

Abstract
Cerebrospinal fluid (CSF) studies consistently show that CSF levels of amyloid-beta 1–42 (Aβ42) are reduced and tau levels increased prior to the onset of cognitive decline related to Alzheimer’s disease (AD). However, the preclinical prediction accuracy for low CSF Aβ42 levels, a surrogate for brain Aβ42 deposits, is not high. Moreover, the pathology data suggests a course initiated by tauopathy contradicting the contemporary clinical view of an Aβ initiated cascade. CSF Aβ42 and tau data from 3 normal aging cohorts (45–90 years) were combined to test both cross-sectional (n = 766) and longitudinal (n = 651) hypotheses: 1) that the relationship between CSF levels of Aβ42 and tau are not linear over the adult life-span; and 2) that non-linear models improve the prediction of cognitive decline. Supporting the hypotheses, the results showed that a u-shaped quadratic fit (Aβ2) best describes the relationship for CSF Aβ42 with CSF tau levels. Furthermore we found that the relationship between Aβ42 and tau changes with age—between 45 and 70 years there is a positive linear association, whereas between 71 and 90 years there is a negative linear association between Aβ42 and tau. The quadratic effect appears to be unique to Aβ42, as Aβ38 and Aβ40 showed only positive linear relationships with age and CSF tau. Importantly, we observed the prediction of cognitive decline was improved by considering both high and low levels of Aβ42. Overall, these data suggest an earlier preclinical stage than currently appreciated, marked by CSF elevations in tau and accompanied by either elevations or reductions in Aβ42. Future studies are needed to examine potential mechanisms such as failing CSF clearance as a common factor elevating CSF Aβxx analyte levels prior to Aβ42 deposition in brain.
Introduction
Improved biomarker characterization during the presymptomatic stages of Alzheimer’s disease (AD) is necessary to adequately assess the risk for cognitive decline and optimize interventions. It is widely believed that in elderly at risk for AD, reductions in the cerebrospinal fluid (CSF) levels of amyloid beta 1–42 (Aβ42), which are associated with brain amyloid deposition [1,2], precede elevations in CSF tau levels, a marker of neurodegeneration [3]. Support for this sequence of CSF biomarker changes comes from preclinical studies showing that lower CSF Aβ42 levels predict cognitive decline [4,5]. Others report that CSF Aβ42 and tau levels configured as a ratio, are superior to univariate predictors of future impairment [1], thus further highlighting the value of CSF Aβ42 reductions. However, this clinical view of early AD lesions conflicts with the neuropathology which identifies tauopathy more commonly than Aβ lesions in younger brains [6]. Moreover, adding to the uncertainty, normal aging studies have been inconsistent, showing that CSF Aβ42 levels increase [7,8] or decrease [9], or do not change with age [10]. Offering a clue to this discrepancy, transgenic animal models show CSF Aβ42 elevations occur prior to Aβ42 reductions and brain deposition [11], a trend also seen in early onset AD [12,13]. We reasoned that these divergent human aging findings could be explained by under sampling a non-linear Aβ42 trajectory with the added complexity of non-standardized cohorts. Utilizing cross-sectional (n = 766) and longitudinal data (n = 651) from three normal elderly cohorts with similar clinical and CSF protocols, we tested and confirm two major hypotheses: 1) CSF Aβ42 levels have a non-linear relationship with both age and CSF biomarkers for tau pathology. We observed during mid to late-adult life, a quadratic or U-function uniquely and significantly described the CSF Aβ42 relationships to age and to CSF tau levels. For CSF Aβ38 or Aβ40, only linear relationships were found with age and tau levels; and 2) the cross-sectional and longitudinal results confirm the hypothesis that elevated Aβ42 levels contribute to the prediction of future cognitive decline.
Methods
Human subjects
The data were derived from three prospective and currently active longitudinal studies that sample aging subjects with normal cognition (NC). Written informed consent was obtained from all subjects. The NYU studies were approved by the NYU School of Medicine Institutional Review Board (IRB) and the ADNI and NACC studies were IRB approved by each of collaborating sites. All of the reported studies and all clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki. The authors were not involved in data collection for the ADNI and NACC studies and both datasets were anonymized prior to being received by the authors. In total there were 766 subjects with LP data in the individual cohort studies. Of those, 651 subjects had a clinical follow-up and were included in the combined cross-sectional and prediction studies of cognitive outcome. For the prediction study, 573 subjects retained the clinical diagnosis of normal (Stable NC) at their last clinical follow-up evaluation and 78 demonstrated future cognitive decline consistent with mild cognitive impairment (MCI) or AD (Fig 1). A second prediction analysis was done with a subset of the Stable NC and Future MCI/AD groups matched 1:1. The matched subjects were selected by having an exact match of a Stable NC subject to a Future MCI/AD subject in terms of cohort, sex, ApoE4 status, race (Caucasion or non-Caucasian), baseline age split at 75y (<75y or >75y); as well as matching age within 5 years, years of education within 2 years, and follow-up time within 2 years. Exact sampling was done without replacement, and for 23 decliners no available matching control was found. Consequently, we examined 55 Future MCI/AD and 55 matching Stable NC.
Fig 1. Subject flow chart.

The patient flow chart shows the inclusion and exclusions of subjects in the analyses conducted for this study.
The NYU cohort of community dwelling volunteers (n = 331), included n = 230 with a clinical follow-up. These data were derived from NIH supported RO1 aging studies between 1997 and 2016 (MdeL, Principal Investigator). All subjects received a standard protocol consisting of medical, neurological, psychiatric, neuropsychological testing, clinical laboratory, magnetic resonance imaging (MRI) examinations, and lumbar puncture (LP). The primary goal of the NYU studies was to examine CSF biomarkers and magnetic resonance imaging (MRI) predictors of cognitive impairment in aging.
The second cohort (n = 335, including n = 326 with a clinical follow-up) was enrolled in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to use serial magnetic resonance imaging (MRI), positron emission tomography (PET), fluid biological markers, and clinical and neuropsychological assessment to describe and predict progressive cognitive impairment as related to Alzheimer’s disease (AD). For up-to-date information, see www.adni-info.org.
The third cohort (n = 100, including n = 95 with a clinical follow-up) was derived from the National Alzheimer’s Coordinating Center (NACC). NACC was established by the NIH National Institute on Aging (NIA) in 1999 to facilitate collaborative research across Alzheimer’s Disease Centers (ADC). NACC has developed and maintains a large relational database of standardized clinical and neuropathological research data in partnership with the Alzheimer's Disease Genetics Consortium and the National Cell Repository for Alzheimer's Disease. Of four participating NACC sites, data from only one site was selected. One dropped site had only two cognitively normal subjects and two dropped sites did not provide batch adjustments for the CSF values. Further, to avoid any possible overlap, three NACC subjects from the selected site that matched the ADNI data by year and month of birth, sex, education and APOE ε4 carrier status, were removed from the NACC data set. The data used was from CSF collected between January 2010 and February 2013.
