

Abstract
Single-molecule fluorescence microscopy enables unrivaled sub-cellular quantitation of genomically encoded fusions of native proteins with fluorescent protein reporters. Fluorescent proteins must undergo in vivo maturation after expression before they become photoactive. Maturation effects must be quantified during single-molecule analysis. Here we present a method to characterise maturation of GFP and mCherry genetic protein fusions in budding yeast Saccharomyces cerevisiae.
Keywords: Single-molecule, Fluorescence, Fluorescent protein maturation, Protein fusion, GFP, mCherry, Yeast
Background
Single-molecule fluorescence microscopy enables sensitive quantification of molecular stoichiometry, mobility and copy number, not only on a cell-by-cell basis but also precisely to individual sub-cellular compartments (Leake, 2012; Wollman and Leake, 2015; Shashkova et al., 2017 ). The technique relies on endogenously expressed fluorescent protein fusions of the wild type protein of interest such that there is one-to-one labelling. However, all fluorescent proteins have an in vivo maturation time varying from a few minutes to several tens of minutes before entering a bright fluorescing state ( Badrinarayanan et al., 2012 ). It is therefore of upmost importance to measure any maturation effects and quantify if there is any immature ‘dark fraction’ of labelled protein. These measurements are also particularly relevant to fluorescence recovery after photobleaching (FRAP). FRAP can be used to study molecular turnover in living cells ( Beattie et al., 2017 ). FRAP is based on photobleaching of a cell region where a fluorescently labelled component is localized, followed by quantification of any fluorescence recovery in that region over time. The measured relation between the fluorescence intensity as a function of time following an initial photobleach can be used to determine molecular mobility and kinetics parameters, such as the rate of dissociation of a particular fluorescent component from a molecular complex ( Leake et al., 2006 ). Therefore, any ‘new’ fluorescence coming from fluorescent protein maturation might affect this apparent result. We present here a protocol to characterise the maturation of Mig1-GFP and Nrd1-mCherry fusion proteins in living yeast Saccharomyces cerevisiae cells used in our single-molecule studies ( Wollman et al., 2017 ).
We blocked protein translation in living cells by adding cycloheximide ( Hartwell et al., 1970 ), and then measured any cellular fluorescence recovery after cells were completely photobleached by continuous illumination. Such fluorescence recovery is then used as a metric for newly matured GFP and/or mCherry in the cell. Our results are broadly consistent with in vivo maturation of GFP and mCherry reported previously ( Badrinarayanan et al., 2012 ; Khmelinskii et al., 2012 ), but since maturation kinetics may be dependent on cell type and the specific extracellular microenvironment, it is important to quantify these maturation effects under the same experimental conditions used for the in vivo microscopy on the actual fusion strains of interest.
Materials and Reagents
Sterile pipette tips, 1 ml, 200 µl, 10 µl (STARLAB, catalog numbers: S1111-6801, S1111-0806, S1111-3800)
14 ml conical tubes (Corning, Falcon®, catalog number: 352059)
Petri dishes 92 mm diameter (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 172931)
Microscopy slides (Fisher Scientific, catalog number: FB58622)
Cover slips (Scientific Laboratory Supplies, catalog number: MIC3124)
Yeast S. cerevisiae YSH2348 with Mig1-GFP and Nrd1-mCherry genetically integrated protein fusions, MATa MIG1-GFP-HIS3 NRD1-mCherry-hphNT1 MET LYS (Hohmann lab, University of Gothenburg, Sweden)
D(+)-Glucose (VWR, catalog number: 101176K)
Bacto-yeast extract (BD, BactoTM, catalog number: 212750)
Peptone from meat (Merck, catalog number: 1072241000)
Agar-agar (Merck, catalog number: 1016141000)
MilliQ water
Yeast nitrogen base without amino acids, without (NH4)2SO4 (Sigma-Aldrich, catalog number: Y1251)
Ammonium sulfate, (NH4)2SO4 (Merck, catalog number: 1012171000)
