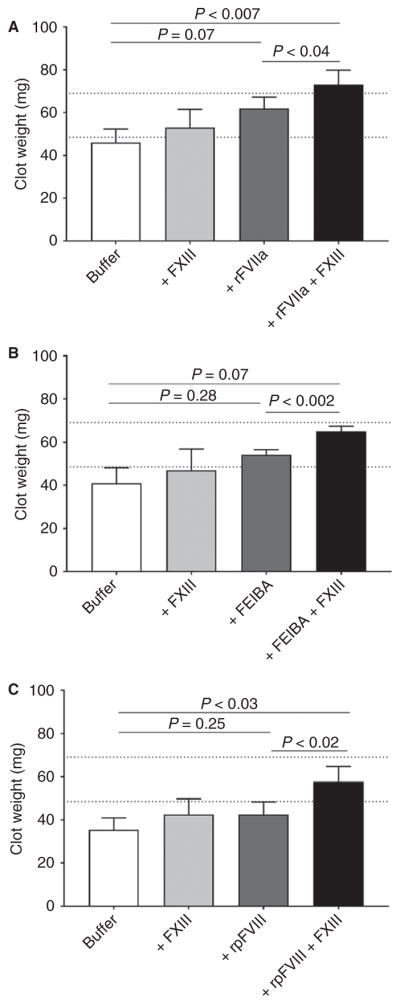
As compared with hemostatic therapies alone, FXIII cotreatment increases whole blood clot weight. Hemophilic whole blood was treated with protein buffer (0.75% w/v bovine serum albumin in 0.9% sodium chloride solution), FXIII (2 IU mL−1) or (A) recombinant activated FVII (rFVIIa) (25 nM), (B) plasma-derived anti-inhibitor coagulant complex (FEIBA) (1 IU mL−1), or (C) recombinant B-domain-deleted porcine FVIII (rpFVIII) (1 IU mL−1), with or without FXIII, and clotted with tissue factor and recalcification. Contracted clots were weighed. Dashed lines indicate ranges of clot weights for n = 4 non-hemophilic donors. Bars show the mean ± standard error of them mean for n = 5–8 individual hemophilic donors per treatment. Treatments were compared by the use of repeated measures ANOVA with the Holm–Sidak multiple comparisons test.
Discussion
Due to insufficient thrombin generation, hemophilic patients form an unstable initial clot with an abnormal fibrin structure [1–7]. Hemostatic agents increase thrombin generation, leading to increased fibrin formation, increased fibrin network density, and thinner fibrin fibers [4,6,7]. However, despite hemostatic therapies, some hemophilic patients experience prolonged bleeding. Given the well-established ability of FXIIIa to stabilize clots, FXIII treatment in hemophilia has been previously evaluated in vitro and in vivo [28–31]. However, these studies did not directly assess the effects of FXIII cotreatment on fibrin or α2-antiplasmin crosslinking, or address the contribution of FXIIIa to contracted whole blood clot properties. Therefore, we sought to extend the published literature by determining the effects of FXIII cotreatment on FXIII activation, crosslinking, and clot weight. Herein, we demonstrated that FXIII cotreatment accelerated FXIII activation, which increased fibrin and α2-antiplasmin crosslinking. We additionally showed that, in hemophilic samples with inhibitors, FXIII cotreatment increased whole blood clot weight. Collectively, these data provide a further mechanistic rationale for FXIII cotreatment in hemophilia.
Our data show that the effects of FXIII on clot crosslinking and composition occur in the presence, but not in the absence, of conventional hemostatic agents. This finding probably reflects the indispensable role of thrombin in both the generation of fibrin and the activation of FXIII, and previous observations that both fibrin formation and FXIII activation are delayed in hemophilia [3]. Thus, in the absence of sufficient procoagulant activity, the addition of even supraphysiologic concentrations of FXIII, alone, to hemophilic plasma fails to improve clot formation. However, in the presence of conventional hemostatic agents that stimulate thrombin generation, both fibrin formation and FXIII activation are increased, and the addition of supraphysiologic concentrations of FXIII further accelerates FXIII activation, and consequently enhances fibrin and α2-antiplasmin crosslinking.
