

Abstract
Few replicable genetic associations for Major Depressive Disorder (MDD) have been identified. Recent studies of MDD have identified common risk variants by using a broader phenotype definition in very large samples, or by reducing phenotypic and ancestral heterogeneity. We sought to ascertain whether it is more informative to maximize the sample size using data from all available cases and controls, or to use a sex or recurrent stratified subset of affected individuals. To test this, we compared heritability estimates, genetic correlation with other traits, variance explained by MDD polygenic score, and variants identified by genome-wide meta-analysis for broad and narrow MDD classifications in two large British cohorts - Generation Scotland and UK Biobank. Genome-wide meta-analysis of MDD in males yielded one genome-wide significant locus on 3p22.3, with three genes in this region (CRTAP, GLB1, and TMPPE) demonstrating a significant association in gene-based tests. Meta-analyzed MDD, recurrent MDD and female MDD yielded equivalent heritability estimates, showed no detectable difference in association with polygenic scores, and were each genetically correlated with six health-correlated traits (neuroticism, depressive symptoms, subjective well-being, MDD, a cross-disorder phenotype and Bipolar Disorder). Whilst stratified GWAS analysis revealed a genome-wide significant locus for male MDD, the lack of independent replication, and the consistent pattern of results in other MDD classifications suggests that phenotypic stratification using recurrence or sex in currently available sample sizes is currently weakly justified. Based upon existing studies and our findings, the strategy of maximizing sample sizes is likely to provide the greater gain.
Introduction
Major Depressive Disorder (MDD) is a frequently disabling, chronic disorder for which there is substantial evidence of a genetic contribution to its liability1. Until recently, the largest international mega-analysis of clinically diagnosed MDD (9240 MDD cases and 9519 controls) yielded no genome-wide significant findings2. Given the success of similarly sized studies for other adult psychiatric disorders3,4, this study suggested that MDD is an extensively heterogeneous phenotype. This heterogeneity, in addition to the relatively high prevalence and low heritability of MDD, impacts substantially on the statistical power to detect genetic effects1,5. Possible means of improving statistical power include stratifying the phenotype into potentially more homogeneous subtypes, or considerably increasing the sample size whilst accepting a broader phenotype. Both of these approaches have since identified associations between genetic variants and MDD6–9.
A genome-wide association study (GWAS) from the CONVERGE consortium identified two genome-wide significant loci (chromosome 10q21.3 and 10q26.13) using a severe depressive phenotype (5053 cases and 5337 controls) in female Han Chinese individuals, treated in a hospital setting6. A subsequent study by the Psychiatric Genomics Consortium (PGC) stratified the PGC MDD mega-analysis sample2 by age of onset and identified a risk conferring locus at chromosome 3q27.2 in individuals with onset after 27 years of age9.
In contrast to the above study designs, two studies have utilized larger sample sizes with less detailed structured clinical assessments. The first of these studies came from the CHARGE consortium and employed a quantitative assessment of depressive symptoms using the Center for Epidemiological Studies Depression Scale. In a combined dataset of 51 258 individuals, a genome-wide significant locus was identified at chromosome 5q21.27. More recently, Hyde et al8 conducted a GWAS using 23andMe data of self-reported depression in 45,773 cases and 106,354 controls, revealing 15 genome-wide significant loci. The genetic correlation (rG) between the 23andMe depression phenotype and the clinical phenotype reported by the PGC was rG(SE) = 0.73(0.09), suggesting a strong association between the additive genetic components of each trait. Previous work comparing self-reported depression and clinically defined MDD in Generation Scotland: Scottish Family Health Study (GS:SFHS) also provides evidence that these traits have substantially overlapping common genetic architectures10.
The findings from these four studies support both phenotypic stratification and increased sample size as strategies which may help reveal the underlying architecture of MDD. The international collaborative efforts by groups such as the PGC and the development of large-scale biobanks with genetic and extensive phenotypic information will ensure ever increasing sample numbers. It is therefore timely to investigate the contrasting strategies that may be employed in the analysis of these emerging datasets.
In the current study, we sought to compare these strategies by conducting a suite of genetic analyses for depression and stratified subtypes in two UK-based cohorts: GS:SFHS11,12 and UK Biobank (UKB)13,14. To maximize the sample size, an unstratified analysis was initially conducted. This used MDD diagnostic information obtained at structured clinical interview15 in GS:SFHS (2603 cases, 16,122 controls), and a probable MDD phenotype obtained from a touchscreen questionnaire16, previously validated by Smith et al17, in UKB (8248 cases, 16,089 controls). Subsequent analyses stratified the phenotype on the basis of recurrence or sex. Each approach was evaluated using several metrics: the successful identification of variants reaching genome-wide significance in GWAS meta-analysis, an increased SNP-based heritability estimate, identification of significant genetic correlations with other traits using LD score regression, and increased variance explained by polygenic profile scores for MDD derived from three independent cohorts. These metrics aim to test whether basic stratification of the MDD phenotype improves etiological insight.
Materials and methods
This study analyzed data from Generation Scotland: The Scottish Family Health Study (GS:SFHS), (data available on request: http://www.generationscotland.co.uk) and UKB, (data available on request: http://www.ukbiobank.ac.uk). GS:SFHS received ethical approval from the NHS Tayside Committee on Medical Research Ethics (REC Reference Number: 05/S1401/89). UK Biobank received ethical approval from the Research Ethics Committee (REC Reference Number: 11/NW/0382). The present analyses were conducted under UK Biobank data application number 4844. All participants provided informed consent.
Data and code availability
Data are available to qualified researchers on a cost-recovery basis via online application processes, accessible via www.gsaccess.org and www.ukbiobank.ac.uk/register-apply/. The code used in these analyses is available on request from the lead author.
Participants
Generation Scotland: The Scottish Family Health Study (GS:SFHS)
GS:SFHS is a family and population-based study consisting of 23,690 participants recruited via general medical practices across Scotland. The recruitment protocol and sample characteristics are described in detail elsewhere11,12. Briefly, participants were over 18 years old, and not ascertained on the basis of having any particular disorder. A diagnosis of depression (MDD) was made using the structured clinical interview for DSM-IV disorders (SCID)15. Participants who answered yes to either of the two screening questions were invited to continue the interview, which provided information on the presence or absence of a lifetime history of MDD, age of onset and number of depressive episodes. Participants who answered no to both screening questions or who completed the SCID but did not meet the criteria for depression were assigned control status. Case definition was further refined through NHS data linkage. Controls with a history of antidepressants or who had been referred to a secondary psychiatric care centre (n = 1 072) were excluded, as were cases who had received a previous diagnosis of schizophrenia or bipolar disorder (n = 47). This resulted in 2603 depression cases (of which 1289 were recurrent) and 16,122 controls. Stratification by sex resulted in 1859 female cases, 9159 female controls, 770 male cases and 6958 male controls.