Complete supplement mixture (ForMedium, catalog number: DCS0019)
Cycloheximide (Sigma-Aldrich, catalog number: C7698)
EtOH (VWR, catalog number: VWRC20821.330)
Glucose 50% w/v (see Recipes)
YPD agar with 4% glucose (see Recipes)
YPD liquid medium with 4% glucose (see Recipes)
YNB (Yeast Nitrogen Base) liquid medium without glucose (see Recipes)
YNB (Yeast Nitrogen Base) liquid medium with 4% glucose (see Recipes)
100 mg/ml cycloheximide solution in EtOH (see Recipes)
Equipment
Pipettes (STARLAB, model: ErgoOne® Single-Channel Pipette, 2-20 µl, 20-200 µl and 100-1,000 µl)
pH-meter (Scientific & Chemical Supplies, catalog number: PHM975050)
Two timers (Fisher Scientific, catalog number: 15177414)
Autoclave (Getinge, model: 400/500LS-E Series Steam Sterilizers (533LS-E))
Magnetic stirrer (Chemtech Scientific, model: C-MAG HS7)
30 °C incubator (Eppendorf, New Brunswick ScientificTM, model: Innova® 4000)
Spectrophotometer (Biochrom, model: WPA S800)
Centrifuge (Eppendorf, model: 5810 R)
Mercury-arc excitation fluorescence microscope Zeiss Axiovert 200M (Carl Zeiss, model: Axiovert 200 M) with an AxioCamMR3 camera with separate filter sets: 38HE for GFP and 43HE for mCherry excitation; Plan-Apochromat 1.40-numerical-aperture oil immersion, 100x objective
Software
AxioVision
ImageJ 1.50g
Excel
MATLAB 2017a
Procedure
-
Cell preparation
Streak cells from a frozen stock, using a sterile pipette tip on a freshly-prepared YPD agar plate (see Recipes), and incubate at 30 °C for at least 24 h.
Set an overnight culture in a 14 ml tube by inoculating 3 ml of YPD with cells grown on a YPD plate. Single colonies are not needed for genomically integrated strains. Incubate at 30 °C, 180 rpm.
-
In the morning exchange the YPD medium (see Recipes) to YNB medium (see Recipes) supplemented with 4% glucose:
Pellet the cells by centrifugation at 1,000 x g for 3 min, remove the supernatant.
Resuspend the cells in 3 ml of YNB medium without any carbon source.
Pellet the cells by centrifugation at 1,000 x g for 3 min, remove the supernatant.
Suspend the cells in 3 ml of YNB supplemented with 4% glucose and incubate at 30 °C, 180 rpm, for ~4 h.
Wash the culture by centrifugation (1,000 x g, 3 min) and re-suspend in 2 ml of YNB with 4% glucose. Incubate at 30 °C, 180 rpm, for about 10 min.
Add 2 μl of 100 mg/ml cycloheximide solution (see Recipes) to the final concentration of 100 μg/ml. Incubate for 1 h at room temperature, without shaking, protect from light.
Place 5 μl of the culture on a microscope slide and cover with a 22 x 22 mm coverslip. Avoid any air under the coverslip.
-
Data acquisition
Place the sample under the microscope, coverslip on the objective and find a region of interest containing 5-10 cells (100x magnification) which appear stationary and firmly anchored to the glass surface.
Optimize exposure times for in vivo imaging of both GFP and mCherry fluorescent proteins to be able to detect a clear signal without saturating the detector. Under our microscope: GFP exposure time–22 sec, mCherry–7 sec.
Find another region with 5-10 cells positioned far away from the previous one to avoid any potential bleaching from previous illumination exposure.
Take a brightfield and a fluorescence image, by pressing the ‘snap’ button, with both channels using chosen exposure times, opening the mercury lamp shutter for only the length of exposure.
Photobleach GFP or mCherry by continuous illumination of the appropriate wavelength until the region appears completely dark. Continue for 1 min longer. With our settings the total exposure time is: 3 min 40 sec for GFP and 4 min for mCherry. Immediately after, begin timing and acquire a picture of the bleached fluorescent protein with an appropriate channel and a brightfield image. This is denoted time point 0 min.
Continue acquiring both fluorescent and brightfield pictures at the following time points after bleaching: 7.5, 15, 25, 30, 40, 60, 90 and 120 min.
As simultaneous photobleaching of GFP and mCherry is not possible under this microscope, the time points were staggered for GFP and mCherry as listed in Table 1.