Study enrollment criteria
All included subjects were clinically evaluated as having normal cognition (NC) and were between the ages 45 to 90y at the time of the baseline LP. All subjects had 12 or more years of education, APOE genotyping, a Mini-Mental State Examination (MMSE) [14,14] score of 28 or greater, a Clinical Dementia Rating (CDR) [15] score of 0 and/or a Global Deterioration Scale (GDS) [4] score of 1 or 2. In total, 131 subjects were excluded—30 subjects were dropped due to missing data and an additional 101 subjects did not meet the above screening characteristics despite having a diagnosis of normal cognition.
Lumbar puncture, CSF collection and assays
The NYU procedures for the lumbar puncture and CSF handling are published [16] and are consistent with the recommendations of Vanderstichle et al [17]. In brief, CSF Amyloid beta (Aβ42) levels and Total tau (T-tau) were measured using a standard ELISA protocol (Innotest®, Innogenetics, Ghent, Belgium). An Innogenetics sandwich ELISA assay was used to detect tau phosphorylated at threonine 181 (P-tau181). In this assay a phospho-dependent capture antibody, AT270 (P176PAPKTpP132), and a human specific tau detection antibody, HT7 (P159PGQK163), were utilized [18]. Batch wise rescaling of Aβ42 was performed using linear regression with a single reference batch of 236 NC subjects. For a subset of 233 NYU subjects, the Aβ42, Aβ38, and Aβ40 levels were additionally examined with the MSD Abeta Triplex (Meso Scale Discovery, Rockville, Maryland). All assays were conducted at the Sahlgrenska University Hospital in Sweden by board-certified laboratory technicians who were blinded to clinical data.
The procedures used by ADNI for assay technology, validation, quality control and adjustments for batch variations are found online in the ADNI data primer for CSF analyses (http://adni.info.org/). Briefly, the CSF AD biomarkers Aβ42, P-tau181 and T-tau were measured on the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) with Innogenetics immunoassay kit–based reagents (INNO-BIA AlzBio3, Ghent, Belgium [19]). The biomarker data file (UPENNBIOMK_MASTER.csv) was downloaded from the ADNI website on January 2017.
Unlike NYU and ADNI, the published NACC reports do not state CSF collection methods. However, the included NACC site used an ELISA analytic protocol similar to the method used at NYU with all provided data assayed in one batch. When referring to P-tau181 and T-tau collectively, we use the term X-tau; and when referring to multiple Aβ fragments we use Aβxx.
Statistical analyses
Demographic differences between cohorts and Outcome Group were assessed using ANOVA with Tukey post hoc correction for pairwise comparisons for continuous variables. Chi-square tests with Bonferroni correction for pairwise comparisons were used to test nominal variables for group differences. The standard confounds of age, sex and ApoE4 status were added to all statistical models as covariates with the exception of models with demographically matched outcome groups.
We observed in the individual cohort scatterplots that both high and low levels Aβ42 were associated high Tau levels. From this observation we hypothesized that a quadratic fit of Aβ42 would explain more of the variance in the Tau measures than the linear fit of Aβ42 alone. Linear regression models, with the T-Tau and P-Tau181 measures as dependent variables, were used to test this hypothesis. The linear Aβ42 term followed by the quadratic Aβ42 term (Aβ2) were added consecutively to test the incremental change in the model with the standard confounds. To account for possible heteroscedasticity of the estimators, robust (Huber-White) sandwich estimators were used to estimate the standard errors. Sensitivity analysis was performed using all available subjects with normal cognition at their baseline LP (no exclusions) from each cohort.
To have sufficient power to investigate possible interaction effects and the relationship of Aβ2 with future cognitive decline, subjects with follow-up clinical data from the three cohorts were combined and analyzed using Generalized Estimating Equations (GEE). The GEE allowed us to account for possible clustering of subjects within cohorts. We assumed an unstructured correlation matrix in all GEE models with cohort included as a within-subjects effect. The GEE models were analyzed first on the raw biomarker values. However, since the cohorts used different assays, the scale of the raw values were different for each cohort making graphing and the interpretation of model coefficients problematic. For this reason, z scores for each cohort were calculated using the mean and standard deviation from the raw biomarker values. The results are reported and graphed using the z scores.
The X-Tau measures were examined as the dependent variables with the identity link function in the GEE, with the standard covariates, Aβ42, Aβ2, and the interactions of the standard confounds with Aβ42 and Aβ2. In the presence of significant interaction terms, the data was subdivided and reanalyzed to explore the interaction.
We hypothesized that both high and low Aβ42 were associated with future cognitive decline and that this effect would be stronger in younger subjects. To test this hypothesis we used the GEE with the logit link and Outcome group as a binary dependent variable. Aβ42 was added to the standard confounds as an independent variable to test a linear association with Future MCI/AD. Subsequently, we added Aβ2 and its interaction with age. Observing a significant interaction with age, the sample was divided at the median age of the Future MCI/AD group (75y) and the GEE models including the standard confounds, Aβ42 and Aβ2 were evaluated separately in young (45-75y) and old age groups (75.1-86y). This analysis was replicated in the matched outcome groups.
In the subset of 233 NYU subjects who additionally had Aβ38, Aβ40, and Aβ42 measured with MSD, we hypothesized that Aβ42 would show quadratic relationships with X-Tau measures, whereas Aβ38 and Aβ40 would show linear relationships. To test this hypothesis we used linear regression models with the X-Tau measures as the dependent variables and the linear and quadratic terms for Aβ38, Aβ40 and Aβ42 as independent variables.
All analyses were checked for violations of the model assumptions and any conflicts are reported. The Box Cox transformation procedure [20] was used to determine the most appropriate power transformation to reconfigure values to a normal distribution. Differences in variances were tested using Levene's Test for Equality of Variances. All variables were centered for the calculation of higher order terms to avoid multicollinearity with the main effects. For all results, statistical significance was defined as a two-sided p-value of less than 5%. Statistical analyses were performed using IBM SPSS (Version 23.0) and figures were rendered in Adobe Illustrator (CC 2015).
Results
Between cohort comparisons
The three cohorts did not differ in education level, MMSE, or proportions of APOE ε4 carriers. The cohorts differed in age (F = 148.2, p < .01) and in the proportions of Caucasian (χ2 = 10.7, p < .01), and female subjects (χ2 = 14.2, p < .01), see Table 1.