UK Biobank (UKB)
UKB13 is a population-based health research resource consisting of approximately 500,000 people, aged between 40 and 69 years, who were recruited between the years 2006 and 2010 from across the UK14. Of these, 152,729 individuals were included in the first genotype data release. In the current study we restricted the sample to individuals of white British ancestry. Participants who were also in GS:SFHS, their relatives and relatives of remaining UKB participants (relatives: up to and including third degree) were identified by a kinship coefficient ≥ 0.0442, using the KING toolset18, and subsequently excluded (n = 7 698). Depression case/control status was assessed in 172,751 of the 500,000 individuals using a self-diagnosed touchscreen questionnaire. Case status was defined as either “probable single lifetime episode of major depression” or “probable recurrent major depression (moderate and severe)”. Control status was defined as “no mood disorder”, as described by Smith et al17. 149,847 individuals had sufficient data to allow an assessment of case/control status. Individuals with probable bipolar disorder (n = 1 615) or mild depressive/manic symptoms (n = 26 847) were excluded. After exclusions outlined above, this resulted in 8248 depression cases (of which 6056 were recurrent) and 16,089 controls. Stratification by sex resulted in 5138 female cases, 7013 female controls, 3110 male cases and 9076 male controls. Further information on sample collection, genotyping and assessment of the depression phenotype in GS:SFHS and UKB are provided in the Supplementary Methods.
Imputation and quality control
GS:SFHS
Autosomal genotype data were available for all GS:SFHS individuals in the present study (n = 18 725). Genotypes were imputed using the Haplotype Reference Consortium reference panel (HRC.r1-1)19 via the Sanger Imputation Server pipeline (https://imputation.sanger.ac.uk). Prior to imputation, individuals with missingness ≥ 3% were excluded, as were SNPs with a call rate of ≤98%, Hardy Weinberg Equilibrium (HWE) P-value ≤ 1 × 10−6, and a minor allele frequency (MAF) ≤ 1%. Phasing of genotype data was performed using the SHAPEIT2 algorithm20 utilizing the duoHMM option, which refines phasing by utilizing pedigree information. Imputation was performed using PBWT software21. Multi-allelic variants, monomorphic variants and SNPs with an imputation INFO score < 0.8 were removed22. Population outliers (more than 6SDs from the mean of the first principal component (PC)) were identified and removed from the sample23, as were one from each of 52 monozygotic twin pairs, identified by IBD (preferentially retaining cases), and 7 individuals who matched samples from the Psychiatric Genomics Consortium, identified using genotype checksums24. After imputation, individuals with missingness ≥ 2%, and genotype with a call rate of ≤98%, MAF ≤ 0.5% and HWE P-value ≤ 1E-05 were excluded using PLINK version 1.9.25,26. Strand ambiguous SNPs with 40% ≤ MAF ≤ 50% were also excluded.
UKB
Autosomal genotypes were available for all UKB individuals in the present study (n = 24 337). Pre-imputation QC, phasing and imputation are described elsewhere27. In brief, prior to phasing, multiallelic SNPs or those with MAF ≤ 1% were removed. Phasing of genotype data was performed using a modified version of the SHAPEIT2 algorithm28. Imputation to a reference set combining the UK10K haplotype and 1000 Genomes Phase 3 reference panels29 was performed using IMPUTE2 algorithms30,31. A further QC protocol was then applied at the Wellcome Trust Centre for Human Genetics before the data was released, as described elsewhere32. The analyses presented here were restricted to autosomal variants with an imputation INFO score ≥ 0.9 and MAF ≥ 0.5%.
Of the SNPs which passed QC in each dataset, only SNPs in common between both datasets were used in subsequent analyses, with allele and strand in GS:SFHS harmonized to be consistent with UKB, resulting in 7,105,178 autosomal SNPs.
Statistical analysis
All analyses present here were performed on four subsets of the data: all available cases and controls (MDD), all controls and recurrent cases (rMDD), female controls and cases (fMDD), and male controls and cases (mMDD). The total sample size for each depression subgroup was n = 43 062 for MDD, n = 39 556 for rMDD, n = 23 169 for fMDD and n = 19 886 for mMDD. The number of cases and controls and demographic information for these subsets are shown in Supplementary Table 1.
Association analysis
GS:SFHS
GWAS of MDD, rMDD, fMDD and mMDD in GS:SFHS were conducted using mixed linear model based association (MLMA) analysis33, implemented in GCTA (v1.25.)34. To account for population structure, two genomic relationship matrices (GRMs) were used, as this method allows the inclusion of closely and distantly related individuals in genetic analyses35. The first GRM included pairwise relationship coefficients for all individuals. The second GRM had off-diagonal elements < 0.05 set to 0. GRMs were created using the mixed linear model with candidate marker excluded (MLMe) approach, where GRMs are calculated excluding SNPs located on the chromosome under analysis33. No fixed effects covariates were fitted in this analysis as sex was being assessed as a stratifier, and the two GRMs adequately accounted for population stratification (tested using univariate LD Score Regression36). MLMA employs restricted maximum likelihood methods on the linear scale. As such, test statistics (betas and their corresponding standard errors) were transformed to Odds Ratios and their corresponding 95% Confidence Intervals on the liability scale using a Taylor transformation expansion series37,38. Further details of GWAS can be found in the Supplementary Methods.
UKB
GWAS of MDD, rMDD, fMDD, and mMDD in UKB were conducted using logistic regression, implemented in PLINK v1.925. Assessment centre, genotype array and batch were fitted as fixed effects. The first 8 PCs (out of 15) supplied by UKB were also fitted, as visual inspection indicated that these PCs resulted in multiple clusters, indicating structure in the data.
Meta-analysis, variant look-up and gene-based analysis
The meta-analysis of GS:SFHS and UKB was conducted using the classical inverse-variance approach, which weights effect sizes by sampling distribution, implemented in the METAL package39. SNPs with a meta-analysis P-value of P ≤ 1E-05 were subjected to clump-based linkage disequilibrium pruning using PLINK25 using an LD r2 cut off of 0.1 and a 500 kb sliding window to create SNP sets of approximately independent “lead SNPs”. All SNPs which surpassed genome-wide significance were entered into the NHGRI-EBI catalog of published GWAS40,41 (www.ebi.ac.uk/gwas/) to observe whether these SNPs had been previously observed in association analysis.
