

Summary
The kappa-opioid receptor (KOP) mediates the actions of opioids with hallucinogenic, dysphoric and analgesic activities. The design of KOP analgesics devoid of hallucinatory and dysphoric effects has been hindered by an incomplete structural and mechanistic understanding of KOP agonist actions. Here we provide a crystal structure of the human KOP in complex with the potent epoxymorphinan opioid agonist MP1104 and an active state stabilizing nanobody. Comparisons between inactive and active state opioid receptor structures reveal substantial conformational changes in the binding pocket, intracellular and extracellular regions. Extensive structural analysis and experimental validation illuminate key residues that propagate larger scale structural rearrangements and transducer binding that, collectively, elucidate the structural determinants of KOP pharmacology, function, and biased signaling. These molecular insights promise to accelerate the structure-guided design of safer and more effective kappa-opioid receptor therapeutics.
Keywords: GPCR, opioid receptor, addiction, crystallography, active state, nanobody, structure-function, morphinan
ETOC
A crystal structure of the active κ-opioid receptor provides a guide for the development of safe and effective new analgesics
Introduction
The kappa- (KOP) and closely related mu- (MOP) and delta- (DOP) opioid receptors are G protein-coupled receptors (GPCRs) for endogenous opioid peptides (dynorphins, endorphins and enkephalins, respectively). KOP was originally identified as the receptor for hallucinogenic synthetic (Martin et al., 1976; Pfeiffer et al., 1986) and naturally occurring (Roth et al., 2002) opioids. KOP also functions as the principal opioid receptor subtype responsible for mediating the myriad actions of dynorphin and dynorphin-related peptides on stress, addiction, emotion and perception (Bruchas et al., 2010; Chavkin et al., 1982). In fact, KOP has emerged as an alternative molecular target for the creation of safer analgesics (Bruchas and Roth, 2016) given the side effect associated with MOP agonists including respiratory depression, tolerance, dependence and constipation. Recent studies have uncovered further potential therapeutic areas for KOP ligands, such as affective disorders and addiction-related behaviors (Bruchas et al., 2010; Bruchas and Roth, 2016). Consistent with the therapeutic promise of biased agonism (i.e. the selective activation of beneficial pathways over deleterious signaling pathways) (Kenakin and Christopoulos, 2013; Urban et al., 2007), activating Gi/o protein-mediated pathways downstream of KOP, while avoiding arrestin-mediated signaling holds promise for designing safer KOP therapeutics devoid of the dysphoric and hallucinatory actions of conventional KOP agonists (Brust et al., 2016; Spetea et al., 2017; White et al., 2015).
As recently demonstrated for other GPCRs, structural insights from active and inactive receptor states can be leveraged in virtual ligand screening campaigns, and have already yielded new tool compounds ((Wang et al., 2017), some of which show promise as safer analgesics (Manglik et al., 2016; Spahn et al., 2017; Zheng et al., 2017). Even though antagonist-bound inactive state structures of all four opioid receptors [MOP, KOP, DOP, and nociceptin (NOP), (Fenalti et al., 2014; Fenalti et al., 2015; Granier et al., 2012; Manglik et al., 2012; Thompson et al., 2012; Wu et al., 2012)] have provided unprecedented molecular insights into opioid receptor structure, a molecular understanding of KOP activation remains elusive. This is largely due to our insufficient molecular understanding of the active state GPCRs, as obtaining crystal structures of such GPCR states remains challenging.
To facilitate a deeper understanding of KOP function and, more generally, GPCR activation, we report the active state crystal structure of KOP in complex with a high-affinity agonist and an active state stabilizing nanobody (Nb) at 3.1 Å resolution. A comparison with other opioid receptor structures identifies residues critical for KOP activation, and illuminates key molecular determinants of subtype selectivity and signaling bias.
Results
A nanobody-stabilized active state KOP structure
We adopted nanobody technology to obtain an active state-like crystal structure of KOP, as structure determination of signaling complexes remains incredibly challenging. Nbs–small single-chain antibodies – have facilitated the structural characterization of high-affinity agonist states of the β2-adrenergic (β2AR) (Rasmussen et al., 2011a), M2 muscarinic (M2R) (Kruse et al., 2013), and MOP (Huang et al., 2015) receptors, by mimicking the binding of signal transducers. We raised nanobodies by injecting KOP liposomes (Vardy et al., 2015) bound to the agonist salvinorin A (SalA) (Roth et al., 2002) into a llama, and used phage display to identify nine clones from these immune libraries (Pardon et al., 2014). To verify KOP-nanobody binding, we measured nanobody recruitment to unliganded, agonist-bound, and antagonist-bound KOP using Bioluminescence Resonance Energy Transfer (BRET) (Figure 1A). Only the identical clones Nb6 and Nb7 (Nb6/7) from the KOP-SalA library, as well as the MOP active state stabilizing nanobody Nb39 (Huang et al., 2015) bound KOP in our assay (Figure 1B, S1A). Consistent with a preference for inactive KOP, we observed strong basal BRET between Nb6/7 and unliganded KOP (Figure 1B); we also found that the selective KOP agonist SalA reduced the BRET ratio, while the selective KOP antagonist JDTic (Thomas et al., 1998) reversed SalA’s effect (Figure S1B). Remarkably, Nb39 was recruited in an agonist- and efficacy-dependent manner to KOP by the full agonist SalA and partial agonist diprenorphine, while the antagonist JDTic had no effect (Figure S1C). These findings implied that Nb39 stabilizes an active state conformation of KOP as reported for MOP (Huang et al., 2015).
Figure 1. Identification of inactive and active state stabilizing nanobodies and overall structure of the KOP-MP1104-Nb39 complex.
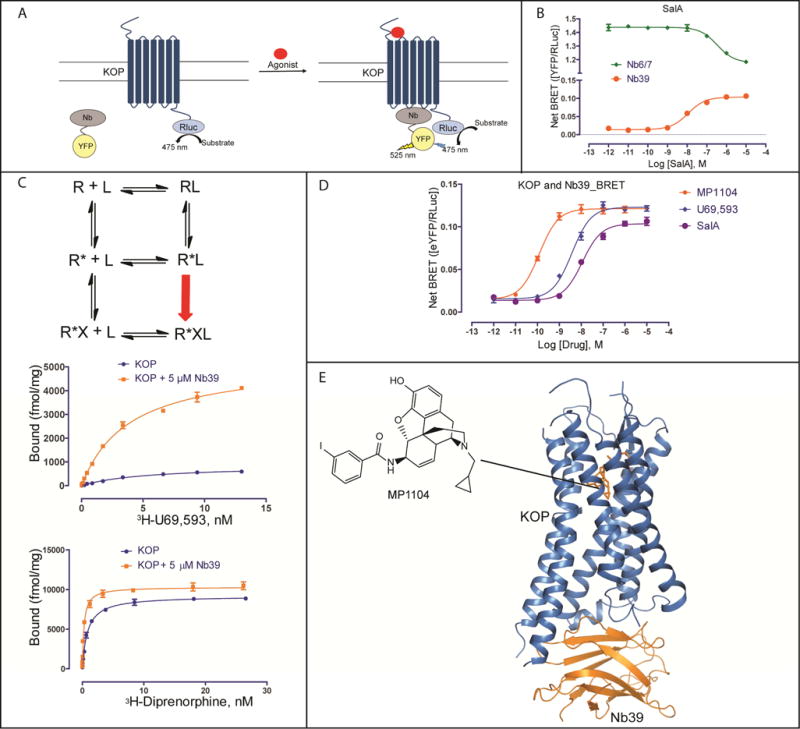
(A) Cartoon of receptor-nanobody (Nb) interaction monitored by bioluminescence resonance energy transfer (BRET). Agonist stimulated GPCR is bound by active state stabilizing Nb causing BRET signal. Nanobodies are tagged with C-terminal YFP and GPCRs are tagged with C-terminal Rluc (Renilla luciferase).
(B) BRET measurement of SalA-mediated Nb recruitment at KOP. The clones Nb6 and Nb7 (EC50 = 341±15 nM), which have the same protein sequence, show dissociation from the receptor upon SalA stimulation, indicating that Nb6/7 is pre-bound to KOP. The BRET signal from the KOP-Nb39 complex (EC50 = 11.3±1.1 nM) increases with increasing SalA concentrations, indicating that Nb39 recognizes active state KOP (N = 3).
(C) (top) Scheme of extended ternary complex model of GPCR activation. R, receptor; L, ligand; R*, active state receptor; X, transducer or transducer mimetics. (middle and bottom) Measurement of saturation binding at KOP with or without Nb39. High-affinity binding sites increase about 6-fold for the full agonist 3H-U69,593 in the presence of Nb39 (Kd = 2.80±0.06 nM, Bmax = 5262±138 fmol/mg) compared to KOP alone (Kd = 5.41±0.05 nM, Bmax = 860±35 fmol/mg) (N = 3). High-affinity binding sites increase by ~10% for the partial agonist 3H-Diprenorphine in the presence of Nb39 (Kd = 0.26±0.02 nM, Bmax = 10315±195 fmol/mg) compared to KOP alone (Kd = 0.84±0.05 nM, Bmax = 9191±120 fmol/mg) (N = 3).
(D) Ligand-mediated Nb39 binding to KOP measured BRET. EC50s for Nb39 recruitment: MP1104, 0.12±0.02 nM; U69,593, 3.78±0.04 nM; SalA, 11.2±0.8 nM (N = 3).
(E) Overall x-ray crystal structure of the KOP-MP1104-Nb39 complex. See also Figure S1.
To further test this hypothesis we used the extended ternary complex model of GPCR activation (Samama et al., 1993), which predicts that high-affinity agonist binding sites should increase in number when a G protein (or its Nb mimic) allosterically stabilizes the activated receptor state (Figure 1C). We confirmed this prediction via saturation binding studies wherein Nb39 increased KOP high-affinity agonist binding sites more than 6-fold for the full agonist 3H-U69,593 and ~10% for the weak partial agonist 3H-diprenorphine (Figure 1C). Additionally, Nb39 attenuated the agonist dissociation rate by approximately 6-fold whereas Nb6 accelerated agonist dissociation >2-fold (Figure S1D). Importantly, BRET studies revealed that Gαi1 dose-dependently inhibits KOP/Nb39 interactions (Figure S1E, Table S1) while β-arrestin2 modestly promotes Nb39 binding to KOP (Figure S1E, Table S1), confirming previous suggestions that β-arrestins and G proteins recognize different receptor conformations (Wacker et al., 2017a).
To identify a ligand suitable for crystallization of a Nb39-stabilized KOP active state, we tested several ligands in the BRET assay, and found that the epoxymorphinan MP1104 (Figure 1D) (Varadi et al., 2015) displayed the highest potency and efficacy for Nb39 recruitment to KOP. MP1104 has picomolar KOP binding affinity (Figure S1F) and is a potent KOP, MOP and DOP agonist (Figure S1G).
As Nb39 and MP1104 cooperatively promote a stable active state KOP, we were able to determine the x-ray crystal structure of a KOP-MP1104-Nb39 complex (Figure 1E). Crystals were obtained using a newly engineered human KOP construct with an N-terminal thermostabilized apocytochrome b562RIL (BRIL) (Chun et al., 2012) to increase receptor expression, and to facilitate crystallization (see Methods for details). Binding and functional assays showed that this KOP crystallization construct retains high affinity for MP1104 and elicits MP1104-mediated Gi activation similar to wt KOP (Figures S2A and S2B). The structure of the KOP-MP1104-Nb39 complex was determined to 3.1 Å resolution in the space group P21 with two monomers per asymmetric unit (Table 1, Figure S2C and S2D). While we observed strong density for KOP, Nb39, and MP1104 (Figure S2E), no electron density was observed for the likely disordered N-terminal BRIL fusion protein as reported for the NOP receptor (Thompson et al., 2012) and the nanobody-stabilized β2AR structures (Rasmussen et al., 2011a).
Table 1.
Data Collection and Refinement Statistics.
| Structure | BRIL – KOP – MP1104 – Nb39 | |
|---|---|---|
| Data Collection | APS, GMCA/CAT 23ID-B/D, 1.033 Å, 10-μm | |
| Crystals | 21 | |
| Resolution (Å) | 46.94–3.10 (3.27–3.10) | |
| Space group | P21 | |
| Complexes/ASU | 2 | |
| Unit cell dimensions a, b, c (Å) | 62.3, 150.8, 100.2 | |
| α, β, γ (°) | 90, 105.7, 90 | |
| No. total reflections | 89325 (10237) | |
| No. unique reflections | 30278 (4159) | |
| Multiplicity | 3.0 (2.5) | |
| Completeness (%) | 93.6 (88.2) | |
| Mean I/σ(I) | 4.0 (0.9) | |
| Rmerge (%) | 18.7 (96.1) | |
| CC1/2 (%) | 98.8 (53.7) | |
| Refinement Statistics | ||
| Resolution used in refinement (Å) | 46.94–3.10 (3.21–3.10) | |
| No. reflections used in refinement | 30255 (2708) | |
| No. reflections used for R-free | 1505 (138) | |
| R-work (%) | 25.4 (24.5) | |
| R-free (%) | 27.5 (28.3) | |
| Number of atoms | Complex A | Complex B |
| KOP | 2117 | 2013 |
| Nb39 | 959 | 917 |
| MP1104 | 33 | 33 |
| Lipids | 42 | 28 |
| Overall B – 2 factors (Å) | Complex A | Complex B |
| KOP | 82.6 | 82.9 |
| Nb39 | 92.0 | 90.7 |
| MP1104 | 80.0 | 82.5 |
| Lipids | 113.4 | 121.0 |
| Model Statistics | ||
| RMSD Bond (Å) | 0.010 | |
| RMSD Bond (°) | 0.95 | |
| Ramachandran Favored (%)a | 94.1 | |
| Ramachandran Allowed (%)a | 5.9 | |
| Ramachandran Outliers (%)a | 0.0 | |
| Rotamer outliers (%)a | 1.1 | |
| Molprobity scorea | 1.8 | |
Highest-resolution shell is shown in parentheses.
As defined in MolProbity.
Large-scale structural changes in active state KOP
We next analyzed overall helical movements by comparing the inactive state KOP-JDTic structure (PDB ID: 4DJH) (Wu et al., 2012) and the active state KOP-MP1104-Nb39 complex (Figure 2A) and observed substantial rearrangements in the relative positions of the helices. These include an overall contraction of the extracellular portion in the active state KOP, with extracellular loop (ECL) 2, and transmembrane helices (TM) 4–6 moving closer to the receptor core (Figure 2B). The orthosteric pocket of the active state structure shows a ~10% reduced volume when compared with that of the inactive state KOP-JDTic complex (~945 Å3 in active KOP vs ~1049 Å3 in inactive KOP) (Figure 2D). Similar inward movements of helices and a contraction of the pocket were also observed in the active state MOP structure (Figure 2E), and other active state GPCR structures (Figure S2F) (Huang et al., 2015; Kruse et al., 2013; Rasmussen et al., 2011b). These conserved structural rearrangements may indicate a general activation mechanism among many Class A GPCRs whereby contraction of the helical bundle on the extracellular side facilitates an opening of the helical bundle on the intracellular side which accommodates the binding of transducers (and vice versa). The larger contraction in the KOP orthosteric site (10%,~104 Å3) compared to MOP (6%, ~58 Å3) is likely due to the more compact positions of TM2, TM3, TM6 and TM7 around the agonist in the active state KOP structure, and the deeper pocket in the KOP inactive state, stabilized by the unique chemotype of JDTic.
Figure 2. Large-scale structural changes between inactive and active KOP.