FXIIIa-mediated crosslinking of fibrin γ-chains and α-chains and α2-antiplasmin is essential for clot biophysical and biochemical stability. FXIIIa-mediated γ-chain crosslinks stabilize fibrin fiber branches, and α-chain crosslinks contribute to clot stiffness and resistance to lysis [23,24]. Therefore, our data showing that cotreatment of hemophilic plasma with FXIII increases γ-chain and α-chain crosslinks complement previous findings that FXIII cotreatment reduces clot permeability and increases whole blood clot stability [28–30]. FXIIIa-mediated α-chain crosslinking also promotes RBC retention in contracted clots [25,26], and we observed here that FXIII cotreatment increased the RBC content of hemophilic clots. Increased RBC retention in clots may further enhance clot stability, as RBCs decrease clot permeability, reduce plasmin generation, and increase resistance to lysis [34,35]. Furthermore, during FXIIIa-mediated clot contraction, FXIII crosslinks α2-antiplasmin to fibrin α-chains [32,33,36]. The FXIIIa-mediated crosslinking of α2-antiplasmin within a contracted clot is essential for clot resistance to fibrinolysis [37,38]. Therefore, our data showing that FXIII cotreatment increases the rate and amount of α2-antiplasmin crosslinking supports previous TEG data showing that, in the presence of tissue-type plasminogen activator, FXIII cotreatment with rFVIIa or rFVIII increases whole blood clot stability [29,30]. Collectively, these data suggest that cotreatment with FXIII augments hemostasis by increasing FXIIIa-mediated fibrin and α2-antiplasmin crosslinking. These data provide a mechanistic rationale for employing FXIII cotreatment strategies to manage refractory bleeding in hemophilic patients.
FXIII cotreatment offers several therapeutic advantages. First, FXIII has a relatively long half-life (9–19 days) that is suitable for prophylactic dosing [39]. Second, FXIII may work for both acute bleeds [40] and in postoperative patients, in whom FXIII levels are often reduced [41]. Third, FXIII treatment does not increase thrombin generation, and has not been associated with thromboembolic events [39], reducing concerns about thrombotic risk if FXIII is used with hemostatic agents (FVIII, rFVIIa, FEIBA, or rpFVIII). Fourth, although FXIII requires intravenous administration, it does not require renal or hepatic metabolism, which is favorable in older patients with comorbidities. Consequently, FXIII cotreatment with hemostatic therapies (FVIII/FIX, rFVIIa, FEIBA, or rpFVIII) is an attractive treatment strategy during refractory bleeding.
Our study has potential limitations. First, FVIII inhibitors have variable inhibitory kinetics based on the targeted FVIII epitope, which may affect clot formation in different conditions [42]. However, our patient cohort was small and heterogeneous with regard to baseline FVIII activity and human and porcine FVIII inhibitor titers, and was therefore underpowered to establish whether interactions exist between inhibitor titers and response to FXIII cotreatment. Second, we were not able to measure thrombin generation in the hemophilic whole blood samples. However, FXIII did not alter thrombin generation in plasma, and the whole blood samples had normal platelet counts and showed no change in the clot time in TEG assays, suggesting that FXIII did not alter thrombin generation in whole blood assays. Third, our experiments focused on the effects of FXIII cotreatment in hemophilia A; however, hemophilia B patients can also develop inhibitors and experience refractory bleeding, despite hemostatic therapy. Accordingly, cotreatment with hemostatic agents and supraphysiologic concentrations of FXIII may also augment hemostasis in these patients. This premise will require direct testing in future studies. Fourth, our study, like previous studies, lacks supportive experiments obtained in preclinical animal models. One previous study used a saphenous vein bleeding model to test the effect of FXIII in FIX-deficient mice, but failed to detect an improved time to clot formation [30]. The lack of effect may reflect insensitivity of this model to FXIIIa-mediated effects on clot composition or resistance to fibrinolysis. Although venous thrombosis models have demonstrated sensitivity to FVIII and FXIII separately [25,43,44], the relevance of intravascular thrombosis models to hemostasis following vascular injury is unclear. Further in vivo study awaits the development of hemostasis models that are sensitive to the effects of both hemostatic agents and FXIII activity.
