

Abstract
Previous studies of a thermophilic alcohol dehydrogenase (ht-ADH) demonstrated a range of discontinuous transitions at 30°C that include catalysis, kinetic isotope effects, protein hydrogen-deuterium exchange rates, and intrinsic fluorescence properties. Using the FRET response from a Trp-NADH donor-acceptor pair in T-jump studies of ht-ADH, we now report microsecond protein motions that can be directly related to active site chemistry. Two distinctive transients are observed: a slow, kinetic process lacking a temperature break, together with a faster transient that is only detectable above 30°C. The latter establishes a link between enzyme activity and microsecond protein motions near the cofactor binding site, in a region distinct from a previously detected protein network that communicates with the substrate binding site. While evidence of direct dynamical links between microsecond protein motions and active site bond cleavage events is extremely rare, these studies highlight the potential of T-jump measurements to uncover such properties.
Graphical Abstract
Increasingly, the dynamical sampling of protein conformational substates is expected to play an important role in controlling the rate of enzyme catalyzed reactions.1–7 stochastic sampling of states may involve motions that range from picoseconds to milliseconds,8 and the microsecond time regime has been predicted to be an especially key component in the creation of productive conformations.9 The microsecond regime can be difficult to access, as it is typically too long for routine molecular dynamics simulations and NMR is not suitable for large protein systems such as the tetrameric thermophilic alcohol dehydrogenase from B. stearothermophilus (ht-ADH), an archetypic system for relating changes in protein dynamics to active site chemistry.
Previous studies of ht-ADH demonstrated numerous anomalous breaks in behavior as the temperature is reduced below 30°C, that include an increase in the enthalpy of activation for the millisecond chemical (hydride transfer) step,10 an increase in the temperature dependence of the kinetic isotope effect (KIE) on hydride transfer,10 and a change in the pattern of time averaged hydrogen deuterium exchange (HDX) within the substrate binding domain.11 These aggregate behaviors can be explained by active conformational sampling at the elevated, physiologically relevant temperatures of B. stearothermophilus, that becomes reduced below the 30 °C break point. In the present study, we follow the protein dynamics of the ternary complex of ht-ADH with NADH and the substrate analog isobutyramide, via time resolved temperature-jump FRET spectroscopy. We observe two relaxation rates, the faster of which disappears below 30 °C. We attribute the absence of the faster rate below 30 °C to a “freezing out” of collective motions that modulate the FRET efficiency. We conclude that these same motions are important in the search for reactive conformations in the Michaelis complex.
Fluorescence spectroscopy is a useful probe for studying enzyme motions because it is environmentally sensitive. Ht-ADH contains three tryptophans (Trp): W87 that resides behind bound substrate, W49 that is proximal to bound cofactor, and W167 located on the protein surface far from the active site (Figure 1). In this work, we focus on W87F, a variant having W87 alone substituted by phenylalanine. Control experiments with W167in show that W167 does not participate in FRET with the cofactor (Figure S1), which simplifies assignment of the FRET signal origin to energy transfer between W49 and NADH.12–13 Furthermore, W87F and wildtype ht-ADH exhibit similar kinetic properties and a break in behavior at 30°C,13 making W87F a suitable surrogate for studying site-specific protein motions.
Figure 1.
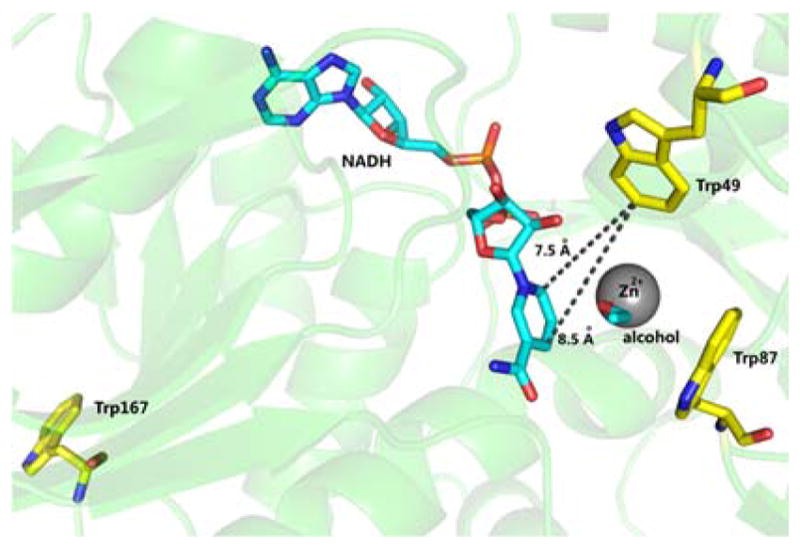
Structure of ht-ADH in green with NADH and alcohol in cyan. The three tryptophan residues are yellow. This structure was created from an overlap of PDB: 1RJW and 3MEQ.