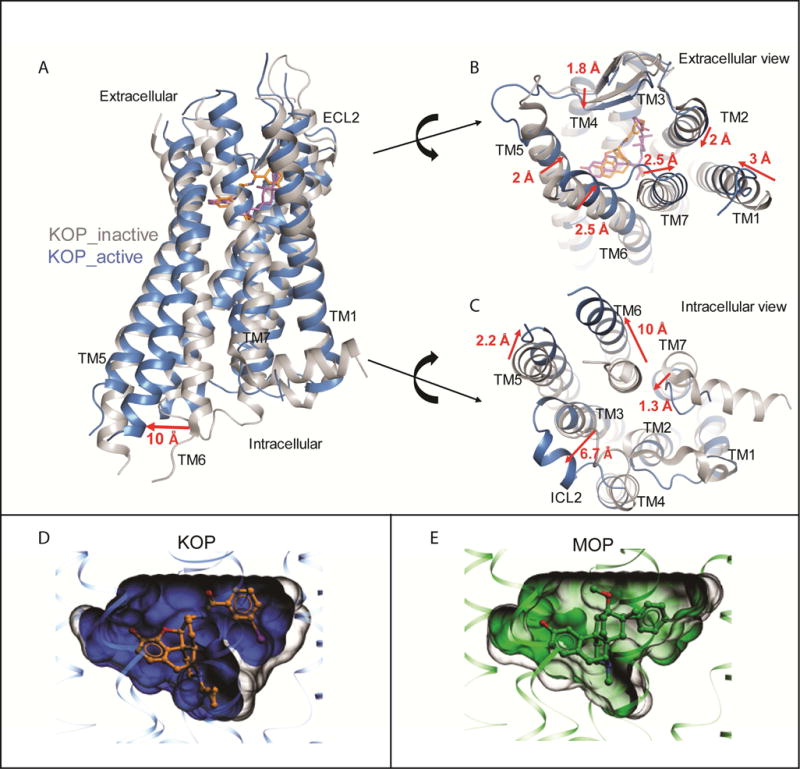
(A) Structural alignment of active (blue) and inactive (PDB ID: 4DJH, grey) KOP shows TM6 outward displacement of ~10 Å.
(B) Extracellular view highlights contraction of the TMs and extracellular loops upon binding to MP1104 (orange) versus JDTic (purple). Distances were measured between Cα atoms of I581.31, Q1152.60, Q213ECL2, D2235.35, L2996.60 and S3107.33.
(C) Intracellular view shows expansion of the 7TM bundle upon binding of MP1104 and Nb39 versus JDTic, with particularly pronounced movements in TM5–7 and ICL2. Distances were measured between the Cα atoms of K2545.66, D2666.27, Y3307.53 and D168ICL2. Nb39 and T4L fusion proteins have been omitted for clarity.
(D) Reduction of orthosteric site volumes in KOP and MOP upon activation. (left) Superimposed pockets for KOP, inactive (PDB ID: 4DJH, grey) 1049 Å3, and active (blue) 945 Å3. (right) Superimposed pockets for MOP, inactive (PDB ID: 4DKL, grey) 1112 Å3 and active (PDB ID: 5C1M, green) 1053 Å3. Volumes were calculated for the pockets of 4 superimposed receptors, and uniformly delimited between the level of extracellular lipid layer boundary (as predicted by OMP database), and the Cα atom of conserved residue W6.48.
See also Figure S2.
At the intracellular surface, the KOP-MP1104-Nb39 structure shows outward movements of TM6 (~10 Å) and ICL2 (6.7 Å), and inward movements of TM5 and TM7 by 2.2 Å and 1.3 Å, respectively, as compared to the inactive state (Figure 2C). These rearrangements are stabilized by several key KOP-Nb39 interactions (Figure S2G) whereby Nb39 is inserted into an intracellular receptor crevice that presumably serves as a binding pocket for key signal transducer residues. This interface is conserved among opioid receptors (Figure S2H), explaining why the MOP-originated Nb39 also stabilizes KOP. We also observed a conformation of KOP that is consistent with a general expansion of the intracellular binding site to accommodate transducers, reminiscent of those described for G protein, arrestin or Nb-stabilized active state structures of β2AR (Rasmussen et al., 2011a; Rasmussen et al., 2011b), M2R (Kruse et al., 2013), rhodopsin (Kang et al., 2015) and MOP (Huang et al., 2015).
Molecular determinants for KOP ligand binding and agonist efficacy
We next characterized ligand-receptor contacts of the agonist MP1104 and the antagonist JDTic bound to the KOP orthosteric pocket. We found that the JDTic and MP1104 core scaffolds assume distinctive poses (Figure 3A), albeit with common features typical for opioid ligands: (i) anchored in the receptor binding pocket through a salt bridge to D1383.32 in TM3; (ii) interact with TM5 through a phenolic group; and (iii) form interactions with TM2/3 via chemically diverse moieties.
Figure 3. MP1104 interactions in the active state KOP binding pocket.
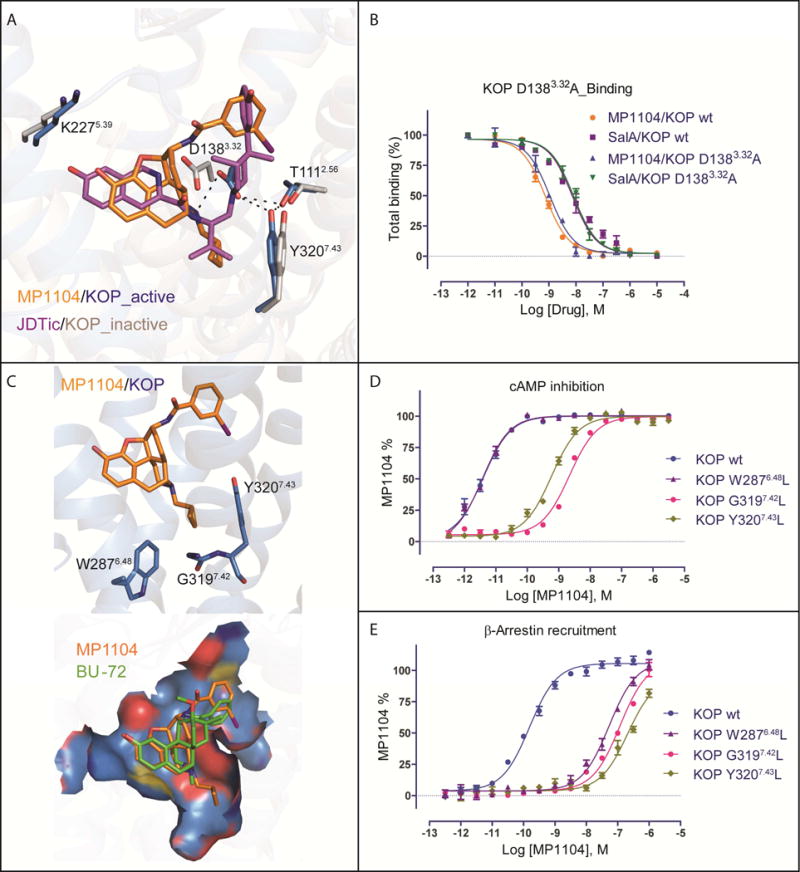
(A) Binding pose comparison of MP1104 (orange) in the active state KOP (blue) compared with JDTic (purple) in the inactive state (grey, PDB ID: 4DJH). Main interactions involved MP1104 and binding pocket residues are shown, with hydrogen bonds depicted as dashed lines (black).
(B) Comparison of MP1104 or SalA binding affinity at KOP wt and KOP D1383.32A mutant using 3H-diprenorphine (N = 3). See Table S2 for values.
(C) (top) Major interactions between the cyclopropylmethyl group of MP1104 (orange) and the hydrophobic pocket of active KOP (blue). (bottom) Comparison of binding pose between MP1104 (orange) and BU-72 (green) in KOP and MOP shows that MP1104 extends into the hydrophobic pocket but BU-72 does not.
(D, E) Mutations of hydrophobic pocket residues (W2876.48L, G3197.42L and Y3207.43L) strongly affect MP1104’s G protein activation (D) and β-arrestin2 recruitment (E), as measured by cAMP inhibition and Tango assay, respectively (N = 3). See Table S3, S4 for values
See also Figure S3.
The antagonist JDTic and the agonist MP1104 both form a salt bridge between their respective amine moieties and D1383.32 of the receptor as observed in many GPCR-ligand complexes (Figure 3A). The larger distance of this salt bridge (3.0 Å) compared to similar interactions in the KOP-JDTic (2.6 Å), and MOP-BU-72-Nb39 (2.7 Å) structures implies a weaker ionic interaction between MP1104 and KOP. Our mutagenesis study confirmed that, similar to SalA which does not contain a basic amine, MP1104 maintains high binding affinity (Figure 3B) but attenuated functional activity at the D1383.32A mutant, whereas Dynorphin A 1–17 binding and functional activity is abolished (Vardy et al., 2015) (Table S2). D1383.32 also forms a hydrogen bond network with T1112.56 and Y3207.43 in KOP-MP1104-Nb39 that is likely critical for full KOP activation as mutation of these residues strongly attenuates or abolishes β-arrestin2 recruitment mediated by MP1104 or Dynorphin A 1–17, respectively (Figure S3A).
The MP1104 and JDTic phenolic groups extend toward TM5 forming water-mediated hydrogen bonds with the backbone carbonyl oxygen of K2275.39 –as seen in other opioid receptor structures (Fenalti et al., 2014; Huang et al., 2015; Manglik et al., 2012; Wu et al., 2012). This interaction has been proposed to mimic that of the N-terminal tyrosine found in endogenous opioid peptides (Fenalti et al., 2015; O’Connor et al., 2015). Because of the lower resolution of the KOP-MP1104-Nb39 structure we do not observe the waters involved in this interaction, but could predict their position by energy-based water modeling algorithms. (Figure S3B). Support for this prediction comes from studies where we replaced the hydroxyl groups (−OH) on the MP1104 scaffold with a methoxy group (−OCH3), resulting in decreased KOP affinity (Figure S3C) as observed for similar JDTic modifications (Urbano et al., 2014). The methoxy substitution, however, affects MOP binding affinity more severely than KOP, and these findings suggest that interactions with TM5 can be exploited for KOP selective ligand design.
MP1104’s cyclopropylmethyl group extends into a hydrophobic pocket at the bottom of the orthosteric site, similar to the isopropyl moiety in JDTic (Figure 3C). This hydrophobic pocket has been proposed to play an important role in determining agonist or antagonist activity at MOP (Huang et al., 2015). Our analysis indicates that the connection between ligand-receptor interactions within this hydrophobic pocket of opioid receptors and corresponding ligand efficacy may not be as straightforward as previously proposed. Compared to MP1104’s cyclopropylmethyl group, BU-72 in the MOP/BU-72 complex has a methyl substituent at its tertiary amine, and thus does not fully extend into this hydrophobic pocket (Figure 3C) (Huang et al., 2015). We observed several contacts between MP1104 and residues of this hydrophobic pocket including hydrophobic interactions between the cyclopropylmethyl group and the aromatic ring of the Y3207.43 side chain, the side chain of W2876.48, and the backbone of G3197.42 (Figure 3C).
The Y3207.43L and G3197.42L mutations strongly reduced MP1104’s potency for both G protein signaling and β-arrestin2 recruitment, while the W2876.48L mutant, selectively reduced MP1104’s β-arrestin2 recruitment potency (Figure 3D, 3E, Table S3, S4). Similar effects were also observed for other tested KOP agonists (Table S3, S4), suggesting that this pocket is a general node in relating structural changes in the binding pocket to the engagement of transducers. Importantly, substituting MP1104’s cyclopropylmethyl with a methyl group resulted in a >15-fold reduction of KOP agonist potency (Figure S3D). Since MP1104 potently activates all three canonical opioid receptors (Figure S1E), we docked MP1104 into the MOP active state and observed a similar orientation as BU-72, with MP1104’s cyclopropylmethyl group extending into MOP’s hydrophobic pocket (Figure S3E).
Collectively our results indicate that the precise orientation of the rigid and bulky morphinan scaffold within the binding pocket is critical for determining ligand efficacy/potency via minor changes in contact forces or tensions generated by substituents. The orientation within the pocket likely depends on i) the hybridization of intramolecular bonds that determines the angles between the functional modules of the compound, and ii) receptor subtype specific interactions. Accordingly, even small modifications to identical scaffolds can subtly affect a compound’s binding mode, and thereby its potency and/or efficacy, as observed for other GPCR ligands (Wacker et al., 2017b).
Potential mechanisms of KOP activation
To clarify molecular mechanisms of KOP activation, we investigated in detail how conformational changes are likely propagated between ligand and transducer binding sites (Figure 4A).
Figure 4. Activation signal propagation within KOP motifs.
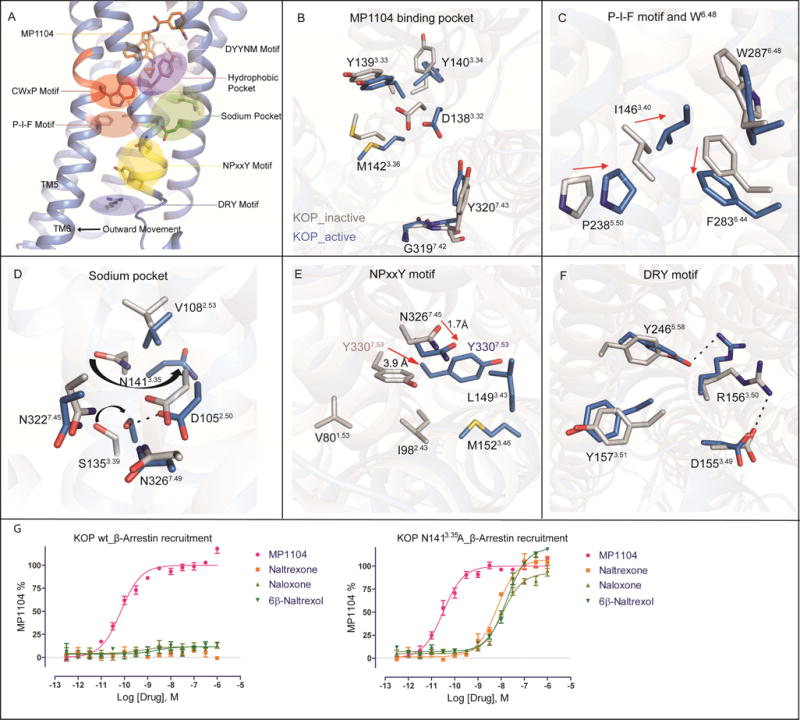
(A) Close ups highlight activation-related conformational changes in key receptor motifs, and show connection of structural changes from orthosteric site to the cytoplasmic transducer binding site. Conformational changes between active KOP (blue) and inactive KOP (grey) are highlighted for (B) MP1104 binding pocket, (C) P-I-F motif, (D) sodium binding pocket, (E) NPxxY motif, and (F) DRY motif.
(G) KOP N1413.35A mutation switches classic opioid receptor antagonists (left) into full agonists (right) in Tango-arrestin recruitment (N = 3). In KOP wt: MP1104 (red), EC50 = 0.071±0.008 nM, Emax = 100±2; in KOP N1413.35A: MP1104 (red), EC50 = 0.027±0.008 nM, Emax = 100±2, naltrexone (orange), EC50 = 6.53±0.90 nM, Emax = 107±3, naloxone (light green), EC50 = 12.75±1.50 nM, Emax = 93±2, 6β-naltrexol (green), EC50 = 22.47±1.8 nM, Emax = 120±2.
We observed several rearrangements in the MP1104-bound orthosteric site, specifically in the anchoring D3.32YYNM3.36 motif (Figure 4B), which is conserved in opioid receptors and formed by TM3 residues that have previously been proposed as a hub of GPCR structural rearrangements (Venkatakrishnan et al., 2013). MP1104’s cyclopropylmethyl group interacts with the conserved W2876.48 residue of the CWxP motif within hydrophobic pocket (Figure 3C), and with M1423.36 of the D3.32YYNM3.36 motif. W2876.48, which is located at the center of an aromatic cluster involved in GPCR activation (Holst et al., 2010; Shi et al., 2002), forms hydrophobic contacts with F2836.44, thus coupling the ligand binding site to the P5.50-I3.40-F6.44 motif, a central “microswitch” for the activation of many GPCRs (Katritch et al., 2013). Facilitated by KOP’s ligand binding site contraction in the active state, P2385.50 moves inward and I1463.40 changes its side chain rotameric state, which likely promotes the rotation of F2836.44 (Figure 4C). This rotation has been linked to the TM6 swivel motion causing receptor opening on the intracellular for transducer binding (Figure 2A) (Katritch et al., 2013; Rasmussen et al., 2011a; Valentin-Hansen et al., 2012; Wacker et al., 2013).
