

Abstract
The extracellular matrix (ECM) is a key acellular structure in constant remodeling to provide tissue cohesion and rigidity. Deregulation of the balance between matrix deposition, degradation and crosslinking results in fibrosis. Bone marrow fibrosis (BMF) is associated with several malignant and nonmalignant pathologies severely affecting blood cell production. BMF results from abnormal deposition of collagen fibers and enhanced lysyl oxidase-mediated ECM crosslinking within the marrow, thereby increasing marrow stiffness. Bone marrow stiffness has been recently recognized as an important regulator of blood cell development, notably by modifying the fate and differentiation process of hematopoietic or mesenchymal stem cells. This review surveys the different components of the ECM and their influence on stem cell development, with a focus on the impact of the ECM composition and stiffness on the megakaryocytic lineage in health and disease. Megakaryocyte maturation and the biogenesis of their progeny, the platelets, are thought to respond to environmental mechanical forces through a number of mechanosensors, including integrins and mechanosensitive ion channels, reviewed here.
Keywords: Extracellular matrix, megakaryocyte development, platelet development, mechanosensors
Composition of the Bone Marrow Extracellular Matrix (ECM)
The bone marrow (BM) ECM is a non-cellular structure that provides physical support for tissue integrity, elasticity and hematopoesis1–3. The ECM is vital in normal hematopoiesis and plays a role in pathological states. It is comprised of various matrix proteins, such as collagens, laminin, fibronectin, and fibrinogen, as well as various soluble proteins, such as cytokines, chemokines and secreted enzymes1,4,5. Of the different ECM structural proteins, the main ones are collagens, which include fibrillary collagen (collagens I, II, III, V and XI) and non-fibrillary collagen, with the former providing stiffness to the ECM1. Cellular adhesion to collagen has been found to promote bone marrow cells and megakaryocyte (MK) expansion6–8. Similar to collagen, fibronectin is important for cellular interactions with the ECM, and plays a role in adhesion, migration, growth and differentiation9–12. The ECM also contains proteoglycans with attached glycosaminoglycan (GAG) side chains. These GAGs bind to and sequester growth factors in the ECM and may act as a reservoir of these cell-impacting proteins1,3. Heparan sulfate is a GAG involved in mobilization of HSCs13, and the GAG hyaluronic acid is important for normal hematopoiesis14–16. Other ECM proteins, such as fibrillin-1, tenascin-C, agrin, thrombospondin and matrilin-4 are important for hematopoietic stem cell (HSC) proliferation and survival during homeostatic and stress conditions, as well as regulation of angiogenesis17–21.
The ECM is in constant remodeling. Among ECM modifying proteins are matrix metalloproteinases (MMPs), tissue inhibitors of MMPs (TIMPs), plasmin, and lysyl oxidase (LOX). MMPs are a family of zinc-dependent endopeptidases that are the main enzymes responsible for ECM breakdown, while TIMPs counteract this breakdown by inhibiting MMPs22. Dysregulation of these counteracting proteins affects the ECM in some pathological states, such as multiple myeloma (MM) and primary myelofibrosis (PMF)5,23. TIMP-3, not only inhibits MMPs in the BM, but it also plays a role in HSC expansion, differentiation and trafficking24,25. Plasmin is a protein activated by urokinase plasminogen activator (uPA), which has been found to be expressed in mouse hematopoietic stem cells and progenitors and by human BM stromal cells26–28. Plasmin degrades fibrin, fibronectin and laminin, while activating MMPs27. Other components of hemostasis are also found in the ECM, such as plasminogen activator inhibitor 1 (PAI-1), tissue plasminogen activator (tPA), and fibrinogen. In the bone marrow, these factors are important for hematopoietic regeneration29,30. PAI-1 inhibits the degradation of plasmin during hemostasis, and it has also been reported to be expressed in MKs31. Altered PAI-1 level is associated with hematological malignancies, including MM and myeloproliferative neoplasms (MPNs)32,33. Fibrinogen is found in the vascular sinusoids in the BM34. LOX is a copper-dependent secreted enzyme, the activity of which eventually leads to the crosslinking of collagen and elastin precursors, resulting in increased ECM stiffness35. Hence, depending on the balance between matrix secretion and deposition, cross-linking and degradation, tissue stiffness may vary under physiological or pathophysiological conditions.