The steady state tryptophan fluorescence of W87F is modulated by ligand binding (Figure 2A). Compared to apoenzyme, the binary complex of ht-ADH with isobutyramide shows a slight increase in Trp fluorescence (~330 nm). Binding NADH to the apoenzyme quenches the Trp fluorescence, while inducing NADH fluorescence (~435 nm), due to Forster energy transfer from W49 to NADH. The FRET signal depends on the distance and relative orientation of the donor and acceptor transition dipoles,14 with W49 expected to lie within ca. 9 Å of the nicotinamide ring of cofactor (Figure 1), well within 23.4 Å, the Forster radius of this FRET pair.14
Figure 2.

A) Equilibrium fluorescence spectra of ht-ADH W87F (2 μM) with ligands NADH (20 μM) and isobutyramide (100 mM) in potassium phosphate buffer (50 mM, pH 7) excited at 280 nm. FRET between W49 and NADH is strongest in the ternary complex. B) Temperature dependent fluorescence spectra of ht-ADH W87F:NADH:isobutyramide. The inset shows the integrated NADH emission (420 – 460 nm) vs temperature.
The ht-ADH W87F•NADH•isobutyramide ternary complex shows a much stronger FRET signal (Figure 2A), indicating that binding of the substrate analog alters the enzyme conformation and brings W49 into better alignment with NADH. Temperature dependent fluorescence spectra show that the FRET signal of the tertiary complex is nearly temperature independent below 30 °C (Figure 2B). The decrease in FRET intensity at higher temperatures may be attributed to an increase in flexibility of W87F ADH allowing for fluctuations that lead to possible changes in the relative orientation of the donor-acceptor transition dipole moments as well as some increase in the average distance between W49 and the nicotinamide ring of the cofactor.
T-jump fluorescence spectroscopy was used to interrogate conformational fluctuations of the W87F ternary complex on the ns to μs timescale. T-jumps of 9 °C were generated by laser heating with a range of final temperatures between 22 – 50 °C (Figure S2). The resulting FRET transients display distinct behaviors as a function of temperature (Figure 3A). In no instance were these found to exhibit a concentration dependence with respect to NADH or isobutyramide, indicating relaxation events that are independent of ligand association/dissociation processes (Figure S3). A number of fits to the experimental traces were tested via a combination of single and/or two exponential decays across the experimental temperature regime. A best fit was identified that corresponds to two exponentials above 30 °C together with a single decay process below this point (Chart S1). It is striking that the slow relaxation shows continuity across the full temperature range, whereas the fast process has been slowed to such an extent below 30 °C that it no longer contributes to the observed transients. These changes in rate constants are mirrored in their amplitudes, with the slow rate being characterized by a constant amplitude (within the noise of the measurement) whereas the fast rate shows a linear increase above 30 °C (Figure S4). The Arrhenius plot (Figure 3B) shows the dramatic decrease in the rate of the fast process with decreasing temperature and its disappearance at 30 °C. Moreover, its activation energy 10.4 ± 1.3 kcal/mol is close to the enthalpy of activation, 11.0 ± 0.6 kcal/mol for the hydride transfer step above 30 °C,13 whereas that of the slow process is weakly temperature dependent with an activation energy of 3.0 ± 0.9 kcal/mol. Thus, the T-jump FRET response, which is sensitive to motions along the reaction coordinate (Figure 1), provides direct support for the view that catalysis is enabled by protein motions that become activated above 30 °C. These are likely collective motions involved in the search for a reactive conformation. Further insight as to the origin of the two phases comes from the temperature dependence of fluorescence anisotropy of the tertiary complex (Figure S5). Below 30 °C, the anisotropy decreases with increasing temperature, indicating the relative orientation of the donor and acceptor transition dipole moments is changing with temperature. Therefore, the slow T-jump FRET response observed in this temperature regime is due to a reorientation of the donor and acceptor with respect to one another. The combined anisotropy and temperature jump FRET results lead to assignment of the slow phase to a local motion that predominately changes the orientation of the dipole moments. Above 30 °C, the fluorescence anisotropy is relatively temperature independent, either because the dipole orientation is no longer changing, or more likely, the average dipole orientation factor is 2/3 due to conformational sampling. Thus, the trend in the fast T-jump FRET, dominant above 30 °C, corresponds to collective motions that modulate the distance between the FRET pair.
Figure 3.