Changes in TM3 residues also appear to connect the orthosteric and sodium pocket, the NPxxY motif, and larger scale activation-related changes in TM7. The sodium pocket is located between TM2, TM3 and TM7 and accommodates a single sodium ion, which acts as a negative allosteric modulator at opioid receptors (Fenalti et al., 2014; Pasternak et al., 1975; Pert et al., 1973) and most other GPCRs (Katritch et al., 2014). In KOP, D1052.50, N1413.35 and S1453.39 form the sodium pocket, which collapses upon activation and instead D1052.50 forms a hydrogen bond with S1453.39 (Figure 4D). The N1413.35 side chain changes its rotameric state in the active state of KOP (Figure 4D) similar to MOP (Huang et al., 2015), and directly connects changes in the orthosteric site to the sodium pocket. As in DOP (Fenalti et al., 2014), N3.35 forms a water-mediated hydrogen bond with D3.32 and directly coordinates sodium. Through a hydrogen bond between D1052.50 and N3267.49, the sodium pocket is further directly coupled to the NPxxY motif at the intracellular tip of TM7, which has been implicated in propagating structural changes in the orthosteric site through a sequence of conformational changes leading to larger scale helical movements (Katritch et al., 2014; Valentin-Hansen et al., 2012). Furthermore, upon KOP activation, the NPxxY motif residues N3227.45 and Y3307.53 move toward the receptor core by 1.7 Å and 3.9 Å, respectively, with the hydroxyl group of Y3307.53 moving as much as 7.8 Å (Figure 4E), which is consistent with similar activation-related changes in other class A GPCRs (Katritch et al., 2013).
At the intracellular end of TM3, we observed additional structural rearrangements in the conserved DRY motif, which has been shown to directly interact with signal transducers through R1563.50. Specifically, we observed that the salt bridge between R1563.50 and D1553.49, which is characteristic for many inactive state structures, is broken in the KOP active state, and instead R1563.50 points toward the receptor core, forming a hydrogen-bond with Y2465.58 (Figure 4F). A similar configuration of R3.50 has previously been observed in the structures of β2AR bound to a heterotrimeric G protein (Rasmussen et al., 2011b), adenosine A2A receptor bound to a thermostabilized “mini-Gs” (Carpenter et al., 2016), opsin bound to a C-terminal peptide of transducin (Scheerer et al., 2008), and rhodopsin bound to visual arrestin (Kang et al., 2015).
Together, our observations confirm and extend previous suggestions that structural changes in TM3 residue interfaces are critical for coupling ligand-mediated changes in the orthosteric site and the transducer interface (Venkatakrishnan et al., 2013). This hypothesis is further strengthened by the finding that a N1413.35A mutation converts several antagonists into full agonists (Figure 4G).
Structural determinants for biased signaling and subtype selectivity
Pathway selective KOP ligands are not only important tools for elucidating receptor mechanisms and uncovering novel receptor physiology, they are also therapeutically desirable. It is becoming clear that KOP’s analgesic and anti-pruritic effects appear to be G protein-mediated while many of the undesirable actions of KOP agonists may be mediated by arrestin-ergic and other non-canonical pathways (Bruchas and Roth, 2016; Brust et al., 2016; White et al., 2015). We thus performed structure-activity relationship (SAR) and docking experiments to identify and further characterize the molecular determinants for biased signaling at KOP.
Clues regarding the structural features responsible for biased signaling at KOP come from studying IBNtxA (Majumdar et al., 2011), a close MP1104 analog. Compared to MP1104, IBNtxA has a substituted hydroxyl group at position C14, and a fully saturated cyclohexanyl C-ring (Figure 5A). Although MP1104 is a full and unbiased agonist at both KOP and MOP, IBNtxA displays G protein signaling bias at MOP (Figure 5B and 5C, Figure S4A, Table S5). To understand the molecular basis of these functional differences we docked IBNtxA into the active state structures of KOP and MOP. We observed substantial conformational differences within the orientation of the iodobenzamide moiety of the compound (Figure S4B) likely due to MP1104’s boat configuration and IBNtxA’s chair configuration, caused by their respective unsaturated and saturated C-rings (Figure 5A). IBNtxA’s iodobenzamide moiety is pointed toward the extracellular region in both receptors, instead of towards TM2 and TM3 as for MP1104. In KOP, MP1104’s iodobenzamide moiety appears stabilized by a weak, water-mediated hydrogen bond between the ligand’s carbonyl and Y3127.35 (3.6 and 4.1 Å distance in molecules A and B respectively), while MOP contains W3207.35 at this position (Figure 5A). Since the IBNtxA docking pose in KOP suggests a similar receptor interaction through a stronger direct or water-mediated bond to Y3127.35 (3.4 Å distance) (Figure 5A), we hypothesized that differences at position 7.35 could explain IBNtxA’s functional dissimilarities between MOP and KOP. Indeed, when we mutated KOP’s Y3127.35 to a tryptophan to mimic MOP’s configuration, we observed a pathway-selective attenuation of IBNtxA’s arrestin recruitment potency, while MP1104 was only slightly affected (Figure 5D). Moreover, we observe similar functional profiles for several analogs that all contain saturated C-rings (Figure S4C).
Figure 5. Structural insights for the design of biased and selective KOP ligands.
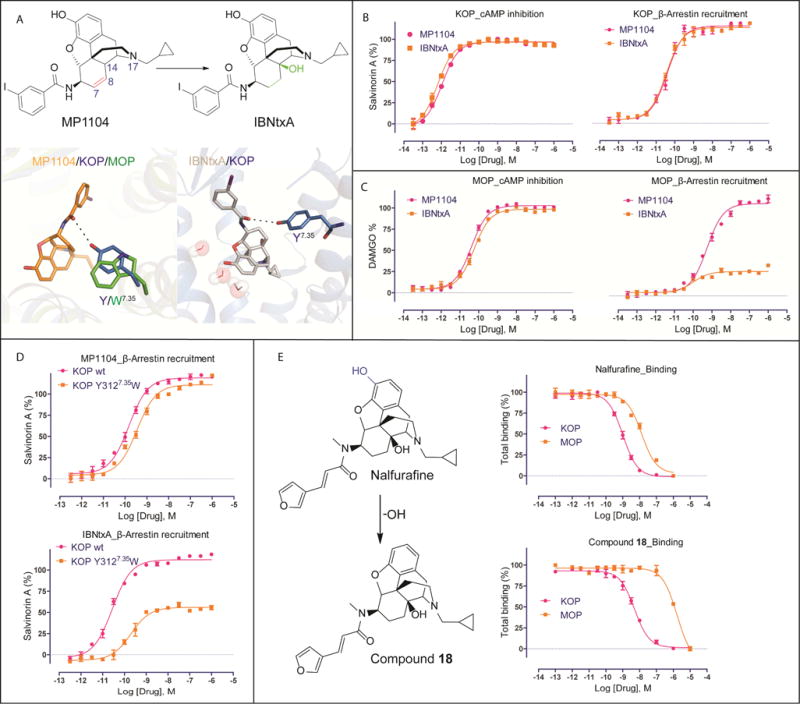
(A) Chemical structures of MP1104 and IBNtxA. Chemical differences are highlighted in color. Comparison of iodobenzamide binding pose between MP1104 (orange, top) and docked IBNtxA (white, bottom) in KOP (blue). Y3127.35 in KOP forms a hydrogen bond with amide oxygen of both compounds.
(B) MP1104 (red) and IBNtxA (orange) are balanced full agonists in KOP measuring in cAMP inhibition and Tango-arrestin recruitment. Gi: MP1104, EC50 = 0.003±0.001 nM, Emax = 97±1; IBNtxA, EC50 = 0.002±0.001 nM, Emax = 96±1. Arrestin: MP1104, EC50 = 0.035±0.010 nM, Emax = 115±8; IBNtxA, EC50 = 0.032±0.010 nM, Emax = 113±12. Bias factor toward G protein: 0.6 and 1.5 for MP1104 and IBNtxA, respectively (N = 3).
(C) IBNtxA (orange) displays G protein biased activity in MOP, while MP1104 (red) appears balanced in cAMP inhibition and Tango-arrestin recruitment. Gi: MP1104, EC50 = 0.04±0.010 nM, Emax = 103±6; IBNtxA, EC50 = 0.056±0.012 nM, Emax = 99±6. Arrestin: MP1104, EC50 = 0.55±0.03 nM, Emax = 126±7; IBNtxA, EC50 = 0.10±0.03 nM, Emax = 23±4. Bias factor toward G protein: 1.6 and 12 for MP1104 and IBNtxA, respectively (N = 3).
(D) The KOP Y3127.35W mutant (orange) shows slightly reduced MP1104- and strongly reduced IBNtxA-mediated arrestin recruitment compared to KOP wt (red) (N = 3). MP1104/KOP wt: EC50 = 0.035±0.010 nM, Emax = 119±9; MP1104/KOP Y3127.35W: EC50 = 0.055±0.02 nM, Emax = 111±4. IBNtxA/KOP wt: EC50 = 0.03±0.01 nM, Emax = 112±10; IBNtxA/KOP Y3127.35W: EC50 = 0.20±0.06 nM, Emax = 52±4.
(E) Binding affinity of Nalfurafine (Ki = 0.32±0.02 nM in KOP and 4.20±0.21 nM in MOP) and compound 18 (Ki = 1.50±0.05 nM in KOP and 533±65 nM in MOP) (N = 3). See also Figure S4, S5.
In addition to the desired signaling properties, subtype selectivity is critical for developing safer therapeutics with reduced side effects. To identify features responsible for selective KOP actions we next docked representative agonists U69,593, U50,488 and SalA into the current active state structure. U69,593 and U50,488 (Figure S5A) show similar binding poses, forming ionic interactions between the pyrrolidinyl/amide nitrogen and D1383.32, while exhibiting steric overlap with the MP1104 crystallographic pose. One major difference of U69,593 or U50,488 from peptides and morphinan opioids is the lack of phenol moiety, which interacts with H2916.52 and Y1393.33 residues in the classical opioids. Accordingly, mutations in these residues to Ala had no significant impact on ligand binding to U69,593, while Dynorphin A binding was reduced by >10-fold and ~5-fold respectively (Vardy et al., 2013). At the opposite side of the pocket, the phenyl ring of both U69,593 and U50,488 occupies the same pocket as the iodo-phenyl moiety in the MP1104-KOP structure and the linker amide of both also forms a hydrogen bond with Q1152.60. SalA’s docking mode (Figure S5B) is consistent with previous studies (Cunningham et al., 2011) (Vardy et al., 2013; Wu et al., 2012; Yan et al., 2009). In our current model, Y3127.35 forms a hydrogen bond with the carbonyl oxygen atom of SalA at the 1-position (See Figure S5B for atom numbering). The SalA 2-position acetyl group forms hydrophobic interactions with the aromatic ring of Y3127.35 and may hydrogen bond with the neighboring Y3137.36 hydroxyl group. The 4-position methyl ester of SalA is situated in a small, largely enclosed pocket where it is stabilized by an H-bond with the C210ECL2 backbone NH group. The furan ring of SalA engages in edge-face aromatic interactions with Y1393.33. It is noteworthy that these docking poses based on the active state KOP structure are somewhat different from those derived from the inactive state KOP-JDTic structure (Vardy et al., 2013), highlighting the unique conformation of the active-state binding pocket.
Efforts to improve KOP selectivity by modifying MP1104’s or IBNtxA’s iodobenzamide group were unsuccessful (Figure S5C). Instead, we adopted a novel strategy which emerged from our earlier structure-guided observation that modifications of the morphinan phenolic group affects binding affinity for KOP less than for MOP (Figure S3C).
The ligands’ phenol groups interact with a complex water network that is different between opioid receptor subtypes due to non-conserved residues in positions 5.36, 6.58, and 7.35, which form part of the “address” domain in opioid receptors (Larson et al., 2000) (Figure S3B). In MOP, the phenolic group of BU-72 forms strong water-mediated interactions with the side chain of K3036.58 (Huang et al., 2015). In contrast, in KOP the shorter E2976.58 side chain precludes formation of this water-mediated interaction with MP1104’s phenolic group. This suggests that removal of the phenolic moiety can differentially reduce MOP ligand affinity without impacting KOP binding. As mentioned above, the KOP-selective agonists U69,593 and U50,488 lack the phenolic group. Moreover, comparison of Nalfurafine, an approved IBNtxA-related analog for chronic itch (Endoh et al., 2001), and its analog compound 18, which lacks the hydroxyl moiety of the phenolic group (Nagase et al., 2012) (Figure 5E), revealed a large reduction in MOP affinity, while retaining high KOP affinity (Figure 5E). Thus, modification of the phenol group represents a viable path toward generating KOP selective ligands with improved pharmacological profiles.
Discussion
Here we present the nanobody-stabilized active state structure of KOP, and provide detailed molecular insights into KOP activation, opioid receptor selectivity, and biased signaling.
While we observed large-scale conformational changes in the KOP active state reminiscent of those seen in other GPCRs, we also find distinct rearrangements in the KOP’s ligand binding pocket. A detailed analysis of these differences highlights limitations of prior “message-address”-based hypotheses (Larson et al., 2000), which postulated that opioid receptor selectivity and efficacy are determined by distinctive compound moieties. Previous studies suggested that larger hydrophobic amine substituents on prototype morphinans conferred opioid antagonism and thereby provided a road-map for the design of selective agonists and antagonists. Instead, our findings indicate that combinatorial interactions with conserved and non-conserved residues specify ligand selectivity, pharmacology, efficacy and signaling bias.
Using a combination of structural analysis, molecular docking, binding and functional studies we identified important opioid receptor residues involved in conferring different patterns of biased signaling between opioid receptor subtypes. For instance, we showed that replacing Y7.35 in the binding pocket of KOP with W7.35 found in MOP, transforms the balanced KOP agonist IBNtxA into a G protein biased ligand thereby mimicking its activity at MOP. The finding that residues at the same position confer differential signaling patterns is consistent with observations that a ligand may elicit very different patterns of activity between two receptors. Molecular insights into these mechanisms may ultimately be exploited for the design of polypharmacological ligands with “customized” activities at each target.
Taken together these findings not only expand our general mechanistic framework of GPCR activation, but also provide molecular insights into KOP structure and function. Given the urgent need to develop safer opioid medications in an effort to battle the growing opioid epidemic, these molecular insights could greatly accelerate the design of novel KOP ligands through structure-enabled technologies and large-scale virtual ligand screening.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Bryan L. Roth (bryan_roth@med.unc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For KOP expression, we used the sf9 insect cells derived from the parental Spodoptera frugiperda cell line IPLB-Sf-21-AE (Expression systems). Cells were grown in ESF 921 medium (Expression systems) at 27°C and 125 rpm. Na nobodies were expressed at 27°C in E.coli WK6 (su-) cells in TB medium (Terrific Broth, Sigma). Nanobodies were induced with 1 mM IPTG (final concentration, Isopropyl β-D-1-thiogalactopyranoside) when the bacteria density reached an OD600 of 0.6–0.8 and bacteria cells were grown overnight at 170 rpm. For KOP functional assays, Human embryonic kidney (HEK) 293T (ATCC CRL-11268) cells were cultured in DMEM (Dulbecco’s Modified Eagle Medium). Wild type or mutant KOP plasmids were transfected into HEK 293T cells using the calcium precipitation method.
METHOD DETAILS
Generation of human KOP receptor crystallization construct
Crystallization of the human KOP complex was done using an engineered receptor construct that was modified based on the KOP-T4L sequence (Wu et al., 2012). The final construct a) lacks N-terminal residues 1–53, b) lacks C-terminal residues 359–380, c) contains M1-L106 of the thermostabilized apocytochrome b562 RIL (BRIL) from E. coli (M7W, H102I, R106L) in place of receptor N-terminus residues M1-H53, a glycine-serine linker was inserted between BRIL and receptor to facilitate crystallization. Further modifications are I135L mutation was introduced to increase expression; a haemagglutinin (HA) signal sequence followed by a FLAG tag at the N terminus, then a 10X His tag followed by a TEV protease site to enable purification by immobilized metal affinity chromatography.