In conclusion, our results demonstrate that FXIII cotreatment accelerates FXIII activation, resulting in increased α-chain-rich HMW crosslinked fibrin species and α2-antiplasmin crosslinking, and increases the weight of contracted hemophilic whole blood clots. Demonstration of these functional effects is an important next step in determining the operant mechanism. Moreover, demonstration of these effects in whole blood from inhibitor patients – the individuals who are most likely to receive this therapy during refractory bleeding episodes – fills an important gap in the clinical development of this approach. Collectively, these findings provide further a mechanistic rationale for the use of FXIII with hemostatic agents in hemophilia.
Essentials.
Factor XIII (FXIII)-mediated fibrin crosslinking is delayed in hemophilia.
We determined effects of FXIII cotreatment with hemostatic agents on clot parameters.
FXIII cotreatment accelerated FXIII activation and crosslinking of fibrin and α2-antiplasmin.
These data provide biochemical rationale for FXIII cotreatment in hemophilia.
Acknowledgments
This study was supported by research funding from the National Institutes of Health (R01HL126974 to A. S. Wolberg and T32HL007149 to the University of North Carolina and J. D. Beckman) and a 2016 Mentored Research Award from the Hemostasis and Thrombosis Research Society by CSL Behring to J. D. Beckman.
The authors thank J. R. Byrnes, S. Kattula, J. Mickelson and D. M. Monroe for helpful discussions, and A. Ma, N. S. Key and the staff of the University of North Carolina Hemophilia Treatment Center for their assistance in obtaining patient samples.
Footnotes
Addendum
J. D. Beckman designed and performed experiments, analyzed results, and wrote the manuscript. L. H. Holle performed experiments and reviewed the manuscript. A. S. Wolberg supervised experiments and data analysis, and wrote the manuscript.
Disclosure of Conflict of Interests
CSL Behring provided funds to the Hemostasis and Thrombosis Research Society for the Mentored Research Award program, but had no input into the selection of awardees. CSL Behring had no direct interaction with the authors on the study design or results. The authors state that they have no other conflict of interest.
References
- 1.Cawthern KM, van’t Veer C, Lock JB, Di Lorenzo ME, Branda RF, Mann KG. Blood coagulation in hemophilia A and hemophilia C. Blood. 1998;91:4581–92. [PubMed] [Google Scholar]
- 2.Butenas SS, Brummel KE, Branda RF, Paradis SG, Mann KG. Mechanism of factor VIIa-dependent coagulation in hemophilia blood. Blood. 2002;99:923–30. doi: 10.1182/blood.v99.3.923. [DOI] [PubMed] [Google Scholar]
- 3.Brummel-Ziedins KE, Branda RF, Butenas S, Mann KG. Discordant fibrin formation in hemophilia. J Thromb Haemost. 2009;7:825–32. doi: 10.1111/j.1538-7836.2009.03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dargaud Y, Lienhart A, Negrier C. Prospective assessment of thrombin generation test for dose monitoring of bypassing therapy in hemophilia patients with inhibitors undergoing elective surgery. Blood. 2010;116:5734–7. doi: 10.1182/blood-2010-06-291906. [DOI] [PubMed] [Google Scholar]
- 5.Sixma JJ, van den Berg A. The haemostatic plug in haemophilia A: a morphological study of haemostatic plug formation in bleeding time skin wounds of patients with severe haemophilia A. Br J Haematol. 1984;58:741–53. doi: 10.1111/j.1365-2141.1984.tb06121.x. [DOI] [PubMed] [Google Scholar]
