

Abstract
Haploinsufficiency of ANKRD11 due to deletion or truncation mutations causes KBG syndrome, a rare genetic disorder characterized by intellectual disability, autism spectrum disorder, and craniofacial abnormalities. However, little is known about the neurobiological role of ANKRD11 during brain development. Here we show that ANKRD11 regulates pyramidal neuron migration and dendritic differentiation in the developing moue cerebral cortex. Using an in utero manipulation approach, we found that Ankrd11 knockdown delayed radial migration of cortical neurons. ANKRD11-deficient neurons displayed markedly reduced dendrite growth and branching as well as abnormal dendritic spine morphology. Ankrd11 knockdown suppressed acetylation of epigenetic molecules such as p53 and Histone H3. Furthermore, the mRNA levels of Trkb, Bdnf, and neurite growth-related genes were downregulated in ANKRD11-deficient cortical neurons. The Trkb promoter region was largely devoid of acetylated Histone H3 and p53, and was instead occupied with MeCP2 and DNMT1. Overexpression of TrkB rescued abnormal dendrite growth in these cells. Our findings demonstrate a novel role for ANKRD11 in neuron differentiation during brain development and suggest an epigenetic modification as a potential key molecular feature underlying KBG syndrome.
Keywords: ANKRD11, dendrite, arborization, dendritic spine, TrkB, BDNF, histone acetylation, autism, intellectual disability
Introduction
KBG syndrome is a rare developmental disorder that affects several body systems. The initials “KBG” are the first letters of the surnames of the first families in which this disorder was diagnosed (Herrmann et al., 1975). A key feature of KBG syndrome is neurological symptoms that include intellectual disability, autistic behavior, and seizure (Herrmann et al., 1975; Sirmaci et al., 2011; Skjei et al., 2007; Willemsen et al., 2010). KBG syndrome is caused by haploinsufficiency of the Ankyrin repeat domain-containing protein 11 (ANKRD11) gene resulting from either loss-of-function mutations in the ANKRD11 gene or microdeletions in chromosome 16q24.3, which includes ANKRD11 (Isrie et al., 2012; Lo-Castro et al., 2013; Sirmaci et al., 2011; Walz et al., 2015; Youngs et al., 2011). Although ANKRD11 loss-of-function results in KBG syndrome, the neural function of ANKRD11 is poorly understood.
As a chromatin regulator, ANKRD11 has two transcription repression domains and one activation domain (Zhang et al., 2004). Accordingly, ANKRD11 displays a dual function in transcriptional activation and suppression. ANKRD11 participates in transcription inhibition by interacting with histone deacetylases (HDACs) and histone molecules, resulting in the inhibition of ligand-dependent transcription (Zhang et al., 2004). In a different cellular context, ANKRD11 interacts with histone acetyltransferases (HATs) such as p160 coactivator family members and activates gene transcription (Zhang et al., 2004). Similarly, ANKRD11 associates with other histone modifiers, p53 acetyltransferase and its cofactors including P/CAF and hADA3, and acetylates p53, which activates the transcription of p53 target genes (Neilsen et al., 2008). Epigenetic regulation of histones and their associated molecules is important for proper neuronal differentiation during brain development. HDACs and HATs control histone acetylation and thus the gene transcription necessary for nervous system development and synaptic plasticity (Guan et al., 2009; Lilja et al., 2013; Siebzehnrubl et al., 2007; Trakhtenberg and Goldberg, 2012). For example, HDAC inhibition has been shown to promote neurite outgrowth and neuron survival by altering BDNF gene expression (Gaub et al., 2010; Hasan et al., 2013).
Dendritic arborization and spine morphogenesis are critical in establishing proper neural circuitry during brain development. Accordingly, dendritic abnormalities are the most obvious hallmark for developmental disorders associated with intellectual disability and autism spectrum disorder (ASD) (Granato and De Giorgio, 2014; Kaufmann and Moser, 2000; London and Hausser, 2005). Thus, we aimed to characterize the molecular function of ANKRD11 in pyramidal neuron development including migration, dendrite arborization, and spine morphogenesis during brain development. We found that Ankrd11 knockdown disrupted dendrite and spine formation in developing pyramidal neurons. Additionally, altered BDNF/TrkB signaling and neurite-associated gene transcription were associated with Ankrd11 knockdown phenotypes as was the acetylation of histones and histone modifiers. Our findings indicate a novel function of ANKRD11 during brain development.
Materials and Methods
Plasmids
To generate shAnkrd11, we targeted a sequence (5′- GCAGCTTCCTACACAGCAATC -3′) and its complement, and then cloned them into a modified pSuper-Basic vector as previously described (Ka et al., 2014a; Kim et al., 2006) and pLUX-shRNA2 vector (Takara Bio Inc, Mountain View, CA, USA). For control, non-silencing shRNAs were generated using scrambled targeting sequences (5′ GCTTACTCCCCGAGCAACATA -3′ and 5′- GCCCAGACTACTCTCACAGAT -3′). pCDNA-TrkB was acquired from Addgene (Cambridge, MA, USA).
Immunostaining
Immunostaining of brain sections or dissociated neural cells was performed as described previously (Ka et al., 2017a; Kim et al., 2005). The following primary antibodies were used: Chicken anti-GFP (A10262; Thermo Fisher Scientific Inc., Waltham, MA, USA), rabbit anti-GFP (A11122; Thermo Fisher Scientific Inc., Waltham, MA, USA), and mouse anti-ANKRD11 (SC-81049; Santa Cruz Biotechnology Inc., Dallas, TX, USA). Appropriate secondary antibodies conjugated with Alexa Fluor dyes (A11039, A11008; Thermo Fisher Scientific Inc., Waltham, MA, USA) were used to detect primary antibodies.
In utero electroporation
In utero electroporation was performed as described previously (Ka et al., 2016a; Ka and Kim, 2016). Briefly, timed pregnant female mice at E14.5 were deeply anesthetized, and the lateral ventricles of embryonic brains were injected with plasmid DNA (2 mg/μl) and 0.001% fast green (Sigma-Aldrich Corporation, St. Louis, MO, USA) using a Picospritzer II (Parker Hannifin, Hollis, NH, USA). Electroporation was achieved using a BTX ECM830 elecroporator (5 100 ms pulses separated by 900 ms intervals were applied at 45V). Embryos were allowed to develop in utero for the indicated time. For hippocampal gene delivery, the electrodes were placed at an angle opposite to the one used in cortical targeting, as described previously (Ka et al., 2014b).
Morphometry
For quantifying neurons in cortical layers, images of brain sections at periodic distances along the rostro-caudal axis were taken with the Zeiss LSM710 confocal microscope with a 40X or 20X objective and ZEN software. We counted GFP and DAPI-positive cells in a field of 0.5 mm2 throughout the rostro-caudal extent of layers I–VI of the cerebral cortex. Ten mice were used for each experiment (control mice, N= 5; mutant mice, N= 5). Stereological analysis of immunostained cells was performed by analyzing one-in-six series of 40 μm coronal sections (240 μm apart) (Jung et al., 2016). For the quantification of length, number, or thickness of primary dendrites and branches, confocal images containing layer II/III of the cerebral cortex from more than 5 mice were analyzed by using ImageJ (NIH). For analyzing cultured cells, more than 20 fields scanned horizontally and vertically were examined in each condition. Cell numbers were described in figure legends. The calculated values were averaged, and some results were recalculated as relative changes versus control.
Primary neuron cultures
Primary neuronal culture was performed as described previously (Ka et al., 2017b; Kim et al., 2009a). Briefly, cerebral cortices from E13.5–16.5 mice were isolated and dissociated with trituration after trypsin/EDTA treatment. The cells were then plated onto poly-D-lysine/laminin-coated coverslips and cultured in medium containing neurobasal medium, B27 supplements and N2 supplements.
Cell transfection
Mouse cortical neurons were transfected with various plasmids as described in a previous publication (Ka et al., 2014a). Embryonic cortices were dissociated and suspended in 100 μl of Amaxa electroporation buffer containing 1–10 μg of plasmid DNA. Then, suspended cells were electroporated with an Amaxa Nucleofector apparatus. After electroporation, cells were plated onto coated coverslips and the medium was changed 4 hours later to remove the remnant transfection buffer. For transfecting DNA constructs into attached cells, Lipofectamine-mediated (Thermo Fisher Scientific Inc., Waltham, MA, USA) transfection was performed according to the manufacturer’s protocol.
Lentivirus preparation
The same oligomers that were used to generate shAnkrd11 were subcloned into the BamHI and EcoRI sites of pLUX-shRNA2 vector (Takara Bio Inc, Mountain View, CA, USA), according to the manufacturer’s protocol. For efficient packaging, to decrease the risk of recombination, and to produce replication competent viruses, pMDLg/pRRE and pRSV-Rev were used. Viral particles were pseudotyped with pMD2.G plasmid encoding vesicular stomatitis virus G-protein. The lentivirus was generated by co-transfection of HEK 293T cells with four plasmids using a liposome mediated gene transfer. After 48 hours, the supernatant was collected and filtered through 0.45 μm cellulose acetate filters. Higher titer stocks were obtained using an ultrafiltration device (EMD Millipore, Billerica, MA, USA) and stored frozen at −80°C. Lentiviral titers were determined by infecting HEK 293T cells at the density of 5 × 105 cells/well with a serial dilution of the stock virus in the presence of polybrene (8 μg/ml). The cells were incubated for 4 days. High titers of engineered lentiviruses were produced (1 × 108 infectious unit/ml), as measured by green-fluorescence colony formation on HEK 293T cells.
Western Blotting
Western blotting was performed as described previously (Kim et al., 2004; Kim et al., 2009b). Cellular lysates from cultured neurons were prepared using RIPA buffer and the protein content was determined using a Bio-Rad Protein Assay system. Proteins were separated on 4–12% SDS-PAGE gradient gel and transferred onto a nitrocellulose membrane. Then the membrane was incubated with mouse anti-ANKRD11 (SC81049; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti-acetyl-Histone H3 (ab4441; Abcam, Cambridge, MA, USA), rabbit anti-acetyl-p53 (ab62376; Abcam, Cambridge, MA, USA), rabbit anti-p-AKT (9271; Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit anti-p-GSK3β antibody (9336S; Cell Signaling Technology, Inc., Danvers, MA, USA) rabbit anti-p-S6 (4858S; Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit anti-p-S6K (9205S; Cell Signaling Technology, Inc., Danvers, MA, USA) or mouse anti-Actin (ab3280; Abcam, Cambridge, MA, USA) at 4°C overnight. Appropriate secondary antibodies conjugated to HRP were used (Cell Signaling Technology, Inc., Danvers, MA, USA) and the ECL reagents (Thermo Fisher Scientific Inc., Waltham, MA, USA) were used for immunodetection. For quantification of band intensity, blots from 3 independent experiments for each molecule of interest were used. Signals were measured using Image J and represented by relative intensity versus control. Actin was used as an internal control to normalize band intensity.