Gene-based analysis was performed for MDD, rMDD, fMDD, and mMDD using MAGMA42. The gene-based statistics were derived using the summary statistics from each meta-analysis. Genetic variants were assigned to genes based on their position according to the NCBI 37.3 build, with a gene boundary defined by an extended region between 20 kb upstream of transcript start site and 20 kb downstream of transcript end site for each of the genes. This resulted in a total of 18 111 genes for MDD, fMDD, and mMDD, and 17,225 genes for rMDD being analyzed. The European panel of the 1000 Genomes data (phase 1, release 3) was used as a reference panel to account for linkage disequilibrium43. A genome-wide significance threshold for gene-based associations was calculated using the Bonferroni method (α = 0.05/18 111; P < 2.76 × 10−6 for MDD, fMDD and mMDD; α = 0.05/17 225; P < 2.90 × 10−6 for rMDD).
Pathway and functional genomic analyses
Pathway and functional genomic analyses were performed using the GWAS results for each of the MDD meta-analyses. These included DEPICT analyses44, reference to RegulomeDB45 (http://www.regulomedb.org/) and to the Genotype-Tissue Expression Portal (http://www.gtexportal.org) for independent SNPs with P < 1.0 × 10−5 and all genome-wide significant SNPs (P < 5.0 × 10−8, nSNPs = 6). Further information on pathway and functional genomic analysis can be found in the Supplementary Methods.
Heritability, polygenicity and cross-trait genetic correlations
Univariate GCTA-GREML46 analyses were used to estimate the proportion of variance explained by all common (MAF > 1%) SNPs for each of the depression phenotypes. A relatedness cutoff of 0.05 was used in the generation of the genetic relationship matrix, as including close relatives inflates heritability estimates47. This did not alter the sample size in UKB due to previous sample filtering, however in GS:SFHS this reduced the sample size by 38.5–58.4% (Supplementary Table 15). In GS:SFHS, the first 20 PCs were fitted as fixed effects. In UKB, batch, recruitment centre and the first 8 PCs were fitted. Univariate Linkage Disequilibrium Score regression (LDSR), implemented in LD Score (v1.0.0.)36, was applied to GWAS summary statistics to evaluate the proportion of inflation in the test statistics caused by confounding biases, such as population stratification, relative to genuine polygenicity. This method also provides an estimate of SNP-based heritability. Pre-computed LD Scores were used, estimated from the European-ancestry samples in the 1000 Genomes Project43. To obtain heritability estimates on the liability scale, sample and population prevalence estimates were used. Sample prevalence estimates were calculated as the proportion of cases in each subset. Population prevalence estimates were derived from the literature48–50. Prevalence estimates used in GCTA-GREML and LDSC are given in Supplementary Table 1. Genetic correlations between meta-analyzed depression subgroups and 200 health-related traits were calculated using bivariate LDSR51, implemented in the LD Hub software52. Traits derived from non-Caucasian or mixed ethnicity samples were removed prior to analysis. False discovery rate (FDR) correction was applied across the 800 tests to correct for multiple testing53.
Polygenic profiling analysis
To test the association of MDD-associated alleles with each subtype of MDD in GS:SFHS and UKB, summary statistics for major depressive disorder from the Psychiatric Genomics Consortium (minus GS:SFHS, n = 50 455 cases, 105,411 controls; minus UKB, n = 43 204 cases, 95,680 controls)54, UKB and GS:SFHS (from the current study) were used to provide weights for polygenic profile scores (PGS).
PGS for GS:SFHS and UKB individuals were derived at 5 GWAS P-value thresholds (PT < 0.01, < 0.05, < 0.1, < 0.5 and all SNPs) using PRSice55. Genotyped SNPs (with MAF > 1%) were subjected to clump-based linkage disequilibrium (LD) pruning, using an LD r2 cut off of 0.25 and 200 kb sliding window to create SNP sets in approximate linkage equilibrium. PGS were then standardized to have a mean of zero and a unit standard deviation.
In GS:SFHS, the associations between PGS and MDD, rMDD, fMDD, and mMDD were tested using a mixed linear model, covarying for the first 20 PCs to account for population stratification. Prior to this analysis, the requisite number of PCs was established using a stepwise linear regression approach, adding one PC at a time, and using a likelihood-ratio test (LRT), the output of which was assessed against a mixed 0.5(χ2) + 0.5(0) distribution56. An additive genetic component was fitted as a random effect to account for the increased relatedness within GS:SFHS. To ensure that common environment was adequately modeled, models incorporating shared parent-offspring, sibling, and spousal environmental components as additional random effects were tested using a stepwise LRT approach, however no environmental component improved model fit. Further details of mixed linear model selection are provided in the Supplementary Methods. F-statistics, degrees of freedom, effect sizes, Z-scores and P-values were derived using the Wald Conditional F-test57, in ASReml-R58.
In UKB, the association between PGS and MDD, rMDD, fMDD, and mMDD was tested in a generalized linear model framework by regressing the PGS onto the phenotype, covarying for assessment centre, genotype array and batch and the first eight PCs.
FDR correction was applied across the 80 tests to correct for multiple testing53. For both GS:SFHS and UKB, trait variance explained by the PGS was calculated using: (var(x × β))/var(y), where x was the standardized PGS, β was the corresponding regression coefficient and y was the phenotype59.
Results
Meta-analysis of depression in GS:SFHS and UKB
One genomic region on chromosome 3p22.3 achieved genome-wide significance in the males only case/control (mMDD) analysis (index SNP rs4478037, β(SE) = 0.29(0.05), P = 2.29 × 10−8). None of the SNPs achieving genome-wide significance (nSNPs = 6) associated with any phenotype in currently published GWAS available via the NHGRI-EBI catalog. One variant (rs7613051) within the local genomic region (defined 3:33000000–33200000) has previously shown an association with Atopic dermatitis60, however this SNP is not in LD with the genome-wide significant SNPs (r2 < 0.1). Meta-analysis of MDD, rMDD, and fMDD did not yield any genome-wide significant findings. Manhattan plots are shown for each trait in Fig. 1, and summary statistics for independent loci with a meta-analysis association P ≤ 1 × 10−6 are shown in Table 1. A regional association plot for genome-wide significant index SNP, rs4478037, is shown in Fig. 2. Regional association plots for this SNP in other depression subtypes are shown in Supplementary Figure 4, demonstrating that this locus does not replicate in any other depression subtype (minimum P = 8.05 × 10−4 in MDD, β(SE) = 0.10(0.05)). Full details of all independent loci used in downstream analyses (P ≤ 1 × 10−5) are shown in Supplementary Tables 4-11. The QQ plots (Supplementary Figure 3) demonstrate λGC ranged from 1.02–1.06, comparable to the value (1.056) observed in the PGC mega-analysis of MDD2. Univariate LDSR analyses estimated that meta-analyzed MDD subtypes had mean chi-squared statistic (μχ2) values ranging from 1.018 (mMDD) to 1.062 (MDD) with a Ratio, defined as (Intercept-1)/(μχ2-1), ≤ 0.35 across subtypes, indicating that any inflation in μχ2 can be attributed to polygenicity rather than residual population stratification36.