Bone Marrow ECM Stiffness: Measurement and Natural Contributors
Several ECM components in the BM contribute to the stiffness of the tissue. This property is typically assessed by methods such as rheometry36 and atomic force microscopy37. Rheometry involves measuring the viscoelastic properties of a substance by looking at the relationship between deformations and stresses in order to calculate Young’s modulus, a measurement for stiffness38. The limitation of rheometry is that it looks at the BM as a whole at the macroscopic level and cannot detect regional or cellular heterogeneity of stiffness38. Other studies have used a micropipette aspiration method to measure stiffness by placing a glass capillary micropipette next to the tissue and applying vacuum pressure to measure the aspirated length, which is then used to calculate soft tissue stiffness39. This method may be used in BM to find regional differences in stiffness; however, it faces the same limitations of rheometry in that it does not measure stiffness at the cell level. One way to measure ECM stiffness in BM at the cellular level involves atomic force microscopy (AFM) as it can provide a spatial resolution of almost 1 nm and can be used in solution and in living cells40. AFM can also be used to measure stiffness of collagens and other ECM components41. Another method for measuring ECM stiffness at the cellular level is optical tweezers active microrheology42.
The stiffness of the ECM is heterogeneous throughout the BM38, likely due to heterogeneous distribution of ECM components. Collagens are important contributors to ECM stiffness; Types I, II and III are the most abundant collagens in the ECM43. For example, type IV and type I collagen are found mainly near the endosteal surface in the BM along with fibronectin, and fibronectin is also found in MKs and in central marrow44,45. Type IV collagen is detected in the vasculature of the BM, while type I collagen is not normally found in those areas45. ECM stiffness increases in a non-linear fashion as the concentration of type I collagen increases, while increasing concentrations of type III collagen mixed with type I collagen decreases stiffness41,46. Fibronectin, a high molecular weight glycoprotein, appears to be an important regulator of ECM stiffness via contribution to the organization of collagen fibrils. In vitro experiments showed that fibronectin and integrins α11β1 and α2β1 are necessary to form a collagen matrix47. This study showed that a collagen matrix did not form in a fibronectin-deficient mouse fibroblast cell line cultured in the absence of exogenous fibronectin, while the addition of fibronectin was sufficient for the collagen matrix to form. When this fibronectin-deficient cell line was transfected to express, either, α11 or α2 integrins, a collagen matrix formed even in the absence of fibronectin, however, this matrix was not as well organized. This suggested that both fibronectin and integrins α11β1 and α2β1 are necessary for forming a well-developed collagen matrix47. Mice engineered to lack liver fibronectin developed extensive liver fibrosis following chronic liver injury, compared to matching controls. These null mice had more extensive and disorganized collagen deposition compared to mice with fibronectin48.
LOX is a copper-dependent enzyme that causes oxidative deamination of lysine and hydroxylysine residues on collagen, forming aldehydes that spontaneously cross link, thereby increasing ECM stiffness35,49. LOX has been implicated in the progression of several malignancies, including renal cell carcinoma (RCC), colorectal and breast cancers50–52. Collagen-coated gels were found to be stiffer when treated with conditioned media of cultured primary RCC cells as measured by AFM. This stiffness was reduced when β-aminopropionitrile (BAPN), a LOX inhibitor50. Augmented stiffness was also seen in vivo when colorectal cancer (CRC) cell lines were stably transfected with either vector, LOX or catalytically inactive LOX and implanted in nude mice. The tumors from LOX-transfected cells were stiffer than from cells with catalytically inactive LOX51. LOX is also upregulated in hematopoietic malignancies, including myeloproliferative neoplasms4,53,54.
Effect of ECM Components and Stiffness on Bone Marrow Cell Adhesion and Development
Effect on megakaryocyte development and platelet formation
The effect of type I collagen on (megakaryocyte) MK development and function has been extensively studied. Type I collagen affects MK development by stimulating hematopoietic stem cells (HSCs) to differentiate through the megakaryocytic lineage6,8. Type I collagen, however, inhibits proplatelet formation through activation of integrin α2β1 and downstream Rho/ROCK axis55. Interestingly, MKs on N-acetylated type I collagen, which decreases mechanical tension, produce more proplatelet compared to type I collagen without this modification55. In contrast to type I collagen, type III and IV collagens stimulate proplatelet production via the PI3K/Akt signaling pathway44,56,57. The stiffness of type I collagen is greater than type IV collagen, and MKs plated on type I collagen remain spread after 16 hours of incubation, while MKs plated on type IV collagen extend proplatelets57. MKs cultured on type IV collagen also show increased integrin β1 activation and internalization at 3 and 8 hours of adhesion compared to type I collagen.
Fibronectin is another ECM component that is important to MK function and development58. MKs adhere to fibronectin and proplatelet formation is increased by activation of its receptors, α5β1 (VLA-5) and α4β1 (VLA-4), on MKs59,60. Fibronectin also modulates MK adhesion to type I collagen61. HSCs cultured on fibronectin produced more CFU-MKs as the matrix became stiffer, and blocking the fibronectin receptor integrin α5β1 abolished this increase62.