Discovery and purification of nanobodies
KOP specific nanobodies were generated as described before (Pardon et al., 2014). In brief, one llama (Lama glama) was immunized six times with in total 0.5 mg purified BRIL-KOP DREADD (KOP D1383.32N) bound to SalA [KOP D1383.32N was used here because it has higher affinity with SalA than wild type (Vardy et al., 2015)]. Four days after the final boost, blood was taken to isolate peripheral blood lymphocytes. RNA was purified from these lymphocytes and reverse transcribed by PCR to obtain cDNA. The resulting library was cloned into the phage display vector pMESy4 bearing a C-terminal hexa-His tag and a Glu-Pro-Glu-Ala-tag (EPEA-tag, also called or CaptureSelect C-tag). Selections were performed either on BRIL-KOP in liposomes solid phase coated directly on plates. Six different families were selected by biopanning. After two rounds of selection, periplasmic extracts were made and subjected to ELISA screens. Clones giving a positive signal in ELISA were sequenced and analysed. Plasmids were transformed to E. coli WK6 cells, KOP specific nanobodies i.e. Nb6 and Nb7 were expressed and purified following steps 70–73 described in the previous protocol (Pardon et al., 2014). Nb39 DNA sequence was synthesized (Integrated DNA Technologies, IDT) based on the protein sequence in the active-state MOP structure (PDB ID: 5C1M) (Huang et al., 2015), and was expressed and purified using the same protocols as Nb6/7. Nanobodies were concentrated and desalted to the buffer: 10 mM HEPES, 100 mM NaCl and 10% Glycerol and stored at −80 °C for f uture use.
Expression and purification of KOP
High-titer recombinant baculovirus (>109 viral particles per ml) was generated using the Bac-to-Bac Baculovirus Expression System (Invitrogen). ~5 μg of recombinant bacmid in 50 μl Sf-900 II SFM media (Invitrogen) and 3 μl Cellfectin II Reagent (Invitrogen) in another 50 μl Sf-900 II SFM media (Invitrogen) were incubated for 30 min. Recombinant baculovirus was obtained by transfecting the above mixed solution into 400 μl Sf-900 II SFM media including 5×105 settled Spodoptera frugiperda (Sf9) cells (Expression Systems) in a 12-well plate (Corning). After 5 h, media was exchanged for 1 ml Sf-900 II SFM media (Invitrogen) and incubated for 5 days at 27 °C. P0 viral stock with ~109 virus particles per ml was harvested as the supernatant and used to generate high-titer baculovirus stock by infection of 40 ml of Sf9 cells (cell density: 2–3 × 106 cells/ml) and incubation for 3 days. Viral titers were determined by flow-cytometric analysis of cells stained with gp64-PE antibody (Expression Systems). Expression of KOP was carried out by infection of Sf9 cells at a cell density of 2.5 × 106 cells/ml in ESF921 media (Expression Systems) with P1 or P2 virus at a MOI (multiplicity of infection) of 3. 5% production boost additive (PBA, Expression Systems) was added to maintain cell alive. Final concentration of 10 μM naltrexone was added to help the receptor trafficking. Cells were harvested by centrifugation at 48 h post infection, washed in 1x PBS, and stored at −80 °C until use. Cells were first washed by res uspending frozen cell pellets in a low-salt buffer containing 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl and protease inhibitors (500 μM AEBSF, 1 μM E-64, 1 μM Leupeptin, 150 nM Aprotinin). Membranes purification was followed by 4 repeated centrifugation in a high osmolarity buffer containing 1.0 M NaCl, 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl, to remove soluble and membrane associated proteins. Purified membranes were directly flash-frozen in liquid nitrogen and stored at −80 °C for future use.
Purified membranes were resuspended in buffer containing 10 mM HEPES, pH 7.5, 10 mM MgCl2, 20 mM KCl, 150 mM NaCl, 50 μM MP1104 (synthesized in house), and 1x protease inhibitors (500 μM AEBSF, 1 μM E-64, 1 μM Leupeptin, 150 nM Aprotinin), and incubated at room temperature for 1 h. The sample was then transferred to 4 °C for 30 min. After another 30 min incubation in the presence of 2 mg/ml iodoacetamide (Sigma), membranes were solubilized in 10 mM HEPES, pH 7.5, 150 mM NaCl, 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace), 0.2% (w/v) cholesteryl hemisuccinate (CHS, Sigma), and protease inhibitors for 2 h at 4 °C. The supernatant was obtained by centrifugation at 150,000 × g for 30 min and was incubated with 20 mM imidazole and TALON IMAC resin (Clontech) overnight at 4 °C using approximately 500 μl resin for protein purified from 1 L of cells. The resin was then washed with 10 column volumes (cv) of Wash Buffer I (50 mM HEPES, pH 7.5, 800 mM NaCl, 0.1% (w/v) DDM, 0.02% (w/v) CHS, 20 mM imidazole, 10% (v/v) glycerol, and 25 μM MP1104, followed by 10 cv of Wash Buffer II (25 mM HEPES, pH 7.5, 150 mM NaCl, 0.05% (w/v) DDM, 0.01% (w/v) CHS, 10% (v/v) glycerol, and 25 μM MP1104). Proteins were eluted in 2.5 cv of Wash Buffer II + 250 mM imidazole, concentrated in a 100 kDa molecular weight cut-off Vivaspin 20 concentrator (Sartorius Stedim) to 500 μl, and imidazole was removed by desalting the protein over PD MiniTrap G-25 columns (GE Healthcare). The N-terminal 10× His-tag was removed by addition of His-tagged TEV protease (Homemade) and incubation overnight at 4 °C. Protease, cleaved His-tag and uncleaved protein were removed by passing the suspension through equilibrated TALON IMAC resin (Clontech) and collecting the flow-through. Excessive Nb39 (KOP/Nb39 m/m: 1:2) was then added to the protein sample and incubated for 3 h. KOP-MP1104-Nb39 complexes were then concentrated to ~30 mg/ml with a 100 kDa molecular weight cut-off Vivaspin 500 centrifuge concentrator (Sartorius Stedim). Protein purity and monodispersity were tested by analytical size-exclusion chromatography.
Lipidic cubic phase crystallization
KOP-MP1104-Nb39 complexes were reconstituted into lipidic cubic phase (LCP) by mixing protein solution and a monoolein/cholesterol (10:1 w/w) mixture in a ratio of 2:3 v/v (protein solution/lipid) using the twin-syringe method (Caffrey and Cherezov, 2009). Crystallization was set up in 96-well glass sandwich plates (Marienfeld GmbH) using 50 nl LCP drops dispensed from a 10 μl gas-tight syringe (Hamilton) using a handheld dispenser (Art Robbins Instruments) and overlaid with 1 μl of precipitant solution. Upon optimization, KOP-MP1104-Nb39 crystals were obtained in 100 mM Bis-tris pH 6.5–7.0, 140–200 mM magnesium sulfate hydrate, 100 mM sodium citrate tribasic dehydrate, 10 mM Manganese(II) chloride tetrahydrate, 28–30% PEG400. Crystals grew to a maximum size of 50 μm ×30 μm ×20 μm within three days and were harvested directly from the LCP matrix using MiTeGen micromounts before flash-freezing and storage in liquid nitrogen.
Data collection, structure solution and refinement
X-ray data were collected at the 23ID-B and 23ID-D beamline (GM/CA CAT) at the Advanced Photon Source, Argonne, IL using a 10 μm minibeam at a wavelength of 1.0330 Å and a Dectris Eiger-16m detector and Dectris Pilatus3–6m detector, respectively. Diffraction data were collected by exposing the crystals for 0.2 s to an unattenuated beam using 0.2° oscillation per frame. 315 frames collected from 21 crystals were indexed, integrated, scaled, and merged using HKL3000 (Minor et al., 2006). Initial phases were obtained by molecular replacement in PHASER (McCoy et al., 2007) using 3 independent models of a truncated 7TM portion of KOP (PDB ID: 4DJH), a nanobody Nb39 from the MOP-Nb39-BU-72 complex (PDB ID: 5C1M), and the thermostabilized apocytochrome b562RIL protein (PDB ID: 1M6T) (Chu et al., 2002). Two copies of the 7TM portion of each the receptor and the nanobody Nb39 but no BRIL were found in asymmetric unit. Refinement was performed with PHENIX (Adams et al., 2010), REFMAC5 (Murshudov et al., 1997) and autoBUSTER (Smart et al., 2012) followed by manual examination and rebuilding of the refined coordinates in the program COOT (Emsley et al., 2010) using 2mFo – DFc and mFo – DFc maps. Final refinement was performed using autoBUSTER with four TLS groups (two KOP and two Nb39 TLS groups), a total of 882 residues (307 KOP and 134 Nb39), and two MP1104 ligands and two cholesterols molecules. The data collection and refinement statistics are shown in Table 1.
cAMP inhibition assay
To measure KOP Gαi-mediated cAMP inhibition, HEK 293T (ATCC CRL-11268) cells were co-transfected with human KOP along with a luciferase-based cAMP biosensor (GloSensor; Promega) and assays were performed similar to previously described (Fenalti et al., 2014). After 16 h, transfected cells were plated into Poly-lysine coated 384-well white clear bottom cell culture plates with DMEM + 1% dialysed FBS at a density of 15,000–20,000 cells per 40 μl per well and incubated at 37 °C with 5% CO2 overnight. The next day, drug solutions were prepared in fresh drug buffer [20 mM HEPES, 1X HBSS, 0.3% bovine serum album (BSA), pH 7.4] at 3X drug concentration. Plates were decanted and received 20 μl per well of drug buffer (20 mM HEPES, 1X HBSS) followed by addition of 10 μl of drug solution (3 wells per condition) for 15 min in the dark at room temperature. To stimulate endogenous cAMP via β adrenergic-Gs activation, 10 μl luciferin (4 mM final concentration) supplemented with isoproterenol (400 nM final concentration) were added per well. Cells were again incubated in the dark at room temperature for 15 min, and luminescence intensity was quantified using a Wallac TriLux microbeta (Perkin Elmer) luminescence counter. Results (relative luminescence units) were plotted as a function of drug concentration, normalized to % SalA stimulation, and analyzed using “log(agonist) vs. response” in GraphPad Prism 5.0.
Tango arrestin recruitment assay
The KOP Tango constructs were designed and assays were performed as previously described (Kroeze et al., 2015; Liu et al., 2013). HTLA cells expressing TEV fused-β-Arrestin2 (kindly provided by Dr. Richard Axel, Columbia Univ.) were transfected with the KOP Tango construct. The next day, cells were plated in DMEM supplemented with 1% dialyzed FBS in poly-L-lysine coated 384-well white clear bottom cell culture plates at a density of 10,000–15,000 cells/well in a total of 40 μl. The cells were incubated for at least 6 h before receiving drug stimulation. Drug solutions were prepared in drug buffer (20 mM HEPES, 1X HBSS, 0.3% BSA, pH 7.4) at 3X and added to cells (20 μlper well) for overnight incubation. Drug solutions used for the Tango assay were exactly the same as used for the cAMP assay. The next day, media and drug solutions were removed and 20 μl per well of BrightGlo reagent (purchased from Promega, after 1:20 dilution) was added. The plate was incubated for 20 min at room temperature in the dark before being counted using a luminescence counter. Results (relative luminescence units) were plotted as a function of drug concentration, normalized to % SalA stimulation, and analyzed using “log(agonist) vs. response” in GraphPad Prism 5.0.
GTPγ[35S] assay
KOP-Gαi1, KOP-Gαi1 Y3127.35W and MOP-Gαi1 fusion constructs were transfected into HEK 293T cells and membrane was prepared 48 hrs later. The GTPγ[35S] assay was conducted in assay buffer (20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, pH 7.4). In a 96-well plate, 20 μL of 30 μM GDP, 20 μL of 100 μM GTPγS (for Non-specific) or buffer (for Total), 20 μL of 3 nM GTPγ[35S], 20 μL of a serial dilution of KOP agonist and 120 μL of premixed membrane and 2.1mg/mL WGA-SPA PVT beads (Perkin Elmer) were added sequentially to each well (200 μL/well). The plate was sealed and agitated for 20–120 min at RT, and counted in SPA mode in a TriLux microbeta (Perkin Elmer). Results (CPM) were plotted as a function of drug concentration, normalized to % SalA or DAMGO stimulation, and analyzed using “log(agonist) vs. response” in GraphPad Prism 5.0.
Bioluminescence Resonance Energy Transfer (BRET) assay
To measure KOP-nanobody recruitment, HEK293T cells were co-transfected in a 1:3 ratio with human KOP containing C-terminal Renilla luciferase (RLuc8) and nanobody containing a C-terminal YFP. After at least 16 hours, transfected cells were plated in poly-lysine coated 96-well white clear bottom cell culture plates in plating media (DMEM + 1% dialyzed FBS) at a density of 40–50,000 cells in 200 μl per well and incubated overnight. The next day, media was decanted and cells were washed twice with 60 μL of drug buffer (20 mM HEPES, 1X HBSS, pH 7.4), then 60 μL of the RLuc substrate, coelenterazine h (Promega, 5 μM final concentration in drug buffer) was added per well, incubated an additional 5 minutes to allow for substrate diffusion. Afterwards, 30 μL of drug (3X) in drug buffer (20 mM HEPES, 1X HBSS, 0.1% BSA, pH 7.4) was added per well and incubated for another 5 minutes. Plates were immediately read for both luminescence at 485 nm and fluorescent eYFP emission at 530 nm for 1 second per well using a Mithras LB940 multimode microplate reader. The ratio of eYFP/RLuc was calculated per well and the net BRET ratio was calculated by subtracting the eYFP/RLuc per well from the eYFP/RLuc ratio in wells without Nb-YFP present. The net BRET ratio was plotted as a function of drug concentration using Graphpad Prism 5 (Graphpad Software Inc., San Diego, CA).
Radioligand binding and ligand dissociation assays
Binding assays were performed using Sf9 membrane fractions expressing the crystallization construct BRIL-KOP or HEK293 T membrane preparations transiently expressing KOP wt or KOP mutants. Binding assays were set up in 96-well plates in the standard binding buffer (50 mM Tris, 0.1 mM EDTA, 10 mM MgCl2, 0.1% BSA, pH 7.40). Saturation binding assays with 0.1–20 nM [3H]-Diprenorphine or [3H]-U69,593 in standard binding buffer were performed to the determine equilibrium dissociation constant (Kd) and Bmax, whereas 10 uM final concentration of JDTic was used to define nonspecific binding. For the competition binding, 50 μL each of 3H-Diprenorphine (final 1 nM), drug solution (3X) and homogenous membrane solution was incubated in 96-well plate in the standard binding buffer. Reactions (either saturation or competition binding) were incubated for 2 h at room temperature in the dark, and terminated by rapid vacuum filtration onto chilled 0.3% PEI-soaked GF/A filters followed by three quick washes with cold washing buffer (50 mM Tris HCl, pH 7.40) and read. Results (with or without normalization) were analyzed using GraphPad Prism 5.0 using one-site or allosteric IC50 shift models where indicated.
Radioligand dissociation assays were performed in 96-well plates in the standard binding buffer (50 mM Tris, 0.1 mM EDTA, 10 mM MgCl2, 0.1% BSA, pH 7.40). All assays utilized 2 concentrations of radioligand ([3H]-U69,593 = 0.5–2.0 nM) (PerkinElmer). For dissociation assays, membranes were incubated with radioligand for at least 2 hours at 37 °C in the absence or presenc e of Nb6 or Nb39 before the addition of 10 μL of 10 μM excess cold ligand to the 200 μL membrane suspension at designated time points. Time points spanned 2 minutes to 2 hours. Non-specific binding was determined by addition of 10 μM JDTic for KOP. Immediately at time = 0 min, plates were harvested by vacuum filtration onto 0.3% polyethyleneimine pre-soaked 96-well filter mats (Perkin Elmer) using a 96-well Filtermate harvester, followed by three washes of cold wash buffer (50 mM Tris pH 7.4). Scintillation (Meltilex) cocktail (Perkin Elmer) was melted onto dried filters and radioactivity was counted using a Wallac Trilux MicroBeta counter (PerkinElmer). Data were analyzed using “Dissociation – One phase exponential decay” in Graphpad Prism 5.0.
Molecular modeling
The structure of kappa opioid receptor complex co-crystallized with MP1104 was prepared for docking experiments by addition and optimization of hydrogen atoms, and optimization of side chain residues. The ligand docking box for potential grid docking was defined as the whole extracellular half of the protein, including co-crystallized MP1104 ligand. Energy minimized structures of MP1104, IBNtxA, U-69,593, U-50,488 and Salvinorin A (SalA) were docked unto the kappa opioid receptor orthosteric site with a thoroughness value of 30, and top scored docking solutions were retained. The retained top scored docking poses were further optimized by several rounds of minimization and Monte Carlo sampling of ligand conformation and surrounding side chain residues (within 4 Å if the ligand) in the orthosteric ligand pocket. All the above molecular modeling operations were performed in ICM-Pro v3.8–5 molecular modeling package (Abagyan et al., 1994).