Fig. 1.

Manhattan plot of P-values from SNP-based association meta-analysis of all depression cases and controls (MDD, n = 43 062), recurrent only cases and all controls (rMDD, n = 39 556), females only cases and controls (fMDD, n = 23 169) and males only cases and controls (mMDD, n = 19 886). The blue line indicates the threshold for genome-wide significance (P < 5 × 10-8), the red line indicates the threshold for suggestive significance (P < 1 × 10-5)
Table 1.
Summary statistics for SNPs with association P-value ≤ 1 × 10−6 for depression (MDD), recurrent depression (rMDD), depression in females only (fMDD) and depression in males only (mMDD), sorted within phenotype by genomic positions according to UCSC hg19/NCBI Build 37
| Trait | SNP | CHR | POS | A1/A2 | Freq | β(se) | P | Direction | Genes |
|---|---|---|---|---|---|---|---|---|---|
| MDD | rs56390503 | 4 | 187552576 | T/C | 0.11 | 0.13 (0.03) | 9.08E–07 | ++ | FAT1 |
| rs2964802 | 5 | 10820843 | T/C | 0.28 | 0.09 (0.02) | 6.73E–07 | ++ | - | |
| rs11033303 | 11 | 35871266 | A/G | 0.37 | 0.09 (0.02) | 2.37E–07 | ++ | - | |
| rMDD | rs2291479 | 3 | 178174944 | A/C | 0.4 | −0.10 (0.02) | 9.65E–07 | -- | - |
| rs10959631 | 9 | 11220986 | T/C | 0.2 | −0.12 (0.02) | 8.34E-07 | -- | - | |
| rs11033303 | 11 | 35871266 | A/G | 0.37 | 0.11 (0.02) | 6.02E–07 | ++ | - | |
| rs4438172 | 13 | 111448658 | A/T | 0.25 | 0.11 (0.02) | 8.72E–07 | ++ | - | |
| fMDD | rs9648182 | 7 | 13794849 | A/T | 0.12 | −0.17 (0.03) | 9.14E–08 | -- | - |
| rs17176546 | 7 | 81880914 | A/G | 0.04 | 0.26 (0.05) | 7.93E–07 | ++ | CACNA2D1 | |
| mMDD | rs115736167 | 1 | 155266609 | C/G | 0.02 | −0.46 (0.09) | 1.54E–07 | -- | PKLR |
| rs4478037 | 3 | 33160407 | A/G | 0.08 | 0.29 (0.05) | 2.29E–08 | ++ | CRTAP | |
| rs113485090 | 11 | 73572495 | A/G | 0.04 | -0.32 (0.06) | 2.05E-07 | -- | MRPL48 | |
| rs1380551 | 15 | 24124704 | A/G | 0.15 | -0.18 (0.04) | 3.96E-07 | -- | - |
Column A1/A2 contains the reference and alternate alleles for the index SNP, respectively. The meta-analysis minor allele frequency (Freq) and regression coefficient (β) columns pertain to the reference allele (A1). Chr and Position denote the location of the index SNP. SE is the standard error for β. The direction of effect of the index SNP in GS:SFHS and UKB is shown in the Direction column. The final column, Genes, indicates protein-coding reference sequence genes within 10 kb of the associated loci
Fig. 2.

Regional association plot for rs4478037, an intronic SNP in CRTAP, and the top ranking SNP (rs4478037, P = 2.37 × 10-8) in GWAS of depression in males only
Gene based analysis of MDD subtypes
Three genes at chromosome 3p22.3 (CRTAP, GLB1, and TMPPE) were significantly associated with mMDD after Bonferroni correction (Supplementary Table 12). Whilst CRTAP and GLB1 have not previously shown association with psychiatric disorders, both genes are members of the CNTN1 PPI subnetwork. This subnetwork contains CNTN1, which encodes a protein that may play a role in the formation of axon connections in the developing nervous system61. Furthermore, the CNTN1 PPI subnetwork also contains HTR1A, which encodes a serotonin (5-HT) receptor subtype that binds endogenous 5-HT62. To assess whether significant association of these 3 genes was due to LD in the region, the meta-analysis of MDD in males was re-run conditional on SNPs with an R2 > 0.9 with the top ranking SNP, rs4478037. This analysis, implemented in GCTA (v1.25)34, indicated that the signal was being driven by LD across the region (Supplementary Figure 5). There were no significant gene-based associations with MDD, rMDD or fMDD.
Pathway and functional genomic analyses
Gene set enrichment analysis of SNPs with meta-analysis P ≤ 1 × 10−5, as implemented in DEPICT, indicated a role for two gene sets at FDR < 0.05 in mMDD (Supplementary Table 13): GO terms carboxylic acid binding and CNTN1 PPI subnetwork. No other significant results were observed for tissue enrichment or gene prioritization across MDD definitions.
Using the GTEx database (http://www.broadinstitute.org/gtex/), 25 multi-tissue cis-eQTL associations were identified for 16 independent lead SNPs with meta-analysis P < 1 × 10−5 (Supplementary Tables 4, 6, 8, 10, and 14). The 5 genome-wide significant SNPs identified for mMDD show eQTL evidence for the genes GLB1 and CRTAP. Random effect meta-analysis of multi-tissues for the most significant mMDD SNP, rs447803, yielded P = 3.58 × 10−9 and 8.45 × 10−29 for GLB1 and CRTAP, respectively. For this study, data mining of regulatory elements was restricted to normal cell lines/tissues. There was evidence of regulatory elements (Regulome DB score < 4) for 6 of the lead SNPs with meta-analysis P ≤ 1 × 10−5 (MDD: rs10736455, rs73249855, rs8050755; rMDD: rs60716536; fMDD: rs11613048; mMDD: rs74002781). Of the six SNPs which achieved genome-wide significance in the meta-analysis of mMDD, 2 SNPs (rs11558338 and rs6809511) showed evidence of transcription factor binding, position weight matrix, histone modification, DNase hypersensitivity, and FAIRE regulatory elements. Evidence of regulatory evidence for all independent SNPs with meta-analysis P ≤ 1 × 10−5 are shown in Supplementary Table 14.