Reverse transcription RCR
RNA was extracted from cultured neurons using TRIZOL reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA), and cDNA was synthesized from 1 μg of total RNA using oligo-dT and random hexamers using the Verso cDNA synthesis kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). A measure of 1 μl of cDNA was used in reverse transcription PCR using Master Mix (Promega Life Sciences, Madision, WI, USA). Following primer sequences were used: Ankrd11 forward 5′-GACGCTGCCTTTGATGACTA-3′ and reverse 5′-GATAAGTTTGGCCACCAGGT-3′; Gap43 forward 5′-GGGTCCTCCAAGGCTGAAGAT3′ and reverse 5′-CGGGCACTTTCCTTAGGTTTG-3′; Coronin 1b forward 5′-AGGAGCTGGATTCAAGCAAT-3′ and reverse 5′-GCAGTTTGTAGAACCTGGCA-3′; Rab13 forward 5′-TGCTGGGAAACAAGTGTGAC-3′ and reverse 5′-TGCTTGAGGGCTTACTGTTG-3′; Bdnf forward 5′-GCGGCAGATAAAAAGACTGC-3′ and reverse 5′-CCCGAACATACGATTGGGTA-3′; Trkb forward 5′-TGGTGCATTCCATTCACTGT-3′ and reverse 5′-CTTGGCCATCAGGGTGTAGT-3′; Gapdh forward 5′-AAGGTCATCCCAG AGCTGAA-3′ and reverse 5′-AGGAGACAACCTGGTCCTCA-3′. All primers were initially tested for their specificity by running RT-PCR samples on an agarose gel.
ChIP assay
The ChIP assay was performed using the Magna-CHIP™ A chromatin immunoprecipitation kit (EMD Millipore, Billerica, MA, USA) according to the manufacturer’s protocol. Briefly, chromatin proteins were cross-linked to DNA by addition of formaldehyde to the culture medium to a final concentration of 1%. After a 10 minute incubation at room temperature, the cells were washed two times, and scraped off in ice-cold phosphate-buffered saline (PBS) containing Protease Inhibitor Cocktail II. Cells were pelleted and then resuspended in lysis buffer containing Protease Inhibitor Cocktail II. The resulting lysate was subjected to sonication to reduce the size of DNA to approximately 200–1000 base pairs in length. The sample was centrifuged to remove cell debris and diluted 10-fold in ChIP dilution buffer containing Protease Inhibitor Cocktail II. A 5 μl sample of the supernatant was retained as “Input” and stored at 4°C. Then 5 μg of mouse anti-IgG antibody (negative control), mouse anti-ANKRD11 antibody (SC81049; Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti-acetyl-Histone H3 antibody (ab4441; Abcam, Cambridge, MA, USA), rabbit anti-MeCP2 antibody (3456; Cell Signaling Technology, Inc., Danvers, MA, USA) or rabbit anti-DNMT1 antibody (SC20701; Santa Cruz Biotechnology, Dallas, TX, USA) and 20 μl protein magnetic beads were added to the chromatin solution and incubated overnight at 4°C with rotation. The protein/DNA complexes were then washed with wash buffer 4 times and eluted using ChIP Elution Buffer. Cross-links were then reversed to free DNA by the addition of 1μl proteinase K and incubation at 62°C for 2 hours and incubation at 95°C for 10 minutes. The DNA was purified according to the manufacturer’s protocol. 50 μl of DNA was obtained for each treatment. A measure of 1 μl of DNA was used in PCR using Master Mix (Promega Life Sciences, Madision, WI, USA). The following primer sequences were used: Trkb forward 5′-AGCAGCGGCCCGCGATGTCCC-3′ and reverse 5′- CCCGAGCTGCCAGTGCCAGTC-3′; Exon II CpG forward 5′-AGAGCCCTCGAAAGTGTCAG-3′ and reverse 5′-GGGGACACGTGAGATGATTC-3′. All primers were initially tested for their specificity by running PCR samples on an agarose gel.
Statistical analysis
Normal distribution was tested using the Kolmogorov–Smirnov test, and variance was compared. Unless otherwise stated, statistical significance was determined by two-tailed unpaired Student’s t-test for two-population comparison and one-way or two-way analysis of variance (ANOVA) followed by the Bonferonni post hoc test for multiple comparisons. Data were analysed using GraphPad Prism (GraphPad Software, Inc. La Jolla, CA, USA) and presented as mean (+/−) SEM. P values were indicated in figure legends.
Results
Ankrd11 knockdown delays radial migration of cortical pyramidal neurons
To investigate the role of ANKRD11 in pyramidal neuron development, we knocked down Ankrd11 expression in pyramidal neural progenitors using an shAnkrd11 construct. A plasmid encoding non-silencing shRNA was used as a control. The validity of shAnkrd11 was tested by Western blotting. The level of ANKRD11 in shAnkrd11-transfected cell lysates was 63% lower than the control level (Fig. 1A, B). Immunostaining on shAnkrd11-electroporated tissues or dissociated cells confirmed these results (Fig. S1). By using in utero electroporation of shAnkrd11 into the E14.5 mouse brain, we introduced the shRNA construct to pyramidal neural progenitors and migrating neurons as previously described (Ka et al., 2016a; Ka and Kim, 2016) (Fig. 1C). Between P5 and P7, migrating pyramidal neurons settle in their appropriate cortical layers and start projecting dendrites and axons (Kasper et al., 1994). To determine the regulatory role of ANKRD11 in early neuronal positioning, we collected brain tissues at P7. Both the shAnkrd11 and control shRNA constructs encode GFP in a separate reading frame, allowing tracing of electroporated cells in the cerebral cortex. GFP-labeled control neurons were mostly positioned in layers II–IV of the cerebral cortex with the highest number in layers II/III at P7 (Fig. 1D, E). Neurons expressing shAnkrd11, however, were found throughout layers II–V of the cerebral cortex. We examined whether this positioning defect in ANKRD11-deficient neurons is transient in nature or persists into later postnatal stages. Neurons expressing shAnkrd11 were mostly localized within layers II–IV of the cortical plate at P21, similar to the pattern observed in control neurons (Fig. 1F). The numbers of GFP-positive neurons in each layer were not significantly different between control and shAnkrd11 brain samples (Fig. 1G). These results indicate that ANKRD11 deficiency delays neuronal migration and positioning during cortical development.
Fig. 1. Ankrd11 knockdown effects on neuronal migration and positioning in the developing cerebral cortex.
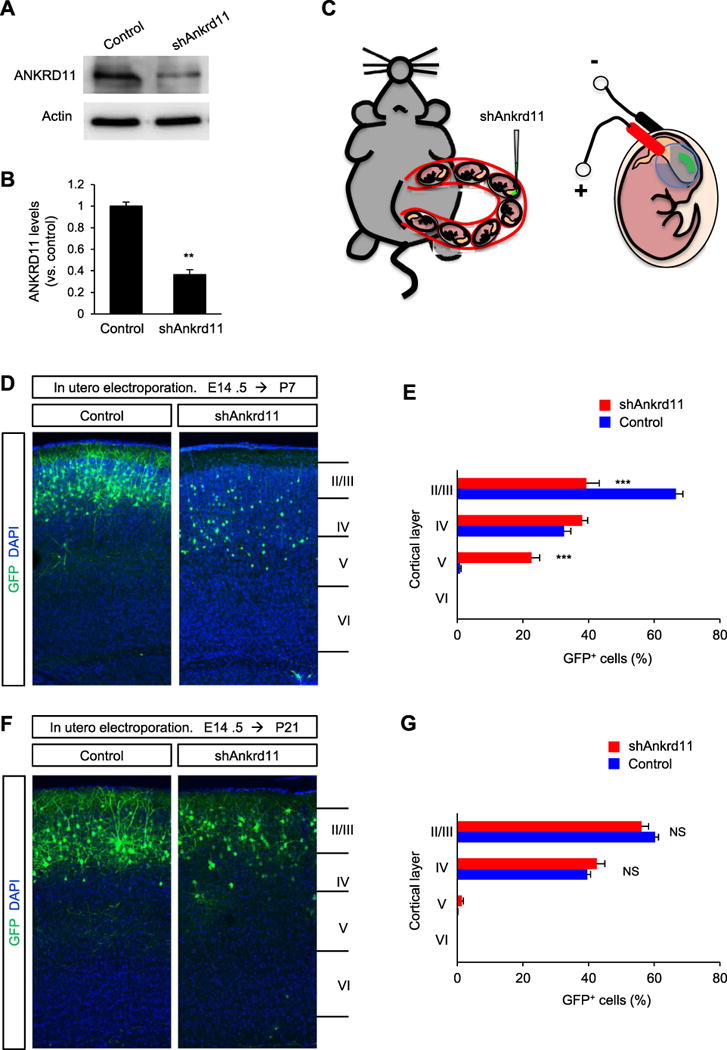
(A) shAnkrd11 knocks down endogenous ANKRD11 in cortical neurons. Isolated cortical cells from E14.5 mice were transfected with non-silencing shRNA (control) or shAnkrd11, and cultured for 7 days. Cellular lysates were then subjected to Western blotting. (B) Quantification of (A). The band intensities were measured using ImageJ. N= 3 independent cultures using 3 mice. Statistical significance was determined by two-tailed Student’s t-test. **p < 0.01. (C) A cartoon showing the procedure of in utero electroporation of shRNAs into the embryonic brain. (D) shRNA-mediated Ankrd11 knockdown results in abnormal positioning of pyramidal neurons during brain development. E14.5 mouse brains were electroporated in utero with the control or shAnkrd11 construct. The in utero electroporated mice were sacrificed at P7 and the brain samples were collected. GFP-positive neurons were assessed in the cerebral cortex. Scale bar: 50 μm. (E) Quantification of neuron positions in cortical layers. N= 5 mice for each condition. Statistical significance was determined by multiple t-tests with the Bonferroni correction test. Data shown are mean (+/−) SEM. ***p< 0.001. (F) Positioning of shAnkrd11-electroporated neurons was restored at P21. E14.5 mouse brains were electroporated in utero with control or shAnkrd11 construct. Neuronal positioning was assessed in the P21 cerebral cortex. Scale bar: 50 μm. (G) Quantification of neuron positions shown in (F). N= 5 mice for each condition. Statistical significance was determined by multiple t-tests with the Bonferroni correction test. NS: no significance.