- 6.He S, Blomback M, Jacobsson Ekman G, Hedner U. The role of recombinant factor VIIa (FVIIa) in fibrin structure in the absence of FVIII/FIX. J Thromb Haemost. 2003;1:1215–19. doi: 10.1046/j.1538-7836.2003.00242.x. [DOI] [PubMed] [Google Scholar]
- 7.Wolberg AS, Allen GA, Monroe DM, Hedner U, Roberts HR, Hoffman M. High dose factor VIIa enhances clot stability in a model of hemophilia B. Br J Haematol. 2005;131:645–55. doi: 10.1111/j.1365-2141.2005.05820.x. [DOI] [PubMed] [Google Scholar]
- 8.Peyvandi F, Mannucci PM, Garagiola I, El-Beshlawy A, Elalfy M, Ramanan V, Eshghi P, Hanagavadi S, Varadarajan R, Karimi M, Manglani MV, Ross C, Young G, Seth T, Apte S, Nayak DM, Santagostino E, Mancuso ME, Sandoval Gonzalez AC, Mahlangu JN, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374:2054–64. doi: 10.1056/NEJMoa1516437. [DOI] [PubMed] [Google Scholar]
- 9.Franchini M, Santoro C, Coppola A. Inhibitor incidence in previously untreated patients with severe haemophilia B: a systematic literature review. Thromb Haemost. 2016;116:201–3. doi: 10.1160/TH16-02-0116. [DOI] [PubMed] [Google Scholar]
- 10.Kessler CM, Ma AD, Al-Mondhiry HA, Gut RZ, Cooper DL. Assessment of acquired hemophilia patient demographics in the United States: the Hemostasis and Thrombosis Research Society Registry. Blood Coagul Fibrinolysis. 2016;27:761–9. doi: 10.1097/MBC.0000000000000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He S, Ezban M, Bark N, Persson E, Hedner U. Fibrin gel structure obtained with a FVIIa analogue with enhanced FX-activating potential in haemophilia. Thromb Haemost. 2009;102:790–2. doi: 10.1160/TH09-02-0135. [DOI] [PubMed] [Google Scholar]
- 12.Kruse-Jarres R, St-Louis J, Greist A, Shapiro A, Smith H, Chowdary P, Drebes A, Gomperts E, Bourgeois C, Mo M, Novack A, Farin H, Ewenstein B. Efficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Haemophilia. 2015;21:162–70. doi: 10.1111/hae.12627. [DOI] [PubMed] [Google Scholar]
- 13.Croteau SE, Abajas YL, Wolberg AS, Nielsen BI, Marx GR, Baird CW, Neufeld EJ, Monahan PE. Recombinant porcine factor VIII for high-risk surgery in paediatric congenital haemophilia A with high-titre inhibitor. Haemophilia. 2017;22:e93–8. doi: 10.1111/hae.13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varadi K, Tangada S, Loeschberger M, Montsch P, Schrenk G, Ewenstein B, Turecek PL. Pro- and anticoagulant factors facilitate thrombin generation and balance the haemostatic response to FEIBA((R)) in prophylactic therapy. Haemophilia. 2016;22:615–24. doi: 10.1111/hae.12873. [DOI] [PubMed] [Google Scholar]
- 15.Zhou ZY, Hay JW. Efficacy of bypassing agents in patients with hemophilia and inhibitors: a systematic review and meta-analysis. Clin Ther. 2012;34:434–45. doi: 10.1016/j.clinthera.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Ingerslev J, Sorensen B. Parallel use of by-passing agents in haemophilia with inhibitors: a critical review. Br J Haematol. 2011;155:256–62. doi: 10.1111/j.1365-2141.2011.08854.x. [DOI] [PubMed] [Google Scholar]
- 17.Valentino LA, Allen G, Gill JC, Hurlet A, Konkle BA, Leissinger CA, Luchtman-Jones L, Powell J, Reding M, Stine K. Case studies in the management of refractory bleeding in patients with haemophilia A and inhibitors. Haemophilia. 2013;19:e151–66. doi: 10.1111/hae.12095. [DOI] [PubMed] [Google Scholar]