Estimating SNP-based heritability and polygenicity
Using GCTA-GREML methods46, the SNP-based heritability (h2SNP) estimates in UKB were consistent and significant across MDD subtypes, with h2SNP(SE) estimates of MDD = 0.20(0.04); rMDD = 0.20(0.03); fMDD = 0.22(0.06) and mMDD = 0.18(0.06). Due to the unrelated subset of individuals in GS:SFHS being markedly smaller than the full sample (nmax = 7 795), the heritability estimates were non-significant across all MDD definitions. Results from GCTA-GREML are shown for MDD subtypes in Supplementary Table 15. LDSR yielded lower h2SNP estimates than GCTA-GREML methods (Supplementary Table 16). This is to be expected as LDSR utilizes summary scores, which have usually been subjected to genomic control, as opposed to full SNP data.
Genetic correlation with health-related traits
Bivariate LDSR showed nominally significant (P < 0.05) genetic correlations (rG) between meta-analyzed MDD and 28 of the 200 health-related traits assessed. Of these, 8 traits survived multiple testing correction: neuroticism (rG(SE) = 0.67(0.07); P = 7.06 × 10−21), depressive symptoms (rG(SE) = 0.81(0.09); P = 1.72 × 10−19), subjective wellbeing (rG(SE) = −0.56(0.08); P = 9.12 × 10−13), age at first birth (rG(SE) = −0.35(0.05); P = 1.92 × 10−10), major depressive disorder (rG(SE) = 0.67(0.12); P = 4.57 × 10−8), PGC cross-disorder analysis (rG(SE) = 0.46(0.09); P = 8.60 × 10−8), bipolar disorder (rG(SE) = 0.32(0.08); P = 4.35 × 10−5) and systemic lupus erythematosus (rG(SE) = 0.28(0.08); P = 8.00 × 10−4). These findings are consistent with previously reported, well-established relationships between MDD and neuroticism63–65, bipolar disorder51,66, PGC cross-disorder66, depressive symptoms7,67 and subjective well-being68. Relationships between MDD and age at first birth, and SLE have been previously reported although these have been based on phenotypic correlations69–71. The majority of these traits (with the exception of age at first birth and systemic lupus erythematosus) demonstrated rG of a similar magnitude, direction and significance with recurrent and female MDD. In contrast to these results, bivariate genetic correlations between mMDD and health-related traits were all non-significant after adjustment for multiple testing (Supplementary Table 17). Summary statistics from univariate and bivariate LDSR (Supplementary Tables 14 and 17) indicate that the lack of association between mMDD and other health-related traits is due to reduced statistical power, rather than a genuine sex difference. Univariate LDSR of mMDD returned a mean χ2 = 1.018, indicating low power (as a minimum mean χ2 = 1.02 is deemed appropriate for LDSR72). In addition, the univariate LDSR h2SNP(SE) = 0.05(0.03) for mMDD. The LDSR rG is calculated as rG = ρg/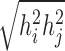 where ρg is the genetic covariance between traits,
where ρg is the genetic covariance between traits, 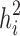 is the heritability of trait i and
is the heritability of trait i and 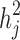 is the heritability of trait j36. Near-zero h2 estimates can therefore cause the rG estimate to become out of bounds (rG > 1), as observed in three out of five nominally significant traits, with large standard errors72, as observed in all nominally associated traits. Fig. 3 shows the genetic correlation of meta-analyzed depression subtypes with significantly correlated health traits.
is the heritability of trait j36. Near-zero h2 estimates can therefore cause the rG estimate to become out of bounds (rG > 1), as observed in three out of five nominally significant traits, with large standard errors72, as observed in all nominally associated traits. Fig. 3 shows the genetic correlation of meta-analyzed depression subtypes with significantly correlated health traits.
Fig. 3.
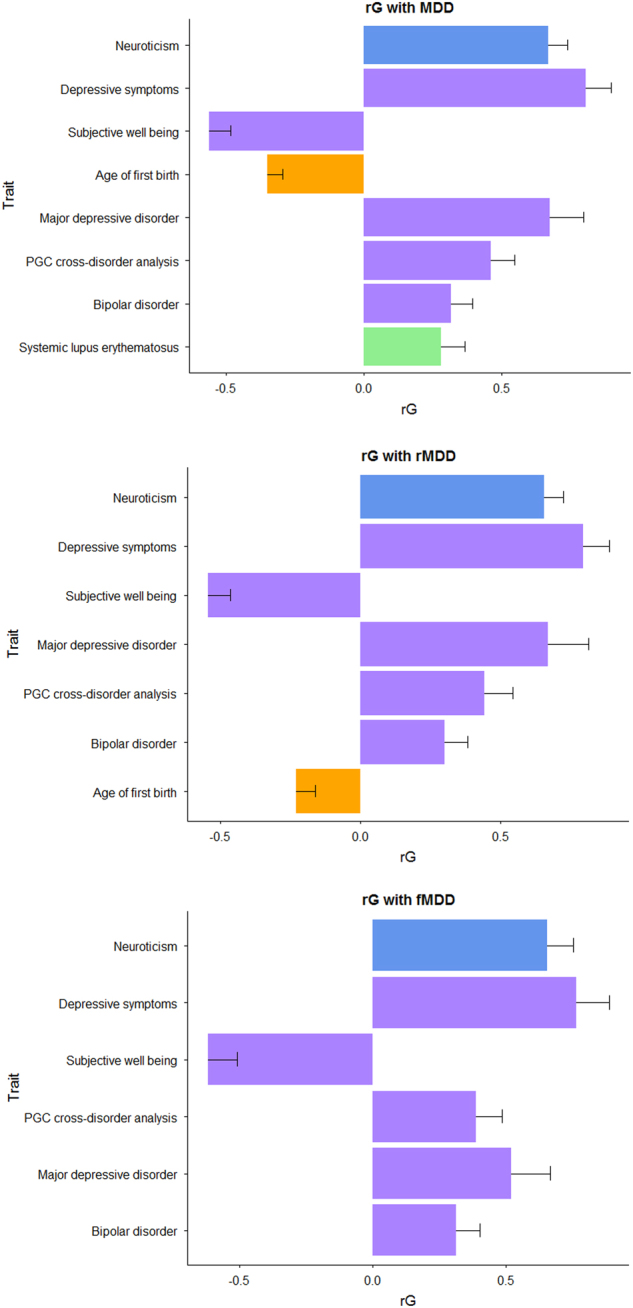
Genetic correlation (rG) between meta-analyzed MDD subsets and other health-related traits, derived using GWAS summary statistics and LD score regression. Traits presented showed a significant rG with MDD subsets after multiple testing correction (FDR p ≤ 0.05) and are coloured by category (personality, psychiatric, reproductive and autoimmune). No rG between mMDD and other health-related traits survived multiple testing correction
Polygenic profiling analysis
The GWAS results from the MDD phenotype in 3 discovery samples (PGC MDD29, UKB and GS:SFHS) were used to build polygenic profile scores (PGS) in GS:SFHS and UKB, incorporating SNPs with a discovery sample association P-value cut-off of PT ≤ 0.01, PT ≤ 0.05, PT ≤ 0.1, PT ≤ 0.5, and all SNPs (PT ≤ 1). Results from PT ≤ 0.05 are shown in Fig. 4, as this PT explained the most variance in the target datasets. Results from all PT are shown in Supplementary Tables 18-21. PGS derived using information from the PGC MDD29 GWAS yielded significant associations between depression phenotype and PGS across almost all thresholds in both GS:SFHS and UKB (with the exception of mMDD at PT ≤ 0.01 in GS:SFHS). PGS derived using information from the UKB MDD GWAS yielded significant associations with MDD, rMDD, and mMDD phenotypes in GS:SFHS at all thresholds, however fMDD did not survive multiple testing correction at any PT. PGS derived using information from the GS:SFHS MDD GWAS yielded significant associations with MDD, rMDD, and fMDD phenotypes in UKB at all thresholds, except PT ≤ 0.01—presumably due to the low number of SNPs contributing to the score. However, mMDD did not survive multiple testing correction at any PT. Across all associations, the largest proportion of variance explained in GS:SFHS was 0.66% for fMDD using the MDD polygenic score derived using SNPs at PT ≤ 0.5 using weights from the PGC MDD29 GWAS. The largest proportion of variance explained in UKB was 0.72% for fMDD, again using SNPs at PT ≤ 0.5 using weights from the PGC MDD29 GWAS.