Ankrd11 knockdown impairs leading process formation and multipolar-bipolar transition in migrating neurons
The leading process functions as a “compass” in migrating neurons and determines the direction of migration during brain development (Marin et al., 2010). Thus, we investigated leading process development in radially migrating ANKRD11-deficient pyramidal neurons. We in utero electroporated control or shAnkrd11 constructs into E14.5 embryos, and assessed leading processes at E17.5. While control neurons displayed a single leading process, ANKRD11-deficient neurons had multiple shorter processes (Fig. 2A, B). The length of leading processes was decreased in ANKRD11-deficient neurons by 58%, compared with control neurons (Fig. 2C). However, the number of leading processes was increased in ANKRD11-deficient neurons by 190%. These results show that ANKRD11 regulates leading process formation in radially migrating pyramidal neurons, and suggest that abnormal leading processes contribute to the delayed migration in ANKRD11-deficient pyramidal neurons. During radial migration, pyramidal neurons undergo a transition from multipolar to bipolar morphology in the intermediate zone, where they rapidly extend leading processes. In E17.5 control brains, 73% of neurons displayed bipolar morphology while 27% of neurons in the intermediate zone exhibited multipolar morphology. ANKRD11 deficiency decreased the number of bipolar neurons by 41% compared with the control condition (Fig. 2D), demonstrating that pyramidal neurons require ANKRD11 for a multi-to bipolar transition in the developing brain.
Fig. 2. ANKRD11 is necessary for leading process development.
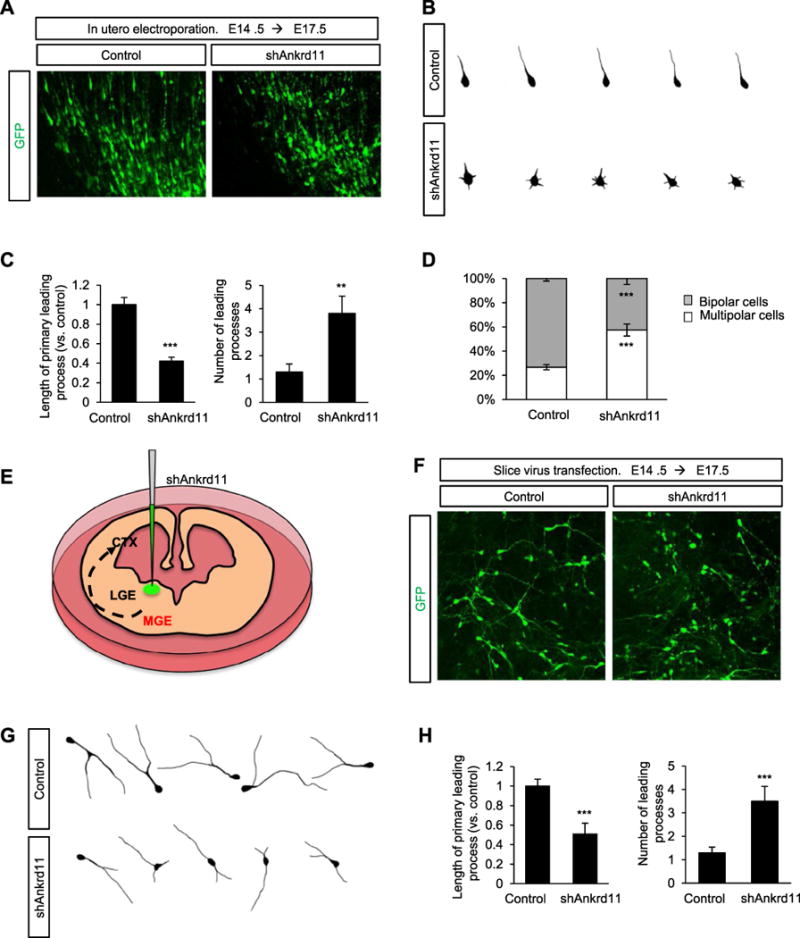
(A) Ankrd11 knockdown induces multiple shorter leading processes in radially migrating pyramidal neurons. A construct encoding control or shAnkrd11 was electroporated in utero into E14.5 mice. Migrating neurons expressing GFP were visualized at E17.5. Scale bar: 25 μm. (B) Representative morphologies of control and shAnkrd11-expressing pyramidal neurons with their leading processes. (C) Quantification of the length and number of leading processes in control and ANKRD11-deficient neurons. N= 163 cells from 5 mice for control, and N= 168 cells from 5 mice for shAnkrd11. Statistical significance was determined by two-tailed Student’s t-test. **p < 0.01, ***p < 0.001. (D) Ankrd11 knockdown disrupts multipolar to bipolar transition. After electroporation of control or shAnkrd11 construct into E14.5 mice, multipolar or bipolar morphology of pyramidal neurons was assessed in the intermediate zone of E17.5 brains. The numbers of neurons with multipolar or bipolar morphologies were quantified. N= 284 cells from 5 mice for control, and N= 324 cells from 5 mice for shAnkrd11. Statistical significance was determined by two-tailed Student’s t-test. ***p < 0.001. (E) A cartoon showing a lentiviral injection of shRNAs onto the MGE of cultured brain slices. (F) Ankrd11 knockdown induces abnormal leading processes in migrating interneurons. Control or shAnkrd11 lentivirus was infected into the MGE region of the cultured E14.5 brain slice, and leading process morphology was assessed after 3 days. Scale bar: 25 μm. (G) Representative morphologies of control and shAnkrd11 lentivirus-infected interneurons in the cerebral cortex. (H) Quantification of the length and number of leading processes. N= 78 cells from 3 mice for control, and N= 84 cells from 3 mice for shAnkrd11. Statistical significance was determined by two-tailed Student’s t-test. ***p < 0.001.
Next, we investigated leading processes in tangentially migrating ANKRD11-deficient interneurons. Unlike radially migrating pyramidal neurons, tangentially migrating interneurons generally have a single leading process with a long branch (Martini et al., 2009; Moffat et al., 2015; Rakic, 1972). We injected a lentivirus containing shAnkrd11 into the medial ganglionic eminence (MGE) region of E14.5 brain slices and assessed leading process morphology in the cerebral cortex 3 days later (Fig. 2E). Most control interneurons formed a typically branched single leading process. However, ANKRD11-deficient neurons developed multiple short leading processes (Fig. 2F, G). The length of the primary leading process was reduced in ANKRD11-deficient interneurons by 49%, compared with control interneurons. However, the number of leading processes was increased in ANKRD11-deficient interneurons by 169%, compared with controls (Fig. 2G). These results revealed the importance of ANKRD11 in leading process morphogenesis in migrating pyramidal neurons and interneurons.
ANKRD11 is required for dendrite arborization in cortical pyramidal neurons
After being positioned in appropriate cortical layers, the leading processes of migrating pyramidal neurons differentiate into primary apical dendrites and generate branches (Polleux and Snider, 2010). Between P7 and P14, the apical and basal dendrites of pyramidal neurons rapidly grow and develop branches (Kasper et al., 1994; Ramaswamy and Markram, 2015). The abnormal leading processes in ANKRD11-deficient neurons led us to examine their dendrite morphology. We injected the control or shAnkrd11 construct into the lateral ventricles of E14.5 mice. After electroporation, we collected brain tissues at P14 and assessed dendrite outgrowth and arborization patterns. In control brains, pyramidal neurons developed primary apical and basal dendrites, and also extended secondary and tertiary apical dendrites, as well as secondary basal dendrites (Fig. 3A, B). However, ANKRD11-deficient neurons generated fewer numbers of dendrites with a shorter and thinner morphology. The numbers of primary, secondary and tertiary dendrites in ANKRD11-deficient neurons were decreased by 21%, 72%, and 96%, respectively, compared with dendrite numbers in control neurons (Fig. 3 C). The lengths of primary apical, total apical and overall total dendrites were decreased by 20%, 68% and 63%, respectively, as well. Moreover, ANKRD11-deficient neurons did not develop tertiary apical dendrites. The thickness of primary dendrites in ANKRD11-deficient neurons was reduced by 59%, compared with control dendrite thickness. This abnormal pattern of dendrite outgrowth and dendritic arborization in ANKRD11-deficient pyramidal neurons persisted into the later postnatal stages, P21 and P42 (Fig. S2A, B). Additionally, we confirmed the role of ANKRD11 in dendrite arborization in cultured neurons. Control or shAnkrd11 construct was transfected into cultured cortical neurons at DIV3 (day in vitro 3). Dendritic morphology was assessed in cultures fixed at DIV7 (Fig. S3A). Similar to the in vivo findings, the numbers of secondary and tertiary dendrites were decreased in ANKRD11-deficient neurons, by 50% and 62%, respectively, compared with control neurons (Fig. S3B), suggesting that the shAnkrd11 effect is cell autonomous. The length of total dendrites was also reduced in ANKRD11-deficient neurons by 66%. Together, these results suggest a requirement for ANKRD11 in dendrite outgrowth and arborization in developing pyramidal neurons.
Fig. 3. Ankrd11 knockdown suppresses dendrite differentiation.