For SalA, an additional round of simulations was performed by molecular dynamics (MD) methods. Gromacs 5.0.4 (Van der Spoel et al., 2005) was used to perform all MD simulations. All input files for MD simulation of the docked conformation of SalA and kappa opioid receptor complex, and parameter files for SalA were generated using CHARMM-GUI server (Kim et al., 2017; Lee et al., 2016). The orientation of helices within the membrane was derived after overlapping the complex with orientation of 4DJH (the JDTic co-crystallized kappa opioid receptor structure) obtained from the OPM server (Lomize et al., 2006). Two MD runs of membrane embedded and water boxed SalA-kappa opioid receptor complex (including 218 POPC lipid molecules, 19206 water molecules, 56 sodium ions and 61 chloride ions) were simulated with the CHARMM forcefield (Best et al., 2012) at 310K temperature with a step size of 2 femtoseconds using 6 GPU-enabled nodes with 16 processors for a period of 650 ns and 500 ns, after minimization and equilibrations. During the MD runs the hydrogen atoms were constrained using LINCS and cut-off of 12 Å was used for Van der Waals and short range electrostatic interactions, along with PME conditions. MD-derived receptor–ligand complexes were then subjected to an iterative refinement process guided by experimental data from mutagenesis studies and SalA structure–activity relationships (SAR). In the first step, rotatable bonds of the ligand and/or amino acid side chains of KOP were modified either manually (for the ligand) or algorithmically with a rotamer library using SCWRL4 (Krivov et al., 2009) (for KOP) so as to maximize the stereoelectronic complementarity of the interacting partners. An energy minimization step was then performed on the complex in SYBYL-X 2.1.1 (Certara USA, Inc., Princeton, NJ) using the Tripos Force Field (Gasteiger–Hückel charges, distance-dependent dielectric constant = 4.0 D/Å, termination criteria: energy gradient cut-off = 0.05 kcal (mol×Å)−1 or 100,000 iterations). This was followed by a HINT (Eugene Kellogg and Abraham, 2000) analysis to assess the energetic favourability of the receptor–ligand complex. The iterative refinement process terminated when no further energetically favourable structural modifications could be identified. The stereochemical quality of the final models was assessed using PROCHECK (Laskowski et al., 1993).
MP1104 analog synthesis
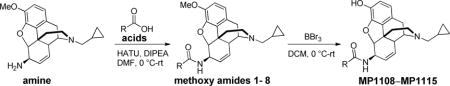
Chemicals were purchased from Sigma-Aldrich Chemicals, Fisher scientific, Alfa Aesar, and were used without further purification. Reaction mixtures were purified by silica gel flash chromatography on E. Merck 230–400 mesh silica gel 60 using a Teledyne ISCO CombiFlash Rf instrument with UV detection at 280 and 254 nm. RediSep Rf silica gel normal phase columns were used with a gradient range of 0–10% MeOH in DCM. The yields reported are isolated yields. NMR spectra were recorded on Bruker Avance III 600 with DCH CryoProbe instruments. Chemical shifts are reported in parts per million (ppm) relative to residual solvent peaks at the nearest 0.01 for proton and 0.01 for carbon (CDCl3 1H: 7.26, 13C: 77.10). Peak multiplicity is reported as the NMR spectra were processed with MestReNova software, namely s – singlet, d – doublet, t – triplet, q – quartet, m – multiplet for examples. Coupling constant (J) values are expressed in Hz. Mass spectra were obtained at the MSKCC Analytical Core Facility using The Waters Acuity SQD LC MS by electrospray (ESI) ionization. High resolution mass spectra were obtained on a Waters Acuity Premiere XE TOF LC-MS by electrospray ionization. Accurate masses are reported for the molecular ion [M+H]+. Purity of the products (≥95%) was confirmed by Waters Acquity UPLC: equipped with a binary solvent manager system, Waters XBridge C18 column (1.7 m × 2.1 × 100mm), PDA, ELS and QDa mass detectors; mobile phase: solvent A: water with 0.1% TFA; solvent B: acetonitrile with 0.1% TFA. Gradient: 5–95% acetonitrile/water with a flow rate of 1 ml/min in a reversed-phase was used. MP903, amine, methoxy amides 1–8, and MP1108–MP1115 were synthesized according to the previous protocols (Majumdar et al., 2012; Váradi et al., 2015).
Reagents/solvents
R = phenyl, m-chlorophenyl, m-bromophenyl, o-iodophenyl, p-iodophenyl, m-trifluoromethyl phenyl, m-fluorophenyl, and m-methyl phenyl; HATU – 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, DIPEA – N,N-Diisopropylethylamine, DMF – N,N-Dimethylformamide, and DCM – dichloromethane.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-methoxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)benzamide 1
(20 mg scale, Yield: 51%); 1H NMR (600 MHz, Chloroform-d) δ = 7.88 – 7.71 (m, 2H), 7.54 – 7.46 (m, 1H), 7.43 (t, J = 7.6 Hz, 2H), 6.75 (d, J = 8.2 Hz, 1H), 6.62 (d, J = 8.2 Hz, 1H), 6.26 (s, 1H), 6.06 – 5.88 (m, 1H), 5.65 (dd, J = 9.8, 1.9 Hz, 1H), 4.97 (s, 1H), 4.70 (s, 1H), 4.20 (s, 1H), 3.88 (s, 3H), 3.54 (s, 1H), 3.38 (d, J = 12.6 Hz, 1H), 3.14 – 2.90 (m, 2H), 2.80 (br, 2H), 2.41 (td, J = 13.3, 4.7 Hz, 1H), 2.08 – 1.88 (m, 1H), 1.19 – 1.06 (m, 1H), 0.76 (d, J = 8.0 Hz, 2H), 0.50 – 0.31 (m, 2H). 13C NMR (151 MHz, CDCl3) δ = 167.54, 146.16, 143.25, 133.66, 131.96, 130.62, 129.72, 128.95, 128.78, 128.14, 127.19, 119.55, 114.84, 91.98, 60.12, 58.69, 56.84, 49.88, 46.67, 43.55, 21.85, 6.93, 4.53, 4.51. ESI-MS m/z: 443.41 [M+H]+.
3-Chloro-N-((7R,12bS)-3-(cyclopropylmethyl)-9-methoxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)benzamide 2
(21 mg scale, Yield: 50%); 1H NMR (600 MHz, Chloroform-d) δ = 7.77 (t, J = 1.9 Hz, 1H), 7.64 (dt, J = 7.7, 1.3 Hz, 1H), 7.47 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H), 7.37 (t, J = 7.9 Hz, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.58 (d, J = 8.2 Hz, 1H), 6.12 (s, 1H), 6.03 – 5.84 (m, 1H), 5.70 (dd, J = 9.8, 1.9 Hz, 1H), 4.93 (t, J = 1.2 Hz, 1H), 4.66 (t, J = 6.6 Hz, 1H), 3.95 (d, J = 5.5 Hz, 1H), 3.88 (s, 3H), 3.32 (s, 1H), 3.05 (dd, J = 42.3, 15.6 Hz, 2H), 2.70 (d, J = 36.3 Hz, 2H), 2.61 – 2.45 (m, 2H), 2.25 (td, J = 13.0, 4.5 Hz, 1H), 2.00 – 1.78 (m, 1H), 1.01 (t, J = 6.5 Hz, 1H), 0.65 (dd, J = 7.0, 3.5 Hz, 2H), 0.28 (d, J = 4.9 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ = 165.95, 146.10, 142.75, 135.74, 134.88, 134.26, 131.87, 130.08, 129.84, 129.57, 127.54, 125.15, 123.49, 119.24, 114.23, 92.16, 60.02, 57.46, 56.81, 50.16, 46.04, 44.21, 34.97, 21.30, 8.23, 4.32, 4.22. ESI-MS m/z: 477.41 [M+H]+.
3-Bromo-N-((7R,12bS)-3-(cyclopropylmethyl)-9-methoxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)benzamide 3
(23 mg scale, Yield: 50%); 1H NMR (600 MHz, Chloroform-d) δ = 7.93 (t, J = 1.8 Hz, 1H), 7.69 (dt, J = 7.9, 1.3 Hz, 1H), 7.64 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H), 7.33 (t, J = 7.9 Hz, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.58 (d, J = 8.2 Hz, 1H), 6.08 – 5.85 (m, 2H), 5.70 (dd, J = 9.7, 2.0 Hz, 1H), 4.93 (t, J = 1.2 Hz, 1H), 4.66 (d, J = 6.8 Hz, 1H), 3.89 (s, 4H), 3.24 (s, 1H), 3.05 – 2.99 (m, 2H), 2.66 (d, J = 32.3 Hz, 2H), 2.53 (d, J = 20.2 Hz, 2H), 2.29 – 2.14 (m, 1H), 1.92 (d, J = 12.8 Hz, 1H), 1.00 (d, J = 7.1 Hz, 0H), 0.73 – 0.57 (m, 2H), 0.27 (d, J = 5.0 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ = 165.80, 146.18, 142.70, 135.93, 134.83, 130.40, 130.35, 129.91, 129.49, 125.59, 123.50, 122.93, 119.19, 114.20, 92.19, 60.19, 57.48, 56.83, 50.12, 46.04, 44.30, 39.60, 21.21, 8.43, 4.27, 4.20. ESI-MS m/z: 521.38 [M+H]+.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-methoxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-2-iodobenzamide 4
(15 mg scale, Yield: 30%); 1H NMR (600 MHz, Chloroform-d) δ = 7.85 (dd, J = 8.0, 1.1 Hz, 1H), 7.51 – 7.32 (m, 2H), 7.18 – 7.02 (m, 1H), 6.70 (d, J = 8.2 Hz, 1H), 6.57 (d, J = 8.2 Hz, 1H), 5.99 (ddd, J = 9.6, 4.9, 1.9 Hz, 1H), 5.68 (dt, J = 14.6, 7.3 Hz, 2H), 5.07 (s, 1H), 4.67 (s, 1H), 3.89 (s, 4H), 3.16 (s, 1H), 2.99 (d, J = 18.7 Hz, 2H), 2.61 – 2.51 (m, 4H), 2.26 (s, 1H), 1.93 (d, J = 12.7 Hz, 1H), 0.95 (s, 1H), 0.61 (s, 2H), 0.24 (s, 2H). 13C NMR (151 MHz, CDCl3) δ = 169.04, 146.26, 142.58, 141.61, 139.88, 131.45, 129.05, 128.58, 128.41, 119.12, 114.31, 92.54, 91.92, 60.03, 57.20, 56.92, 50.08, 45.94, 44.37, 40.39, 35.17, 21.15, 8.52, 4.27, 4.07. ESI-MS m/z: 569.32 [M+H]+.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-methoxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-4-iodobenzamide 5
(18 mg scale, Yield: 36%); 1H NMR (600 MHz, Chloroform-d) δ = 7.85 – 7.74 (m, 2H), 7.53 – 7.39 (m, 2H), 6.69 (d, J = 8.2 Hz, 1H), 6.55 (d, J = 8.1 Hz, 1H), 6.00 – 5.86 (m, 2H), 5.71 (dd, J = 9.7, 2.0 Hz, 1H), 4.91 (d, J = 1.2 Hz, 1H), 4.64 (t, J = 6.6 Hz, 1H), 3.88 (s, 3H), 3.75 (s, 1H), 3.16 (s, 1H), 2.97 (d, J = 18.5 Hz, 1H), 2.88 (d, J = 12.4 Hz, 1H), 2.49 (dd, J = 16.3, 6.5 Hz, 2H), 2.41 – 2.27 (m, 2H), 2.08 (d, J = 49.8 Hz, 1H), 1.87 (d, J = 12.6 Hz, 1H), 0.92 (s, 1H), 0.58 (dd, J = 8.0, 2.7 Hz, 2H), 0.19 (d, J = 5.2 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ = 166.31, 146.22, 142.36, 137.89, 137.17, 133.67, 133.54, 131.38, 130.48, 129.06, 128.67, 123.42, 118.98, 113.86, 98.79, 92.39, 59.97, 56.82, 56.65, 50.09, 45.56, 44.69, 40.29, 36.02, 20.81, 9.12, 4.15, 3.99. ESI-MS m/z: 569.32 [M+H]+.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-methoxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-3-(trifluoromethyl)benzamide 6
(30 mg scale, Yield: 66%); 1H NMR (600 MHz, Chloroform-d) δ = 8.06 (s, 1H), 7.96 (d, J = 7.8 Hz, 1H), 7.80 – 7.68 (m, 1H), 7.59 (t, J = 7.8 Hz, 1H), 6.72 (d, J = 8.2 Hz, 1H), 6.58 (d, J = 8.2 Hz, 1H), 6.08 (s, 1H), 5.98 (ddd, J = 9.9, 6.1, 3.4 Hz, 1H), 5.72 (dd, J = 9.7, 1.9 Hz, 1H), 4.95 (d, J = 1.3 Hz, 1H), 4.76 – 4.48 (m, 1H), 3.89 (s, 4H), 3.27 (s, 1H), 3.11 – 2.91 (m, 2H), 2.67 (d, J = 34.3 Hz, 2H), 2.54 (d, J = 24.2 Hz, 2H), 2.23 (s, 1H), 1.93 (d, J = 12.9 Hz, 1H), 1.00 (s, 1H), 0.65 (d, J = 8.0 Hz, 2H), 0.34 – 0.20 (m, 3H). 13C NMR (151 MHz, CDCl3) δ = 165.83, 146.14, 142.74, 134.75, 131.39, 131.18, 130.24, 129.41, 128.45, 124.65, 124.37, 124.35, 122.84, 119.23, 114.19, 92.14, 60.19, 57.52, 56.81, 50.22, 46.06, 44.28, 39.57, 35.18, 21.21, 8.38, 4.27, 4.21. ESI-MS m/z: 511.41 [M+H]+.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-methoxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-3-fluorobenzamide 7
(27 mg scale, Yield: 66%); 1H NMR (600 MHz, Chloroform-d) δ = 7.52 (ddt, J = 7.7, 4.2, 1.6 Hz, 2H), 7.42 (td, J = 8.0, 5.4 Hz, 1H), 7.20 (ddd, J = 8.3, 2.6, 1.0 Hz, 1H), 6.72 (d, J = 8.2 Hz, 1H), 6.58 (d, J = 8.2 Hz, 1H), 6.03 – 5.84 (m, 2H), 5.70 (dd, J = 9.8, 2.0 Hz, 1H), 4.94 (t, J = 1.2 Hz, 1H), 4.67 (s, 1H), 3.89 (s, 4H), 3.26 (s, 1H), 3.12 – 2.89 (m, 2H), 2.68 (d, J = 34.7 Hz, 2H), 2.53 (s, 2H), 2.23 (s, 1H), 1.94 (d, J = 13.0 Hz, 1H), 1.05 – 0.96 (m, 0H), 0.66 (d, J = 8.0 Hz, 2H), 0.28 (s, 2H). 13C NMR (151 MHz, CDCl3) δ = 165.97, 163.67, 162.02, 146.21, 142.73, 136.24, 136.19, 130.49, 130.44, 129.59, 122.49, 122.47, 119.20, 118.97, 118.83, 114.68, 114.53, 114.27, 92.15, 60.22, 57.58, 56.84, 50.04, 46.09, 44.25, 35.34, 20.17, 8.34, 4.27, 4.22. ESI-MS m/z: 461.43 [M+H]+.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-methoxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-3-methylbenzamide 8
(21 mg scale, Yield: 51%); 1H NMR (600 MHz, Chloroform-d) δ = 7.58 (s, 1H), 7.56 – 7.44 (m, 1H), 7.40 – 7.29 (m, 2H), 6.69 (d, J = 8.2 Hz, 1H), 6.55 (d, J = 8.1 Hz, 1H), 6.00 – 5.84 (m, 2H), 5.71 (dd, J = 9.7, 2.0 Hz, 1H), 4.93 (d, J = 1.2 Hz, 1H), 4.66 (s, 1H), 3.89 (s, 3H), 3.77 (s, 1H), 3.16 (d, J = 15.1 Hz, 1H), 2.98 (d, J = 18.5 Hz, 1H), 2.92 (br, 1H), 2.52 (d, J = 13.1 Hz, 2H), 2.41–2.30 (m, 5H), 2.14 (s, 1H), 1.93 – 1.80 (m, 1H), 0.94 (t, J = 5.1 Hz, 1H), 0.59 (d, J = 7.6 Hz, 2H), 0.27 – 0.12 (m, 2H). 13C NMR (151 MHz, CDCl3) δ = 167.30, 146.28, 142.40, 138.60, 134.08, 132.53, 130.48, 129.41, 128.58, 127.74, 124.02, 118.93, 113.98, 92.49, 60.06, 56.87, 49.97, 45.68, 44.64, 40.20, 35.91, 21.44, 20.90, 9.06, 4.16, 4.04. ESI-MS m/z: 457.47 [M+H]+.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-hydroxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)benzamide MP1108
(3.7 mg scale, Yield: 24%); 1H NMR (600 MHz, Chloroform-d) δ = 7.80 – 7.67 (m, 2H), 7.57 – 7.48 (m, 1H), 7.43 (t, J = 7.6 Hz, 2H), 6.70 (d, J = 8.1 Hz, 1H), 6.51 (d, J = 8.1 Hz, 1H), 6.05 (s, 1H), 5.91 (ddd, J = 9.4, 5.9, 3.1 Hz, 1H), 5.79 – 5.63 (m, 1H), 4.97 – 4.80 (m, 1H), 4.63 (t, J = 6.5 Hz, 1H), 3.80 (s, 1H), 3.30 – 3.08 (m, 1H), 2.95 (d, J = 18.6 Hz, 2H), 2.54 (d, J = 35.2 Hz, 2H), 2.47 – 2.27 (m, 2H), 2.15 (s, 1H), 1.86 (t, J = 14.2 Hz, 1H), 0.90 – 0.74 (m, 1H), 0.58 (d, J = 7.1 Hz, 2H), 0.29 – 0.14 (m, 2H). 13C NMR (151 MHz, CDCl3) δ = 167.42, 144.56, 138.37, 133.90, 131.91, 129.90, 128.74, 127.09, 119.47, 116.79, 93.03, 59.91, 56.86, 50.36, 45.74, 44.69, 30.65, 30.12, 29.80, 27.01, 20.85, 8.85, 4.20, 4.11. ESI-MS m/z: 429.41 [M+H]+. HRMS calcd for C27H29N2O3 [M+H]+, 429.2178; found, 429.2167.