Fig. 4.
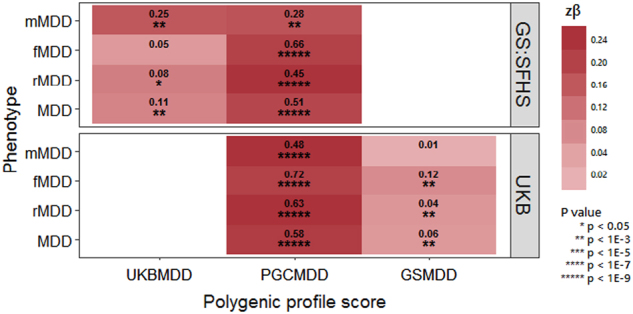
Heat map of associations between the polygenic profiles scores (PGS) for major depressive disorder (MDD), derived from Psychiatric Genomics Consortium (PGC) MDD29, UK Biobank (UKB) and Generation Scotland: The Scottish Family Health Study (GS:SFHS), and MDD subsets in UKB and GS:SFHS. Stronger associations are indicated by darker shades. The amount of variance (%) explained by PGS is indicated for each association. Further information can be found in Supplementary Tables 15-18
Discussion
For many years, the depressed phenotype has been refractory to genetic inquiry due to issues regarding statistical power. Recently, studies have successfully identified loci associated with depression by either substantially increasing the sample size7,8 or by refining the phenotype by illness course9, recurrence and sex6. In this study we used techniques designed to interrogate complex traits to ascertain whether maximizing the sample size (nmax = 43,062) or phenotypic stratification by recurrence or sex was more advantageous for investigating the genetic architecture of MDD, using data from two large UK cohorts. Each MDD definition was evaluated using several metrics: the successful identification of variants reaching genome-wide significance in GWAS meta-analysis, an increased SNP-based heritability estimate, identification of significant genetic correlations with other traits using LD score regression, and increased variance explained by polygenic profile scores for MDD derived from three independent cohorts.
For all analyses, MDD, recurrent MDD and MDD in females returned similar results: overlapping SNP-based heritability estimates; genetic correlations with consistent magnitude of effect, direction, and significance with six health-related traits; low trait variance explained (<1%) and overlapping effect size estimates in polygenic profiling analysis, and no genome-wide significant findings from GWAS meta-analysis (akin to similarly sized published GWAS of these phenotypes2,9).
With the exception of polygenic profiling analysis, MDD in males generally did not conform to the pattern of results demonstrated by other MDD definitions. Some of these differences, such near-zero SNP-based heritability estimate and subsequently no genetic correlations with other traits surviving multiple testing correction, can be attributed to reduced statistical power in this MDD definition. Interestingly, genome-wide meta-analysis yielded a single genome-wide significant locus in depression in males. The locus at chromosome 3p22.3 includes TMPPE, CRTAP, and GLB1 genes; all of which were significant in gene-based testing. Conditional GWAS on the lead signal demonstrated that the signal which spanned these three genes was due to high LD in the region. However, functional genomic analysis of the lead SNP returned eQTL evidence for GLB1 and CRTAP, suggesting that the causal variant is more likely to affect the expression of these genes rather than TMPPE. The lack of replication of this signal in other published GWAS is unfortunate, but unsurprising given that the current study is the largest GWAS of depression in males to date (relative to published GWAS). Ever-increasing sample sizes from international consortia will provide much-needed larger replication datasets for corroborating or dispelling this finding. The lack of replication also highlights the importance of moving towards linking results at the functional level.
There are several limitations to this study. Firstly, the sample size for MDD and rMDD groups are very similar (n = 43,062 and n = 39,556, respectively), therefore perhaps rMDD is not the best stratifier in this sample. The sample size of all depressed cases could have been increased by including individuals with mild depressive/manic symptoms (n = 26,847), however as case classification was based on very few items (two symptoms and help-seeking behavior), it wasn’t possible to determine whether mild symptoms should be classified as cases or controls17, therefore including these individuals could introduce further phenotypic heterogeneity. Whilst the differential model selection in GS:SFHS and UKB used to adequately account for differential family and population structure introduces analytical heterogeneity, the genetic correlation of MDD GWAS summary statistics from the two samples was rG(SE) = 0.997(0.26). Lastly, the higher prevalence of females in GS:SFHS caused a gender imbalance in the sample sizes, resulting in lower statistical power for the male only analysis.
Overall our results suggest that there was little benefit to stratifying depression by either sex or recurrence for currently available data sizes. Extreme differences between sexes, such as opposite directions of effect in the two sexes, would have to exist to necessitate their analysis separately. The power implications of stratifying on these traits is likely to out-weigh the identification of such loci. In situations where the effect of a SNP is only found in one sex and zero effect in the other, such as rs4778037 in this study, it is still better to analyze sexes together to reduce the multiple testing burden of separate analyses. The increased trait variance explained demonstrated by using the largest available training and discovery datasets (PGC MDD29 and UKB MDD, respectively) in polygenic profiling supports increasing the sample size over phenotypic refinement. Similarly, the lack of discernible difference between h2SNP and rG estimates between depression subtypes suggests that the best approach, currently, is to maximize the sample size in order to reduce sampling error and obtain more accurate point estimates. However, addressing recurrence, sex and ancestral heterogeneity in a large ascertained cohort does have intrinsic merit, as demonstrated previously by CONVERGE6, with the implication that addressing several sources of heterogeneity has more utility than implementing recurrence and sex separately6.