-
Data analysis
Images are converted into open standard tiff files from zvi by AxioVision software.
-
Further analysis is performed using ImageJ.
Open the first unbleached brightfield image.
By choosing an ‘oval’ selection tool, define a region of interest (ROI), an area around a cell as shown in Figure 1. It does not matter how much of non-cell area is included as every cell will be background-correct during the analysis.
Open a fluorescence image of the same set, and define the same area of the same cell by simultaneously choosing ‘Shift’ and ‘E’ keys on the keyboard (Selection → Restore).
From the menu bar select: Analyze → set measurements. Pick ‘area’ (represents a number of pixels, N) and ‘integrated density’ (sum intensity for the cell, Scell ). Press ‘OK’.
To obtain numeric values press ‘Ctrl’ + ‘M’ (Analyse → Measure). Record the result in Excel.
Repeat throughout the entire data set for both channels keeping the same ROI.
Repeat the entire procedure for all cells.
Background correction: Choose random background areas around cells (Figure 2) and obtain numerical results for sum intensity ( Sbg ).
-
Find the average ( SAbg ) and multiply by the number of pixels from cell (N) measurements. This is the intensity of the background represented within the cell area ( Ibg ).
;
; -
Subtraction of the average background sum intensity ( Ibg ) from the total intensity of the cell ( Scell ) represents Icell , the cellular fluorescence intensity with background correction.
. -
The average of fluorescence intensity of all cells analysed within the data set gives the final value of the fluorescence intensity ( Ifinal ) with appropriate estimation of SD and/or SE.
. Plot the final fluorescence intensity ( Ifinal ) vs. experimental time (Figures 3A and 3B) for fluorescently labelled cells and wild type autofluorescent cells. Any signal above autofluorescence is due to fluorescent protein maturation. For GFP (Figure 3A), no maturation was detected so it can be assumed that all of the fluorescent protein was mature in the cells and there is no ‘dark’ fraction. For mCherry (Figure 3B), some fluorescence recovery was measured. The following steps outline quantification of the maturation time and dark fraction.
Subtract the autofluorescence from the mean fluorescent protein intensity at each time point (Figure 3C).
-
Export the intensity and time values after the bleach by copying and pasting into two new variables in MATLAB, called x (for the time values ) and y (intensity values):
Right click on the Workspace → New. Name it x or y. Press ‘Enter’ on the keyboard.
Double click on this new variable opens a table in the Editor where values of time (for x) or intensity (for y) can be pasted.
Open the curve fitting toolbox from the Apps menu.
Select x for ‘X data’ and y for ‘Y data’.
-
Choose custom equation and type:
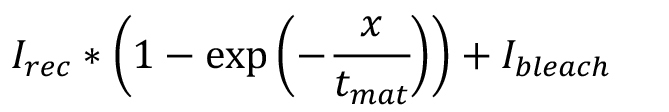
where, Ibleach is the remaining intensity after the bleach, Irec is the recovered intensity above Ibleach and tmat is the maturation time.
If ‘Auto fit’ is ticked, fitting will be automatic.
If the fit has not converged correctly, adjust the ‘Start point’ parameters in ‘Fit Options’ to reasonable estimates from the data i.e., y at x = 0 for Ibleach and y at x = end minus Ibleach for Irec .
If the fit has converged record the fit and goodness of fit parameters from the ‘Results’ panel. For Figure 3C, Irec = 4.3 x 104 ± 3 x 104 counts, Ibleach = 5.2 x 104 ± 2.5 x 104 counts, tmat =17 ± 10 min with R2 = 0.7.
To calculate the proportion dark, immature protein; divide Irec by the initial, autofluorescence corrected pre-bleach intensity. Here give ~5%.
Table 1. Order of photobleaching and data acquisition using two channels.
| GFP | mCherry | |
|---|---|---|
| 1 | Bleaching for 3 min 40 sec | |
| 2 | Timer start, Time point 0 min | |
| 3 | Time point 7.5 min | |
| 4 | Bleaching for 4 min | |
| 5 | Timer start, Time point 0 min | |
| 6 | Time point 15 min | |
| 7 | Time point 7.5 min | |
| 8 | Time point 25 min | |
| 9 | Time point 15 min | |
| 10 | Time point 30 min | |
| 11 | Time point 25 min | |
| 12 | Time point 30 min | |
| 13 | Time point 40 min | |
| 14 | Time point 40 min | |
| 15 | Time point 60 min | |
| 16 | Time point 60 min | |
| 17 | Time point 90 min | |
| 18 | Time point 90 min | |
| 19 | Time point 120 min | |
| 20 | Time point 120 min |
Figure 1. Selection of the region of interest for cell measurements.