Table 1. Cross sectional study demographics and descriptive variables by cohort.
| NYU | ADNI | NACC | |
|---|---|---|---|
| N | 331 | 335 | 100 |
| Age a | 64.7 ± 9.1 (45-88) b,c | 73.7 ± 5.8 (56-90) b,d | 61.9 ± 8.9 (45-83) c,d |
| Education a | 16.8 ± 2.1 (12-20) | 16.7 ± 2.4 (12-20) | 16.4 ± 2.3 (12-20) |
| % Female | 65% c | 54% b,d | 71% c |
| % Caucasian | 92% b | 90% b | 100% c,d |
| % E4+ | 30% | 28% | 36% |
| MMSE a | 29.5 ± 0.7 (28-30) | ± 0.7 (28-30) | 29.4 ± 0.7 (28-30) |
a. Values are mean ± standard deviation (range).
b. Statistically significant difference from NACC cohort (p < .05).
c. Statistically significant difference from ADNI cohort (p < .05).
d. Statistically significant difference from NYU cohort (p < .05).
The X-tau and Aβ42 measures were not normally distributed in any of the cohorts. The X-tau values were positively skewed and the natural log transformation effectively normalized the distributions in all three cohorts. Aβ42 was platykurtic with a high degree of negative excess kurtosis (NYU: kurtosis = -.54; ADNI: kurtosis = -.99; NACC: kurtosis = -.32). There was no evident power transformation that normalized the Aβ42 distributions.
Cross-sectional CSF biomarker relationships in three cohorts
The scatter plots of Aβ42 with the X-tau show in each cohort that Aβ42 bifurcates as the X-tau measures increase, with high and low values of Aβ42 being associated with high X-Tau values (Fig 2). In all three cohorts, the Aβ2 term (quadratic), was a significant addition to the linear Aβ42 models predicting either P-tau181 or T-tau. Specifically, this was found for the prediction of P-tau181 (NYU: R2 change = 3%, F change = 12.2, p < .01; ADNI: R2 change = 2%, F change = 8.2, p < .01; NACC: R2 change = 8%, F change = 11.0, p < .01) and for the prediction of T-tau (NYU: R2 change = 2%, F change = 7.3, p < .01; ADNI: R2 change = 4%, F change = 14.0, p < .01; NACC: R2 change = 6%, F change = 9.1, p < .01; Table 2). The data also showed that Aβ42 was not suitable as a dependent variable because all cohorts showed heteroscedasticity in the variances of the residuals in the models with Aβ42 as the dependent variable. The results in all three cohorts were confirmed with sensitivity analyses including all normal subjects.
Fig 2. Relationship between Aβ42 and tau in 3 cohorts.

Scatter plots for each cohort with Aβ42 on the x-axis and Tau with the natural log transformation (and the associated raw values) on the y-axis. Individual subjects are shown as circles for NYU, triangles for ADNI and squares for NACC. The outcome groups are indicated by color with the cross-sectional NL in blue, Stable NC in gray, and Future MCI/AD in orange. The quadratic fit is shown as a solid red line. The F statistic is from additive value of the quadratic fit to the model including the linear fit and standard confounds. Published cut offs for each biomarker [19,21] are depicted with dashed lines. The cohorts were combined after Z scoring.
Table 2. Linear regression models predicting X-tau (log transformed) by cohort.
| Dependent variable | P-tau 181 | ||||||||||||
| Model a | Independent Variables | NYU (n = 331) | ADNI (n = 335) | NACC (n = 100) | |||||||||
| AIC | βb | 95% CIc | p | AIC | βb | 95% CIc | p | AIC | βb | 95% CIc | p | ||
| Model 1 | Age d | 275 | 0.25 | [.15,.34] | < .01 | 466 | 0.13 | [.03, .22] | < .01 | 64 | 0.35 | [.16, .54] | < .01 |
| Sex | 0.04 | [-.06, .14] | 0.42 | 0.04 | [-.06, .14] | 0.45 | -0.01 | [-.19, .16] | 0.87 | ||||
| ApoE4 | 0.07 | [-.04, .17] | 0.22 | 0.30 | [.19, .40] | < .01 | 0.07 | [-.11, .26] | 0.44 | ||||
| Model 2 | model 1+ Aβ42 linear | 247e | 0.28 | [.18,.38] | < .01 | 445d | -0.27 | [-.38, -.17] | < .01 | 54d | 0.33 | [.11, .55] | < .01 |
| Model 3 | model 2 + Aβ42 quadratic | 236e | 0.13 | [.06, .21] | < .01 | 439d | 0.15 | [.05, .25] | < .01 | 45d | 0.25 | [.09, .41] | < .01 |
| Dependent variable | T-tau | ||||||||||||
| Model a | Independent Variables | NYU (n = 331) | ADNI (n = 335) | NACC (n = 100) | |||||||||
| AIC | βb | 95% CI | p | AIC | βb | 95% CI | p | AIC | βb | 95% CI | p | ||
| Model 1 | Age d | 406 | 0.36 | [.27, .46] | < .01 | 382 | 0.25 | [.15, .36] | < .01 | 121 | 0.43 | [.25, .61] | < .01 |
| Sex | 0.02 | [-.09, .12] | 0.77 | 0.08 | [-.02, .19] | 0.12 | 0.00 | [-.18, .19] | 0.98 | ||||
| ApoE4 | 0.12 | [.02, .23] | < .05 | 0.20 | [.09, .32] | < .01 | 0.07 | [-.11, .25] | 0.45 | ||||
| Model 2 | model 1+ Aβ42 linear | 376e | 0.28 | [.18, .38] | < .01 | 382 | -0.09 | [-.20, .03] | 0.14 | 110d | 0.32 | [.09, .56] | < .01 |
| Model 3 | model 2 + Aβ42 quadratic | 371e | 0.10 | [.04, .17] | < .01 | 370d | 0.20 | [.09, .31] | < .01 | 103d | 0.22 | [.05, .39] | < .05 |
a. All models are cumulative with respect to the variables included in the prior models.
b. Standardized β coefficients give a consistent scale of the slope across variables with 1 unit = 1 standard deviation for each variable.
c. The 95% CI of the β coefficients are calulated using robust (Huber-White) sandwich estimators of the standard error.
d. Values are in years.
e. Significant likelihood ratio test compared to previous model.
The combined sample: Relationships between CSF biomarkers
The quadratic Aβ42 term predicting the X-tau measures was also significant in the combined sample, n = 651: (P-tau181: β = 0.19, 95%CI: 0.12 to 0.26, p < .01; T-tau: β = 0.18, 95%CI: 0.12 to 0.24, p < .01). No significant interactions were found between the quadratic Aβ42 term and the standard confounds, indicating that the strength of the quadratic association did not change with age, between ApoE4 carriers and non-carriers, nor between males and females.