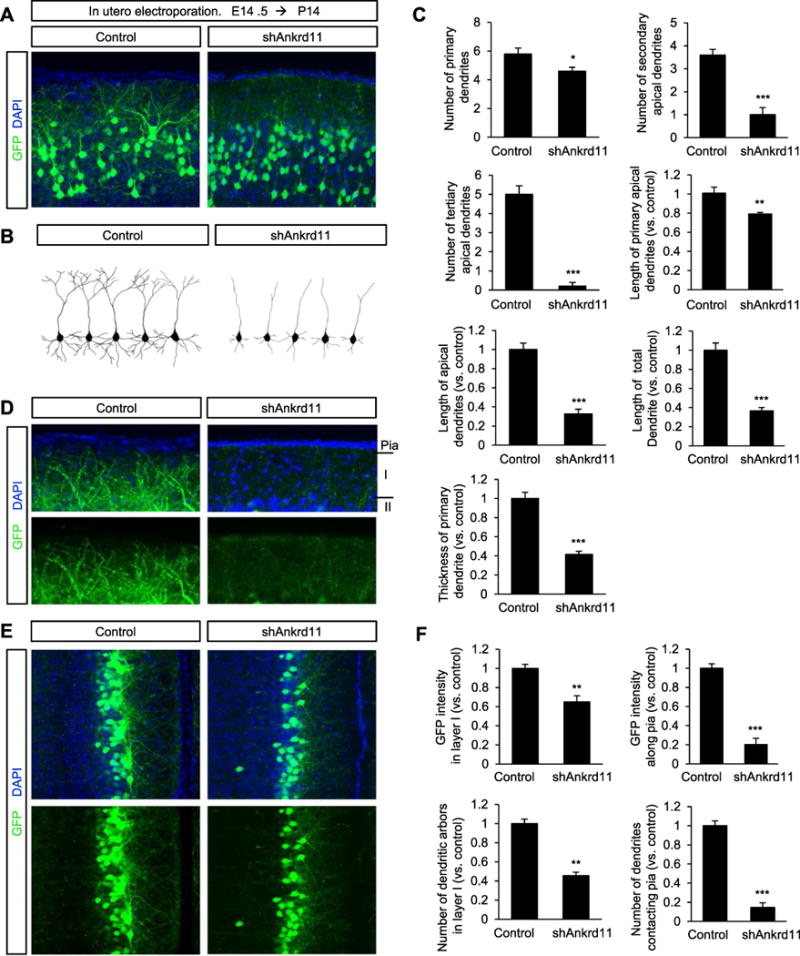
(A) Elimination of Ankrd11 inhibits dendrite outgrowth and arborization in developing pyramidal neurons. Mouse brains were electroporated in utero with control or shAnkrd11 at E14.5 and collected at P14. Dendrites in pyramidal neurons expressing GFP were assessed. Scale bar: 25 μm. (B) Representative morphologies of control and shAnkrd11-expressing pyramidal neurons. (C) The dendrite numbers and lengths were quantified. N= 70 pyramidal neurons from 5 mice for each condition. Statistical significance was determined by two-tailed Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001. (D,E) Dendrite innervation patterns in cortical layer I at P14. (D) Lateral cortex. Scale bar: 10 μm. Arrowheads show the pial surface where dendritic tufts make contacts. (E) Medial cortex. Scale bar: 50 μm. (F) Quantification of dendrite innervation in control layer I. The dendrite numbers and GFP intensity within layer I and along the pia were measured using Image J. N= 5 mice for each condition. Statistical significance was determined by two-tailed Student’s t-test. **p < 0.01. ***p < 0.001.
Cortical layer I originates from the marginal zone during development and receives long-range axon inputs from subcortical regions, which are important for cognitive processing (Baluch and Itti, 2011; Larkum, 2013; Shipp, 2007; van Gaal and Lamme, 2012; Wyss et al., 1990). We further examined dendrite arborization in cortical layer I. After in utero electroporation of shAnkrd11 into E14.5 mice, we collected brain tissues at P14 and assessed dendrites within cortical layer I in the lateral cerebral cortex. In control brains, many dendritic branches from layer II/III pyramidal neurons were elaborated into layer I and attached to the pial surface (Fig. 3D). In contrast, there was a marked reduction in ANKRD11-deficient dendritic branches within layer I. We observed similar innervation abnormalities in ANKRD11-deficient dendrites in the medial cerebral cortex (Fig. 3E). The GFP-positive areas in layer I and along the pial surface in shAnkrd11-electroporated brains were decreased by 35% and 80%, respectively, compared with control brains (Fig. 3F). The numbers of dendrites in layer I and contacting the pial surface were decreased by 55% and 86%, respectively, in shAnkrd11-electroporated brains.
ANKRD11 deficiency results in abnormal spine morphology and number
Dendrites are a major platform on which spines form to receive synaptic inputs. Thus, we sought to determine whether ANKRD11 plays a role in the formation and morphology of dendritic spines. We transfected cultured cortical neurons with shAnkrd11 at DIV5 and fixed them at DIV14, when spines develop in culture. ANKRD11-deificent neurons displayed a 69% decrease in the number of spines compared with control neurons (Fig. 4A, B). Interestingly, ANKRD11-deficient spines appeared longer with smaller heads and narrow necks in comparison to control spines (Fig. 4C). Spine length was increased by 25% in ANKRD11-deficient neurons (Fig. 4D). However, the sizes of spine head and neck were reduced by 64% and 32%, respectively, in ANKRD11-deficient neurons. Next, we sought to investigate whether ANKRD11 could alter the remodeling of dendritic spine morphology. Dendritic spine morphology can be classified into 4 different categories; filopodia, thin, mushroom, or stubby spines (Ka et al., 2016b). Filopodia are typically longer (>2 μm) without a clear head; thin spines have a thin and long neck (>1 μm) and a small head; mushroom spines have a short and narrow neck (<1 μm) and a large head (>0.6 μm) while the stubby spines have a head but no neck (Fig. 4E). ANKRD11-deificent neurons displayed significant reductions in mushroom, thin and stubby spines compared with control neurons (Fig. 4F). The numbers of thin mushroom and stubby spines were decreased by 40%, 72% and 67%, respectively, in ANKRD11-deficient neurons. However, the number of filopodia was increased by 480% in ANKRD11-deficient neurons. Together, these results show that ANKRD11 deficiency leads to dendritic spine malformation.
Fig. 4. Role of ANKRD11 in spine morphogenesis.
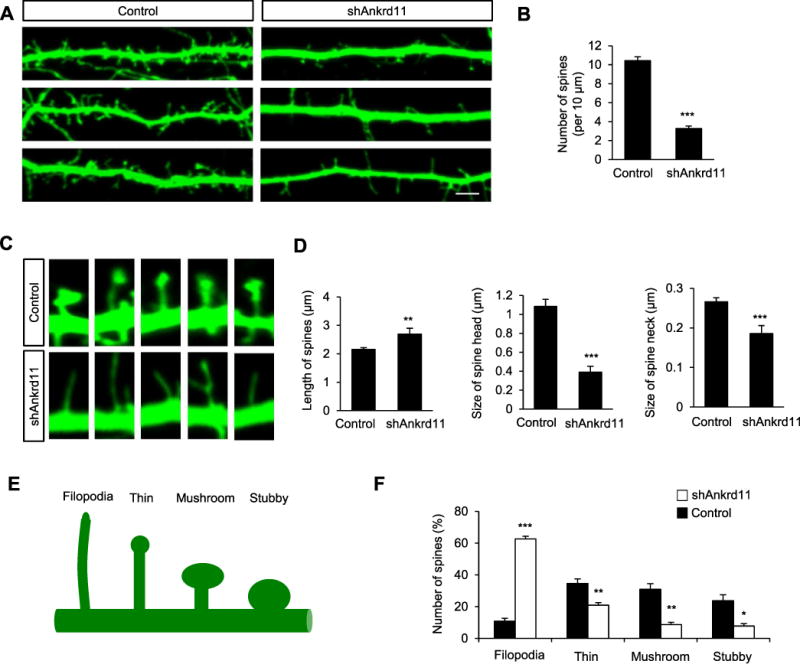
(A) Ankrd11 knockdown decreases the number of dendritic spines in cultured cortical neurons. Cortical neurons were cultured from E16.5 mice, and transfected with control or shAnkrd11 at DIV5. GFP-positive dendrites and spines were assessed at DIV14. Scale bar: 5 μm. (B) The number of dendritic spines was quantified. N= 100 dendrites from 30 neurons using 3 mice for each condition. Statistical significance was determined by two-tailed Student’s t-test. ***p < 0.001. (C) Higher magnification images of dendritic spines. Elimination of Ankrd11 leads to abnormal spine morphology. (D) Quantification of spine length, head and neck sizes. (E,F) Different spine types were also quantified. N=884 spines from 30 neurons using 3 mice for control, and N=825 spines from 30 neurons using 3 mice for shAnkrd11. Statistical significance was determined by two-tailed Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
ANKRD11 is required for acetylation of Histone H3 and p53
Histone acetylation plays an important role in neurite growth (Siebzehnrubl et al., 2007; Trakhtenberg and Goldberg, 2012). Given its role as a chromatin modifier, we investigated whether or not ANKRD11 regulates histone acetylation in cortical neurons. We knocked down Ankrd11 in cultured neurons using a lentivirus-mediated shRNA system, and examined the level of acetylated-Histone H3 in culture lysates by Western blotting. ANKRD11-deficient neurons demonstrated a 68% decrease in the level of acetylated-Histone H3 compared with control neurons (Fig. 5A, B). p53 is part of the histone acetylation complex, and p53 acetylation enhances transcription of genes that are associated with neurite growth (Gaub et al., 2010). Thus, we examined whether ANKRD11 interacts with p53 in cortical neurons. P1 cortical lysates were co-immunoprecipitated with an ANKRD11 antibody, and subsequently immunoblotted with ANKRD11 and p53 antibodies. We found that ANKRD11 was indeed physically associated with p53 in neurons (Fig. 5C). The level of acetylated-p53 was decreased by 54% in ANKRD11-deficient neurons (Fig. 5D, E). These results show that ANKRD11 is required for Histone H3 and p53 acetylation in cortical neurons.
Fig. 5. Ankrd11 knockdown disrupts Histone H3 and p53 acetylation in cortical neurons.