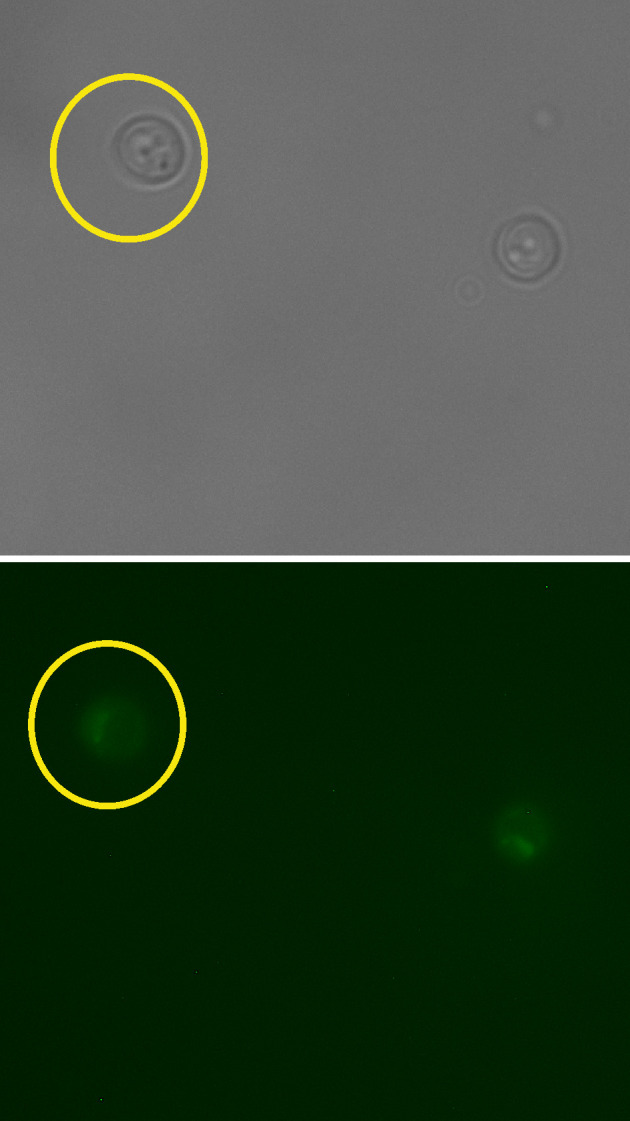
Scale bar = 20 µm.
Figure 2. Selection of the region of interest for the background measurements.

Scale bar = 20 µm.
Figure 3. Characterisation of GFP and mCherry maturation times in vivo.

GFP maturation within genomically integrated protein fusion (A). Maturation of a genomically integrated mCherry fusion (B) and its’ exponential recovery fit (C). Time normalized to bleach time at t = 0.
Data analysis
Data was analyzed as outlined in section C of the Procedure. Statistical methods are outlined in Wollman et al. (2017) but also briefly outlined here. In imaging experiments, each cell can be defined as a biological replicate sampled from the cell population. Sample sizes of ~10 cells were used to generate reasonable estimates of fluorescent protein maturation and are similar to previous studies ( Badrinarayanan et al., 2012 ). Technical replicates are not possible with irreversible photobleaching however noise is characterized by the autofluorescent of wild type control cell measurements.
Notes
Autofluorescence is calculated as indicated in the protocol above but using a wild type yeast strain (i.e., without any fluorescent proteins present).
