

Abstract
Breast cancer is the most commonly diagnosed cancer in women. There is a continued interest for the development of more efficacious treatment regimens for breast carcinoma. Recombinant human tumor necrosis factor–related apoptosis-inducing ligand (rhTRAIL) shows potential as a potent anticancer therapeutic for the treatment of breast cancer, whereas displaying minimal toxicity to normal cells. However, the promise of rhTRAIL for the treatment of breast cancer is dismissed by the resistance to rhTRAIL-induced apoptosis exhibited by many breast cancers. Thus, a cotreatment strategy was examined by applying the natural compound quercetin (Q) as a sensitizing agent for rhTRAIL-resistant breast cancer BT-20 and MCF-7 cells. Quercetin was able to sensitize rhTRAIL-resistant breast cancers to rhTRAIL-induced apoptosis as detected by Western blotting through the proteasome-mediated degradation of c-FLIPL and through the upregulation of DR5 expression transcriptionally. Overall, these in vitro findings establish that Q is an effective sensitizing agent for rhTRAIL-resistant breast cancers.
Keywords: Apoptosis, breast cancer, c-FLIP, DR5, quercetin, rhTRAIL
Introduction
Breast cancer is the most commonly diagnosed cancer in women.1–4 The development of more effective treatment regimens against the different forms of breast carcinoma is being explored.2,4,5 Patients with hormone-dependent and human epidermal growth factor receptor 2 (HER2) overexpression breast cancers often have a better prognosis because of the availability of hormone-targeted therapies.2,5,6 The triple-negative breast cancers (TNBCs) are more challenging to treat because there is no specific hormone to target; hence, TNBC is the deadliest form of breast cancer.2,4
One promising anticancer therapeutic of interest is recombinant human tumor necrosis factor–related apoptosis-inducing ligand (rhTRAIL)—the optimized form of the endogenous death ligand TRAIL (tumor necrosis factor–related apoptosis-inducing ligand). Recombinant human tumor necrosis factor–related apoptosis-inducing ligand consists of the extracellular C-terminus of TRAIL amino acids 114-281 lacking exogenous sequence tags.7–11 Recombinant human tumor necrosis factor–related apoptosis-inducing ligand has shown great potential as an effective anticancer therapeutic due to its ability to induce apoptosis in cancer cells, whereas exhibiting minimal toxicity to normal, nontransformed cells.7–11 Recombinant human tumor necrosis factor–related apoptosis-inducing ligand initiates the extrinsic pathway of apoptosis by binding to the extracellular death receptors (DRs) DR4 and DR5 leading to trimerization of the receptors followed by the activation of caspase 8; the subsequent activation of the executioner caspases 3, 6, and 7; and the eventual cleavage of poly (adenosine diphosphate-ribose) polymerase or PARP (hallmark of apoptosis).12–16 In addition, rhTRAIL can activate the intrinsic pathway of apoptosis independently of p53 through the caspase 8–mediated cleavage of Bid to truncated Bid (tBid) facilitating the release of cytochrome c from the mitochondria followed by the activation of caspase 9 and the subsequent activation of the executioner caspases.13–15,17–19 Despite this, most of the breast cancer cells exhibit resistance to TRAIL treatment due to the upregulation of antiapoptotic proteins such as cellular FLICE-like inhibitory protein (c-FLIP) and the downregulation of DR5. Clinical trials have been completed with TRAIL, but further trials have since been terminated due to a limited therapeutic efficacy.7,20–22 Consequently, many studies have focused on determining sensitizing agents that have the capability to overcome rhTRAIL resistance.
One potential sensitizing agent is Quercetin (Q); it is a naturally occurring flavonol found in different vegetables, fruits, tea, red wine, and coffee.23–25 Quercetin has been shown to produce antiproliferative and proapoptotic effects in cancer cells such as prostate, cervical, lung, breast, and colon.23,24,26–30 Quercetin can induce apoptosis in some cancer cell lines through the downregulation of antiapoptotic proteins, survivin, Bcl-xL, and Bcl-2, and through the upregulation of proapoptotic proteins, Bad and Bax.29,31–33 Investigations involving human hepatoma and prostate cancer cells have demonstrated that Q can enhance TRAIL-induced apoptosis through the upregulation of DR5.28,34,35 In addition, nontumorigenic breast epithelial MCF-10A cells were not affected by Q treatment after 24, 48, and 72 hours supporting Q’s safety for systemic application.29,36 Furthermore, no major cytotoxic effects have been observed in different in vivo studies, and clinical trials have administered Q with no major cytotoxic effects cited.24,31,37–39 Therefore, these findings suggest that Q has the potential to be an effective sensitizing agent.
The intention of this study was to investigate the capability of Q to sensitize rhTRAIL-resistant TNBC BT-20 cells and hormone-dependent breast cancer MCF-7 cells and to elucidate the underlying mechanism for Q’s sensitization. Our study demonstrates that Q has the ability to induce the proteasome-mediated degradation of c-FLIPL and to induce the upregulation of DR5 facilitating the execution of the extrinsic pathway and thereby sensitizing breast cancers to rhTRAIL-induced apoptosis. Thus, the presented evidence reveals that Q is a good sensitizing agent for rhTRAIL-resistant breast cancers.
Methods
Drugs and chemicals
Recombinant human tumor necrosis factor–related apoptosis-inducing ligand was produced according to well-defined and previously detailed protocols.8–10 Recombinant human tumor necrosis factor–related apoptosis-inducing ligand was aliquoted and stored at −80°C. Quercetin dihydrate (lot no. D00166146, molecular weight of 338.3 g/mol; Calbiochem, San Diego, CA, USA) was dissolved in 7.5 mg/mL of polyethylene glycol (molecular weight of 400 g/mol; Fisher Scientific, Hampton, NH, USA) and then filtered, aliquoted, and stored at −20°C.39 MG132 proteasome inhibitor (molecular weight of 457.6 g/mol; Calbiochem) was dissolved in dimethyl sulfoxide to produce a 10 mM stock that was filtered, aliquoted, and stored at −20°C.