Other BM ECM components, such as fibrinogen, plasmin, PAI-1 and vitronectin, and thrombospondin-2 (TSP-2) have also been reported to affect MK function and proplatelet production34,63,64. MKs adhere to fibrinogen via αIIbβ360. Fibrinogen is mainly found in the vascular sinusoids in the BM, and upon binding to it, MKs have increased proplatelet formation34. PAI-1 inhibited MK adhesion to vitronectin, but its effect was reverse in the presence of tPA or uPA65. Thrombospondin-2 (TSP-2) is a matricellular protein that is expressed by MKs but not platelets and has been found to reduce proplatelet formation and MK differentiation in vitro66.
Glycosaminoglycans (GAGs), other important components of the ECM, play an important role in MK development67. The GAG hyaluronic acid appears to be an important inhibitor of platelet production as depolymerazation of hyaluran is necessary for thrombopoesis; MKs with hyaluronidase-2 deficiency are less mature and make fewer proplatelets than hyaluronidase-2 containing MKs68. However, in normal MKs, high molecular weight hyaluronic acid does not effect MKs and, therefore, may make a good ex vivo 3D scaffold69. This is in contrast to the GAG dermatan sulfate, which increases proplatelet production in thrombopoetin (TPO)-stimulated MKs, compared to MKs cultured without this GAG70.
LOX is a non-structural component of the ECM that affects MK proliferation, adhesion and function. LOX is expressed in immature normal MKs and downregulated as MKs mature53. As mentioned above, the stiffness of the ECM is increased by LOX via cross-linking of collagen fibers35, and LOX expression is upregulated in MKs of mouse or human primary myelofibrosis. LOX can also potentiate platelet derived growth factor (PDGF)-mediated MK proliferation by oxidizing and activating the PDGF receptor53,71. Another role of LOX is the activation of the collagen receptor α2β1 on platelets, which leads to increased adhesion to collagen72. Reducing the stiffness of the ECM by inhibiting LOX crosslinking of collagen in mouse bone marrow, increased platelet level, supporting the notion that a less stiff matrix favors platelet biogenesis57. LOX may affect MK development by mechanisms other than catalytic oxidation of receptors or the ECM. LOX is secreted as a 50-kDa pro-enzyme that is cleaved by BMP-1 (also expressed by MKs) to release the mature LOX enzyme and the 18-kDa propeptide (LOX-PP) that can enter cells73,74. LOX-PP decreases MK polyploidy, possibly by decreasing the expression of cell cycle regulators75.
Stiffness also plays a role in MK function and development. MKs cultured in 3D media that mimic BM ECM stiffness had higher ploidy levels than MKs cultured in liquid media76. MKs cultured inside a 3D methylcellulose (MC) hydrogel of medium rigidity (30–60 Pa) had higher ploidy, higher demarcation membrane development and more proplatelet formation than MKs cultured in 2D on top of the gel or in liquid culture76or in a 3D stiffer rigidity (300–600 Pa)76. A separate study looked at MKs cultured on collagen-coated soft gels (300 Pa) or stiff gels (34 kPa) and found that MKs had higher ploidy on softer gels compared to stiffer gels, independent of the collagen concentration77. However, this effect was abolished when the MKs were treated with a non-muscle myosin inhibitor77. Ex vivo studies show that MKs forms more proplatelets on low to medium stiffness silk films than on high stiffness silk films, regardless if the silk films were coated with type I or type IV collagen57,78. Increased stiffness also led to increased β1 integrin activation and internalization in MKs57. Although some stiffness is needed for MK maturation, it appears that a stiff ECM is detrimental to MK development and proplatelet formation. ECM stiffness appears to control proplatelet formation via PI3K/Akt signaling pathways with decreased stiffness causing increased Akt phosphorylation while MKs had decreased proplatelet formation when treated with Akt inhibitor57. Figure 1 includes an illustrative summary of the main ECM components that have the potential to affect stiffness.
Figure 1. Schematic illustration of megakaryocyte adhesion and fragmentation into platelets in the context of normal ECM stiffness.