A) Temperature-jump (ΔT = 9 °C) FRET transients of the highest and lowest final temperatures. The transients are shown as normalized FRET intensity to highlight the difference in shape. The highest temperature fits best to a double exponential and the lowest temperature fits best to a single exponential. B) Arrhenius plot of observed relaxation rates. The slower relaxation rate is present at all temperatures (2.6 × 105 s−1 at 34 °C) and appears to be slightly temperature dependent. Conversely, the faster rate (5.7 × 105 s−1 at 34 °C) is present only at temperatures above 30°C and is strongly temperature dependent.
The disappearance of the fast process below 30 °C is suggestive of a glass transition, and it has been hypothesized that thermophilic enzymes may have much higher glass transition temperatures than mesophilic enzymes.15 Ht-ADH catalysis still occurs below this temperature, but the activation enthalpy is much higher, 17.0 ± 0.4 kcal/mol,10, 13 which has been previously attributed to an impairment of the facilitating protein motions.16 The rate of catalysis for a related dehydrogenase, lactate dehydrogenase (LDH), has also been shown to be controlled by the search for a reactive conformation in the formation of binary and ternary complexes through collective motions.1, 9 Fast and slow kinetics have been observed for LDH on similar timescale as observed here for ADH.17 It has been shown that psychrophilic, mesophilic, and thermophilic LDH have the same enzymatic efficiency at the host organism’s optimal growth temperature.10 Furthermore, pig heart LDH demonstrates a structural change and similar break in enzyme activity around 35°C, indicating that elevated glass transitions may not be restricted to thermophilic enzymes.18
Previous studies of ht-ADH show strong structural anisotropy with regard to a network of protein motions that connects a solvent exposed dimer (Interface I, Figure S6) with the substrate binding pocket ca. 17 Å from W87.19 This interface connects subunits in which the primary contact is a Tyr25-Tyr25 stacking interaction that has been correlated with temperature breaks in kcat, the kinetic isotope effect, and the fluorescent lifetime of W87in.12, 19 The structure of ADH is a dimer of dimers, generating both the symmetrical dimer Interface I, and a second asymmetrical Interface, II, characterized by a contact between W49 and Phe272 from the adjacent subunit (Figure S6). A T-jump study of apoW78F showed microsecond fluorescence transients, which were attributed to opening and closing of this contact; the apoprotein does not adopt the structure of the ternary complex, and no 30 °C break was observed in the rates.13
The remarkable T-jump results reported here implicate a second network of motions within ht-ADH that arises at Interface II and modulates the relative position of the reactive nicotinamide ring of the cofactor, as reported by FRET. Thus, positioning of the hydride donor and acceptor for reaction, as well as other key residues that engage in the stabilization of dipolar and electrostatic changes accompanying the net hydride transfer reaction, appears to be facilitated by two structurally orthogonal networks, directed at the cofactor on one hand and the substrate on the other (Figure S6).
A general theory for hydrogen transfer in enzymatic C-H cleavage reactions has emerged in which hydrogen transfer occurs via a tunneling reaction dictated by protein motions that create both reactant-product ground state degeneracies and close hydrogen donor acceptor distances.20–21 This process has been represented by ktun within a full rate equation that incorporates a prior protein conformational search for catalytically relevant protein substates defined as Fconf:22
| (1) |
As discussed, the enthalpic barrier for C-H cleavage reactions that occur via tunneling will be comprised of a combination of local rapid protein motions (ps-ns) together with slower more remote motions (μs-ms) that can be considered to arise primarily from Fconf and the outer sphere reorganization term, λout, in ktun. Independent of time scale, all of the motions that precede the tunneling event are considered to be equilibrated, stochastic processes.1, 23
In a recently completed study of the enzyme soybean lipoxygenase, the enthalpic barriers for rates of hydrogen deuterium exchange (HDX) within a remote loop at the protein surface were found to correlate with mutation-induced trends in the enthalpic barriers for the rate of H-tunneling at the active site.22 The physical picture that emerged involves a well-defined motional network that connects the SLO protein-solvent interface with the active site ca. 15–30 Å away. For this earlier study, the absolute magnitude of the enthalpies from HDX and catalysis were distinct, as anticipated from the different physical processes controlling a thermally averaged HDX vs. the chemical hydride transfer step. In contrast, the present T-jump study of ht-ADH provides a microsecond time constant and activation energy for protein reorganization that can be directly compared to kcat. Of note, although the time scales for kcat and FRET in ht-ADH differ by three orders of magnitude, the energies of activation are almost identical. While the latter could be fortuitous, the data strongly implicate a link between thermally equilibrated, microsecond protein motions and the creation of active site configurations that support hydrogen transfer via tunneling. Although evidence for such direct dynamical links between microsecond protein motions and active site bond cleavage events is extremely rare, T-jump measurements appear ideally suited both to the detection of such motions and to the resolution of their relationship to networks for motional communication between protein-solvent interfaces and enzyme active sites.