Cell culture
Human breast cancer MCF-7 (ATCC HTB-22) and BT-20 (ATCC HTB-19) cells were cultured in Dulbecco’s Modified Eagle Medium (Cleveland Clinic Cell Services Media Core, Cleveland, OH, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Gaithersburg, MD, USA), 1% antibiotics-antimycotics (Gibco), 1% l-glutamine (Gibco), 1% nonessential amino acids (Gibco), and 1% sodium pyruvate (Gibco). Human nontumorigenic breast epithelial MCF-10A (ATCC CRL-10317) cells were cultured in HuMEC Ready Medium (Gibco). Cells were maintained in a humidified atmosphere with 5% CO2 at 37°C. Cells were treated with drugs 24 hours after plating, incubated with drugs for an additional 72 hours, and collected for the different assays described below.36,40
Annexin V/propidium iodide assays-flow cytometry
Cells were typsinized, spun at 1000 rpm for 3 minutes, and washed with phosphate-buffered saline (PBS). Cells were incubated with Annexin V-FITC and propidium iodide (PI) solution (Annexin V: FITC Apoptosis Detection Kit I; BD Life Sciences, Sparks, MD, USA) for 15 minutes at room temperature in the dark. Apoptosis was detected using the BD FACSCanto II and applying FACSDiva software and Flowing Software 2. Each experiment was performed in triplicate, and 3 independent experiments were conducted for each cell line to obtain the mean percent of apoptotic cells ± SEM.
Determination of apoptotic-associated protein levels by Western blotting
Cells were collected and washed with PBS. Total cell lysates were prepared by lysing with radioimmunoprecipitation assay (RIPA) buffer (AMRESCO, Dallas, TX, USA) and a protease inhibitor cocktail (Calbiochem). The lysates were placed on ice for 30 minutes, were spun at 10 000 rpm for 10 minutes, and were quantified by applying the Pierce BCA Protein Assay (Thermo Fisher Scientific, Waltham, MA, USA). Aliquots of 35 μg of protein were prepared, denatured with 4× Laemmli sample buffer (250 mM Tris-HCl, 8% sodium dodecyl sulfate [SDS], 40% glycerol, 8% β-mercaptoethanol, and 0.02% bromophenol blue), and separated on 12% SDS-polyacrylamide gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Temecula, CA, USA) employing the semidry transfer method (Bio-Rad, Richmond, CA, USA). Each membrane was blocked with 5% nonfat milk at room temperature for 1 hour and incubated overnight at 4°C with a diluted primary antibody: anti-PARP, anti-caspase 3, anticleaved caspase 3, anti-caspase 7, anti-caspase 8, anticleaved caspase 8, anti-Bid (Cell Signaling Technology, Danvers, MD, USA), or anti-FLIP (Enzo Life Sciences, Farmingdale, NY, USA). The membranes were washed in Tris-buffered saline (TBS)-Tween (0.15 M NaCl, 0.02 M Tris, and 0.3% Tween 20 with a pH of 7.4), incubated with a horse radish peroxidase–conjugated secondary antibody either goat anti-mouse IgG or goat anti-rabbit IgG (Bio-Rad) diluted in 5% nonfat milk for 1 hour at room temperature, and washed in TBS-Tween. The membranes were then exposed on HyBlot CL Autoradiography Film (Denville Scientific, Holliston, MA, USA); the protein bands were detected using chemiluminescence via Pierce ECL2 Western Blotting Substrate (Thermo Fisher Scientific). The membranes were then probed for β-actin (Promega, Madison, WI, USA) as the internal loading control.
Determination of cytochrome c release
Cells were collected, washed, and resuspended in permeabilization buffer (400 µg/mL digitonin, 75 mM KCl, 1 mM NaH2PO4, 8 mM Na2HPO4, 250 mM sucrose) with a protease inhibitor cocktail. All of the samples were kept on ice for 10 minutes and spun at 16 000g for 5 minutes at 4°C; the supernatants were kept as the cytosolic fractions. Protein quantification was executed as described above, and aliquots of 60 µg of protein were prepared, denatured, and separated on 15% SDS-polyacrylamide gels. Proteins were transferred to PVDF membranes and blocked as above. The membranes was incubated overnight at 4°C with anti-cytochrome c (Cell Signaling Technology) diluted in 5% nonfat milk. The membrane was washed and developed as above.
Western blot analysis of DR4 and DR5 expression
Cells were collected with cell dissociation buffer (Gibco) and lysed with RIPA buffer as described above. After the membranes were blocked, the membranes were incubated overnight at 4°C with anti-DR4 (Imgenex, Littleton, CO) in 5% nonfat milk or anti-DR5 (Cell Signaling Technology) in 5% bovine serum albumin (Fisher Scientific). The membranes were washed and developed as described above. Densitometry was calculated from ImageJ software.
Flow cytometry analysis of DR4 and DR5 expression
Cells were collected with cell dissociation buffer and spun at 1000 rpm for 3 minutes. Cells were resuspended in staining buffer (2% FBS, 0.02% sodium azide, and PBS) and incubated with anti-DR4-PE or anti-DR5-PE (eBioscience, San Diego, CA, USA) for 1 hour in the dark at 4°C; a mouse IgG1 K isotype control (eBioscience) was used to compensate for any nonspecific binding. Cells were washed twice with staining buffer and resuspended in staining buffer for analysis. DR4 and DR5 membrane expressions were analyzed on a BD FACSCanto II flow cytometer using FACSDiva software. Histograms were prepared employing Flowing Software 2. Each experiment was performed in triplicate, and 3 independent experiments were conducted for each cell line to obtain the fold increase in DR4 or DR5 cell surface expression relative to the vehicle-treated control ± SEM.