Phenotypic stratification still has plenty of scope for aiding the tractability of genetic analysis in depression. There are many traits which could be used to create subgroups, including treatment response and physical comorbidities, and perhaps these will be more successful than sex and recurrence. However, it is worth noting that due to the limitations of statistical power with current sample sizes, the performance of the stratified phenotypes presented here is a lower bound of the stratification strategy. Recently, age-at onset9 and use of polygenic risk scores derived from health-related traits73 have been shown to result in subsets of depression with improved heritability. Our study suggests that, until a better understanding of the determinants of genetic heterogeneity in depression exist—increasing sample number remains the optimal strategy.
Electronic supplementary material
Acknowledgements
Generation Scotland received core funding from the Chief Scientist Office of the Scottish Government Health Directorate CZD/16/6 and the Scottish Funding Council HR03006. Genotyping of the GS:SFHS samples was carried out by the Genetics Core Laboratory at the Wellcome Trust Clinical Research Facility, Edinburgh, Scotland and was funded by the UK’s Medical Research Council and the Wellcome Trust (Wellcome Trust Strategic Award “STratifying Resilience and Depression Longitudinally” (STRADL) (Reference 104036/Z/14/Z). YZ acknowledges support from China Scholarship Council. IJD is supported by the Centre for Cognitive Ageing and Cognitive Epidemiology which is funded by the Medical Research Council and the Biotechnology and Biological Sciences Research Council (MR/K026992/1). AMMcI and T-KC acknowledges support from the Dr Mortimer and Theresa Sackler Foundation. This work has made use of the resources provided by the Edinburgh Compute and Data Facility (ECDF). (http://www.ecdf.ed.ac.uk/).
We are grateful to all the families who took part, the general practitioners and the Scottish School of Primary Care for their help in recruiting them, and the whole Generation Scotland team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, healthcare assistants and nurses. Ethics approval for the study was given by the NHS Tayside committee on research ethics (reference 05/S1401/8). This research has been conducted using the UK Biobank Resource—application number 4844.
Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium
For a full list of MDD working group of the PGC investigators, see the Supplementary Material
Competing interests
The authors declare that they have no competing financial interests.
Footnotes
Members of Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium are listed above the References
For a full list of MDD working group of the PGC investigators, see the Supplementary Material
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41398-017-0034-1.
References
- 1.Flint J, Kendler KS. The genetics of major depression. Neuron. 2014;81:484–503. doi: 10.1016/j.neuron.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Major Depressive Disorder Working Group of the Psychiatric GC. et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry. 2013;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ripke S, et al. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011;43:969–U77. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sklar P, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 2011;43:977–U162. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wray NR, Maier R. Genetic Basis of Complex Genetic Disease: The Contribution of Disease Heterogeneity to Missing Heritability. Curr. Epidemiol. Rep. 2014;1:220–227. doi: 10.1007/s40471-014-0023-3. [DOI] [Google Scholar]
- 6.CONVERGE consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–591. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hek K, et al. A genome-wide association study of depressive symptoms. Biol. Psychiatry. 2013;73:667–678. doi: 10.1016/j.biopsych.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyde CL, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016;48:1031. doi: 10.1038/ng.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Power RA, et al. Genome-wide association for major depression through age at onset stratification. The Psychiatric Genomics Consortium Major Depressive Disorder Working Group. Biol. Psychiatry. 2017;81:325–335. doi: 10.1016/j.biopsych.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng Y, et al. Shared genetics and couple-associated environment are major contributors to the risk of both clinical and self-declared depression. EBioMedicine. 2016;14:161–167. doi: 10.1016/j.ebiom.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith BH, et al. Cohort Profile: Generation Scotland: Scottish Family Health Study (GS:SFHS). The study, its participants and their potential for genetic research on health and illness. Int. J. Epidemiol. 2013;42:689–700. doi: 10.1093/ije/dys084. [DOI] [PubMed] [Google Scholar]
- 12.Smith BH, et al. Generation Scotland: the Scottish Family Health Study; a new resource for researching genes and heritability. BMC Med. Genet. 2006;7:74. doi: 10.1186/1471-2350-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen NE, Sudlow C, Peakman T, Collins R, Biobank UK. UK biobank data: come and get it. Sci. Transl. Med. 2014;6:224ed4. doi: 10.1126/scitranslmed.3008601. [DOI] [PubMed] [Google Scholar]
- 14.Sudlow C, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.First M. B., Spitzer R. L., Gibbon M., Williams J. B. Structured Clinical Interview for DSM-IV Axis I Disorders, Clinician Version (SCID-CV). Washington, DC, USA: American Psychiatric Publishing, Inc. 1997.
- 16.UK Biobank. Touchscreen questionnaire 2012. http://www.ukbiobank.ac.uk/wp-content/uploads/2011/06/Touch_screen_questionnaire.pdf?phpMyAdmin=trmKQlYdjjnQIgJ%2CfAzikMhEnx6.
- 17.Smith DJ, et al. Prevalence and Characteristics of Probable Major Depression and Bipolar Disorder within UK Biobank: Cross-Sectional Study of 172,751 Participants. PLoS One. 2013;8:e75362. doi: 10.1371/journal.pone.0075362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manichaikul A, et al. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCarthy S, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat. Methods. 2012;9:179–181. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 21.Durbin R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT) Bioinformatics. 2014;30:1266–1272. doi: 10.1093/bioinformatics/btu014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagy R, et al. Exploration of haplotype research consortium imputation for genome-wide association studies in 20,032 Generation Scotland participants. Genome Med. 2017;9:23. doi: 10.1186/s13073-017-0414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amador C, et al. Recent genomic heritage in Scotland. BMC Genomics. 2015;6:437. doi: 10.1186/s12864-015-1605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ripke S. GWAS genotypic overlap test without sharing genotypes 2015. https://personal.broadinstitute.org/sripke/share_links/checksums_download/readme_0415b.txt.
- 25.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell S, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.UK Biobank. Genotype imputation and genetic association studies using UK Biobank data 2015. http://biobank.ctsu.ox.ac.uk/crystal/refer.cgi?id=157020.