However, for the linear Aβ42 term significant interactions with age were found: (P-tau181: β = -0.025, 95%CI: -0.033 to -0.016, p < .01; T-tau: β = -0.019, 95%CI: -0.028 to -0.010, p < .01). To investigate the interaction of age on the relationship between Aβ42 and X-tau, we broke the combined sample into age quartiles. The younger two quartiles showed positive linear correlations between Aβ42 and the X-tau biomarkers (Fig 3 and Table 3). Whereas in the two older quartiles, Aβ42 was negatively associated with P-tau181, but the negative association with T-tau did not reach significance (Fig 3). Interactions of Aβ42 with sex and ApoE4 status failed to reach statistical significance.
Fig 3. Relationship between age quartiles, Aβ42 and X-tau in 3 cohorts combined.

Scatter plots by age quartiles with the x-axis showing for Aβ42 the combined z scores and raw values by cohort. The y-axis shows the log transformed P-tau181 and T-tau raw scores by cohort and the combined z scores. Individual subjects are shown as circles for NYU, triangles for ADNI and squares for NACC. The outcome groups are indicated by color with the cross-sectional NL in blue, Stable NC in gray, and Future MCI/AD in orange. For each quartile, the linear fit is shown as a solid light blue line and the quadratic fit as a solid red line. The shaded area represents the area outside the 95% CI of the Aβ42 values for each quartile and the vertical dotted line is at the mean Aβ42 for each quartile.
Table 3. Aβ42 in the prediction of X-tau by age quartiles.
| Quartile | N | age range | Aβ42 variance a | Dependent variable | linear fit Aβ42 | quadratic fit Aβ42 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| β b | 95% CI c | p value | β b | 95% CI c | p value | |||||
| Q1 | 162 | 45.1–62.9 | 0.80 | P-tau181 | 0.42 | 0.25, 0.59 | < .01 | 0.18 | 0.06, 0.30 | < .01 |
| Q2 | 162 | 63.0–70.31 | 0.96 | 0.20 | 0.01, 0.39 | < .05 | 0.28 | 0.17, 0.39 | < .01 | |
| Q3 | 164 | 70.33–75.9 | 1.03 | -0.23 | -0.41, -0.04 | < .05 | 0.19 | 0.01, 0.36 | < .05 | |
| Q4 | 163 | 76.0–89.7 | 1.18 | -0.18 | -0.32, -0.03 | < .05 | 0.14 | 0.03, 0.25 | < .05 | |
| Q1 | same as above | T-tau | 0.44 | 0.27, 0.60 | < .01 | 0.14 | 0.02, 0.25 | < .05 | ||
| Q2 | 0.30 | 0.11, 0.49 | < .01 | 0.25 | 0.15,0.35 | < .01 | ||||
| Q3 | -0.11 | -0.29, 0.07 | 0.22 | 0.17 | 0.01, 0.33 | < .05 | ||||
| Q4 | -0.07 | -0.22, 0.08 | 0.34 | 0.18 | 0.07, 0.29 | < .01 | ||||
a. Significant increase in variance across quartiles.
b. β coefficients are unstandardized values from the GEE with CSF z scores.
c. The 95% CI of the β coefficients are calulated using robust (Huber-White) sandwich estimators of the standard error.
Increase in the variance of Aβ42 with age
Interestingly, the variance of Aβ42 significantly increased across the age quartiles (Levene's Test of Equality of Error Variances F = 5.1, p < .01), despite the absence of significant differences in the means between the age quartiles (Fig 3). In other words, with increasing age there are increased numbers of subjects with high and low CSF Aβ42 levels. Since older age, low CSF Aβ42, and high tau levels are associated with a greater risk of AD, these changes in the variance of Aβ42 suggested that both high and low Aβ42 values could be associated with future decline. This hypothesis was tested in the GEE model predicting Outcome group.
Outcome group descriptions
The association of the CSF biomarkers and future clinical decline was examined in the combined sample of 78 Future MCI/AD and 573 Stable NC, (Table 4). Demographically, the Future MCI/AD were older (t = 7.5, p < .01), had less education (t = -2.5, p < .05), and a higher proportion of males (χ2 = 4.6, p < .05). By design, there were no differences in the matched group in terms of the demographic variables.
Table 4. Demographics and baseline descriptive variables for outcome groups.
| Stable NC | Future MCI/AD | Macthed sample age 51-75y | Matched sample age 75-86y | |||
|---|---|---|---|---|---|---|
| Stable NCb | Future MCI/ADb | Stable NCb | Future MCI/ADb | |||
| n (NYU,ADNI,NACC) | 573 (209,271,93) | 78 (21,55,2) | 30 (11,19,0) | 30 (11,19,0) | 25 (4,21,0) | 25 (4,21,0) |
| Age a | 68.2 ± 8.9 (45–90) | 74.5 ± 6.7 (51–86)c | 71.0 ± 4.2 (55.3–75.0) | 69.7 ± 5.4 (50.9–75.0) | 81.2 ± 3.2 (75.1–86.3) | 79.6 ± 2.9 (75.1–85.8) |
| Education a | 16.8 ± 2.2 (12–20) | 16.1 ± 2.4 (12–20) c | 16.7 ± 1.7 (12–20) | 16.9 ± 1.9 (12–20) | 16.3 ± 2.4 (12–20) | 16.0 ± 2.6 (12–20) |
| % Female | 61% | 49% c | 50% | 50% | 40% | 40% |
| % Caucasian | 94% | 90% | 90% | 90% | 100% | 100% |
| % E4+ | 30% | 31% | 23% | 23% | 16% | 16% |
| MMSE a | 29.4 ± 0.7 (28–30) | 29.2 ± 0.8 (28–30) | 29.3 ± 0.7 (28–30) | 29.3 ± 0.7 (28–30) | 29.4 ± 0.8 (28–30) | 29.1 ± 0.8 (28–30) |
| Project follow-up time a | 3.6 ± 2.4 (0.5–15.3) | 4.4 ± 2.8 (0.5–10.2) | 4.0 ± 2.6 (0.9–10.1) | 4.1 ± 2.3 (1.2–9.0) | 3.7 ± 2.4 (0.5–9.2) | 3.6 ± 2.6 (0.5–10.1) |
a. Values are mean ± standard deviation (range).
b. A subset of the Stable NC and Future MCI/AD were matched 1:1 on demographic variables.
c. Statistically significant difference from Stable NC (p < .05).