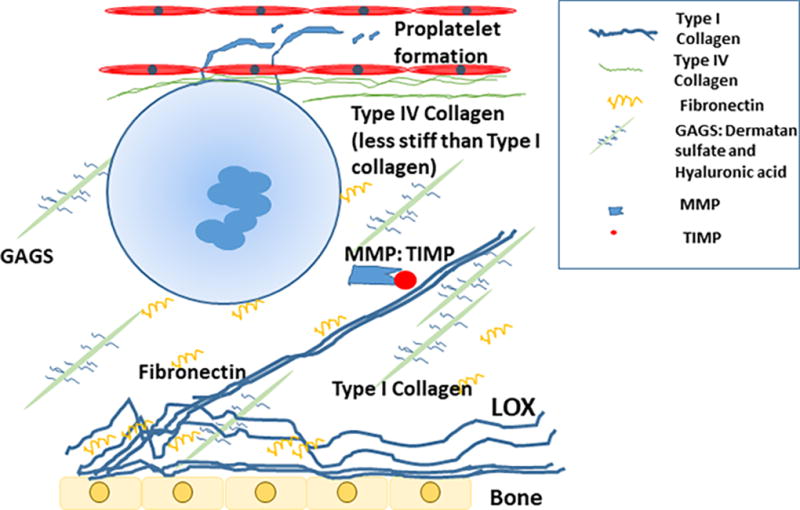
As depicted here, type I collagen is normally found near the periosteum, and other collagen types, such as collagen IV, Glycosaminoglycans (GAGS) and fibronectin are spread throughout the niche. Metalloproteinases (MMPs) and tissue inhibitors of MMPs (TIMP) are present to remodel and turnover the ECM. Collagen I is less stiff than collagen IV and the rigidity of both is impacted by the level of a cross-linking enzyme, lysyl oxidase (LOX) secreted from low ploidy megakaryocytes and osteoblasts. Other components of hemostasis found in the ECM (and not illustrated here), such as plasminogen activator inhibitor 1 (PAI-1), tissue plasminogen activator (tPA) and fibrinogen were too found to impact megakaryocyte development through mechanisms outlined in the review.
Effect on blood stem cells and other bone marrow cells
The stiffness of the ECM is also important for HSC fate and differentiation62,79–81. HSCs cultured on stiffer gels coated with fibronectin (mimicking the endosteal region of the BM) promoted the maintenance of myeloid progenitors, while laminin-coated gels promoted erythroid differentiation62. Laminin also increased the number of mature red blood cell (RBC) progenitors derived from human HSCs82. Further, culturing murine HSCs on three dimensional collagen matrices of different stiffness revealed that HSCs on stiffer matrices were more quiescent81. HSCs cultured on collagen gels showed increased viability when cultured on less stiff gels (44 Pa) than on stiffer gels (3.48 kPa)83. Similarly, on collagen-coated stiff PA gels (136 kPa), HSCs showed decreased viability as the collagen concentration coating the gels was increased83. Culturing cord blood derived-CD34+ HSCs on collagen gels led to decreased expansion but increased myeloid differentiation, compared to HSCs cultured in suspension84. In a gene-expression assay, several growth factors, chemokines and cytokines were upregulated in HSCs cultured on collagen gels compared to suspension culture, including interleukins 8, 6 and 1β, TNFα, CXCL2, and CCL2 which are involved in HSC proliferation and mobilization84. Similar to HSCs, human mesenchymal stem cells (MSCs) cultured on a type I collagen-coated gel matrix show different phenotypes depending on the rigidity of the matrix. MSCs cultured on a softer matrix appear more neurogenic and express more neurogenic genes, and those cultured on more rigid surfaces exhibit an osteogenic phenotype79.80
Fibronectin is another ECM component that is important for hematopoiesis. Fibronectin was found to induce apoptosis and decrease proliferation of human HSC lines via the fibronectin receptor, VLA-5, however it promotes erythropoiesis via VLA-485–87. Erythrocyte progenitors lose their attachment to fibronectin as they mature through down-regulation of VLA-5, which may allow them to detach from the ECM to be released into circulation88.
GAGs appear to play an important role in hematopoiesis. In mice treated with 5-fluorouracil (5-FU), injection of hyaluronic acid (HA) increased BM recovery as well as augmented IL-1 and IL-6 production by HSC, which support hematopoiesis15. Heparan sulfate is another GAG that is important for hematopoiesis89. HSCs express Mac-1 (CD11b/CD18) and CD45 which mediate their adhesion to stromal heparan sulfate90.
The remodeling of the ECM and the balance between MMPs and TIMPs is important for maintaining hematopoiesis. TIMP-3 is an endogenous inhibitor of MMPs and important regulator of HSC proliferation and trafficking, as well as bone turnover. Up-regulation of TIMP-3 in mice via BM transplantation of retroviral transduced HSCs increased BM and blood myeloid cell counts,, while it decreased lymphocyte levels24. There was also increased HSC trafficking to the blood and spleen and late onset of fatal osteosclerosis in mice with up-regulated TIMP-3, compared to matching controls24. The role of TIMP-3 in stimulating quiescent HSCs to proliferate appears to be independent of its MMP-inhibiting activity and may be due to direct inhibition of angiopoietin-1 signaling, which mediates HSC quiescence25.