Reverse transcription-polymerase chain reaction analysis for DR5 and c-FLIPL
Total RNA was extracted from cells using TRIzol Reagent (Ambion, Pittsburgh, PA, USA). Reverse transcription-polymerase chain reaction (RT-PCR) was performed following the manufacturer’s protocol (Invitrogen SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase; Thermo Fisher Scientific). Human DR5 messenger RNA (mRNA) was amplified using the forward primer 5′-GGGAGCCGCT-CATGAGGAAGTTGG-3′ and the reverse primer 5′-GG-CAAGTCTCTCTCCCAGCGTCTC-3′ (182-bp [base pairs] product). For c-FLIPL, forward primer 5′-CTTGGCC-AATTTGCCTGTAT-3′ and the reverse primer 5′-CCCATGAACATCCTCCTGAT-3′ were used (149-bp product). For β-actin, the forward primer 5′-TGACGGGGTCACCCACA-CTGTGCC-3′ and the reverse primer 5′-CTGCATCCT-GTCGGCAATGCCAG-3′ were used (570-bp product). Complementary DNA synthesis was performed at 60°C for 30 minutes using the Applied Biosystems GeneAmp PCR System 9700. The PCR cycling conditions (40 cycles) were chosen as follows: denature for 2 minutes at 94°C, anneal for 30 seconds at 55°C for c-FLIPL and 65°C for DR5 and β-actin, extend for 1 minute and 30 seconds at 68°C, and execute a final extension for 10 minutes at 68°C. Reaction products were analyzed on 1.2% agarose gels. The bands were visualized by ethidium bromide (Invitrogen, Carlsbad, CA, USA) and a UV illuminator (UVP, Upland, CA, USA).
Examining posttranslational effects of Q
Cells were treated with 0.25 µM MG132 alone and in combination with 50 µM Q along with a vehicle-treated control. Cells were collected, washed, lysed, and quantified as above. Western blot analysis was performed as above probing for c-FLIP.
Co-immunoprecipitation
Columns were prepared according to the manufacturer’s instructions (Pierce Co-IP Kit) with 5 µg of anti-c-FLIP. Cells were collected, washed, lysed, and quantified. Co-immunoprecipitation (Co-IP) was preformed overnight at 4°C with 500 µg of precleared lysate. Proteins were eluted according to the manufacturer’s instructions and analyzed by Western blotting probing for ubiquitin (Cell Signaling Technology) and c-FLIP on 15% SDS-polyacrylamide gels.
Statistical analysis
Data were analyzed using Student t test and analysis of variance, and the differences between experimental and control groups were considered statistically significant at P values less than .05.
Results
Fluorescence-activated cell sorting analysis of rhTRAIL-induced apoptosis
Fluorescence-activated cell sorting (FACS) analysis was completed on breast cancer cells treated with increasing concentrations of Q (12.5, 25, and 50 µM) in the presence or absence of 100 ng/mL rhTRAIL to ascertain Q’s sensitizing effects on rhTRAIL-induced apoptosis (Figure 1). Quercetin enhanced rhTRAIL-induced apoptosis in both breast cancer cell lines. However, for breast cancer MCF-7 cells, Q did not have a considerable impact on promoting rhTRAIL-induced apoptosis when compared with Q-mediated rhTRAIL-induced apoptosis in breast cancer BT-20 cells. For example, breast cancer MCF-7 and BT-20 cells treated with 50 µM Q produced on average about 15% and 20% apoptotic cells, respectively (P < .05), whereas the cotreatment of 50 µM Q and 100 ng/mL rhTRAIL on MCF-7 and BT-20 cells produced on average about 25% and 45% apoptotic cells, respectively (P < .05). It should be noted that 100 ng/mL rhTRAIL alone did not produce a significant amount of apoptotic breast cancer cells when compared with the vehicle-treated control.
Figure 1.

Q enhances rhTRAIL-induced apoptosis in breast cancer cells. Cells were treated with increasing concentrations of Q in the presence and absence of 100 ng/mL rhTRAIL for 72 hours. (A) For the representative FACS plots, the left top quadrants represent Annexin V−/PI+ dead cells, the right top quadrants represent Annexin V+/PI+ late apoptotic dead cells, the left bottom quadrants represent Annexin V−/PI− viable cells, and the right bottom quadrants represent Annexin V+/PI− early apoptotic cells. Bar graphs (B) BT-20 and (C) MCF-7 display the average total percent of apoptotic cells (Annexin V+ cells) ± SEM, and the average was calculated from 3 independent experiments performed in triplicate (n = 9). P < .05 except *. PI indicates propidium iodide; Q, quercetin; rhTRAIL, recombinant human tumor necrosis factor–related apoptosis-inducing ligand.
Detection of the pathway of apoptosis
Breast cancer cells were treated the same as the FACS analysis. The protein levels of caspase 8, cytosolic cytochrome c, caspase 3 (only for BT-20 cells because MCF-7 cells lack procaspase 3 expression), caspase 7, and cleaved PARP were all upregulated with the cotreatment of Q and rhTRAIL, whereas Bid expression was downregulated when compared with the vehicle-treated controls and single-agent treatments in both breast cancer cell lines (Figure 2). Densitometry results are illustrated in Figure 3. These results indicate that the extrinsic pathway was induced as marked by caspase 8 activation, executioner caspase 3 and/or caspase 7 activation, and PARP cleavage. In addition, Q alone did exhibit minimal proapoptotic effects on breast cancer cells as illustrated by PARP cleavage. Furthermore, cytochrome c was released from the mitochondria in both breast cancer cells treated with 50 μM Q demonstrating the initiation of the intrinsic pathway of apoptosis. Also, it should be noted that PARP fragmentation was not observed with single-agent rhTRAIL treatment at 100 ng/mL for both breast cancer cell lines supporting data gathered from other researchers that breast cancer BT-20 and MCF-7 cells are TRAIL resistant.41 In addition, TRAIL did not induce apoptosis in MCF-10A cells.42 These data together with the data from FACS shown in Figure 1 strongly suggest that Q sensitized breast cancer cells to rhTRAIL-induced apoptosis through the induction of the extrinsic pathway of apoptosis.