Aβ42 and Aβ2 associations with clinical outcome
Our combined sample findings replicate prior observations of lower levels of Aβ42 being associated with Future MCI/AD (Wald Chi-square = 7.3, OR = 1.4, p < .01). However, the addition of the quadratic (Aβ2) term and it’s interaction with age (Wald Chi-square = 3.9, OR = 1.03, p < .05) confirmed the hypothesis that both high and low Aβ42 values contribute to the prediction of future clinical decline particularly at a younger age. Splitting the full sample at the median age of the decline group (75y) and fixing the specificity at 75%, we found that in the younger group (stable NC n = 434 vs. Future MCI/AD n = 39) the quadratic Aβ42 has the greater sensitivity (72%) compared to the linear Aβ42 term (sensitivity = 61%). In the >75y sample (stable NC n = 139 vs. Future MCI/AD n = 39), neither the quadratic nor the linear Aβ42 term demonstrated sensitivity (31% and 33% respectively).
In the matched sample study the linear Aβ42 term did not reach significance. However, the quadratic (Aβ2) term was a significant predictor of outcome group (Wald Chi square = 5.9, OR = 1.6, p < .05). This result was driven by the strong quadratic effect in the younger (<75y) sample (Wald Chi-square = 4.2, OR = 1.6, p < .05). Moreover, like the larger unmatched sample, the quadratic (Aβ2) term did not reach significance in the older (>75y) subjects.
MSD analyses: The relationships between Aβ42, Aβ38, and Aβ40 with age and X-tau
As the relationship between Aβ42 to age and to X-tau can be affected by brain Aβ deposition, we also examined Aβ38 and Aβ40 which like Aβ42 are sensitive to production but unlike Aβ42, are known to have minimal brain accumulations [22]. We observed that Aβ38 and Aβ40 had strong positive linear relationships with age: (R2 change = 5%, F change = 11.0, p < .01 and R2 change = 5%, F change = 13.2, p < .01, respectively), whereas Aβ42 did not show a significant linear age effect (replicating the finding for the total sample).
Both Aβ38 and Aβ40 show strong positive linear effects with X-tau. This is clearly seen for T-tau, (R2 change = 35%, F change = 147, p < .01, and R2 change = 31%, F change = 118, p < .01, respectively), as well as for P-tau181, (R2 change = 40%, F change = 168, p < .01, and R2 change = 35%, F change = 135, p < .01, respectively, Fig 4). As in the total sample, Aβ2 uniquely shows the quadratic relationship to P-tau181 (R2 change = 2%, F change = 4.2, p < .05) and to T-tau (R2 change = 2%, F change = 4.6, p < .05).
Fig 4. Relationship of X-tau with Aβ38 and Aβ40.
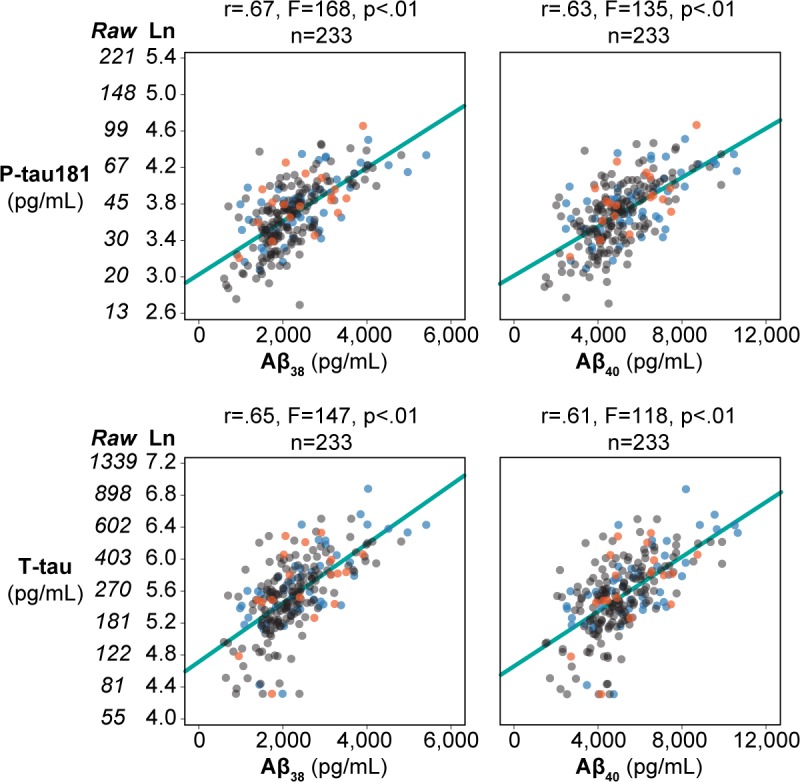
Scatter plots for n = 233 depicting Aβ38 and Aβ40 on the x-axis and the natural log transformation of X-Tau (and the associated raw values) on the y-axis. The outcome groups are indicated by color with the cross-sectional NL in blue, Stable NC in gray, and Future MCI/AD in orange. The linear fit is shown as a solid light blue line.
Discussion
The present study of cognitively normal elderly describes for the first time, quadratic relationships between Aβ42 and both P-tau181 and T-tau. We observed a U-function, such that both higher and lower levels of Aβ42 were associated with increasing X-tau levels. This effect was observed in three independent subject cohorts and for each age quartile ranging from 45–90 years. Only linear effects were found for either Aβ38 or Aβ40 with respect to X-Tau levels. Most importantly, we observed the quadratic Aβ42 term (Aβ2) was superior to the linear Aβ42 term as a predictor of future clinical decline, an effect most pronounced in younger subjects. Three important observations were made in this study, all centered on the non-linearity of the Aβ42 level.
First, both elevations and reductions in the CSF Aβ42 level are associated with increased CSF levels of P-tau181 and T-tau, known biomarkers markers of brain degeneration. These data suggest that Braak’s observation that in aging, tau pathology precedes Aβ42 pathology [6], may be under appreciated. CSF biomarker studies that rely on decreased CSF Aβ42 levels to identify brain amyloid sequestration and mark the onset of preclinical AD [23] will have missed an earlier stage of elevated Aβ42 also accompanied by elevated tau levels. Our data suggest that the use of ratio approaches to identify subjects at risk for AD are not optimal as individuals with higher Aβ42 levels would not be identified as at increased risk. Specifically, we observed that in subjects under 75y, adding the quadratic term to the prediction model significantly increased the model from an accuracy of 61% to 72%. For subjects older than 75y, neither the linear nor the quadratic terms predicted outcome.
Of the 78 the Future MCI/AD group, there were 5 subjects (3 from ADNI and 2 from NYU) with a non-AD diagnosis (2 with Parkinson’s, 2 with cortical basal degeneration and 1 with undetermined etiology) at their last visit (1.3 to 6 years after baseline). Removal of these subjects did not affect the quadratic prediction model, and further, none of these 5 subjects had extreme Aβ42 values. Rather, their z scores ranged from -1.6 to 1.1 and their mean (z = -.43) did not differ from the mean of the rest of the Future MCI/AD group (z = -.29). We therefore conclude that the quadratic Aβ42 measure is a predictor of MCI/AD outcome.