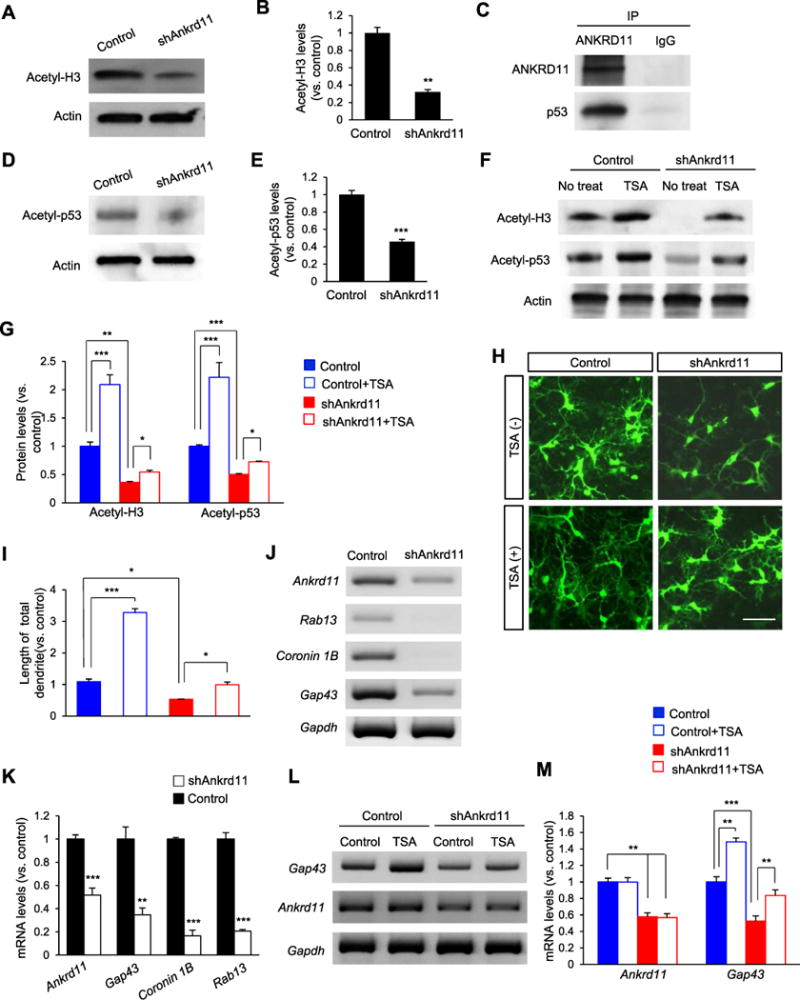
(A) Ankrd11 knockdown decreases Histone H3 acetylation. Cortical neurons from E16.5 mice were cultured and infected with a lentivirus expressing control or shAnkrd11 at DIV3. Cellular lysates from the cultures at DIV6 were subjected to Western blotting. (B) Quantification of acetyl-Histone H3. The acetylation level was normalized to Actin expression. The band intensities were measured using Image J. N= 3 independent cortical cultures using 3 mice for each condition. Statistical significance was determined by two-tailed Student’s t-test. **p < 0.01. (C) ANKRD11 interacts with p53. E16.5 brain lysates were immunoprecipitated with an ANKRD11 antibody and subsequently subjected to Western blotting using either a p53 or an ANKRD11 antibody. (D) Ankrd11 knockdown reduces p53 acetylation. Western blotting was performed to measure the level of acetyl-p53 using control and ANKRD11-deficent neuron lysates. (E) Quantification of (D). N= 3 independent cortical cultures using 3 mice for each condition. Statistical significance was determined by two-tailed Student’s t-test. ***p < 0.001. (F) TSA treatment increases the acetylation levels of Histone H3 and p53 in ANKRD11-deficient neurons. E16.5 cortical neurons were cultured and infected with control or shAnkrd11 lentivirus. Neurons were treated with TSA at DIV3. Cellular lysates were collected at DIV6 and subjected to Western blotting. (G) Quantification of acetyl-Histone H3 and acetyl-p53 levels. Statistical significance was determined by two-way ANOVA followed by the Bonferroni post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001. (H) TSA increases dendrite outgrowth in ANKRD11-deficient neurons. Cortical neurons from E16.5 mice were cultured and infected with control or shAnkrd11 lentivirus. Neurons were treated with TSA at DIV3 and fixed at DIV6. Scale bar: 25 μm. (I) Quantification of dendrite length. N= 50 neurons from 3 independent cultures using 3 mice. Statistical significance was determined by two-way ANOVA followed by the Bonferroni post hoc test. *p < 0.05, ***p < 0.001. (J) Expression of p53 target genes. RT-PCR was performed to measure mRNA levels of Gap43, Coronin 1b and Rab13, using cortical lysates from control and shAnkrd11 lentivirus cultures. (K) Quantification of (J). The mRNA levels were normalized to Gapdh expression. N= 3 independent cortical cultures using 3 mice. Statistical significance was determined by two-tailed Student’s t-test. **p < 0.01, ***p < 0.001. (L) TSA increases the level of Gap43 mRNA in ANKRD11-deficient neurons. RT-PCR was performed using cortical lysates. (M) Quantification of mRNA levels shown in (L). The levels of mRNA were normalized to Gapdh expression. N= 3 independent cortical cultures using 3 mice. Statistical significance was determined by two-way ANOVA followed by the Bonferroni post hoc test. **p < 0.01, ***p < 0.001..
Next, we sought to determine whether histone deacetylases (HDACs) are involved in ANKRD11-mediated histone and p53 acetylation. We treated control and ANKRD11-deficient neurons with a histone deacetylase inhibitor TSA, and measured the acetylation levels of Histone H3 and p53. As expected, ANKRD11-deficiency decreased acetylation of Histone H3 and p53 by 64% and 53%, respectively, compared with the control condition (Fig. 5F, G). In control neurons, TSA treatment increased acetylated Histone H3 and p53 by 115% and 122%, respectively, compared with no treatment condition. TSA treatment also increased the levels of acetylated Histone H3 and p53 in ANKRD11-deficient neurons, however, the acetylation levels increased by TSA in ANKRD11-deficient neurons were not comparable to the increases seen in TSA-treated control neurons. These results suggest that ANKRD11 regulates histone acetylation by modulating not just HDACs but other molecules including histone acetyltransferases. To examine whether ANKRD11-mediated histone acetylation is functionally relevant, we assessed dendritic outgrowth in ANKRD11-deficient cortical neurons after TSA treatment. Cultured cortical neurons were infected with control or shAnkrd11 lentivirus at DIV3, and treated with TSA at DIV5 for 2 days. While Ankrd11 knockdown decreased dendrite outgrowth, TSA treatment suppressed the inhibitory effect of Ankrd11 knockdown on dendrite outgrowth (Fig. 5H). In control neurons, TSA treatment increased the length of dendrite by 237% compared with the untreated condition (Fig. 5I). In ANKRD11-deficient neurons, the dendritic length following TSA treatment was increased by 41%, compared the length in untreated neurons.
Finally, using RT-PCR we measured the mRNA levels of p53 target genes Gap43, Coronin 1B and Rab13 in ANKRD11-deficient neurons., We found that the levels of Gap43, Coronin 1B and Rab13 were decreased by 65%, 83% and 79%, respectively, in ANKRD11-deficient neurons compared with those in control neurons (Fig. 5J, K). We also examined whether inhibition of HDAC activity by TSA rescues the level of Gap43 mRNA in ANKRD11-deficient neurons. TSA treatment increased the Gap43 level by 59% in ANKRD11-deficient neurons, compared with untreated condition (Fig. 5L, M). TSA treatment had no effect on Ankrd11 expression. Together, these results indicate that ANKRD11 regulates the acetylation of Histone H3 and p53 and the expression of p53-mediated neural target genes in cortical neurons, and suggest that this acetylation is associated in part with HDAC activity.
ANKRD11 regulates BDNF/TrkB signaling in cortical neurons
BDNF/TrkB signaling plays a key role in dendrite outgrowth and spine differentiation. Thus, we sought to investigate if BDNF/TrkB signaling is altered in ANKRD11-deficient neurons. Lentivirus-mediated knockdown of Ankrd11 was performed in cultured cortical neurons. Using RT-PCR, we first measured the levels of Bdnf and Trkb mRNAs in cell lysates. We found that Bdnf and Trkb levels were reduced by 49% and 62%, respectively, in ANKRD11-deficient neurons, compared with the levels in control neurons (Fig. 6A, B), suggesting a potential role of ANKRD11 in BDNF/TrkB signaling. Thus, we examined BDNF/TrkB signaling in ANKRD11-deficient neurons by measuring the activities of its downstream kinases and effectors. Western blotting showed that phosphorylation levels of AKT, GSK-3, S6K, and S6 were reduced in ANKRD11-deficient neurons by 35%, 35%, 41%, and 46%, respectively, compared with the levels in control neurons (Fig. 6C, D). Similarly, the level of phospho-S6 was reduced in ANKRD11-deficient neurons. These results indicate that Ankrd11 knockdown suppresses BDNF/TrkB signaling.
Fig. 6. Ankrd11 knockdown induces abnormal BDNF/TrkB signaling.
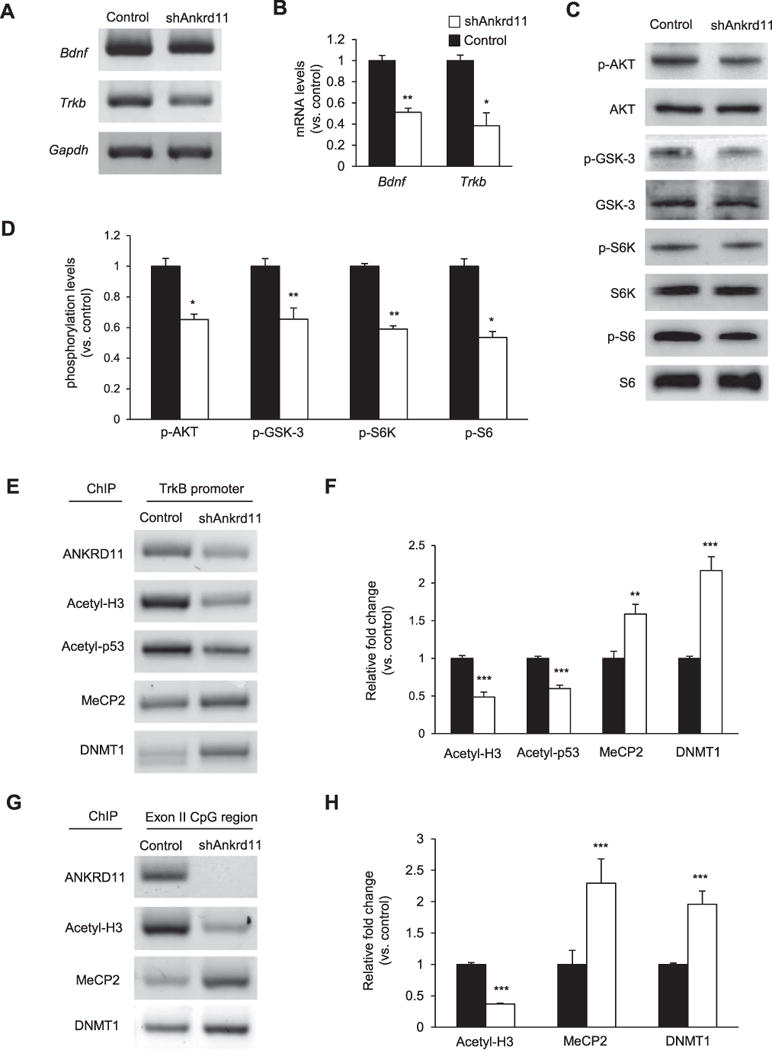
(A) Ankrd11 knockdown decreases the levels of bdnf and Trkb mRNAs. E16.5 cortical neurons were cultured and infected with control or shAnkrd11 lentivirus. RT-PCR was performed using total RNAs from cellular lysates (B) Quantification of mRNA levels shown in (A). The relative mRNA levels were normalized to Gapdh expression. N= 3 independent cortical cultures using 3 mice. Statistical significance was determined by two-tailed Student’s t-test. *p < 0.05, **p < 0.01. (C) Ankrd11 knockdown decreases the phosphorylation levels of BDNF/TrkB signal transducers. Western blots shows phosphorylation of AKT, GSK-3, S6K, and S6 in control and ANKRD11-deficient neurons. (D) Quantification of (C). The protein levels were normalized to total protein levels. N= 3 independent cultures using 3 mice. Statistical significance was determined by two-tailed Student’s t-test. *p < 0.05, **p < 0.01. (E) Levels of epigenetic regulators on the Trkb promoter. ChIP assays were performed using neuronal lysates and an antibody to ANKRD11, acetyl-Histone H3, acetyl-p53, MeCP2, or DNMT1, followed by PCR to measure the levels of the Trkb promoter associated with the epigenetic proteins. (F) Quantification of (E). The relative levels were normalized to the input fraction of the acetyl-Histone H3. N= 3 independent cortical cultures using 3 mice. Statistical significance was determined by two-tailed Student’s t-test. **p < 0.01, ***p < 0.001. (G) Levels of epigenetic regulators on the CpG region of the Trkb exon II. ChIP assays were performed and the CpG region was amplified using PCR. (H) Quantification of (G). The relative levels were normalized to the input fraction of acetyl-Histone H3. N= 3 independent cortical cultures using 3 mice. Statistical significance was determined by two-tailed Student’s t-test. ***p < 0.001.