Figure 2.

Q and rhTRAIL induce the extrinsic pathway of apoptosis. Both breast cancer cell lines (A) BT-20 and (B) MCF-7 were sensitized to rhTRAIL-induced apoptosis via Q cotreatment as marked by the activation of caspase 8, the activation of executioner caspases 3 and 7, and the cleavage of PARP. β-actin was used as loading control and probed for each blot; the β-actins shown are representative results for each cell line. PARP indicates poly (adenosine diphosphate-ribose) polymerase; PI, propidium iodide; Q, quercetin; rhTRAIL, recombinant human tumor necrosis factor–related apoptosis-inducing ligand.
Figure 3.

Densitometry results for apoptotic-associated proteins. Densitometry results for immunoblots were calculated for breast cancer (A) BT-20 and (B) MCF-7 cells using ImageJ software and graphs were generated applying GraphPad Prism. PARP indicates poly (adenosine diphosphate-ribose) polymerase.
Synergism of Q and rhTRAIL
The Chou-Talalay method was applied to calculate the combination index (CI) for the cotreatment of Q and rhTRAIL in both breast cancer cell lines (Figure 4). The CIs for both breast cancer cell lines were less than 1 indicating that a synergic effect was observed and not an additive effect (CI = 1) or an antagonistic effect (CI > 1).
Figure 4.
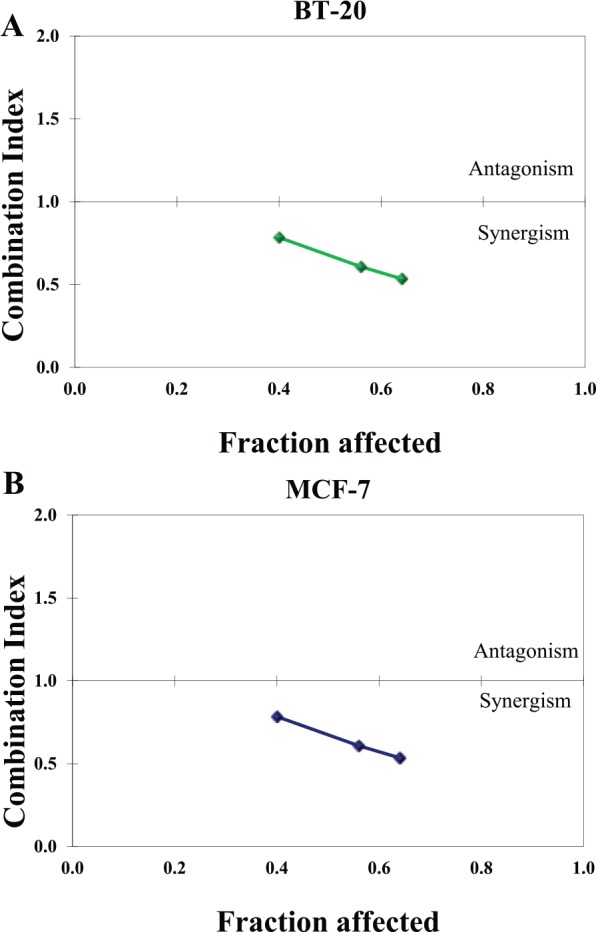
Combination indexes (CIs) for the cotreatment of Q and rhTRAIL indicate synergism. Combination indexes were calculated and the cotreatment of Q and rhTRAIL displayed synergic effect (all CIs < 1.0) in both breast cancer cell lines (A) BT-20 and (B) MCF-7. Q indicates quercetin; rhTRAIL, recombinant human tumor necrosis factor–related apoptosis-inducing ligand.
Q induces the proteasome-mediated degradation of c-FLIPL
Considering that Q was shown to sensitize breast cancer to rhTRAIL-induced apoptosis through the extrinsic pathway by enhanced activation of caspase 8, Q’s impact on the expression of the caspase 8 inhibitor c-FLIP was assessed. Results gathered from Western blot analysis demonstrate that Q downregulated the expression of the long form of c-FLIP (c-FLIPL) in breast cancer in a dose-dependent manner after 72 hours (Figure 5A). Preliminary assays were performed with 24-, 48-, and 72-hour Q-treated breast cancer cells, and Q’s impact on c-FLIPL was not displayed until after 72 hours. At the highest concentration of Q used in our study (50 µM), expression of c-FLIPL was downregulated by about 9-fold and 3-fold in comparison with the vehicle-treated control for breast cancer BT-20 and MCF-7 cells, respectively. To observe whether Q-induced downregulation occurred through the proteasome-mediated degradation of c-FLIPL, a proteasome inhibitor MG132 was used. MG132 was used at a low concentration of 0.25 µM in presence and absence of 50 µM Q along with a vehicle-treated control. Inhibition of the proteasome resulted in the prevention of c-FLIPL downregulation through Q treatment. In addition, c-FLIPL expression did not differ between the vehicle-treated control and the breast cancer cells treated with only the proteasome inhibitor MG132 (Figure 5B). Furthermore, Co-IP was performed probing for ubiquitin demonstrating that Q promoted the ubiquitination of c-FLIPL in breast cancer (Figure 5C and D). Hence, for breast cancer, Q enhances rhTRAIL-induced apoptosis through the proteasome-mediated degradation of c-FLIPL via increased ubiquitination.
Figure 5.