- 18.Valentino LA, Holme PA. Should anti-inhibitor coagulant complex and tranexamic acid be used concomitantly? Haemophilia. 2015;21:709–14. doi: 10.1111/hae.12723. [DOI] [PubMed] [Google Scholar]
- 19.Neufeld EJ, Negrier C, Arkhammar P, Benchikh el Fegoun S, Duelund Simonsen M, Rosholm A, Seremetis SV. Safety update on the use of recombinant activated factor VII in approved indications. Blood Rev. 2015;29(Suppl 1):S34–41. doi: 10.1016/S0268-960X(15)30006-0. [DOI] [PubMed] [Google Scholar]
- 20.Walsh CE, Soucie JM, Miller CH. Impact of inhibitors on hemophilia A mortality in the United States. Am J Hematol. 2015;90:400–5. doi: 10.1002/ajh.23957. [DOI] [PubMed] [Google Scholar]
- 21.Lorand L, Gray AJ, Brown K, Credo RB, Curtis CG, Domanik RA, Stenberg P. Dissociation of the subunit structure of fibrin stabilizing factor during activation of the zymogen. Biochem Biophys Res Commun. 1974;56:914–22. doi: 10.1016/s0006-291x(74)80275-5. [DOI] [PubMed] [Google Scholar]
- 22.Collet JP, Shuman H, Ledger RE, Lee S, Weisel JW. The elasticity of an individual fibrin fiber in a clot. Proc Natl Acad Sci USA. 2005;102:9133–7. doi: 10.1073/pnas.0504120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collet JP, Moen JL, Veklich YI, Gorkun OV, Lord ST, Montalescot G, Weisel JW. The alphaC domains of fibrinogen affect the structure of the fibrin clot, its physical properties, and its susceptibility to fibrinolysis. Blood. 2005;106:3824–30. doi: 10.1182/blood-2005-05-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duval C, Allan P, Connell SD, Ridger VC, Philippou H, Ariens RA. Roles of fibrin alpha- and gamma-chain specific cross-linking by FXIIIa in fibrin structure and function. Thromb Haemost. 2014;111:842–50. doi: 10.1160/TH13-10-0855. [DOI] [PubMed] [Google Scholar]
- 25.Aleman MM, Byrnes JR, Wang JG, Tran R, Lam WA, Di Paola J, Mackman N, Degen JL, Flick MJ, Wolberg AS. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. 2014;124:3590–600. doi: 10.1172/JCI75386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byrnes JR, Duval C, Wang Y, Hansen CE, Ahn B, Mooberry MJ, Clark MA, Johnsen JM, Lord ST, Lam W, Meijers JC, Ni H, Ariens RA, Wolberg AS. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain crosslinking. Blood. 2015;126:1940–8. doi: 10.1182/blood-2015-06-652263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muszbek L, Bereczky Z, Bagoly Z, Komaromi I, Katona E. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol Rev. 2011;91:931–72. doi: 10.1152/physrev.00016.2010. [DOI] [PubMed] [Google Scholar]
- 28.Rea CJ, Foley JH, Sorensen B. Factor XIII in the treatment of hemophilia A. N Engl J Med. 2012;366:281–3. doi: 10.1056/NEJMc1113270. [DOI] [PubMed] [Google Scholar]
- 29.Rea CJ, Foley JH, Ingerslev J, Sorensen B. Factor XIII combined with recombinant factor VIIa: a new means of treating severe hemophilia A. J Thromb Haemost. 2011;9:510–16. doi: 10.1111/j.1538-7836.2010.04171.x. [DOI] [PubMed] [Google Scholar]
- 30.Rea CJ, Foley JH, Okaisabor O, Sorensen B. FXIII: mechanisms of action in the treatment of hemophilia A. J Thromb Haemost. 2014;12:159–68. doi: 10.1111/jth.12478. [DOI] [PubMed] [Google Scholar]
- 31.Ng C, Silliman CC, Pearl G, Smith W, Manco-Johnson M, Wang M. Treatment of refractory hemorrhage with factor XIII in a patient with hemophilia A with inhibitor. Pediatr Blood Cancer. 2013;60:E23–5. doi: 10.1002/pbc.24478. [DOI] [PubMed] [Google Scholar]