3-Chloro-N-((7R,12bS)-3-(cyclopropylmethyl)-9-hydroxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)benzamide MP1109
(6.8 mg scale, Yield: 37%); 1H NMR (600 MHz, Chloroform-d) δ = 7.76 (t, J = 1.9 Hz, 1H), 7.64 (dt, J = 7.8, 1.3 Hz, 1H), 7.45 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H), 7.33 (t, J = 7.8 Hz, 1H), 6.71 (d, J = 8.1 Hz, 1H), 6.51 (d, J = 8.1 Hz, 1H), 6.35 (s, 1H), 5.96 – 5.80 (m, 1H), 5.71 (dd, J = 9.8, 1.9 Hz, 1H), 4.86 (d, J = 1.2 Hz, 1H), 4.61 (t, J = 6.5 Hz, 1H), 3.88 (d, J = 5.3 Hz, 1H), 3.33 (s, 1H), 2.98 (dd, J = 27.6, 15.3 Hz, 2H), 2.64 – 2.57 (m, 2H), 2.44 (ddd, J = 37.6, 16.1, 8.6 Hz, 2H), 2.23 (s, 1H), 1.81 (dt, J = 12.5, 2.7 Hz, 1H), 1.01 (s, 1H), 0.60 (ddd, J = 11.9, 8.5, 5.1 Hz, 2H), 0.24 (p, J = 4.4 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ = 166.22, 144.48, 138.72, 135.67, 134.84, 131.87, 130.02, 129.67, 128.75, 127.57, 125.28, 119.60, 117.26, 92.77, 59.75, 56.94, 50.60, 45.91, 44.44, 39.33, 34.94, 20.99, 8.43, 4.33, 4.21. ESI-MS m/z: 463.35 [M+H]+.HRMS calcd for C27H28ClN2O3 [M+H]+, 463.1788; found, 463.1778.
3-Bromo-N-((7R,12bS)-3-(cyclopropylmethyl)-9-hydroxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)benzamide MP1110
(4.2 mg scale, Yield: 22%); 1H NMR (600 MHz, Chloroform-d) δ = 7.93 (t, J = 1.8 Hz, 1H), 7.71 (dd, J = 7.9, 1.4 Hz, 1H), 7.62 (ddd, J = 7.9, 2.0, 1.0 Hz, 1H), 7.29 (t, J = 7.9 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.53 (d, J = 8.1 Hz, 1H), 6.31 (s, 1H), 5.89 (ddd, J = 9.5, 5.9, 3.2 Hz, 1H), 5.76 – 5.59 (m, 1H), 4.96 – 4.80 (m, 1H), 4.63 (t, J = 6.5 Hz, 1H), 3.93 (s, 1H), 3.46 (d, J = 38.6 Hz, 1H), 3.01 (dd, J = 54.0, 14.6 Hz, 2H), 2.65 (d, J = 37.4 Hz, 2H), 2.54 – 2.45 (m, 2H), 2.30 (s, 1H), 1.90 – 1.80 (m, 1H), 1.07 (s, 1H), 0.64 (d, J = 8.0 Hz, 2H), 0.39 – 0.16 (m, 2H). 13C NMR (151 MHz, CDCl3) δ = 166.11, 144.41, 138.79, 135.80, 134.86, 130.45, 130.30, 129.50, 128.93, 125.78, 122.89, 119.67, 117.31, 92.80, 59.68, 57.03, 50.59, 46.05, 44.33, 38.93, 34.65, 30.12, 21.10, 8.18, 4.46, 4.32. ESI-MS m/z: 507.32 [M+H]+. HRMS calcd for C27H28BrN2O3 [M+H]+, 507.1283; found, 507.1259.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-hydroxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-2-iodobenzamide MP1111
(3.6 mg scale, Yield: 34%); 1H NMR (600 MHz, Chloroform-d) δ = 7.85 (d, J = 7.9 Hz, 1H), 7.47 – 7.34 (m, 2H), 7.11 (td, J = 7.7, 1.7 Hz, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.50 (d, J = 8.1 Hz, 1H), 5.98 – 5.87 (m, 2H), 5.81 – 5.57 (m, 1H), 5.05 (s, 1H), 4.63 (t, J = 6.6 Hz, 1H), 3.79 (s, 1H), 3.22 – 3.05 (m, 1H), 2.95 (d, J = 18.5 Hz, 2H), 2.39 (t, J = 83.9 Hz, 5H), 1.89 (d, J = 11.7 Hz, 1H), 1.02 – 0.74 (m, 1H), 0.58 (s, 2H), 0.20 (s, 2H). 13C NMR (151 MHz, CDCl3) δ = 169.19, 144.62, 141.42, 139.90, 138.31, 131.52, 128.78, 128.36, 128.01, 119.50, 116.80, 92.49, 59.81, 56.70, 50.35, 45.73, 44.70, 30.12, 20.88, 8.71, 4.26, 4.10. ESI-MS m/z: 555.32 [M+H]+. HRMS calcd for C27H28IN2O3 [M+H]+, 555.1145; found, 555.1147.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-hydroxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-4-iodobenzamide MP1112
(3.1 mg scale, Yield: 22%); 1H NMR (600 MHz, Chloroform-d) δ = 7.76 (d, J = 8.3 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H), 6.70 (d, J = 8.1 Hz, 1H), 6.52 (d, J = 8.1 Hz, 1H), 6.11 (s, 1H), 5.88 (ddd, J = 9.5, 6.1, 3.2 Hz, 1H), 5.72 (dd, J = 9.8, 1.9 Hz, 1H), 4.85 (d, J = 1.2 Hz, 1H), 4.60 (t, J = 6.5 Hz, 1H), 3.83 (s, 1H), 3.21 (d, J = 39.8 Hz, 1H), 2.95 (d, J = 18.6 Hz, 2H), 2.64 – 2.47 (m, 2H), 2.47 – 2.28 (m, 2H), 2.18 (s, 1H), 1.89 – 1.78 (m, 1H), 1.04 – 0.88 (m, 1H), 0.59 (d, J = 6.5 Hz, 2H), 0.26 – 0.16 (m, 2H). 13C NMR (151 MHz, CDCl3) δ = 166.68, 144.53, 138.46, 137.93, 133.29, 129.81, 128.73, 128.62, 119.57, 116.96, 98.98, 92.88, 59.86, 56.85, 50.42, 45.78, 44.63, 39.70, 35.38, 30.13, 20.88, 8.71, 4.25, 4.16. ESI-MS m/z: 555.25 [M+H]+. HRMS calcd for C27H28IN2O3 [M+H]+, 555.1145; found, 555.1125.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-hydroxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-3-(trifluoromethyl)benzamide MP1113;
(6.1 mg scale, Yield: 24%); 1H NMR (600 MHz, Chloroform-d) δ = 8.04 (s, 1H), 7.96 (d, J = 7.8 Hz, 1H), 7.84 – 7.67 (m, 1H), 7.55 (t, J = 7.8 Hz, 1H), 6.70 (d, J = 8.1 Hz, 1H), 6.51 (d, J = 8.1 Hz, 1H), 6.27 (s, 1H), 5.90 (ddd, J = 9.5, 6.0, 3.1 Hz, 1H), 5.78 – 5.63 (m, 1H), 4.88 (t, J = 1.1 Hz, 1H), 4.63 (t, J = 6.5 Hz, 1H), 3.84 (s, 1H), 3.22 (d, J = 51.5 Hz, 1H), 2.95 (d, J = 18.2 Hz, 2H), 2.56 (d, J = 36.6 Hz, 2H), 2.46 – 2.26 (m, 2H), 2.19 (s, 1H), 1.88 – 1.73 (m, 1H), 0.97 (s, 1H), 0.63 – 0.53 (m, 2H), 0.25 – 0.16 (m, 2H). 13C NMR (151 MHz, CDCl3) δ = 166.07, 144.45, 138.49, 134.70, 131.37, 131.15, 130.41, 129.78, 129.37, 128.47, 124.61, 124.28, 124.25, 122.80, 119.60, 117.02, 92.81, 59.84, 56.87, 50.66, 45.79, 44.60, 35.26, 30.12, 20.89, 8.68, 4.25, 4.13. ESI-MS m/z: 497.42 [M+H]+. HRMS calcd for C28H28F3N2O3 [M+H]+, 497.2052; found, 497.2060.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-hydroxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-3-fluorobenzamide MP1114
(4.8 mg scale, Yield: 22%); 1H NMR (600 MHz, Chloroform-d) δ = 7.57 – 7.43 (m, 2H), 7.38 (td, J = 8.1, 5.6 Hz, 1H), 7.23 – 7.10 (m, 1H), 6.70 (d, J = 8.1 Hz, 1H), 6.51 (d, J = 8.1 Hz, 1H), 6.22 (s, 1H), 5.88 (ddd, J = 9.8, 4.7, 2.0 Hz, 1H), 5.72 (dd, J = 9.8, 1.9 Hz, 1H), 4.87 (d, J = 1.2 Hz, 1H), 4.61 (t, J = 6.5 Hz, 1H), 3.97 – 3.75 (m, 1H), 3.28 (s, 1H), 2.96 (d, J = 18.6 Hz, 2H), 2.58 (d, J = 38.4 Hz, 2H), 2.51 – 2.35 (m, 2H), 2.21 (s, 1H), 1.83 (dt, J = 12.8, 2.8 Hz, 1H), 1.04 – 0.94 (m, 1H), 0.60 (t, J = 6.7 Hz, 2H), 0.23 (q, J = 4.5 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ = 166.23, 163.62, 161.98, 144.53, 138.57, 136.18, 130.42, 130.37, 129.74, 128.69, 122.61, 122.59, 119.57, 118.96, 118.82, 117.10, 114.71, 114.56, 92.79, 59.80, 56.89, 50.52, 45.83, 44.54, 39.52, 30.12, 20.93, 8.57, 4.28, 4.17. ESI-MS m/z: 447.43 [M+H]+. HRMS calcd for C27H28 FN2O3 [M+H]+, 447.2084; found, 447.2076.
N-((7R,12bS)-3-(cyclopropylmethyl)-9-hydroxy-2,3,4,4a,7,7a-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-3-methylbenzamide MP1115
(3.5 mg scale, Yield: 38%); 1H NMR (600 MHz, Chloroform-d) δ = 7.58 (s, 1H), 7.54 (dt, J = 4.2, 2.1 Hz, 1H), 7.32 (dd, J = 4.6, 2.2 Hz, 2H), 6.69 (d, J = 8.1 Hz, 1H), 6.51 (d, J = 8.1 Hz, 1H), 5.97 (s, 1H), 5.94 – 5.87 (m, 1H), 5.73 (dd, J = 9.8, 1.9 Hz, 1H), 4.89 (d, J = 1.2 Hz, 1H), 4.62 (t, J = 6.6 Hz, 1H), 3.78 (s, 1H), 3.14 (s, 1H), 2.93 (t, J = 19.1 Hz, 2H), 2.51 (d, J = 27.9 Hz, 2H), 2.40 (b, 5H), 2.12 (s, 1H), 1.91 – 1.78 (m, 1H), 1.02 – 0.88 (m, 1H), 0.58 (d, J = 7.0 Hz, 2H), 0.20 (s, 2H). 13C NMR (151 MHz, CDCl3) δ = 167.57, 144.55, 138.63, 138.28, 133.87, 132.66, 129.98, 128.74, 128.62, 127.75, 124.08, 119.43, 116.68, 93.12, 59.94, 56.82, 50.31, 45.70, 44.76, 40.02, 35.69, 21.46, 20.81, 8.99, 4.17, 4.07. ESI-MS m/z: 443.41 [M+H]+. HRMS calcd for C28H31N2O3 [M+H]+, 443.2335; found, 443.2321.
N-((4aS,7R,12bS)-3-allyl-4a,9-dihydroxy-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-4-fluoro-3-iodobenzamide MP903
(Yield: 68%); 1H NMR (600 MHz, Chloroform-d) δ = 8.23 (dd, J = 5.9, 2.2 Hz, 1H), 7.78 (ddd, J = 8.5, 4.8, 2.2 Hz, 1H), 7.35 (d, J = 9.0 Hz, 1H), 7.07 (dd, J = 8.5, 7.5 Hz, 1H), 6.68 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 5.81 (ddt, J = 16.7, 10.1, 6.4 Hz, 1H), 5.27 – 5.09 (m, 2H), 4.58 (d, J = 5.6 Hz, 1H), 4.11 (dq, J = 9.5, 4.7 Hz, 1H), 3.20 – 3.03 (m, 3H), 2.97 (d, J = 6.0 Hz, 1H), 2.63 (dd, J = 18.4, 6.1 Hz, 1H), 2.55 (d, J = 7.5 Hz, 1H), 2.20 (d, J = 8.1 Hz, 2H), 1.91 (dtd, J = 14.1, 9.1, 5.1 Hz, 1H), 1.68 (dt, J = 14.1, 5.4 Hz, 1H), 1.61 – 1.43 (m, 3H). 13C NMR (151 MHz, CDCl3) δ = 164.57, 164.48, 162.82, 142.94, 139.18, 138.86, 135.23, 132.34, 130.60, 129.24, 129.19, 124.92, 119.46, 118.24, 117.56, 115.65, 115.49, 92.76, 81.58, 81.40, 70.31, 62.46, 57.88, 50.49, 47.30, 43.63, 31.75, 29.08, 23.18, 22.95, 22.78. HRMS calcd for C26H27FN2O3 [M+H]+, 577.1000; found, 577.0988.