Q promotes the proteasome-mediated degradation of c-FLIPL. Western blotting revealed that Q decreases c-FLIPL expression in (A) BT-20 and MCF-7 cells in a dose-dependent manner. (B) BT-20 and MCF-7 cells were treated with a proteasome inhibitor MG132 alone and in combination with Q for 72 hours, and the cotreatment of MG132 and Q recovered c-FLIPL protein expression in breast cancer. Co-IP was performed on (C) BT-20 and (D) MCF-7 cells treated in the presence and absence of 50 µM Q for 72 hours. Q enhanced the ubiquitination of c-FLIPL in breast cancer. Blots were also probed for c-FLIPL to confirm that the Co-IP was properly executed. Co-IP indicates co-immunoprecipitation; Q, quercetin.
Q’s impact on DR4 and DR5 expression
Western blot and flow cytometry analyses were performed to discover Q’s effects on DR4 and DR5 protein and cell surface expression in breast cancer. DR5 protein expression was upregulated in BT-20 cells with Q treatment dose dependently only after 72 hours, but Q did not influence DR4 protein expression in BT-20 cells (Figure 6A). DR5 and DR4 protein expressions did not change significantly in Q-treated MCF-7 cells (Figure 7A). Moreover, FACS analysis revealed that BT-20 cells express both receptors on the cell surface (Figure 6B to D), and DR4 expression was not upregulated significantly with Q (P > .05) for all treatments when compared with control), whereas Q did induce the upregulation of DR5 membrane expression. The FACS analysis also showed that MCF-7 cells express both receptors on the cell surface (Figure 7B to D). DR4 cell surface expression was not upregulated significantly with single-agent Q treatment (P > .05 for all treatments when compared with control), but 50 μM Q slightly upregulated DR5 membrane expression when compared with the vehicle-treated control. The results obtained from FACS analysis agree with the data derived from Western blot analysis for both cell lines. Therefore, Q-induced DR5 upregulation is an additional factor for the heightened rhTRAIL sensitivity observed in TNBC cells.
Figure 6.

DR4 and DR5 expression in Q-treated breast cancer BT-20 cells. (A) DR4 and DR5 (mature form) protein levels were assessed after 72 hours of treatment; Q upregulated DR5 expression in a dose-dependent manner. (B) DR5 and (C) DR4 cell surface expression levels for Q-treated BT-20 cells were analyzed by flow cytometry. For the representative histograms (B) and (C), vehicle-treated control = green, 12.5 µM Q = yellow, 25 µM Q = black, and 50 µM Q = blue. (D) The bar graphs represent the average fold increase in DR4 or DR5 cell surface expressions relative to the vehicle-treated control ± SEM from 3 independent experiments performed in triplicate (n = 9). P > .05 except *. Q indicates quercetin.
Figure 7.

DR4 and DR5 expression for Q-treated breast cancer MCF-7 cells. (A) DR4 and DR5 (mature form) protein levels were analyzed after 72 hours of treatment; Q slightly upregulated DR5 protein expression. Cell surface expression of (B) DR5 and (C) DR4 for Q-treated breast cancer MCF-7 cells was evaluated. For the representative histograms (B) and (C), vehicle-treated control = green, 12.5 µM Q = yellow, 25 µM Q = black, and 50 µM Q = blue. (D) The bar graphs represent the fold increase in DR4 or DR5 cell surface expression relative to the vehicle-treated control ± SEM from 3 independent experiments performed in triplicate (n = 9). P > .05 except *. Q indicates quercetin.
Comparing DR4 and DR5 expression in breast cells
Western blot and flow cytometry analyses were performed to compare the expression of DR4 and DR5 in breast cancer BT-20 and MCF-7 cell lines to DR4 and DR5 expression in nontumorigenic breast epithelial MCF-10A cells. MCF-10A had the highest DR4 and DR5 membrane and protein expression, and BT-20 cells had the lowest DR4 and DR5 protein and membrane expression levels (Figure 8). In addition, Q did not affect the expression of DR4 and DR5 in MCF-10A cells. Overall, the data demonstrate that Q specifically upregulates DRs in malignant breast cancer cell lines only (Figure 9).
Figure 8.

Comparing DR4 and DR5 expression among breast cancer cells and normal breast cells. (A) DR4 and DR5 (mature form) protein levels were compared among breast cancer cells (BT-20 and MCF-7) and nontumorigenic breast epithelial cells (MCF-10A). (B) DR5 and (C) DR4 cell surface expression for cells were also evaluated. For the representative histograms (B) and (C), BT-20 = blue, MCF-7 = red, MCF-10A = green. (D) The bar graphs represent the DR4 or DR5 cell surface expression ± SEM from 3 independent experiments performed in triplicate (n = 9). (E) MCF-10A cells were treated with Q for 72 hours, and there was no change in DR4 and DR5 expression.
Figure 9.

Densitometry results for death receptors and c-FLIPL. Densitometry results for Western blots for DR4, DR5, and c-FLIPL were calculated using ImageJ software and graphs were generated applying GraphPad Prism. (A) BT-20; (B) MCF-7; (C) BT-20, MCF-7, and MCF-10A; and (D) MCF-10A.
Q’s effects on DR5 and c-FLIPL expression at the transcriptional level
Quercetin was shown to increase the expression of DR5 and decrease the expression of c-FLIPL in breast cancer, but it was unknown whether Q acts at the transcriptional level to affect DR5 and c-FLIPL. Therefore, RT-PCR was executed applying β-actin as a positive control (Figure 10). Through RT-PCR analysis, we show that Q did not induce any change in c-FLIPL mRNA expression in breast cancer cells. The combined data suggest that Q-induced c-FLIPL downregulation must occur at the posttranslational level. Finally, Q did increase DR5 mRNA levels dose dependently in TNBC BT-20 cells correlating with the increase in the protein expression observed through Western blot and FACS analyses. Therefore, Q-induced DR5 upregulation in TNBC occurs transcriptionally.
Figure 10.