- 28.O’Connell J, et al. Haplotype estimation for biobank-scale data sets. Nat. Genet. 2016;48:817–820. doi: 10.1038/ng.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J, et al. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nat. Commun. 2015;6:8111. doi: 10.1038/ncomms9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howie B, Marchini J, Stephens M. Genotype Imputation with Thousands of Genomes. G3-Genes. Genom. Genet. 2011;1:457–469. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biobank UK. UK Biobank Phasing and Imputation Documentation (2015) https://biobank.ctsu.ox.ac.uk/crystal/docs/impute_ukb_v1.pdf.
- 32.UK Biobank. Genotyping and quality control for UK Biobank (2015) http://biobank.ctsu.ox.ac.uk/crystal/refer.cgi?id=155580.
- 33.Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 2014;46:100–106. doi: 10.1038/ng.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaitlen N, et al. Using Extended Genealogy to Estimate Components of Heritability for 23 Quantitative and Dichotomous Traits. PLoS Genet. 2013;9:e1003520. doi: 10.1371/journal.pgen.1003520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visscher PM, et al. Statistical Power to Detect Genetic (Co)Variance of Complex Traits Using SNP Data in Unrelated Samples. PLoS Genet. 2014;10:e1004269. doi: 10.1371/journal.pgen.1004269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cortes A, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. 2013;45:730. doi: 10.1038/ng.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacArthur J, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids. Res. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids. Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS. Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.1000 Genomes Project Consortium. et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pers TH, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 2015;6:5890. doi: 10.1038/ncomms6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boyle AP, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia C, et al. Pedigree- and SNP-Associated Genetics and Recent Environment are the Major Contributors to Anthropometric and Cardiometabolic Trait Variation. PLoS. Genet. 2016;12:e1005804. doi: 10.1371/journal.pgen.1005804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altamura AC, et al. Lifetime prevalence of brief recurrent depression (results from a community survey) Eur. Neuropsychopharmacol. 1995;5:99–102. doi: 10.1016/0924-977X(95)00037-P. [DOI] [PubMed] [Google Scholar]
- 49.Kessler RC, et al. The epidemiology of major depressive disorder - Results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 50.Weissman MM, et al. Cross-national epidemiology of major depression and bipolar disorder. JAMA. 1996;276:293–299. doi: 10.1001/jama.1996.03540040037030. [DOI] [PubMed] [Google Scholar]
- 51.Bulik-Sullivan B, et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015;47:1236. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng J, et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2016;33:272–279. doi: 10.1093/bioinformatics/btw613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. Roy. Stat. Soc. B Met. 1995;57:289–300. [Google Scholar]
- 54.MDD Working Group of the PGC. Identifying the genetic contribution to depression. BIPOLAR DISORDERS: WILEY-BLACKWELL 111 RIVER ST, HOBOKEN 07030-5774, NJ USA, p 36. 2016.
- 55.Euesden J, Lewis CM, O’Reilly PF. PRSice: Polygenic Risk Score software. Bioinformatics. 2015;31:1466–1468. doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Visscher PM. A note on the asymptotic distribution of likelihood ratio tests to test variance components. Twin. Res. Hum. Genet. 2006;9:490–495. doi: 10.1375/twin.9.4.490. [DOI] [PubMed] [Google Scholar]
- 57.Kenward MG, Roger JH. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics. 1997;53:983–997. doi: 10.2307/2533558. [DOI] [PubMed] [Google Scholar]
- 58.Butler, D., Cullis, B., Gilmore, A., & Gogel, B. ASReml-R reference manual, release 3. Tech report, Dep PrimInd Queensl. 2007, https://www.vsni.co.uk/downloads/asreml/release3/asreml-R.pdf.
- 59.Nakagawa S, Schielzeth H. A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol. Evol. 2013;4:133–142. doi: 10.1111/j.2041-210x.2012.00261.x. [DOI] [Google Scholar]
- 60.Hirota T, et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat. Genet. 2012;44:1222–1226. doi: 10.1038/ng.2438. [DOI] [PubMed] [Google Scholar]
- 61.Gennarini G. et al. The role of Gpi-anchored axonal glycoproteins in neural development and neurological disorders. Mol. Cell Neurosci. 2016. [DOI] [PubMed]
- 62.Rojas PS, Fiedler JL. What Do We Really Know About 5-HT1A Receptor Signaling in Neuronal Cells? Front Cell Neurosci. 2016;10:272. doi: 10.3389/fncel.2016.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fanous A, Gardner CO, Prescott CA, Cancro R, Kendler KS. Neuroticism, major depression and gender: a population-based twin study. Psychol. Med. 2002;32:719–728. doi: 10.1017/S003329170200541X. [DOI] [PubMed] [Google Scholar]
- 64.Hettema JM, Prescott CA, Kendler KS. Genetic and environmental sources of covariation between generalized anxiety disorder and neuroticism. Am. J. Psychiatry. 2004;161:1581–1587. doi: 10.1176/appi.ajp.161.9.1581. [DOI] [PubMed] [Google Scholar]
- 65.Jardine R, Martin NG, Henderson AS. Genetic covariation between neuroticism and the symptoms of anxiety and depression. Genet. Epidemiol. 1984;1:89–107. doi: 10.1002/gepi.1370010202. [DOI] [PubMed] [Google Scholar]
- 66.Lee SH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013;45:984. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foley DL, Neale MC, Kendler KS. Genetic and environmental risk factors for depression assessed by subject-rated symptom check list versus structured clinical interview. Psychol. Med. 2001;31:1413–1423. doi: 10.1017/S0033291701004755. [DOI] [PubMed] [Google Scholar]
- 68.Okbay A, et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 2016;48:624–633. doi: 10.1038/ng.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mirowsky J, Ross CE. Depression, parenthood, and age at first birth. Soc. Sci. Med. 2002;54:1281–1298. doi: 10.1016/S0277-9536(01)00096-X. [DOI] [PubMed] [Google Scholar]
- 70.Palagini L, et al. Depression and systemic lupus erythematosus: a systematic review. Lupus. 2013;22:409–416. doi: 10.1177/0961203313477227. [DOI] [PubMed] [Google Scholar]
- 71.Zhang L, Fu T, Yin R, Zhang Q, Shen B. Prevalence of depression and anxiety in systemic lupus erythematosus: a systematic review and meta-analysis. BMC Psychiatry. 2017;17:70. doi: 10.1186/s12888-017-1234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bulik-Sullivan BK. LD Score documentation. https://github.com/bulik/ldsc/wiki/Heritability-and-Genetic-Correlation.
- 73.Milaneschi Y, et al. Polygenic dissection of major depression clinical heterogeneity. Mol. Psychiatry. 2016;21:516–522. doi: 10.1038/mp.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available to qualified researchers on a cost-recovery basis via online application processes, accessible via www.gsaccess.org and www.ukbiobank.ac.uk/register-apply/. The code used in these analyses is available on request from the lead author.