QUANTIFICATION AND STATISTICAL ANALYSIS
Dose response, log(τ/KA) calculation and ligand bias quantification
Bias factor toward G protein was calculated with SalA or DAMGO as a reference agonist. In detail, dose-response data with respect to reference ligands were fit using the Black and Leff operational model in Graphpad Prism 5.0, where EMAX represents the maximum response of the system and was set to 100, KA is the functional dissociation constant for the agonist, and τ is the efficacy of the agonist in the given pathway, and n is the slope of the response. Data for MP1104 or IBNtxA was fit globally with reference ligand responses such that EMAX and n are shared parameters and KA and τ are then fit individually for MP1104 or IBNtxA. Transduction coefficients (log (τ/KA)) were calculated using the Black and Leff operational model (Black and Leff, 1983) in Graphpad Prism 5.0. Using SalA or DAMGO as the full agonist reference, transduction coefficients for Gi cAMP inhibition and Tango β-Arrestin2 translocation were calculated and averaged across experiments (N = 3). Calculation of bias factors utilized the method by Kenakin et al.(Kenakin et al., 2012), where the Δlog(τ/KA) was calculated relative to the reference ligand and the ΔΔlog(τ/KA) was calculated by subtracting the Gi transduction coefficient from the β-Arrestin2 transduction coefficient.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The accession number for the coordinates and structures factors of KOP-MP1104-Nb39 complex reported in this paper is PDB: 6B73.
Supplementary Material
(A) Screening of nanobodies from the KOP-SalA library using BRET assay. As can be seen, Nb6/7 (blue) shows dose-response upon SalA stimulation (N = 3). Note that concentration at −12 represents “no drugs” added.
(B) The antagonist JDTic reverses SalA-induced dissociation in a concentration-dependent manner. Cells expressing KOP-Rluc and Nb6-YFP were treated with 0.1 μM SalA for 5 min and then stimulated by increasing concentrations of JDTic for 5 min and BRET signal was measured.
(C) Recruitment of Nb39 to KOP in the presence of full agonist SalA (EC50 = 10.2±0.8 nM, Emax = 100±2, orange), partial agonist diprenorphine (EC50 = 5.20±0.03 nM, Emax = 32±2, blue), or antagonist JDTic (no activity detected, red) (N = 3).
(D) Comparison of U69,593 dissociation rate at KOP only (blue), KOP+Nb6 (green triangle) and KOP+Nb39 (yellow) shows a slow U69,593 off-rate with Nb39 and a faster off-rate with Nb6 (N = 3).
(E) Interaction of KOP and Nb39 in the presence of Gαi1 or β-arrestin2. Using BRET assay, KOP-Rluc and Nb39 were co-transfected with different amounts of Gαi1 or β-arrestin2 (N = 3). See Table S1 for values.
(F) Measurement of MP1104 binding affinity at MOP (Ki = 0.18±0.06 nM, red), KOP (Ki = 0.20±0.08 nM, orange) and DOP (Ki = 1.38±0.05 nM, light green) using radioligand 3H-Diprenorphine (N = 3).
(G) Measurement of MP1104-mediated Gi activation in KOP (MP1104, EC50 = 0.003±0.001 nM, Emax = 100±2, orange; SalA, EC50 = 0.033±0.008 nM, Emax = 98±2, red), MOP (MP1104, EC50 = 0.034±0.008 nM, Emax = 104±1, orange; DAMGO, EC50 = 3.45±0.06 nM, Emax = 100±2 red) and DOP (MP1104, EC50 = 0.67±0.07 nM, Emax = 97±2 orange; DADLE, EC50 = 1.34±0.05 nM, Emax = 100±2 red) (N = 3).
(A) Confirmation of KOP crystallization construct functionality in binding assays. MP1104 (orange), Ki = 0.33±0.02 nM; SalA (light green), Ki = 25.3±1.40 nM (N = 3).
(B) Confirmation of KOP crystallization construct functionality in cAMP inhibition assays. MP1104 (orange), EC50 = 0.006±0.001 nM; SalA (light green), EC50 = 0.26±0.02 nM (N = 3).
(C) Crystal packing of KOP-MP1104-Nb39 complex. KOP (blue), Nb39 (orange), MP1104 (orange sphere).
(D) Crystal contacts within anti-parallel monomers (top) or parallel dimers (bottom). The KOP-MP1104-Nb39 complex displays an anti-parallel receptor dimer in the asymmetric unit through interactions between ECL2 and intracellular loop 2 (ICL2) of the two monomers (D218ECL2—H1623.56/P163ICL2). Parallel dimers are observed within the crystal lattice with dimer interfaces formed by TM1 and TM2 residues.
(E) Electron density of MP1104 and ligand binding pocket residues. (left) Different angles showing structure of receptor-bound MP1104 (orange stick model) with mFo-DFc omit density map contoured at 3σ (green mesh). (middle) Different angles showing structure of receptor-bound MP1104 (orange stick model) with 2mFo-DFc electron density map contoured at 1σ (blue mesh). (right) Different angles showing structure of ligand binding pocket residues with 2mFo-DFc electron density map contoured at 1σ (blue mesh). All density maps were generated in Refmac5. Interactions between ligand and receptor residues result in continuous 2mFo-DFc density that appears “broken” when only receptor or ligand density is displayed. Grey dots indicate Hydrogen-bonds between receptor and ligand.
(F) Comparison of inactive and active-state β2AR or M2R indicates a common contraction during activation. Distances were measured between the Cα atoms of M401.39, S2035.42, and F2906.52, N3127.39 in β2AR; I261.35, V4076.55, I417ECL3, and N4197.32 in M2R.
(G and H) Residues within KOP-Nb39 interface (G) indicate they are highly conserved among the three canonical opioid receptors (H).
(A) Mutagenesis studies indicate that KOP T1112.56 and Y3207.43 residues are crucial for full activation of KOP. KOP wt (MP1104, EC50 = 0.035±0.005 nM, Emax = 100±2; Dynorphin A, EC50 = 12.60±1.15 nM, Emax = 75±3). KOP T1112.56 A (MP1104, EC50 = 3.50±0.23 nM, Emax = 93±3; Dynorphin A, EC50 = 12.30±1.05 nM, Emax = 30±2). KOP Y3207.43A (MP1104, EC50 = 10.20±0.89 nM, Emax = 91±2; Dynorphin A, no activity detected). (N = 3).
(B) Comparison of water-mediated interactions with the phenol group between KOP and MOP. Active state KOP residues are shown in blue sticks, and blue cartoon representation. Active state MOP (PDB ID: 5C1M) residues are shown in green sticks. MP1104 and BU-72 are shown in orange and green colored carbon sticks respectively. Crystallographic waters in active MOP (from 5C1M) are shown in green balls, and calculated waters in KOP are represented by blue balls. Water mediated hydrogen bonds common to both KOP and MOP are shown in black broken lines, while water mediated hydrogen bonds specific for MOP are shown in red broken lines.
(C) The MP1104 analog, MP1107, causes more severe reduction of binding affinity in MOP than in KOP. Chemical differences between compound structures are highlighted in red. MP1104 (red), Ki = 0.20±0.02 nM in KOP (top) and 0.16±0.03 nM in MOP (bottom); MP1107 (orange), Ki = 1.07±0.06 nM in KOP (top) and 5.85±0.08 nM in MOP (bottom) (N = 3).
(D) Removal of the cyclopropyl group reduces MP1104’s functional activity in KOP. Chemical differences between compound structures are highlighted in red. Gi activation (top): MP1104 (red), EC50 = 0.003±0.001 nM, Emax = 97±1; MP1101 (orange), EC50 = 0.055±0.010 nM; Emax = 98±1. β-Arrestin2 recruitment (bottom): MP1104 (red), EC50 = 0.043±0.010 nM, Emax = 113±2; MP1101 (orange), EC50 = 0.41±0.08 nM, Emax = 111±2 (N = 3).
(E) Docking of MP1104 (orange) in the active structure of MOP (green) indicates the cyclopropylmethyl groups also extends into the hydrophobic pocket.
(A) GTPγ[35S] (top panel) and BRET (arrestin recruitment) (bottom panel) activities indicates that KOP Y3127.35W recapitulates IBNtxA’s biased activity in MOP. Bias factor towards G protein in MOP was calculated with DAMGO as a reference agonist based on the Black and Leff operational model and defined as 10ΔΔLog(τ/KA): 0.8 and 9 for MP1104 and IBNtxA, respectively (N = 3). See Table S5 for values.
(B) Comparison of IBNtxA (grey) and MP1104 (orange) docking in MOP indicates that the iodobenzamide group adopts different orientations.
(C) Chemical structures and arrestin recruitment activity of IBNtxA analogs with a single bond in the saturated ring (highlighted in red). (left) Arrestin activity in KOP: MP903 (red), EC50 = 0.074±0.012 nM, Emax = 108±10; IBNtxA (orange), EC50 = 0.030±0.009 nM, Emax = 95±8; MP970 (light green), EC50 = 0.16±0.04 nM, Emax = 92±6; MP1104 (green), EC50 = 0.037±0.008 nM, Emax = 100±2. (right) Arrestin activity in MOP: MP903 (red), EC50 = 0.12±0.05 nM, Emax = 33±5; IBNtxA (orange), EC50 = 0.075±0.008 nM, Emax = 25±6; MP970 (light green), EC50 = 0.54±0.10 nM, Emax = 21±5; MP1104 (green), EC50 = 0.55±0.08 nM, Emax = 100±2 (N = 3). Bias factor towards G protein: 10.3 and 9 for MP903 and MP970, respectively.
(A) Docking poses of U69,593 and U50,488 in the active state KOP-MP1104-Nb39 structure.
(B) Putative binding mode for SalA in the KOP-MP1104-Nb39 structure.
(C) Chemical structures and binding affinity of MP1104 analogs with modifications in the iodobenzamide group. Kis in KOP (left) and MOP (right) respectively: MP1104, 0.20±0.06 and 0.16±0.05 nM; MP1108, 0.42±0.05 and 0.42±0.06 nM; MP1109, 0.13±0.03 and 0.16±0.05 nM; MP1110, 0.12±0.04 and 0.18±0.04 nM; MP1111, 0.27±0.06 and 0.20±0.05 nM; MP1112, 0.12±0.03 and 0.17±0.03 nM; MP1113, 0.14±0.07 and 0.24±0.06 nM; MP1114, 0.16±0.07 and 0.27±0.08 nM; MP1115, 0.18±0.08 and 0.25±0.06 nM (N = 3).
Highlights.
κ-opioid receptor active-state crystal structure disclosed
κ-opioid receptor structure reveals features involved in biased signaling
Structure provides template for creation of safe and effective analgesics
Acknowledgments
This work was supported by NIH Grants RO1MH61887, U19MH82441, the NIMH Psychoactive Drug Screening Program Contract (all to B.L.R.), and the Michael Hooker Distinguished Chair of Pharmacology (B.L.R.), as well as DA035764 (B.L.R., R.C.S., V.C., V.K) and DA038858 (V.K). This research was also funded, in part, by grants from the National Institute on Drug Abuse (DA0624), the Mayday Foundation and the Peter F. McManus Trust (G.W.P.), DA034106 (S.M.) and an NIH/NCI Cancer Center Support Grant P30 CA008748 to Memorial Sloan Kettering Cancer Center. We thank INSTRUCT, part of the European Strategy Forum on Research Infrastructures (ESFRI), and the Research Foundation – Flanders (FWO) for their support to the nanobody discovery and thank Nele Buys and Katleen Willibal for the technical assistance. We also thank R.H.J. Olsen for providing the KOP-Gαi1 and MOP-Gαi1 constructs. We gratefully acknowledge M. J. Miley and the UNC macromolecular crystallization core for advice and use of their equipment for crystal harvesting and transport, which is supported by the National Cancer Institute under award number P30CA016086. We thank J. Smith, R. Fischetti, and the staff of GM/CA@APS, which has been funded with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
T.C designed experiments, expressed and screened nanobodies, receptor constructs, and receptor ligands, crystallized nanobody-receptor complex, collected diffraction data, performed ligand binding and functional assays, analyzed all data, and prepared the manuscript. S.M designed the MP1104 analogs and edited the manuscript. S.Z analyzed the structure and performed the docking experiments. C.K synthesized MP1104. J.D.M helped with binding assay, analyzed the data and prepared the manuscript. S.W helped with protein expression and crystal optimization. P.D.M conducted the SalA modeling. R.U synthesized MP1104 analogs under S.M.’s supervision. E.V generated KOP-SalA liposomes for immunization. B.E.K helped with diffraction data collection and processing. G.W.H refined the structure. M.Y.L helped with crystallization. J.S and E.P immunized llamas, generated nanobody library and selected and identified all nanobodies. X.P.H helped with functional assays. R.T.S developed the GTPγ[35S] assay. A.R.T helped with the kinetic measurement of GPCR signaling. G.W.P edited the manuscript. F.I.C provided the MP1104 compound. R.C.S edited the manuscript. V.C analyzed the data and prepared the manuscript. V.K analyzed the data, supervised the docking experiments and prepared the manuscript. D.W designed experiments, supervised the structure determination strategy, helped with diffraction data collection, processed the diffraction data, solved and refined the structure, analyzed the data and prepared the manuscript. B.L.R designed the experiments, was responsible for the overall project strategy and management, and prepared the manuscript.