Q induces DR5 upregulation transcriptionally. (A) BT-20 and (B) MCF-7 cells were treated with increasing concentrations of Q for 72 hours, and reverse transcription-polymerase chain reaction was executed with β-actin as a positive control. Q upregulated DR5 mRNA expression in TNBC BT-20 dose dependently, but Q did not affect c-FLIPL mRNA expression in breast cancer. mRNA indicates messenger RNA; Q, quercetin.
Discussion
Breast cancer affects women worldwide. Traditional chemotherapy and radiation treatments for breast cancer rely on p53 to induce the intrinsic pathway of apoptosis, but many cancers possess a nonfunctional p53 gene resulting in necrosis rather than apoptosis after chemotherapy and radiation producing adverse side effects in patients. Recombinant human tumor necrosis factor–related apoptosis-inducing ligand possesses the ability to induce apoptosis through the induction of the extrinsic pathway of apoptosis in cancer cells and induce apoptosis through the induction of the intrinsic pathway of apoptosis independent of p53.7–10,43,44 Recombinant human tumor necrosis factor–related apoptosis-inducing ligand has been proposed to be used as an anticancer therapeutic. However, clinical trials using rhTRAIL as a potential anticancer therapeutic were terminated due to a lack of clinical efficacy, and in vitro studies have found that rhTRAIL treatment had limitations due to many cancer cell lines being resistant. This study focused on evaluating the potential of Q as a potent sensitizing agent for rhTRAIL-induced apoptosis in rhTRAIL-resistant TNBC BT-20 and hormone-dependent breast cancer MCF-7 cells; it should be noted that the combinatorial treatment of Q and rhTRAIL has not been examined before in the breast cancer cell lines of interest.28,34,35,45 Through in vitro analysis, we show that Q possesses the capability to act as a sensitizing agent for rhTRAIL-resistant breast carcinoma.
To assess the interaction of Q and rhTRAIL, Western blot analysis and Annexin V/PI assays were performed after 72 hours of treatment. The time course of 72 hours was chosen after preliminary experiments showed that Q’s impact on c-FLIPL and DR5 did not occur until after 72 hours. Other investigations have treated cells with Q at greater concentrations (100, 150, 175, and 200 µM) for 24 and 48 hours, whereas our investigation applies at most 50 M of Q.23,34,40,46 Both assays confirmed that Q augments rhTRAIL-induced apoptosis in breast cancer BT-20 and MCF-7 cells via the execution of the extrinsic pathway and the activation of caspase 8, the activation of executioner caspases 3 and 7, and the cleavage of PARP; furthermore, the results demonstrated that Q had the ability to promote apoptosis as a single agent but rhTRAIL did not. In addition, the cotreatment of Q and rhTRAIL exhibited a synergistic effect in breast BT-20 and MCF-7 cells. Therefore, Q’s mechanism of sensitization needed to be elucidated.
Previous studies have proposed that one reason cancer cell lines are resistant to rhTRAIL-induced apoptosis is through the upregulation of the c-FLIP, a significant inhibitor of the extrinsic pathway.47 The c-FLIPL is structurally similar to caspase 8, and at high-expression levels, it has the ability to prevent caspase 8 activation when bound to the death-inducing signaling complex and thus suppressing the DR signaling pathway.47–49 Consequently, c-FLIPL expression was evaluated and found to be significantly downregulated in Q-treated breast cancer cells.
Considering Q-induced downregulation of c-FLIPL in breast cancer, the underlying mechanism for Q’s sensitization needed to be elucidated. Through RT-PCR analysis, Q was found to not affect the mRNA expression of c-FLIPL in either breast cancer cell line. Analysis was then performed at the posttranslational level through the treatment of both breast cancer cell lines with the 50 µM Q alone and in combination with the proteasome inhibitor MG132, and c-FLIPL expression was assessed. We have established that Q promoted the ubiquitination of c-FLIPL in breast cancer which has not been determined before in cancer cell lines. This is most likely the only pathway for c-FLIPL degradation because if proteasome inhibitor is applied at a greater concentration, then a more pronounced effect of c-FLIPL recovery would be seen but the dead cells could be due to the high concentration of the inhibitor rather than Q. Thus, from these findings we can conclude that Q sensitizes TNBC and hormone-dependent breast cancer cells to rhTRAIL-induced apoptosis through the proteasome-mediated degradation of c-FLIPL because of enhanced ubiquitination.
Previous investigations have proposed that some rhTRAIL-resistant cancer cell lines express low levels DR4 and DR5 and thereby making them less sensitive to rhTRAIL’s proapoptotic effects.28,34 Quercetin has been shown to upregulate DR5 expression level in prostate cancer and hepatoma but not in breast cancer.28,34 Hence, DR4 and DR5 protein expressions were examined by Western blotting, and DR4 and DR5 cell surface expressions were examined by FACS. The data proved that Q had the ability to significantly upregulate DR5 expression in TNBC BT-20 cells, but Q had less of an effect on DR5 expression in hormone-dependent breast cancer MCF-7 cells. When comparing the expression levels of DRs, nontumorigenic breast epithelial MCF-10A cells had the highest expression of DRs, whereas TNBC BT-20 cells had the lowest expression of DRs. These results match another investigation comparing DR4 and DR5 protein expression in breast cancer MCF-7 cells and nontumorigenic breast epithelial MCF-10A cells where MCF-10A cells expressed more DR4 and DR5 in comparison with MCF-7 cells.50 In addition, Q did not affect DR4 and DR5 protein expression in nontumorigenic breast epithelial MCF-10A cells.
Since Q-induced DR5 upregulation in breast cancer, the underlying mechanism for this augmentation of rhTRAIL sensitivity, needed to be elucidated. The RT-PCR analysis demonstrated that Q enhanced DR5 mRNA expression in a dose-dependent manner most significantly in TNBC BT-20 cells. Thus, Q increases rhTRAIL sensitivity in TNBC cells through the upregulation of DR5 transcriptionally.