Further, the impact of ignoring the quadratic relationship could also be compounded in a longitudinal design where normal appearing levels may follow elevations. Should CSF Aβ42 levels first rise and subsequently fall in the preclinical stages of AD, a subject could have presumably normal appearing Aβ42 levels twice, complicating diagnostic examinations and the assessment of Aβ42 positivity often used in clinical prevention trials. Temporal evidence in support of the view that Aβ42 elevations precede brain Aβ42 depositions comes from murine models for AD [11]. To a very limited extent, trends exist in the presenile dementias [12,13].
To address this concern, we examined two longitudinal CSF data points for subjects who maintained normal cognition over 18 to 36 months. On the third clinical exam within 9 years of baseline, we identified decliners and non-decliners. The longitudinal CSF Aβ42 data revealed a polarization of Aβ42 levels for ε4 negative decliners (Fig 5). Specifically, the annual percentile Aβ42 ranking for non-carrier decliners moved to the extremes of the distribution showing either excessive increases or decreases in the longitudinal Aβ42 levels compared to the non-decliners. This effect was not observed in ε4 carriers. We also examined 18 subjects in the NYU cohort with three or more LP’s studied over 6–10 years. We did not observe for any subject evidence for CSF Aβ42 elevations followed by Aβ42 reductions. Presumably, the time course for AD is long and based on our cross-sectional observations, elevations are likely to have peaked years prior to the dementia transition period under surveillance. We conclude that the temporal course of tau and Aβ42 deposition in brain with respect to the CSF levels remains largely unknown and that longitudinal observations on younger subjects are needed to definitively establish that CSF Aβ42 level elevations occur in advance of cognitive decline, before CSF Aβ42 level decreases, and in advance of brain Aβ deposits.
Fig 5. Percentile rank of the annual percent change in Aβ42 (ε4 genotype and outcome group).

The percentile rankings (by cohort) of the annual percent change in Aβ42 are shown on the y-axis. The subjects are split by E4 carrier status on the x-axis then by Outcome group which is indicated with gray markers for stable NC with baseline Aβ42 levels below the 75th percentile and light red for those above and black for Future MCI/AD with baseline Aβ42 levels below the 75th percentile and red for those above. NYU subjects are represented as circles and ADNI as triangles.
It is worth briefly speculating that abnormal CSF clearance of CSF could affect the CSF analyte levels. It is increasingly apparent from animal studies that differential cross membrane transport properties, enzymatic degradation, molecular size, and brain glymphatic physiology contribute to the production and clearance of Aβ42 to the CSF [24]. While human studies have demonstrated reduced Aβ42 clearance to the CSF [25] and that reduced CSF Aβ42 levels are associated with brain Aβ depositions [1,2], it remains to be shown in humans that prior to brain deposition, CSF clearance is reduced and CSF Aβxx levels and X-Tau levels are elevated. Such reduced CSF clearance could potentially predict vulnerability to brain amyloid lesions and even tau aggregation. Recently, we reported using PET, a reduced CSF clearance in AD associated with elevated brain amyloid [26]. Others have shown this relationship in murine AD models [27]. Indirect evidence in support of a generalized clearance defect comes from our data showing strong linear relationships between X-Tau and both Aβ38 and Aβ40, as these Aβ fragments do not demonstrate appreciable brain depositions [22].
Second, we observed that over the 45 year age span examined, the mean CSF Aβ42 was not associated with age. Rather we observed an increased age associated variance that produced both high and low Aβ42 values. On the other hand, Aβ40, Aβ38, and X-tau were significantly and positively age associated. This result is consistent with one prior cross-sectional aging study that also examined a broad age range (47-84y) and similarly observed that the younger subjects (47-62y) showed a positive correlation between age and Aβ42 levels, whereas the older subjects showed a negative correlation [8]. Other studies that examined subjects of advanced age reported either no changes in Aβ42 [28,29] or reductions with age [30].
We offer that our broad age range, large sample size, and standardized clinical exams supports our explanation invoking a non-linear U-shaped quadratic function in the relationship of Aβxx with age and X-tau pathology. We observed a quadratic fit between Aβ42 and X-tau in each age quartile examined. However, the linear direction of the relationships changed from positive to negative with increasing age. Specifically, in the in the two younger age quartiles (between 45 and 70y), we observed a positive linear association between Aβ42 and tau and in the two older quartiles (between 70 and 90y) we observed that both the variance of Aβ42 increased and the slopes with age became negative. Overall, our results suggest that with increasing age, Aβ42 increases with X-tau up until a point where Aβ42 is either bound in plaques [11,31] or its activity dependent production is decreased [32]. However, as this effect is not found for other Aβ fragments which show consistent age dependent elevations, as others have concluded it points towards an Aβ42 deposition effect. We for the first time demonstrate in our longitudinal study that this nonlinear Aβ42 trajectory identifies subjects at risk for cognitive decline.
Third, our CSF biomarker predictions of clinical outcome only partially agree with prior findings of an increased risk of cognitive decline associated with elevations in the X-tau to Aβ42 ratio [33]. Our results demonstrate a new finding, an added risk associated with elevated CSF Aβ42 levels, the contribution of the Aβ2 term to the prediction. Specifically, Fagan et al first reported that only by combining baseline CSF P-tau181 and Aβ42 in a ratio were prediction effects seen for the transition from CDR = 0 to CDR >0.5 [31]. Similarly, others showed that a lower Aβ42 levels and higher P-tau181 levels best predicted future MCI/AD [34,35]. However, univariate CSF Aβ42 measures have not previously been shown to be predictive of cognitive decline. Consequently, our observation that a quadratic Aβ42 measure, one that considers both elevated and reduced levels as significant predictors is novel. This result is likely due to the added power of the large combined sample examining both younger and older decliners, and for the first time, the inclusion of Aβ42 elevations in the prediction models. We caution that the quadratic approach has not been applied to the transition between mild cognitive impairment and AD.
Overall, our results suggest that elevated tau is necessary for the prediction of cognitive decline across all age groups. However, in younger subjects considering Aβ42 elevations is very important as the cumulative evidence suggests Aβ42 elevations are followed by reductions. Future imaging studies are needed to examine the relationship between elevations in CSF Aβ42 and tau levels and brain depositions of these proteins. It is currently believed that CSF Aβ42 levels drop with brain aggregation but the antecedent relationship of elevations in the Aβ42 levels to brain deposition is unknown, but for one animal study [11]. Moreover, PET imaging data have yet to be reported for longitudinal CSF Tau level changes. Our CSF data are consistent with the view that CSF Tau and CSF Aβ42 elevations precede CSF Aβ42 reductions. This finding is not readily translatable to results from post mortem lesion staging that suggest an earlier tau deposition [6] but they do appear to conflict with formulations suggesting that initial CSF Aβ42 reductions drive the AD cascade in late onset AD [36].