Next, we examine whether ANKRD11 accesses the Trkb promoter in cortical neurons. We performed the chromatin immunoprecipitation assay (ChIP) using an ANKRD11 antibody followed by PCR to amplify the Trkb promoter region. We found that ANKRD11 binds to the Trkb promoter (Fig. 6E). As expected, the level of ANKRD11 binding was decreased in the Ankrd11 knockdown condition. The levels of acetylated Histone H3 and p53 within the promoter region were also decreased by 51% and 40% in ANKRD11-deficient neurons (Fig. 6E, F). Additionally, we found altered binding levels of DNA methylation regulators in the Trkb promoter region. Ankrd11 knockdown resulted in increased binding of methyl-CpG-binding protein 2 (MeCP2) and DNA methyltransferase 1 (DNMT1) to the promoter, suggesting suppression of Trkb transcription. Using the ChIP assay, we further investigated the access of ANKRD11 and acetylated Histone H3 to the exon 2 CpG region, which is a site of DNA methylation. We found that ANKRD11 bound to the Trkb exon 2 CpG region while Ankrd11 knockdown markedly decreased the binding (Fig. 6G, H). Acetylated Histone H3 and MeCP2 showed similar binding patterns in this region as in the Trkb promoter, again suggesting transcription suppression of the Trkb gene by Ankrd11 knockdown.
TrkB overexpression suppresses the Ankrd11 knockdown effect on dendrites
The inhibitory role of Ankrd11 knockdown on TrkB signaling led us to test if overexpression of TrkB could attenuate the dendrite abnormalities we observed in ANKRD11-deficient neurons. We performed in utero electroporation using control, shAnkrd11 or shAnkrd11 and TrkB plasmid constructs into E14.5 brains. Brain tissues were collected at P14 and dendrite morphology was assessed in cortical layers. As shown earlier, shAnkrd11 expression decreased the length of dendrites by 67%, compared with the control condition (Fig. 7 A–C). Co-expression of TrkB and shAnkrd11 restored dendritic growth compared with shAnkrd11 alone. The dendrite length was increased by 112% in the co-expression condition compared with shAnkrd11 alone, however it did not reach to the level of the control condition, suggesting a partial contribution of TrkB signaling in ANKRD11-mediated dendritic growth. The number of tertiary dendrites, an indication of dendritic arborization, was also partially rescued by co-expression of TrkB in ANKRD11-deficient neurons. We confirmed these results in cultured cortical neurons (Fig. S4). Our results show that activation of TrkB signaling via TrkB transcription regulation is an important mechanism for ANKRD11-mediated dendritic arborization.
Fig. 7. TrkB overexpression rescues shAnkrd11-mediated inhibition of dendrite outgrowth.
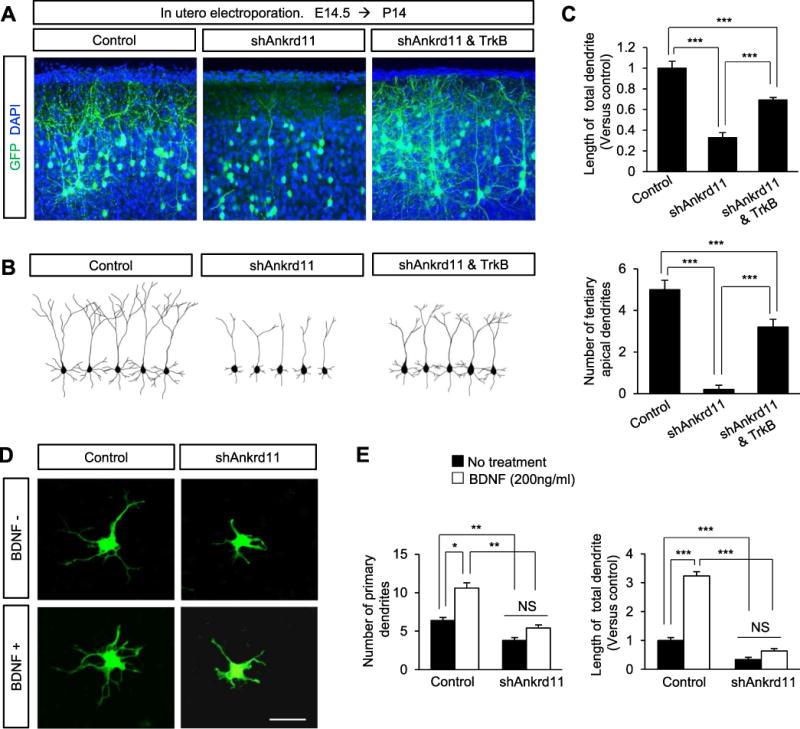
(A) TrkB expression partially restores abnormal dendrites induced by shAnkrd11 in vivo. E14.5 mice were electroporated in utero with control, shAnkrd11 alone, or shAnkrd11 and TrkB constructs. The electroporated brains were collected at P14, and dendrite morphologies in pyramidal neurons expressing GFP were assessed. Scale bar: 25 μm. (B) Representative morphologies of pyramidal neurons and their dendrites shown in (A). (C) The dendrite numbers and lengths of dendrites were quantified. N= 70 neurons from 5 mice for each condition. Statistical significance was determined by one-way ANOVA followed by the Bonferroni post hoc test. ***p < 0.001. (D) BDNF treatment has no effect on dendrite outgrowth in ANKRD11-deficient neurons. Cortical neurons from E16.5 mice were cultured and transfected with control or shAnkrd11. Neurons were treated with BDNF for 3 days before fixation. Scale bar: 10 μm. (E) Quantification of dendrite numbers and lengths in (D). N= 70 cells from 3 independent cultures for each condition. Statistical significance was determined by two-way ANOVA followed by the Bonferroni post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
We also examined whether BDNF treatment rescues abnormal dendrite outgrowth in ANKRD11-deficient neurons. Cultured cortical neurons were treated with 200 ng/ml BDNF (Hoshino et al., 2010; Runge et al., 2012; Sandoval et al., 2011; Suzuki et al., 2007; Suzuki et al., 2004) for 3 days, and dendrite outgrowth was assessed. BDNF treatment increased the number of primary dendrites and the length of total dendrites by 66% and 223%, respectively, in control neurons compared with the untreated condition (Fig. 7D, E). However, while BDNF treatment showed a tendency toward increasing dendrite outgrowth in ANKRD11-deficient neurons, it did not significantly change the number or length of dendrites. We also assessed BDNF concentration-responses on dendrite outgrowth. BDNF treatment at any concentration (0, 50, 100, or 200 ng/ml) increased dendrite numbers in control neurons, but again showed no difference in ANKRD11-deficient neurons (Fig. 5S).
Discussion
In this study, we showed that ANKRD11 regulates dendrite outgrowth, arborization, and spine formation via Trkb transcription and activation of its downstream effectors in the developing mouse brain. We found that ANKRD11 knockdown results in decreased acetylation of Histone H3 at the Trkb promoter locus and altered access for epigenetic modifiers to the same promoter in cortical neurons. We propose a schematic model of epigenetic regulation during dendrite development in Fig. 8. In the presence of ANKRD11, the histone acetylation complex containing ANKRD11, p53 and HATs actively acetylates histone molecules in the promoters of dendrite outgrowth-associated genes including Trkb, resulting in transcription of these genes and the activation of TrkB signaling. Without ANKRD11, however, the promoter regions are devoid of histone acetylation and occupied by DNA methylation-associated proteins such as MeCP2 and DNMT1, resulting in transcriptional inhibition. Our results provide insight into the pathogenic mechanism underlying ANKRD11-associated developmental disability.
Fig. 8. A schematic model illustrating the ANKRD11 role in dendrite outgrowth.
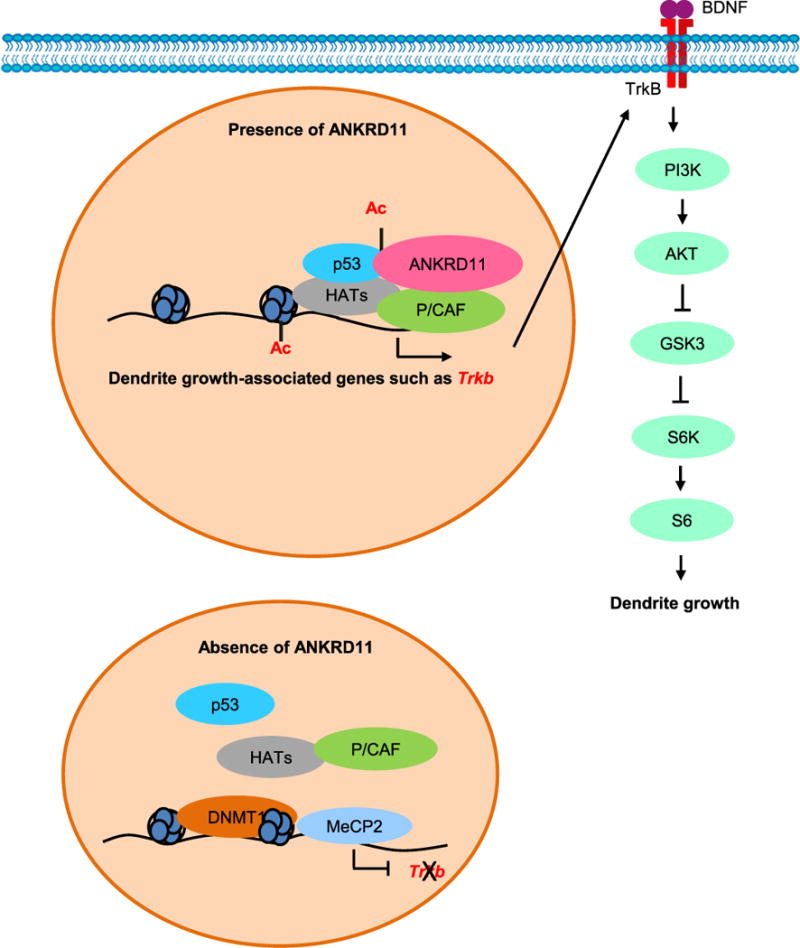
In the presence of ANKRD11, Histone H3 and p53 are acetylated in the promoter regions of dendrite-associated genes such as Trkb, which results in transcription of the genes. Increased Trkb expression activates TrkB signaling leading to the growth and arborization of cortical dendrites. In the absence of ANKRD11, Hisone H3 acetylation is suppressed. Gene expression repressors including MeCP2 and DNMT1 accumulate on dendrite-associated gene promoters and suppress expression of the genes. Thereby, dendritic outgrowth and arborization are inhibited via decreased TrkB signaling.