Conclusions
Overall, the data presented here demonstrate that Q is an effective sensitizing agent for rhTRAIL-resistant breast carcinoma. Through in vitro analysis, the cotreatment of Q and rhTRAIL proved efficacious for hormone-dependent breast cancer and TNBC. Our results suggest that the enhanced ubiquitination of c-FLIPL by Q could be a novel mechanism underlying the downregulation of c-FLIPL facilitating the enhanced rhTRAIL sensitivity. Therefore, this cotreatment should be explored further in vivo to determine its clinical efficacy as a potential breast carcinoma therapeutic regimen especially for the more fatal TNBCs.
Footnotes
Funding:The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the Parker Hannifin Foundation (M.K.).
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: JMK wrote the first draft of the manuscript. JMK, KAT, and MK conceived and designed the experiments; analyzed the data; contributed to the writing of the manuscript; agree with manuscript results and conclusions; jointly developed the structure and arguments for the paper; made critical revisions and approved final version; and reviewed and approved the final manuscript.
References
- 1. Cancer Facts & Figures 2016. Atlanta, GA: American Cancer Society; 2016. http://www.cancer.org. Accessed August 10, 2016. [Google Scholar]
- 2. Rahman M, Davis S, Pumphrey J, et al. TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res Treat. 2009;113:217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oakman C, Viale G, Leo A. Management of triple negative breast cancer. Breast. 2010;19:312–321. [DOI] [PubMed] [Google Scholar]
- 4. Elsamany S, Abdullah S. Triple-negative breast cancer: future prospects in diagnosis and management. Med Oncol. 2014;31:834. [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y, Zhang B. TRAIL resistance of breast cancer cells is associated with constitutive endocytosis of death receptors 4 and 5. Mol Cancer Res. 2008;6:1861–1871. [DOI] [PubMed] [Google Scholar]
- 6. Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28:3271–3277. [DOI] [PubMed] [Google Scholar]
- 7. Kuijlen JM, Bremer E, Mooij JJ, den Dunnen WF, Helfrich W. Review: on TRAIL for malignant glioma therapy? Neuropathol Appl Neurobiol. 2010;36:168–182. [DOI] [PubMed] [Google Scholar]
- 8. Wang D, Shi L. High-level expression, purification, and in vitro refolding of soluble tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Appl Biochem Biotechnol. 2009;157:1–9. [DOI] [PubMed] [Google Scholar]
- 9. Shen YL, Zhang Y, Sun AY, Xia XX, Wei DZ, Yang SL. High-level production of soluble tumor necrosis factor-related apoptosis-inducing ligand (Apo2L/TRAIL) in high-density cultivation of recombinant Escherichia coli using a combined feeding strategy. Biotechnol Lett. 2004;26:981–984. [DOI] [PubMed] [Google Scholar]
- 10. Luo Q, Shen YL, Wei DZ, Cao W. Optimization of culture on the overproduction of TRAIL in high-cell-density culture by recombinant Escherichia coli. Appl Microbiol Biotechnol. 2006;71:184–191. [DOI] [PubMed] [Google Scholar]
- 11. Turner K, Lindner D, Kalafatis M. Recombinant human tumor necrosis factor-related apoptosis-inducing ligand selectively induces apoptosis in malignant melanoma. Int J Cancer Oncol. 2017;4:1–8. [Google Scholar]
- 12. Mahmood Z, Shukla Y. Death receptors: targets for cancer therapy. Experiment Cell Res. 2010;316:887–899. [DOI] [PubMed] [Google Scholar]
- 13. Hellwig C, Rehm M. TRAIL signaling and synergy mechanisms used in TRAIL-based combination therapies. Mol Cancer Therap. 2012;11:3–13. [DOI] [PubMed] [Google Scholar]
- 14. Kimberley F, Screaton G. Following a TRAIL: update on a ligand and its five receptors. Cell Res. 2004;14:359–372. [DOI] [PubMed] [Google Scholar]
- 15. Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2004;12:228–237. [DOI] [PubMed] [Google Scholar]
- 16. Daniels R, Turley H, Kimberley F, et al. Expression of TRAIL and TRAIL receptors in normal and malignant tissues. Cell Res. 2005;15:430–438. [DOI] [PubMed] [Google Scholar]
- 17. Van Geelen CM, de Vries EG, de Jong S. Lessons from TRAIL-resistance mechanisms in colorectal cancer cells: paving the road to patient-tailored therapy. Drug Resist Updat. 2004;7:345–358. [DOI] [PubMed] [Google Scholar]
- 18. Griffith TS, Chin WA, Jackson GC, Lynch DH, Kubin MZ. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J Immunol. 1998;161:2833–2840. [PubMed] [Google Scholar]
- 19. Russo M, Mupo A, Spagnuolo C, Russo G. Exploring death receptor pathways as selective targets in cancer therapy. Biochem Pharmaco. 2010;80:674–682. [DOI] [PubMed] [Google Scholar]
- 20. Herbst R, Eckhardt G, Kurzrock R, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol. 2010;28:2839–2846. [DOI] [PubMed] [Google Scholar]
- 21. Kelley SK, Harris LA, Xie D, et al. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001;299:31–38. [PubMed] [Google Scholar]
- 22. Bellail A, Qi L, Mulligan P, Chhabra V, Hao C. TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials. 2009;4:34–41. [DOI] [PubMed] [Google Scholar]
- 23. Choi E, Bae S, Ahn W. Antiproliferative effects of quercetin through cell cycle arrest and apoptosis in human breast cancer MDA-MB-453 cells. Arch Pharm Res. 2008;31:1281–1285. [DOI] [PubMed] [Google Scholar]