Recipes
-
Glucose 50% w/v
Weigh 500 g of glucose
Bring up to 1 L with MilliQ water
Dissolve by using magnetic stirrer with heating
Autoclave for 20 min at 121 °C
-
YPD agar with 4% glucose
Mix yeast extract 5 g, Bacto-peptone (peptone from meat) 10 g and agar 10 g
Bring up to 460 ml with MilliQ water
Autoclave for 20 min at 121 °C
Add 40 ml of glucose 50% w/v
Cast plates: approximately 25 ml of the medium per plate
Let them solidify, store upside down at 4 °C
-
YPD liquid medium with 4% glucose
Mix yeast extract 5 g and Bacto-peptone (peptone from meat) 10 g
Bring up to 460 ml with MilliQ water
Autoclave for 20 min at 121 °C
Add 40 ml of glucose 50% w/v
-
YNB (Yeast Nitrogen Base) liquid medium without glucose
Mix yeast nitrogen base without amino acids, without (NH4)2SO4 1.7 g, complete supplement 0.79 g, (NH4)2SO4 5 g
Dissolve in 900 ml of MilliQ water, adjust pH 5.8-6.0 using NaOH
Bring up to 1,000 ml with MilliQ water
Autoclave for 20 min at 121 °C
-
YNB (Yeast Nitrogen Base) liquid medium with 4% glucose
Mix yeast nitrogen base without amino acids, without (NH4)2SO4 1.7 g, complete supplement 0.79 g, (NH4)2SO4 5 g
Dissolve in 900 ml of MilliQ water, set pH 5.8-6.0 using NaOH
Bring up to 920 ml with MilliQ water
Autoclave for 20 min at 121 °C
Add 80 ml of glucose 50% w/v
-
100 mg/ml cycloheximide solution in EtOH
Weigh 0.5 g of cycloheximide and dissolve in 5 ml of absolute EtOH
Aliquot and store at -20 °C
Acknowledgments
This work was supported by the Biological Physical Sciences Institute, Royal Society, MRC (grant MR/K01580X/1), BBSRC (grant BB/N006453/1), the European Commission via Marie Curie-Network for Initial Training ISOLATE (Grant agreement No.: 289995), and the Royal Society Newton International Fellowship (NF160208). This protocol was adapted from Wollman et al. (2017).
Conflict of interests: Authors declare no conflict of interest.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Badrinarayanan A., Reyes-Lamothe R., Uphoff S., Leake M. C. and Sherratt D. J.(2012). In vivo architecture and action of bacterial structural maintenance of chromosome proteins . Science 338(6106): 528-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beattie T. R., Kapadia N., Nicolas E., Uphoff S., Wollman A. J., Leake M. C. and Reyes-Lamothe R.(2017). Frequent exchange of the DNA polymerase during bacterial chromosome replication. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hartwell L. H., Culotti J. and Reid B.(1970). Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc Natl Acad Sci U S A 66(2): 352-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khmelinskii A., Keller P. J., Bartosik A., Meurer M., Barry J. D., Mardin B. R., Kaufmann A., Trautmann S., Wachsmuth M., Pereira G., Huber W., Schiebel E. and Knop M.(2012). Tandem fluorescent protein timers for in vivo analysis of protein dynamics . Nat Biotechnol 30(7): 708-714. [DOI] [PubMed] [Google Scholar]
- 5. Leake M. C.(2012). The physics of life: one molecule at a time. Philos Trans R Soc Lond B Biol Sci 368(1611): 20120248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leake M. C., Chandler J. H., Wadhams G. H., Bai F., Berry R. M. and Armitage J. P.(2006). Stoichiometry and turnover in single, functioning membrane protein complexes. Nature 443(7109): 355-358. [DOI] [PubMed] [Google Scholar]
- 7. Shashkova S., Wollman A. J. M., Leake M. C. and Hohmann S.(2017). The yeast Mig1 transcriptional repressor is dephosphorylated by glucose-dependent and-independent mechanisms. FEMS Microbiol Lett 364(14). [DOI] [PubMed] [Google Scholar]
- 8. Wollman A. J. and Leake M. C.(2015). Millisecond single-molecule localization microscopy combined with convolution analysis and automated image segmentation to determine protein concentrations in complexly structured, functional cells, one cell at a time. Faraday Discuss 184: 401-424. [DOI] [PubMed] [Google Scholar]
- 9. Wollman A. J., Shashkova S., Hedlund E. G., Friemann R., Hohmann S. and Leake M. C.(2017). Transcription factor clusters regulate genes in eukaryotic cells. Elife 6: e27451. [DOI] [PMC free article] [PubMed] [Google Scholar]