Genetic studies have shown that ANKRD11 deletion mutations induce neurodevelopmental disorders including ASD, intellectual disability and brain abnormalities (Isrie et al., 2012; Lo-Castro et al., 2013; Sirmaci et al., 2011; Walz et al., 2015; Willemsen et al., 2010; Youngs et al., 2011), suggesting its role in brain development. Indeed, ANKRD11 is expressed in neural precursors and neurons in the developing cortex, and knockdown of Ankrd11 decreases neural progenitor proliferation (Gallagher et al., 2015). Using an in utero gene delivery strategy, we showed that ANKRD11 is required for dendrite outgrowth and arborization in cortical pyramidal neurons at postnatal stages. The morphology of abnormal leading processes has been repeatedly linked to migration deficits and dendrite development (Friocourt et al., 2007; Nasrallah and Golden, 2006). Dendritic abnormality induced by Ankrd11 knockdown appears to originate during embryonic development. For example, ANKRD11-deficient neurons display a disrupted transition from multipolar to bipolar morphology in the developing brain. More specifically, Ankrd11 knockdown leads to the abnormal formation of leading processes that become apical dendrites and arborize in the postnatal brain, which supports our other observations. Many neurodevelopmental disorders are associated with abnormal dendrite architecture. Dendritic and spine abnormalities in cortical pyramidal neurons are the most consistent pathologic correlate in intellectual disability. Individuals with ASD or intellectual disability caused by Down, RETT, and fragile-X syndromes show altered dendritic arborization and spine formation (Huttenlocher, 1974; Kaufmann and Moser, 2000; Marin-Padilla, 1976; Pardo and Eberhart, 2007). The aberrant dendrites and spines in ANKRD11-deficient pyramidal neurons greatly resemble the unbranched dendritic and filopodia-like spine morphology in RETT, Down, and fragile-X syndrome mouse models (Irwin et al., 2002; Jentarra et al., 2010; McKinney et al., 2005). In addition to less dendritic branching, ANKRD11-deficient neurons exhibit markedly reduced dendrite innervation into cortical layer I and fewer attachments of dendritic terminals to the pial surface. Layer I circuitry receives inputs primarily from neurons in higher-order thalamic and cortical areas, and neurons in this layer preferentially increase their activity during attention-demanding processes (Baluch and Itti, 2011; Larkum, 2013; van Gaal and Lamme, 2012). Thus, decreased arborization and innervation of primary dendrites in layer I may play a key role in the generation of abnormal cognitive and social behavior associated with Ankrd11 gene deletions.
Studies have shown that epigenetic regulation plays important roles in neural differentiation (Chen et al., 2014; Guan et al., 2009; Hall et al., 2002; Hao et al., 2004; Hsieh et al., 2004; Lilja et al., 2013; Siebzehnrubl et al., 2007; Trakhtenberg and Goldberg, 2012; Yu et al., 2009). For example, histone acetylation and deacetylation are essential in the regulation of neuronal gene expression (Chen et al., 2014; Yu et al., 2009). HDAC Inhibitors promote neurogenesis, neurite outgrowth, synaptic plasticity and neural protection (Hall et al., 2002; Hao et al., 2004; Hasan et al., 2013; Hsieh et al., 2004). In other studies, HATs have been shown to be required for physiological neuronal outgrowth. Inactivation of HDACs using TSA promotes not only acetylation of Histone H3, but also stimulates Cbp, p300 and Pcaf gene expression by hyperacetylating their promoters, thereby facilitating gene transcription and neuronal outgrowth (Gaub et al., 2010; Hasan et al., 2013). Consistently, our results showed that ANKRD11-deficent neurons with reduced dendrites and branches have lower levels of acetylated Histone H3. p53 is a co-factor of histone acetylation complexes, and acetylated p53 is associated with neurite outgrowth (Di Giovanni et al., 2006; Gaub et al., 2010). The level of acetylated p53 is also decreased in ANKRD11-deficient neurons. TSA treatment partially rescues the levels of acetylated Histone H3 and p53 and the dendritic morphology in ANKRD11-deficient cortical neurons, suggesting that ANKRD11 may regulate dendrite differentiation via Histone H3 and p53 acetylation.
Neurite growth-related genes such as Trkb, Bdnf, Gap43, Coronin 1B, and Rab13 are downregulated in ANKRD11-deficient neurons. Of those, we note reduced levels of bdnf and Trkb mRNAs as well as activities of downstream proteins in ANKRD11-deficient neurons. BDNF/TrkB signaling is a well-established pathway in dendrite growth and differentiation processes such as spine development (Chakravarthy et al., 2006; Tyler and Pozzo-Miller, 2001; Xu et al., 2016; Zhou et al., 2006). Additionally, TrkB downstream effectors, AKT, GSK-3, and mTOR are also involved in neurite and growth cone regulation (Jung et al., 2015; Kim, 2015; Kim and Snider, 2008; Kim and Snider, 2011; Kim et al., 2006; Markus et al., 2002; Read and Gorman, 2009). Our data demonstrated that TrkB overexpression leads to a rescue of the dendrite number and length in ANKRD11-deficient neurons. However, BDNF treatment does not significantly elicit the same effect as TrkB overexpression notwithstanding tendencies toward growth promotion. Thus, the appropriate level of TrkB may act as a rate-limiting step for ANKRD11-mediated dendritic outgrowth and arborization in cortical neurons. Given that Ankrd11 knockdown suppresses acetylation of Histone H3 around the Trkb promoter region and access for acetylated p53 to the promoter, our results suggest a closed chromatin state around the promoter region in ANKRD11-deficient neurons. Interestingly, this unfavorable condition for transcription seems to be potentiated by the increased binding of MeCP2 and DNMT1 to the Trkb promoter. MeCP2 binds to methylated DNA and represses gene transcription (Nan et al., 1998). DNMT1 is a partner of MeCP2 for gene expression regulation (Kimura and Shiota, 2003). Consistently, the levels of MeCP2 and DNMT1 binding to the Trkb exon 2 CpG region are increased in ANKRD11-deficient neurons. These data underscore an important role of ANKRD11 in the regulation of dendrite-associated gene expression and provide insight into the molecular mechanism underlying ANKRD11-assoicated neurodevelopment disorders.
Supplementary Material
Highlights.
ANKRD11 is required for dendrite growth and arborization in cortical neurons.
ANKRD11 regulates acetylation of epigenetic molecules such as p53 and Histone H3 at the Trkb promoter.
Expression of Trkb, Bdnf, and neurite growth-related genes are regulated by ANKRD11.
Overexpression of TrkB rescues abnormal dendrite development in ANKRD11-deficient neurons.
Our findings demonstrate a novel role for ANKRD11 in neuron differentiation during brain development and provide insight into the pathogenic mechanism underlying KBG syndrome.
Acknowledgments
We thank Jeffrey Moffat for critical reading and advice. Research reported in this publication was supported by an award from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R01NS091220 to WYK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contribution
M.K. and W.K. conceived, designed, performed, analyzed, and wrote the study. W.K. supervised the work.