- 24. Russo M, Spagnuolo C, Tedesco I, Bilotto S, Russo G. The flavonoid quercetin in disease prevention and therapy: facts and fancies. Biochem Pharmacol. 2012;83:6–15. [DOI] [PubMed] [Google Scholar]
- 25. Deng XH, Song HY, Zhou YF, Yuan GY, Zheng FJ. Effects of quercetin on the proliferation of breast cancer cells and expression of survivin in vitro. Exp Ther Med. 2013;6:1155–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim Y, Kim WJ, Cha EJ. Quercetin-induced growth inhibition in human bladder cancer cells is associated with an increase in Ca2+-activated K+ channels. Korean J Phys Pharma. 2011;15:279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kandaswami C, Lee LT, Lee PP, et al. The antitumor activities of flavonoids. In Vivo. 2005;19:895–909. [PubMed] [Google Scholar]
- 28. Kim J, Kim E, Park S, Lim J, Kwon T, Choi K. Quercetin sensitizes human hepatoma cells to TRAIL-induced apoptosis via Sp1-mediated DR5 up-regulation and proteasome-mediated c-FLIPS down-regulation. J Cell Biochem. 2008;105:1386–1398. [DOI] [PubMed] [Google Scholar]
- 29. Jeong J, An J, Kwon Y, Rhee J, Lee Y. Effects of low dose quercetin: cancer cell-specific inhibition of cell cycle progression. J Cell Biochem. 2009;106:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pan HC, Jiang Q, Yu Y, Mei JP, Cui YK, Zhao WJ. Quercetin promotes cell apoptosis and inhibits the expression of MMP-9 and fibronectin via the AKT and ERK signalling pathways in human glioma cells. Neurochem Int. 2014;80:60–71. [DOI] [PubMed] [Google Scholar]
- 31. Ferry DR, Smith A, Malkhandi J, Fyfe DW. Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res. 1996;2:659–668. [PubMed] [Google Scholar]
- 32. Ramos S. Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. J Nutr Biochem. 2007;18:427–442. [DOI] [PubMed] [Google Scholar]
- 33. Vargas A, Burd R. Hormesis and synergy: pathways and mechanisms of quercetin in cancer prevention and management. Nutr Rev. 2010;68:418–428. [DOI] [PubMed] [Google Scholar]
- 34. Jung YH, Heo J, Lee Y, Kwon T, Kim YH. Quercetin enhances TRAIL-induced apoptosis in prostate cancer cells via increased protein stability of death receptor 5. Life Sci. 2010;86:351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yi L, Zongyuan Y, Cheng G, Lingyun Z, Guilian Y, Wei G. Quercetin enhances apoptotic effect of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in ovarian cancer cells through reactive oxygen species (ROS) mediated CCAAT enhancer-binding protein homologous protein (CHOP)-death receptor 5 pathway. Cancer Sci. 2014;105:520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Staedler D, Idrizi E, Kenzaoui B, Juillerat-Jeanneret L. Drug combinations with quercetin: doxorubicin plus quercetin in human breast cancer cells. Cancer Chemother Pharmacol. 2011;68:1161–1172. [DOI] [PubMed] [Google Scholar]
- 37. Harwood M, Danielewska-Nikiel B, Borzelleca JF, Flamm GW, Williams GM, Lines TC. A critical review of the data related to the safety of quercetin and lack of evidence of in vivo toxicity, including lack of genotoxic/carcinogenic properties. Food Chem Toxicol. 2007;45:2179–2205. [DOI] [PubMed] [Google Scholar]
- 38. D’Andrea G. Quercetin: a flavonol with multifaceted therapeutic applications? Fitoterapia. 2015;106:256–271. [DOI] [PubMed] [Google Scholar]
- 39. Lamson DW, Brignall MS. Antioxidants and cancer, part 3: quercetin. Altern Med Rev. 2000;5:196–208. [PubMed] [Google Scholar]
- 40. Duo J, Ying GG, Wang GW, Zhang L. Quercetin inhibits human breast cancer cell proliferation and induces apoptosis via Bcl-2 and Bax regulation. Mol Med Rep. 2012;5:1453–1456. [DOI] [PubMed] [Google Scholar]
- 41. Rahman M, Pumphrey JG, Lipkowitz S. The TRAIL to targeted therapy of breast cancer. Adv Cancer Res. 2009;103:43–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dong Y, Yin S, Li J, Jiang C, Ye M, Hu H. Bufadienolide compounds sensitize human breast cancer cells to TRAIL-induced apoptosis via inhibition of STAT3/Mcl-1 pathway. Apoptosis. 2011;16:394–403. [DOI] [PubMed] [Google Scholar]
- 43. Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015;356:197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guan YS, He Q, Zou Q. Status quo of p53 in the treatment of tumors. Anticancer Drugs. 2016;27:811–818. [DOI] [PubMed] [Google Scholar]
- 45. Psahoulia F, Drosopoulos K, Doubravska L, Andera L, Pintzas A. Quercetin enhances TRAIL-mediated apoptosis in colon cancer cells by inducing the accumulation of death receptors in lipid rafts. Mol Cancer Ther. 2007;6:2591–2599. [DOI] [PubMed] [Google Scholar]
- 46. Chou CC, Yang JS, Lu H-F, et al. Quercetin-mediated cell cycle arrest and apoptosis involving activation of a caspase cascade through the mitochondrial pathway in human breast cancer MCF-7 cells. Arch Pharm Res. 2010;33:1181–1191. [DOI] [PubMed] [Google Scholar]
- 47. Safa AR. C-FLIP, a master anti-apoptotic regulator. Exp Oncol. 2012;34:176–184. [PMC free article] [PubMed] [Google Scholar]
- 48. Koff JL, Ramachandiran S, Bernal-Mizrachi L. A time to kill: targeting apoptosis in cancer. Int J Mol Sci. 2015;16:2942–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mishra T, Arya R, Meena S, et al. Isolation, characterization and anticancer potential of cytotoxic triterpenes from Betula utilis bark. PLoS ONE. 2016;11: e0159430. [DOI] [PMC free article] [PubMed] [Google Scholar]