- 32.Sakata Y, Aoki N. Significance of cross-linking of alpha 2-plasmin inhibitor to fibrin in inhibition of fibrinolysis and in hemostasis. J Clin Invest. 1982;69:536–42. doi: 10.1172/JCI110479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamaki T, Aoki N. Cross-linking of alpha 2-plasmin inhibitor to fibrin catalyzed by activated fibrin-stabilizing factor. J Biol Chem. 1982;257:14767–72. [PubMed] [Google Scholar]
- 34.Cines DB, Lebedeva T, Nagaswami C, Hayes V, Massefski W, Litvinov RI, Rauova L, Lowery TJ, Weisel JW. Clot contraction: compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood. 2014;123:1593–603. doi: 10.1182/blood-2013-08-523860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wohner N, Sotonyi P, Machovich R, Szabo L, Tenekedjiev K, Silva MM, Longstaff C, Kolev K. Lytic resistance of fibrin containing red blood cells. Arterioscler Thromb Vasc Biol. 2011;31:2306–13. doi: 10.1161/ATVBAHA.111.229088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ritchie H, Lawrie LC, Crombie PW, Mosesson MW, Booth NA. Cross-linking of plasminogen activator inhibitor 2 and alpha 2-antiplasmin to fibrin(ogen) J Biol Chem. 2000;275:24915–20. doi: 10.1074/jbc.M002901200. [DOI] [PubMed] [Google Scholar]
- 37.Fraser SR, Booth NA, Mutch NJ. The antifibrinolytic function of factor XIII is exclusively expressed through alpha(2)-antiplasmin cross-linking. Blood. 2011;117:6371–4. doi: 10.1182/blood-2011-02-333203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rijken DC, Abdul S, Malfliet JJ, Leebeek FW, Uitte de Willige S. Compaction of fibrin clots reveals the antifibrinolytic effect of factor XIII. J Thromb Haemost. 2016;14:1453–61. doi: 10.1111/jth.13354. [DOI] [PubMed] [Google Scholar]
- 39.Ashley C, Chang E, Davis J, Mangione A, Frame V, Nugent DJ. Efficacy and safety of prophylactic treatment with plasma-derived factor XIII concentrate (human) in patients with congenital factor XIII deficiency. Haemophilia. 2015;21:102–8. doi: 10.1111/hae.12524. [DOI] [PubMed] [Google Scholar]
- 40.Arokszallasi A, Kerenyi A, Katona E, Bereczky Z, Muszbek L, Boda Z, Schlammadinger A. The use of recombinant factor XIII in a major bleeding episode of a patient with congenital factor XIII deficiency – the first experience. Haemophilia. 2015;21:e118–21. doi: 10.1111/hae.12591. [DOI] [PubMed] [Google Scholar]
- 41.Lawrie AS, Green L, Mackie IJ, Liesner R, Machin SJ, Peyvandi F. Factor XIII – an under diagnosed deficiency – are we using the right assays? J Thromb Haemost. 2010;8:2478–82. doi: 10.1111/j.1538-7836.2010.04028.x. [DOI] [PubMed] [Google Scholar]
- 42.Doshi BS, Gangadharan B, Doering CB, Meeks SL. Potentiation of thrombin generation in hemophilia A plasma by coagulation factor VIII and characterization of antibody-specific inhibition. PLoS ONE. 2012;7:e48172. doi: 10.1371/journal.pone.0048172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh II, Smith A, Vanzieleghem B, Collen D, Burnand K, Saint-Remy J-M, Jacquemin M. Antithrombotic effects of controlled inhibition of factor VIII with a partially inhibitory human monoclonal antibody in a murine vena cava thrombosis model. Blood. 2002;99:3235–40. doi: 10.1182/blood.v99.9.3235. [DOI] [PubMed] [Google Scholar]
- 44.Emmerechts J, Vanassche T, Loyen S, Van Linthout I, Cludts K, Kauskot A, Long C, Jacquemin M, Hoylaerts MF, Verhamme P. Partial versus complete factor VIII inhibition in a mouse model of venous thrombosis. Thromb Res. 2012;129:514–19. doi: 10.1016/j.thromres.2011.06.027. [DOI] [PubMed] [Google Scholar]