In summary, we observed that quadratic relationships between Aβ42 and tau were consistently found across age quartiles in three independent normal aging cohorts. Most importantly, the quadratic (Aβ42)2 term improves on the linear Aβ42 in the prediction of future cognitive decline. The data suggest that the Aβ2 term affords an earlier recognition of risk, especially in younger subjects. Overall, our findings support the hypothesis that both high and low Aβ42 levels are associated with elevated Tau levels, and mark the earliest known risk stage for cognitive impairment related to preclinical AD. This observation may prove to be valuable in future secondary prevention trials. Further longitudinal imaging studies are needed to examine the non-linear time course of CSF Aβ42 and brain lesion deposition.
Acknowledgments
Some of the data used in preparation of this article were obtained from the Alzheimer’s disease neuroimaging initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in data analysis or writing of this report. A complete listing of ADNI investigators can be found at: https://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgment_List.pdf. The lead investigator for ADNI is Dr. Michael Weiner (Michael.Weiner@ucsf.edu).
NACC data are contributed by the NIA funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30, AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688, (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133, (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134, (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50, AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Thomas Wisniewski, MD), P30, AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30, AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016570 (PI Marie Francoise Chesselet, MD, PhD), P50 AG005131 (PI Douglas Galasko, MD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P50 AG005136 (PI Thomas Montine, MD, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), and P50 AG047270 (PI Stephen Strittmatter, MD, PhD).
This paper is dedicated to the memories of Dr. Kenneth Rich, who greatly enhanced our studies, to Sidney Aconsky a friend and a 20 year study participant, and to my career long collaborator Dr. Steven H. Ferris.
Data Availability
NYU study data are available from figshare (https://figshare.com/s/16d233d4822b810bcd9b, DOI: 10.6084/m9.figshare.5758554), ADNI data are available from http://adni.loni.usc.edu/data-samples/access-data/, and NACC data are available from https://www.alz.washington.edu/. The owner of the ADNI data is the Alzheimer’s Disease Neuroimaging Initiative and information regarding accessing the data can be found at: http://adni.loni.usc.edu/. The NACC data is owned by the National Alzheimer's Coordinating Center and information regarding accessing the data can be found at: https://www.alz.washington.edu/.
Funding Statement
The NYU study was supported by National Institutes of Health (https://urldefense.proofpoint.com/v2/url?u=https-3A__www.nih.gov_&d=DwIGaQ&c=j5oPpO0eBH1iio48DtsedbOBGmuw5jHLjgvtN2r4ehE&r=pgURDIfnRVfFwMnUonrj4ZXYtKgxaWIxc-LhDyy62ZY&m=OawcrQ4EIao5w1T8t14JaTKdfOlcYRzbq3DM0qkasC8&s=f6hj-5HuGxw-GZ0d2LM6hsZWXmo865oZPLb1OvKjTLs&e= ) grants AG13616 (MdeL), AG022374 (MdeL), AG12101 (MdeL), AG057570 (MdeL), AG032554 (MdeL), HL118624 (RSO), AG049348 (RSO), HL111724 (LG). The Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH 12 2 0012) is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The NACC database is funded by NIA/NIH Grant U01 AG016976. Kaj Blennow and Henrik Zetterberg received funding from the Swedish Research Council grants 2017-00915 (KB) and 2013-2546 (HZ), the Swedish Alzheimer Foundation grant AF-553101 (KB), the Torsten Söderberg Professorship in Medicine at the Royal Swedish Academy of Sciences LUA/ALF VGR project ALFGBG-715986 (KB), the European Research Council grant 681712 (HZ), and the Knut and Alice Wallenberg Foundation grant 2013HZ (HZ). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Fagan AM, Mintun MA, Mach RH, Lee S, Dence CS, Shah AR, et al. (2006) Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid in humans. Ann Neurol 59: 512–519. doi: 10.1002/ana.20730 [DOI] [PubMed] [Google Scholar]
- 2.Blennow K, Mattsson N, Scholl M, Hansson O, Zetterberg H (2015) Amyloid biomarkers in Alzheimer's disease. Trends in Pharmacological Sciences 36: 297–309. doi: 10.1016/j.tips.2015.03.002 [DOI] [PubMed] [Google Scholar]
- 3.Jack CR, Holtzman DM (2013) Biomarker Modeling of Alzheimer's Disease. Neuron 80: 1347–1358. doi: 10.1016/j.neuron.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reisberg B, Ferris SH, Sclan SG (1993) Empirical evaluation of the Global Deterioration Scale for staging Alzheimer's disease. Am J Psychiat 150: 680–681. doi: 10.1176/ajp.150.4.aj1504680 [DOI] [PubMed] [Google Scholar]
- 5.Tijms BM, Bertens D, Slot RE, Gouw AA, Teunissen CE, Scheltens P, et al. (2017) Low normal cerebrospinal fluid A+¦42 levels predict clinical progression in nondemented subjects. Ann Neurol 81: 749–753. doi: 10.1002/ana.24921 [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Zetterberg H, Del TK, Blennow K (2013) Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid-beta changes occur before increases of tau in cerebrospinal fluid. Acta Neuropathol. [DOI] [PubMed] [Google Scholar]
- 7.Shoji M, Kanai M, Matsubara E, Tomidokoro Y, Shizuka M, Ikeda Y, et al. (2001) The levels of cerebrospinal fluid Abeta40 and Abeta42(43) are regulated age-dependently. Neurobiology of Aging 209–215. [DOI] [PubMed] [Google Scholar]
- 8.Lauridsen C, Sando SB, Moller I, Berge G, Pomary PK, Grontvedt GR, et al. (2017) Cerebrospinal Fluid Ab43 Is Reduced in Early-Onset Compared to Late-Onset Alzheimer's Disease, But Has Similar Diagnostic Accuracy to Ab42. Frontiers in Aging Neuroscience 9: 210 doi: 10.3389/fnagi.2017.00210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sutphen CL, Jasielec MS, Shah AR, Macy EM, Xiong C, Vlassenko AG, et al. (2015) Longitudinal Cerebrospinal Fluid Biomarker Changes in Preclinical Alzheimer Disease During Middle Age. JAMA Neurol 72: 1029–1042. doi: 10.1001/jamaneurol.2015.1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y, Potter R, Sigurdson W, Santacruz A, Shih S, Ju YE, et al. (2012) Effects of age and amyloid deposition on abeta dynamics in the human central nervous system. Arch Neurol 69: 51–58. doi: 10.1001/archneurol.2011.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maia LF, Kaeser SA, Reichwald J, Lambert M, Obermuller U, Schelle J, et al. (2015) Increased CSF Abeta during the very early phase of cerebral Abeta deposition in mouse models. EMBO Mol Med 7: 895–903. doi: 10.15252/emmm.201505026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. (2012) Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 367: 795–804. doi: 10.1056/NEJMoa1202753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fagan AM, Xiong C, Jasielec MS, Bateman RJ, Goate AM, Benzinger TLS, et al. (2014) Longitudinal Change in CSF Biomarkers in Autosomal-Dominant Alzheimer's Disease. Science Translational Medicine 6: 226ra30 doi: 10.1126/scitranslmed.3007901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Folstein MF, Folstein SE, McHugh PR (1975) "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: 189–198. [DOI] [PubMed] [Google Scholar]