References
- Baluch F, Itti L. Mechanisms of top-down attention. Trends Neurosci. 2011;34:210–24. doi: 10.1016/j.tins.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Chakravarthy S, et al. Postsynaptic TrkB signaling has distinct roles in spine maintenance in adult visual cortex and hippocampus. Proc Natl Acad Sci U S A. 2006;103:1071–6. doi: 10.1073/pnas.0506305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, et al. 2-Bromopalmitate modulates neuronal differentiation through the regulation of histone acetylation. Stem Cell Res. 2014;12:481–91. doi: 10.1016/j.scr.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S et al. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006;25:4084–96. doi: 10.1038/sj.emboj.7601292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friocourt G, et al. Both doublecortin and doublecortin-like kinase play a role in cortical interneuron migration. J Neurosci. 2007;27:3875–83. doi: 10.1523/JNEUROSCI.4530-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher D, et al. Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev Cell. 2015;32:31–42. doi: 10.1016/j.devcel.2014.11.031. [DOI] [PubMed] [Google Scholar]
- Gaub P, et al. HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 2010;17:1392–408. doi: 10.1038/cdd.2009.216. [DOI] [PubMed] [Google Scholar]
- Granato A, De Giorgio A. Alterations of neocortical pyramidal neurons: turning points in the genesis of mental retardation. Front Pediatr. 2014;2:86. doi: 10.3389/fped.2014.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AC, et al. Valproate regulates GSK-3-mediated axonal remodeling and synapsin I clustering in developing neurons. Mol Cell Neurosci. 2002;20:257–70. doi: 10.1006/mcne.2002.1117. [DOI] [PubMed] [Google Scholar]
- Hao Y, et al. Mood stabilizer valproate promotes ERK pathway-dependent cortical neuronal growth and neurogenesis. J Neurosci. 2004;24:6590–9. doi: 10.1523/JNEUROSCI.5747-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MR, et al. Effect of HDAC inhibitors on neuroprotection and neurite outgrowth in primary rat cortical neurons following ischemic insult. Neurochem Res. 2013;38:1921–34. doi: 10.1007/s11064-013-1098-9. [DOI] [PubMed] [Google Scholar]
- Herrmann J, et al. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig Artic Ser. 1975;11:7–18. [PubMed] [Google Scholar]
- Hoshino N, et al. Brain-derived neurotrophic factor attracts geniculate ganglion neurites during embryonic targeting. Dev Neurosci. 2010;32:184–96. doi: 10.1159/000313902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, et al. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci U S A. 2004;101:16659–64. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR. Dendritic development in neocortex of children with mental defect and infantile spasms. Neurology. 1974;24:203–10. doi: 10.1212/wnl.24.3.203. [DOI] [PubMed] [Google Scholar]
- Irwin SA, et al. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet. 2002;111:140–6. doi: 10.1002/ajmg.10500. [DOI] [PubMed] [Google Scholar]
- Isrie M, et al. Haploinsufficiency of ANKRD11 causes mild cognitive impairment, short stature and minor dysmorphisms. Eur J Hum Genet. 2012;20:131–3. doi: 10.1038/ejhg.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentarra GM, et al. Abnormalities of cell packing density and dendritic complexity in the MeCP2 A140V mouse model of Rett syndrome/X-linked mental retardation. BMC Neurosci. 2010;11:19. doi: 10.1186/1471-2202-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung EM, et al. Loss of GSK-3 Causes Abnormal Astrogenesis and Behavior in Mice. Mol Neurobiol. 2016;53:3954–66. doi: 10.1007/s12035-015-9326-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung EM, et al. Regenerative potential of targeting glycogen synthase kinase-3 signaling in neural tissues. Neural Regen Res. 2015;10:1912–3. doi: 10.4103/1673-5374.169627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka M, et al. Essential Roles for ARID1B in Dendritic Arborization and Spine Morphology of Developing Pyramidal Neurons. J Neurosci. 2016a;36:2723–42. doi: 10.1523/JNEUROSCI.2321-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka M, et al. mTOR regulates brain morphogenesis by mediating GSK3 signaling. Development. 2014a;141:4076–86. doi: 10.1242/dev.108282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka M, et al. MACF1 regulates the migration of pyramidal neurons via microtubule dynamics and GSK-3 signaling. Dev Biol. 2014b;395:4–18. doi: 10.1016/j.ydbio.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka M, Kim WY. Microtubule-Actin Crosslinking Factor 1 Is Required for Dendritic Arborization and Axon Outgrowth in the Developing Brain. Mol Neurobiol. 2016;53:6018–6032. doi: 10.1007/s12035-015-9508-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka M, et al. Transactivation of TrkB by Sigma-1 receptor mediates cocaine-induced changes in dendritic spine density and morphology in hippocampal and cortical neurons. Cell Death Dis. 2016b;7:e2414. doi: 10.1038/cddis.2016.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka M, et al. MACF1 Controls Migration and Positioning of Cortical GABAergic Interneurons. Mice Cereb Cortex. 2017a;27:5525–5538. doi: 10.1093/cercor/bhw319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka M, et al. MTOR controls genesis and autophagy of GABAergic interneurons during brain development. Autophagy. 2017b;13:1348–1363. doi: 10.1080/15548627.2017.1327927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper EM, et al. Pyramidal neurons in layer 5 of the rat visual cortex. III. Differential maturation of axon targeting, dendritic morphology, and electrophysiological properties. J Comp Neurol. 1994;339:495–518. doi: 10.1002/cne.903390404. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–91. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- Kim WY. Brain size is controlled by the mammalian target of rapamycin (mTOR) in mice. Commun Integr Biol. 2015;8:e994377. doi: 10.4161/19420889.2014.994377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, et al. Evidence for sequestration of polyglutamine inclusions by Drosophila myeloid leukemia factor. Mol Cell Neurosci. 2005;29:536–44. doi: 10.1016/j.mcn.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Kim WY, et al. Statins decrease dendritic arborization in rat sympathetic neurons by blocking RhoA activation. J Neurochem. 2009a;108:1057–71. doi: 10.1111/j.1471-4159.2008.05854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, et al. Proteasome inhibitors suppress formation of polyglutamine-induced nuclear inclusions in cultured postmitotic neurons. J Neurochem. 2004;91:1044–56. doi: 10.1111/j.1471-4159.2004.02788.x. [DOI] [PubMed] [Google Scholar]
- Kim WY, Snider WD. Neuroscience. Overcoming inhibitions. Science. 2008;322:869–72. doi: 10.1126/science.1166152. [DOI] [PubMed] [Google Scholar]
- Kim WY, Snider WD. Functions of GSK-3 Signaling in Development of the Nervous System. Front Mol Neurosci. 2011;4:44. doi: 10.3389/fnmol.2011.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, et al. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci. 2009b;12:1390–7. doi: 10.1038/nn.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, et al. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–96. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Shiota K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J Biol Chem. 2003;278:4806–12. doi: 10.1074/jbc.M209923200. [DOI] [PubMed] [Google Scholar]
- Larkum M. A cellular mechanism for cortical associations: an organizing principle for the cerebral cortex. Trends Neurosci. 2013;36:141–51. doi: 10.1016/j.tins.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Lilja T, et al. Like a rolling histone: epigenetic regulation of neural stem cells and brain development by factors controlling histone acetylation and methylation. Biochim Biophys Acta. 2013;1830:2354–60. doi: 10.1016/j.bbagen.2012.08.011. [DOI] [PubMed] [Google Scholar]
- Lo-Castro A, et al. Neurobehavioral phenotype observed in KBG syndrome caused by ANKRD11 mutations. Am J Med Genet B Neuropsychiatr Genet. 2013;162B:17–23. doi: 10.1002/ajmg.b.32113. [DOI] [PubMed] [Google Scholar]
- London M, Hausser M. Dendritic computation. Annu Rev Neurosci. 2005;28:503–32. doi: 10.1146/annurev.neuro.28.061604.135703. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Pyramidal cell abnormalities in the motor cortex of a child with Down’s syndrome. A Golgi study. J Comp Neurol. 1976;167:63–81. doi: 10.1002/cne.901670105. [DOI] [PubMed] [Google Scholar]
- Marin O, et al. Guiding neuronal cell migrations. Cold Spring Harb Perspect Biol. 2010;2:a001834. doi: 10.1101/cshperspect.a001834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus A, et al. Raf and akt mediate distinct aspects of sensory axon growth. Neuron. 2002;35:65–76. doi: 10.1016/s0896-6273(02)00752-3. [DOI] [PubMed] [Google Scholar]
- Martini FJ, et al. Biased selection of leading process branches mediates chemotaxis during tangential neuronal migration. Development. 2009;136:41–50. doi: 10.1242/dev.025502. [DOI] [PubMed] [Google Scholar]
- McKinney BC, et al. Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am J Med Genet B Neuropsychiatr Genet. 2005;136B:98–102. doi: 10.1002/ajmg.b.30183. [DOI] [PubMed] [Google Scholar]
- Moffat JJ, et al. Genes and brain malformations associated with abnormal neuron positioning. Mol Brain. 2015;8:72. doi: 10.1186/s13041-015-0164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–9. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Nasrallah IM, Golden JA. Brain malformations associated with cell migration. Pediatr Dev Pathol. 2006;9:89–97. doi: 10.2350/06-04-0077.1. [DOI] [PubMed] [Google Scholar]
- Neilsen PM, et al. Identification of ANKRD11 as a p53 coactivator. J Cell Sci. 2008;121:3541–52. doi: 10.1242/jcs.026351. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Eberhart CG. The neurobiology of autism. Brain Pathol. 2007;17:434–47. doi: 10.1111/j.1750-3639.2007.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Snider W. Initiating and growing an axon. Cold Spring Harb Perspect Biol. 2010;2:a001925. doi: 10.1101/cshperspect.a001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–83. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Markram H. Anatomy and physiology of the thick-tufted layer 5 pyramidal neuron. Front Cell Neurosci. 2015;9:233. doi: 10.3389/fncel.2015.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read DE, Gorman AM. Involvement of Akt in neurite outgrowth. Cell Mol Life Sci. 2009;66:2975–84. doi: 10.1007/s00018-009-0057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge EM, et al. Neurotrophin-4 is more potent than brain-derived neurotrophic factor in promoting, attracting and suppressing geniculate ganglion neurite outgrowth. Dev Neurosci. 2012;34:389–401. doi: 10.1159/000342996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval R, et al. Homeostatic NMDA receptor down-regulation via brain derived neurotrophic factor and nitric oxide-dependent signalling in cortical but not in hippocampal neurons. J Neurochem. 2011;118:760–72. doi: 10.1111/j.1471-4159.2011.07365.x. [DOI] [PubMed] [Google Scholar]
- Shipp S. Structure and function of the cerebral cortex. Curr Biol. 2007;17:R443–9. doi: 10.1016/j.cub.2007.03.044. [DOI] [PubMed] [Google Scholar]
- Siebzehnrubl FA, et al. Histone deacetylase inhibitors increase neuronal differentiation in adult forebrain precursor cells. Exp Brain Res. 2007;176:672–8. doi: 10.1007/s00221-006-0831-x. [DOI] [PubMed] [Google Scholar]
- Sirmaci A, et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011;89:289–94. doi: 10.1016/j.ajhg.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjei KL, et al. KBG syndrome: report of twins, neurological characteristics, and delineation of diagnostic criteria. Am J Med Genet A. 2007;143A:292–300. doi: 10.1002/ajmg.a.31597. [DOI] [PubMed] [Google Scholar]
- Suzuki S, et al. Brain-derived neurotrophic factor regulates cholesterol metabolism for synapse development. J Neurosci. 2007;27:6417–27. doi: 10.1523/JNEUROSCI.0690-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, et al. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J Cell Biol. 2004;167:1205–15. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakhtenberg EF, Goldberg JL. Epigenetic regulation of axon and dendrite growth. Front Mol Neurosci. 2012;5:24. doi: 10.3389/fnmol.2012.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–58. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gaal S, Lamme VA. Unconscious high-level information processing: implication for neurobiological theories of consciousness. Neuroscientist. 2012;18:287–301. doi: 10.1177/1073858411404079. [DOI] [PubMed] [Google Scholar]
- Walz K, et al. Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum Genet. 2015;134:181–90. doi: 10.1007/s00439-014-1509-2. [DOI] [PubMed] [Google Scholar]
- Willemsen MH, et al. Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. Eur J Hum Genet. 2010;18:429–35. doi: 10.1038/ejhg.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss JM, et al. Dendritic bundling in layer I of granular retrosplenial cortex: intracellular labeling and selectivity of innervation. J Comp Neurol. 1990;295:33–42. doi: 10.1002/cne.902950104. [DOI] [PubMed] [Google Scholar]
- Xu C, et al. Retrolinkin recruits the WAVE1 protein complex to facilitate BDNF-induced TrkB endocytosis and dendrite outgrowth. Mol Biol Cell. 2016;27:3342–3356. doi: 10.1091/mbc.E16-05-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngs EL, et al. ANKRD11 gene deletion in a 17-year-old male. Clin Dysmorphol. 2011;20:170–1. doi: 10.1097/MCD.0b013e328346f6ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu IT, et al. Valproic acid promotes neuronal differentiation by induction of proneural factors in association with H4 acetylation. Neuropharmacology. 2009;56:473–80. doi: 10.1016/j.neuropharm.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Zhang A, et al. Identification of a novel family of ankyrin repeats containing cofactors for p160 nuclear receptor coactivators. J Biol Chem. 2004;279:33799–805. doi: 10.1074/jbc.M403997200. [DOI] [PubMed] [Google Scholar]
- Zhou Z, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–69. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.