References
- Abagyan R, Totrov M, Kuznetsov D. Icm – a New Method for Protein Modeling and Design – Applications to Docking and Structure Prediction from the Distorted Native Conformation. Journal of Computational Chemistry. 1994;15:488–506. [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone phi, psi and Side-Chain chi(1) and chi(2) Dihedral Angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational Models of Pharmacological Agonism. Proc R Soc Ser B-Bio. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain research. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Roth BL. New Technologies for Elucidating Opioid Receptor Function. Trends in pharmacological sciences. 2016;37:279–289. doi: 10.1016/j.tips.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust TF, Morgenweck J, Kim SA, Rose JH, Locke JL, Schmid CL, Zhou L, Stahl EL, Cameron MD, Scarry SM, et al. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci Signal. 2016;9:ra117. doi: 10.1126/scisignal.aai8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter B, Nehme R, Warne T, Leslie AG, Tate CG. Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature. 2016;536:104–107. doi: 10.1038/nature18966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Chu R, Takei J, Knowlton JR, Andrykovitch M, Pei W, Kajava AV, Steinbach PJ, Ji X, Bai Y. Redesign of a four-helix bundle protein by phage display coupled with proteolysis and structural characterization by NMR and X-ray crystallography. J Mol Biol. 2002;323:253–262. doi: 10.1016/s0022-2836(02)00884-7. [DOI] [PubMed] [Google Scholar]
- Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, Kunken J, Xu F, Cherezov V, Hanson MA, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CW, Rothman RB, Prisinzano TE. Neuropharmacology of the naturally occurring kappa-opioid hallucinogen salvinorin A. Pharmacol Rev. 2011;63:316–347. doi: 10.1124/pr.110.003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh T, Tajima A, Izumimoto N, Suzuki T, Saitoh A, Suzuki T, Narita M, Kamei J, Tseng LF, Mizoguchi H, et al. TRK-820, a selective kappa-opioid agonist, produces potent antinociception in cynomolgus monkeys. Jpn J Pharmacol. 2001;85:282–290. doi: 10.1254/jjp.85.282. [DOI] [PubMed] [Google Scholar]
- Eugene Kellogg G, Abraham DJ. Hydrophobicity: is LogP(o/w) more than the sum of its parts? Eur J Med Chem. 2000;35:651–661. doi: 10.1016/s0223-5234(00)00167-7. [DOI] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of delta-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G, Zatsepin NA, Betti C, Giguere P, Han GW, Ishchenko A, Liu W, Guillemyn K, Zhang H, James D, et al. Structural basis for bifunctional peptide recognition at human delta-opioid receptor. Nat Struct Mol Biol. 2015;22:265–268. doi: 10.1038/nsmb.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, Frimurer TM, Schwartz TW. A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J Biol Chem. 2010;285:3973–3985. doi: 10.1074/jbc.M109.064725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WJ, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, et al. Structural insights into mu-opioid receptor activation. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Structure-Function of the G Protein-Coupled Receptor Superfamily. Annu Rev Pharmacol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. Allosteric sodium in class A GPCR signaling. Trends in biochemical sciences. 2014;39:233–244. doi: 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A Simple Method for Quantifying Functional Selectivity and Agonist Bias. Acs Chemical Neuroscience. 2012;3:193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee J, Jo S, Brooks CL, Lee HS, Im W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. Journal of Computational Chemistry. 2017;38:1879–1886. doi: 10.1002/jcc.24829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivov GG, Shapovalov MV, Dunbrack RL., Jr Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguere PM, Sciaky N, Roth BL. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol. 2015;22:362–369. doi: 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DL, Jones RM, Hjorth SA, Schwartz TW, Portoghese PS. Binding of norbinaltorphimine (norBNI) congeners to wild-type and mutant mu and kappa opioid receptors: molecular recognition loci for the pharmacophore and address components of kappa antagonists. Journal of medicinal chemistry. 2000;43:1573–1576. doi: 10.1021/jm000059g. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, S MD, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–191. [Google Scholar]
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi YF, et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput. 2016;12:405–413. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Wacker D, Gati C, Han GW, James D, Wang D, Nelson G, Weierstall U, Katritch V, Barty A, et al. Serial femtosecond crystallography of G protein-coupled receptors. Science. 2013;342:1521–1524. doi: 10.1126/science.1244142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: Orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Majumdar S, Grinnell S, Le Rouzic V, Burgman M, Polikar L, Ansonoff M, Pintar J, Pan YX, Pasternak GW. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc Natl Acad Sci U S A. 2011;108:19778–19783. doi: 10.1073/pnas.1115231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S, Subrath J, Le Rouzic V, Polikar L, Burgman M, Nagakura K, Ocampo J, Haselton N, Pasternak AR, Grinnell S, et al. Synthesis and evaluation of aryl-naloxamide opiate analgesics targeting truncated exon 11-associated mu opioid receptor (MOR-1) splice variants. J Med Chem. 2012;55:6352–6362. doi: 10.1021/jm300305c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hubner H, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. The Journal of pharmacology and experimental therapeutics. 1976;197:517–532. [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nagase H, Imaide S, Yamada T, Hirayama S, Nemoto T, Yamaotsu N, Hirono S, Fujii H. Essential structure of opioid kappa receptor agonist nalfurafine for binding to kappa receptor 1: synthesis of decahydroisoquinoline derivatives and their pharmacologies. Chem Pharm Bull (Tokyo) 2012;60:945–948. doi: 10.1248/cpb.c12-00336. [DOI] [PubMed] [Google Scholar]
- O’Connor C, White KL, Doncescu N, Didenko T, Roth BL, Czaplicki G, Stevens RC, Wuthrich K, Milon A. NMR structure and dynamics of the agonist dynorphin peptide bound to the human kappa opioid receptor. Proc Natl Acad Sci U S A. 2015;112:11852–11857. doi: 10.1073/pnas.1510117112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardon E, Laeremans T, Triest S, Rasmussen SG, Wohlkonig A, Ruf A, Muyldermans S, Hol WG, Kobilka BK, Steyaert J. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc. 2014;9:674–693. doi: 10.1038/nprot.2014.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW, Snowman AM, Snyder SH. Selective enhancement of [3H]opiate agonist binding by divalent cations. Molecular pharmacology. 1975;11:735–744. [PubMed] [Google Scholar]
- Pert CB, Pasternak G, Snyder SH. Opiate agonists and antagonists discriminated by receptor binding in brain. Science. 1973;182:1359–1361. doi: 10.1126/science.182.4119.1359. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011a;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011b;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. The Journal of biological chemistry. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn V, Del Vecchio G, Labuz D, Rodriguez-Gaztelumendi A, Massaly N, Temp J, Durmaz V, Sabri P, Reidelbach M, Machelska H, et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science. 2017;355:966–969. doi: 10.1126/science.aai8636. [DOI] [PubMed] [Google Scholar]
- Spetea M, Eans SO, Ganno ML, Lantero A, Mairegger M, Toll L, Schmidhammer H, McLaughlin JP. Selective kappa receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br J Pharmacol. 2017;174:2444–2456. doi: 10.1111/bph.13854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JB, Mascarella SW, Rothman RB, Partilla JS, Xu H, McCullough KB, Dersch CM, Cantrell BE, Zimmerman DM, Carroll FI. Investigation of the N-substituent conformation governing potency and mu receptor subtype-selectivity in (+)-(3R, 4R)-dimethyl-4-(3-hydroxyphenyl)piperidine opioid antagonists. Journal of medicinal chemistry. 1998;41:1980–1990. doi: 10.1021/jm980063g. [DOI] [PubMed] [Google Scholar]
- Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Urbano M, Guerrero M, Rosen H, Roberts E. Antagonists of the kappa opioid receptor. Bioorg Med Chem Lett. 2014;24:2021–2032. doi: 10.1016/j.bmcl.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Valentin-Hansen L, Holst B, Frimurer TM, Schwartz TW. PheVI:09 (Phe6.44) as a sliding microswitch in seven-transmembrane (7TM) G protein-coupled receptor activation. J Biol Chem. 2012;287:43516–43526. doi: 10.1074/jbc.M112.395137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, flexible, and free. Journal of Computational Chemistry. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Varadi A, Marrone GF, Eans SO, Ganno ML, Subrath JJ, Le Rouzic V, Hunkele A, Pasternak GW, McLaughlin JP, Majumdar S. Synthesis and characterization of a dual kappa-delta opioid receptor agonist analgesic blocking cocaine reward behavior. ACS Chem Neurosci. 2015;6:1813–1824. doi: 10.1021/acschemneuro.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Váradi A, Marrone GF, Eans SO, Ganno ML, Subrath JJ, Le Rouzic V, Hunkele A, Pasternak GW, McLaughlin JP, Majumdar S. Synthesis and Characterization of a Dual Kappa-Delta Opioid Receptor Agonist Analgesic Blocking Cocaine Reward Behavior. ACS chemical neuroscience. 2015;6:1813–1824. doi: 10.1021/acschemneuro.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardy E, Mosier PD, Frankowski KJ, Wu H, Katritch V, Westkaemper RB, Aube J, Stevens RC, Roth BL. Chemotype-selective modes of action of kappa-opioid receptor agonists. J Biol Chem. 2013;288:34470–34483. doi: 10.1074/jbc.M113.515668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardy E, Robinson JE, Li C, Olsen RHJ, DiBerto JF, Giguere PM, Sassano FM, Huang XP, Zhu H, Urban DJ, et al. A New DREADD Facilitates the Multiplexed Chemogenetic Interrogation of Behavior. Neuron. 2015;86:936–946. doi: 10.1016/j.neuron.2015.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Wacker D, Stevens RC, Roth BL. How Ligands Illuminate GPCR Molecular Pharmacology. Cell. 2017a;170:414–427. doi: 10.1016/j.cell.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell. 2017b;168:377–389. doi: 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wacker D, Levit A, Che T, Betz RM, McCorvy JD, Venkatakrishnan AJ, Huang XP, Dror RO, Shoichet BK, et al. D-4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science. 2017;358:381–386. doi: 10.1126/science.aan5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL. The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. The Journal of pharmacology and experimental therapeutics. 2015;352:98–109. doi: 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Bikbulatov RV, Mocanu V, Dicheva N, Parker CE, Wetsel WC, Mosier PD, Westkaemper RB, Allen JA, Zjawiony JK, et al. Structure-based design, synthesis, and biochemical and pharmacological characterization of novel salvinorin A analogues as active state probes of the kappa-opioid receptor. Biochemistry. 2009;48:6898–6908. doi: 10.1021/bi900605n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Huang XP, Mangano TJ, Zou R, Chen X, Zaidi SA, Roth BL, Stevens RC, Katritch V. Structure-Based Discovery of New Antagonist and Biased Agonist Chemotypes for the Kappa Opioid Receptor. J Med Chem. 2017;60:3070–3081. doi: 10.1021/acs.jmedchem.7b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Screening of nanobodies from the KOP-SalA library using BRET assay. As can be seen, Nb6/7 (blue) shows dose-response upon SalA stimulation (N = 3). Note that concentration at −12 represents “no drugs” added.
(B) The antagonist JDTic reverses SalA-induced dissociation in a concentration-dependent manner. Cells expressing KOP-Rluc and Nb6-YFP were treated with 0.1 μM SalA for 5 min and then stimulated by increasing concentrations of JDTic for 5 min and BRET signal was measured.
(C) Recruitment of Nb39 to KOP in the presence of full agonist SalA (EC50 = 10.2±0.8 nM, Emax = 100±2, orange), partial agonist diprenorphine (EC50 = 5.20±0.03 nM, Emax = 32±2, blue), or antagonist JDTic (no activity detected, red) (N = 3).
(D) Comparison of U69,593 dissociation rate at KOP only (blue), KOP+Nb6 (green triangle) and KOP+Nb39 (yellow) shows a slow U69,593 off-rate with Nb39 and a faster off-rate with Nb6 (N = 3).
(E) Interaction of KOP and Nb39 in the presence of Gαi1 or β-arrestin2. Using BRET assay, KOP-Rluc and Nb39 were co-transfected with different amounts of Gαi1 or β-arrestin2 (N = 3). See Table S1 for values.
(F) Measurement of MP1104 binding affinity at MOP (Ki = 0.18±0.06 nM, red), KOP (Ki = 0.20±0.08 nM, orange) and DOP (Ki = 1.38±0.05 nM, light green) using radioligand 3H-Diprenorphine (N = 3).
(G) Measurement of MP1104-mediated Gi activation in KOP (MP1104, EC50 = 0.003±0.001 nM, Emax = 100±2, orange; SalA, EC50 = 0.033±0.008 nM, Emax = 98±2, red), MOP (MP1104, EC50 = 0.034±0.008 nM, Emax = 104±1, orange; DAMGO, EC50 = 3.45±0.06 nM, Emax = 100±2 red) and DOP (MP1104, EC50 = 0.67±0.07 nM, Emax = 97±2 orange; DADLE, EC50 = 1.34±0.05 nM, Emax = 100±2 red) (N = 3).
(A) Confirmation of KOP crystallization construct functionality in binding assays. MP1104 (orange), Ki = 0.33±0.02 nM; SalA (light green), Ki = 25.3±1.40 nM (N = 3).
(B) Confirmation of KOP crystallization construct functionality in cAMP inhibition assays. MP1104 (orange), EC50 = 0.006±0.001 nM; SalA (light green), EC50 = 0.26±0.02 nM (N = 3).
(C) Crystal packing of KOP-MP1104-Nb39 complex. KOP (blue), Nb39 (orange), MP1104 (orange sphere).
(D) Crystal contacts within anti-parallel monomers (top) or parallel dimers (bottom). The KOP-MP1104-Nb39 complex displays an anti-parallel receptor dimer in the asymmetric unit through interactions between ECL2 and intracellular loop 2 (ICL2) of the two monomers (D218ECL2—H1623.56/P163ICL2). Parallel dimers are observed within the crystal lattice with dimer interfaces formed by TM1 and TM2 residues.
(E) Electron density of MP1104 and ligand binding pocket residues. (left) Different angles showing structure of receptor-bound MP1104 (orange stick model) with mFo-DFc omit density map contoured at 3σ (green mesh). (middle) Different angles showing structure of receptor-bound MP1104 (orange stick model) with 2mFo-DFc electron density map contoured at 1σ (blue mesh). (right) Different angles showing structure of ligand binding pocket residues with 2mFo-DFc electron density map contoured at 1σ (blue mesh). All density maps were generated in Refmac5. Interactions between ligand and receptor residues result in continuous 2mFo-DFc density that appears “broken” when only receptor or ligand density is displayed. Grey dots indicate Hydrogen-bonds between receptor and ligand.
(F) Comparison of inactive and active-state β2AR or M2R indicates a common contraction during activation. Distances were measured between the Cα atoms of M401.39, S2035.42, and F2906.52, N3127.39 in β2AR; I261.35, V4076.55, I417ECL3, and N4197.32 in M2R.
(G and H) Residues within KOP-Nb39 interface (G) indicate they are highly conserved among the three canonical opioid receptors (H).
(A) Mutagenesis studies indicate that KOP T1112.56 and Y3207.43 residues are crucial for full activation of KOP. KOP wt (MP1104, EC50 = 0.035±0.005 nM, Emax = 100±2; Dynorphin A, EC50 = 12.60±1.15 nM, Emax = 75±3). KOP T1112.56 A (MP1104, EC50 = 3.50±0.23 nM, Emax = 93±3; Dynorphin A, EC50 = 12.30±1.05 nM, Emax = 30±2). KOP Y3207.43A (MP1104, EC50 = 10.20±0.89 nM, Emax = 91±2; Dynorphin A, no activity detected). (N = 3).
(B) Comparison of water-mediated interactions with the phenol group between KOP and MOP. Active state KOP residues are shown in blue sticks, and blue cartoon representation. Active state MOP (PDB ID: 5C1M) residues are shown in green sticks. MP1104 and BU-72 are shown in orange and green colored carbon sticks respectively. Crystallographic waters in active MOP (from 5C1M) are shown in green balls, and calculated waters in KOP are represented by blue balls. Water mediated hydrogen bonds common to both KOP and MOP are shown in black broken lines, while water mediated hydrogen bonds specific for MOP are shown in red broken lines.
(C) The MP1104 analog, MP1107, causes more severe reduction of binding affinity in MOP than in KOP. Chemical differences between compound structures are highlighted in red. MP1104 (red), Ki = 0.20±0.02 nM in KOP (top) and 0.16±0.03 nM in MOP (bottom); MP1107 (orange), Ki = 1.07±0.06 nM in KOP (top) and 5.85±0.08 nM in MOP (bottom) (N = 3).
(D) Removal of the cyclopropyl group reduces MP1104’s functional activity in KOP. Chemical differences between compound structures are highlighted in red. Gi activation (top): MP1104 (red), EC50 = 0.003±0.001 nM, Emax = 97±1; MP1101 (orange), EC50 = 0.055±0.010 nM; Emax = 98±1. β-Arrestin2 recruitment (bottom): MP1104 (red), EC50 = 0.043±0.010 nM, Emax = 113±2; MP1101 (orange), EC50 = 0.41±0.08 nM, Emax = 111±2 (N = 3).
(E) Docking of MP1104 (orange) in the active structure of MOP (green) indicates the cyclopropylmethyl groups also extends into the hydrophobic pocket.
(A) GTPγ[35S] (top panel) and BRET (arrestin recruitment) (bottom panel) activities indicates that KOP Y3127.35W recapitulates IBNtxA’s biased activity in MOP. Bias factor towards G protein in MOP was calculated with DAMGO as a reference agonist based on the Black and Leff operational model and defined as 10ΔΔLog(τ/KA): 0.8 and 9 for MP1104 and IBNtxA, respectively (N = 3). See Table S5 for values.
(B) Comparison of IBNtxA (grey) and MP1104 (orange) docking in MOP indicates that the iodobenzamide group adopts different orientations.
(C) Chemical structures and arrestin recruitment activity of IBNtxA analogs with a single bond in the saturated ring (highlighted in red). (left) Arrestin activity in KOP: MP903 (red), EC50 = 0.074±0.012 nM, Emax = 108±10; IBNtxA (orange), EC50 = 0.030±0.009 nM, Emax = 95±8; MP970 (light green), EC50 = 0.16±0.04 nM, Emax = 92±6; MP1104 (green), EC50 = 0.037±0.008 nM, Emax = 100±2. (right) Arrestin activity in MOP: MP903 (red), EC50 = 0.12±0.05 nM, Emax = 33±5; IBNtxA (orange), EC50 = 0.075±0.008 nM, Emax = 25±6; MP970 (light green), EC50 = 0.54±0.10 nM, Emax = 21±5; MP1104 (green), EC50 = 0.55±0.08 nM, Emax = 100±2 (N = 3). Bias factor towards G protein: 10.3 and 9 for MP903 and MP970, respectively.
(A) Docking poses of U69,593 and U50,488 in the active state KOP-MP1104-Nb39 structure.
(B) Putative binding mode for SalA in the KOP-MP1104-Nb39 structure.
(C) Chemical structures and binding affinity of MP1104 analogs with modifications in the iodobenzamide group. Kis in KOP (left) and MOP (right) respectively: MP1104, 0.20±0.06 and 0.16±0.05 nM; MP1108, 0.42±0.05 and 0.42±0.06 nM; MP1109, 0.13±0.03 and 0.16±0.05 nM; MP1110, 0.12±0.04 and 0.18±0.04 nM; MP1111, 0.27±0.06 and 0.20±0.05 nM; MP1112, 0.12±0.03 and 0.17±0.03 nM; MP1113, 0.14±0.07 and 0.24±0.06 nM; MP1114, 0.16±0.07 and 0.27±0.08 nM; MP1115, 0.18±0.08 and 0.25±0.06 nM (N = 3).