Supplementary Material
Acknowledgments
We thank Dr. George Blouin, University of Washington, Seattle for helpful discussions and initial trial experiments on this T-jump FRET study.
Funding Sources
This work was supported by National Institutes of Health grants to R.B.D. (GM068036), to J.P.K. (GM118117-015) and to T.G.S. (GM25158).
Footnotes
The Supporting Information contains Materials and Methods, and Figures S1–S6 showing: FRET for W167in (S1), the T-jump transients (S2), relaxation rates vs. ligand concentrations (S3), amplitudes of transients (S4), fluorescence anisotropy (S5) and the topology of ht-ADH (S6). Chart S1 gives the goodness of fit to single vs. double exponential phases. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Klinman JP, Kohen A. Annu Rev Biochem. 2013;82:471–96. doi: 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Callender R, Dyer RB. Acc Chem Res. 2015;48:407–413. doi: 10.1021/ar5002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nashine VC, Hammes-Schiffer S, Benkovic SJ. Curr Opin Chem Biol. 2010;14:644–51. doi: 10.1016/j.cbpa.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hay S, Scrutton NS. Nat Chem. 2012;4:161–8. doi: 10.1038/nchem.1223. [DOI] [PubMed] [Google Scholar]
- 5.Luk LYP, Loveridge EJ, Allemann RK. Phys Chem Chem Phys. 2015;17:30817–30827. doi: 10.1039/c5cp00794a. [DOI] [PubMed] [Google Scholar]
- 6.Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Nature. 2009;462:669–73. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motlagh HN, Wrabl JO, Li J, Hilser VJ. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henzler-Wildman K, Kern D. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 9.Reddish MJ, Peng HL, Deng H, Panwar KS, Callender R, Dyer RB. J Phys Chem B. 2014;118:10854–10862. doi: 10.1021/jp5050546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohen A, Cannio R, Bartolucci S, Klinman JP. Nature. 1999;399:496–499. doi: 10.1038/20981. [DOI] [PubMed] [Google Scholar]
- 11.Liang ZX, Lee T, Resing KA, Ahn NG, Klinman JP. Proc Natl Acad Sci USA. 2004;101:9556–9561. doi: 10.1073/pnas.0403337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meadows CW, Ou R, Klinman JP. J Phys Chem B. 2014;118:6049–6061. doi: 10.1021/jp500825x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meadows CW, Balakrishnan G, Kier BL, Spiro TG, Kinman JP. J Am Chem Soc. 2015;137:10060–10063. doi: 10.1021/jacs.5b04413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. New York: Springer US; 2006. [Google Scholar]
- 15.Ringe D, Petsko GA. Biophys Chem. 2003;105:667–680. doi: 10.1016/s0301-4622(03)00096-6. [DOI] [PubMed] [Google Scholar]
- 16.Nagel ZD, Dong M, Bahnson BJ, Klinman JP. Proc Natl Acad Sci U S A. 2011;108:10520–10525. doi: 10.1073/pnas.1104989108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke AR, Waldman ADB, Hart KW, Holbrook JJ. Biochimica et Biophysica Acta. 1985;829:397–407. doi: 10.1016/0167-4838(85)90250-x. [DOI] [PubMed] [Google Scholar]
- 18.Khrapunov S, Chang E, Callender RH. Biochemistry. 2017;56:3587–3595. doi: 10.1021/acs.biochem.7b00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagel ZD, Cun S, Klinman JP. J Biol Chem. 2013;288:14087–14097. doi: 10.1074/jbc.M113.453951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knapp MJ, Klinman JP. Eur J Biochem. 2002;269:3113–3121. doi: 10.1046/j.1432-1033.2002.03022.x. [DOI] [PubMed] [Google Scholar]
- 21.Hammes-Schiffer S. Acc Chem Res. 2006;39:93–100. doi: 10.1021/ar040199a. [DOI] [PubMed] [Google Scholar]
- 22.Offenbacher AR, Hu S, Poss EM, Carr CAM, Scouras AD, Prigozhin D, Iavarone AT, Palla A, Alber T, Fraser JS, Klinman JP. ACS Cent Sci. 2017;3:570–579. doi: 10.1021/acscentsci.7b00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soudackov AV, Hammes-Schiffer S. Faraday Discuss. 2016;195:171–189. doi: 10.1039/c6fd00122j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.