- 15.Morris JC (1993) The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurol 43: 2412–2414. [DOI] [PubMed] [Google Scholar]
- 16.Spiegel J, Pirraglia E, Osorio RS, Glodzik L, Li Y, Tsui W, et al. (2015) Greater Specificity for Cerebrospinal Fluid P-tau231 over P-tau181 in the Differentiation of Healthy Controls from Alzheimer's Disease. Journal of Alzheimer's Disease 49: 93–100,2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vanderstichele H, Bibl M, Engelborghs S, Le BN, Lewczuk P, Molinuevo JL, et al. (2012) Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer's disease diagnosis: a consensus paper from the Alzheimer's Biomarkers Standardization Initiative. Alzheimers Dement 8: 65–73. doi: 10.1016/j.jalz.2011.07.004 [DOI] [PubMed] [Google Scholar]
- 18.Vanmechelen E, Vanderstichele H, Davidsson P, Van Kerschaver E, Van Der Perre B, Sjogren M, et al. (2000) Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neuroscience Letters 285: 49–52. [DOI] [PubMed] [Google Scholar]
- 19.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. (2009) Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 65: 403–413. doi: 10.1002/ana.21610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Box GEP, Cox DR (1964) An Analysis of Transformations. Journal of the Royal Statistical Society Series B (Methodological) 26: 211–252. [Google Scholar]
- 21.Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, et al. (2009) CSF Biomarkers and Incipient Alzheimer Disease in Patients With Mild Cognitive Impairment. JAMA: the Journal of the American Medical Association 302: 385–393. doi: 10.1001/jama.2009.1064 [DOI] [PubMed] [Google Scholar]
- 22.Kakuda N, Miyasaka T, Iwasaki N, Nirasawa T, Wada-Kakuda S, Takahashi-Fujigasaki J, et al. (2017) Distinct deposition of amyloid-+¦ species in brains with Alzheimer ΓÇÖs disease pathology visualized with MALDI imaging mass spectrometry. Acta Neuropathol Commun 5: 73 doi: 10.1186/s40478-017-0477-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jack CR Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. (2013) Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12: 207–216. doi: 10.1016/S1474-4422(12)70291-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. (2015) Clearance systems in the brain-implications for Alzheimer disease. Nature Reviews Neurology 11: 457–470. doi: 10.1038/nrneurol.2015.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. (2010) Decreased Clearance of CNS Beta-Amyloid in Alzheimer's Disease. Science 330: 1774 doi: 10.1126/science.1197623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Leon MJ, Li Y., Okamura N, Tsui WH, Saint Louis L, Glodzik L, et al. (2017) CSF clearance in Alzheimer Disease measured with dynamic PET. Journal of Nuclear Medicine, online 58: 1471–1476. doi: 10.2967/jnumed.116.187211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng W, Achariyar TM, Li B, Liao Y, Mestre H, Hitomi E, et al. (2016) Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease. Neurobiology of Disease 93: 215–225. doi: 10.1016/j.nbd.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sjogren M, Vanderstichele H, Agren H, Zachrisson O, Edsbagge M, Wikkelso C, et al. (2001) Tau and A-beta42 in Cerebrospinal Fluid from Healthy Adults 21–93 Years of Age: Establishment of Reference Values. Clinical Chemistry 47: 1776–1781. [PubMed] [Google Scholar]
- 29.Glodzik-Sobanska L, Pirraglia E, Brys M, De Santi S, Mosconi L, Rich KE, et al. (2009) The effects of normal aging and ApoE genotype on the levels of CSF biomarkers for Alzheimer's disease. Neurobiology of Aging 30: 672–681. doi: 10.1016/j.neurobiolaging.2007.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paternico D, Galluzzi S, Drago V, Bocchio-Chiavetto L, Zanardini R, Pedrini L, et al. (2012) Cerebrospinal fluid markers for Alzheimer's disease in a cognitively healthy cohort of young and old adults. Alzheimer's & Dementia 8: 520–527. [DOI] [PubMed] [Google Scholar]
- 31.Fagan AM, Roe CM, Xiong C, Mintun M, Morris JC, Holtzman DM (2007) Cerebrospinal Fluid tau/beta-Amyloid42 Ratio as a Prediction of Cognitive Decline in Nondemented Older Adults. Arch Neurol 64: 343–349. doi: 10.1001/archneur.64.3.noc60123 [DOI] [PubMed] [Google Scholar]
- 32.Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, et al. (2011) Neuronal activity regulates the regional vulnerability to amyloid-b deposition. Nature Neuroscience 14: 750–756. doi: 10.1038/nn.2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blennow K, Zetterberg H (2013) The application of cerebrospinal fluid biomarkers in early diagnosis of Alzheimer disease. Med Clin North Am 97: 369–376. doi: 10.1016/j.mcna.2012.12.012 [DOI] [PubMed] [Google Scholar]
- 34.Mattsson N, Insel PS, Donohue M, Jagust W, Sperling R, Aisen P, et al. (2015) Predicting reduction of cerebrospinal fluid beta amyloid 42 in cognitively healthy controls. JAMA Neurology 72: 554–560. doi: 10.1001/jamaneurol.2014.4530 [DOI] [PubMed] [Google Scholar]
- 35.Roe CM, Fagan AM, Grant EA, Hassenstab J, Moulder KL, Maue Dreyfus D, S et al. (2013) Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurol 80: 1784–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jack CR Jr., Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. (2010) Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 8: 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
NYU study data are available from figshare (https://figshare.com/s/16d233d4822b810bcd9b, DOI: 10.6084/m9.figshare.5758554), ADNI data are available from http://adni.loni.usc.edu/data-samples/access-data/, and NACC data are available from https://www.alz.washington.edu/. The owner of the ADNI data is the Alzheimer’s Disease Neuroimaging Initiative and information regarding accessing the data can be found at: http://adni.loni.usc.edu/. The NACC data is owned by the National Alzheimer's Coordinating Center and information regarding accessing the data can be found at: https://www.alz.washington.edu/.