ECM Signaling to Mechanosensors Expressed in Megakaryocytes
Mechanical forces have been shown to be important in MK function and development through a role of shear stress, calcium and mechanosensitive ion channels. For example, shear stress in BM capillaries and sinusoids is important for proplatelet maturation and platelet release91. MKs express several integrins that sense the ECM around them and transduce signals to the cytoskeleton92,93. Among these integrins, α2β1 binds to type I collagen and decreases the production of proplatelets via the Rho/ROCK pathway94. Integrin α2β1 also regulates the assembly of actin stress fibers in MKs, which mediate cell migration94. The Rho/ROCK pathway regulates thrombopoesis by phosphorylating myosin light chain (MLC), which enhances actin-dependent myosin motor activity95. This regulation of thrombopoesis by the Rho/ROCK pathway may be important in controlling the timing of platelet formation by MKs and preventing premature platelet formation while the MKs are still in the osteoblastic niche95. Indeed, patients with MYH9-related diseases (caused by mutations of the heavy chain of the non-muscle myosin) present with macrothrombocytopenia that is thought to be due to premature proplatelet formation in the osteoblastic niche and impaired migration95–97. Given that type I collagen is found primarily in the osteoblastic niche and it is a significant contributor to ECM stiffness, it is logical that areas with large amounts of type I collagen have an unfavorable environment for platelet production8,45,46. Therefore, the sensing of ECM stiffness is important for proper MK function.
Calcium plays an important role in MK function98,99. Increases in cytosolic calcium are due to calcium release from intracellular stores in the endoplasmic reticulum (ER) followed by extracellular calcium influx via the Store-Operated Calcium Entry (SOCE) mechanism100,101. Adenosine diphosphate (ADP) induces an increase in cystolic calcium in MKs, which activates downstream signaling important in MK adhesion an proplatelet formation (FAK, Src, ERK and Akt pathways)98. Intracellular calcium store release into the cytosol appears to be sufficient to sustain proplatelet formation, however extracellular calcium influx is important in the regulation of the interaction between MKs and the ECM98. Extracellular calcium is important in the phosphorylation of MLC, which has been shown to reduce proplatelet formation in MKs55,98. Interestingly, adhesion of MKs to type I collagen increased cytosolic calcium, and inhibition of influx of extracellular calcium by blocking SOCE reduced MK adhesion to type I collagen98,102. Calcium dysregulation has been implicated in MK pathologies, most notably the MPNs essential thrombocythemia (ET) and PMF where mutations in Calreticulin (CALR) have been found in 25% of cases without JAK2 or MPL mutations103.104
Extracellular calcium appears to be important in sensing the ECM in MKs, and MKs express mechano-sensitive ion channels that may play a role in platelet formation. One such mechano-sensitive ion channel is the transient receptor potential cation channel subfamily V member 4 (TRPV4)57. This channel modulated the PI3K/Akt pathway in endothelial cells when physical stimuli were applied to their cell membrane105. In MKs, TRPV4 senses a soft ECM and increased intracellular calcium influx, leading to increased PI3K/Akt pathway activation and proplatelet formation57. Inhibition of TRPV4 in MKs adhered to type IV collagen decreased proplatelet formation57. Mice treated with the LOX inhibitor BAPN had reduced BM ECM stiffness, higher platelet counts and increased TRPV4 activation compared to control, which highlights the importance of this ion channel in sensing soft ECM and increasing platelet formation in vivo57.
Another family of mechano-sensitive ion channels that may play a role in MK and platelet function is the Piezo family. Piezo1 and Piezo2 are mechanically activated ion channels that induce nonselective cationic currents in response to mechanical forces106–108. The Piezo proteins are activated by tension on the lipid membrane itself107,108. Piezo2 appears to be important in mechanosensation and in light touch and pain perception, and is expressed in a subset of somatosensory neurons and Merkel cells106,109. Loss of Piezo2 causes a loss in vibration detection, joint proprioception and touch discrimination, as well as congenital hip dysplasia in humans, while gain of function mutations in Piezo2 are associated with distal arthrogryposis type 5, which presents with congenital contractures, ophthalmopleiga and restrictive lung disease109–111. In chondrocytes, both Piezo1 and TRPV4 are important for ECM mechanosensing, however, Piezo1 is responsible for calcium influx in response to cell membrane stretching112. Piezo1 is expressed in human erythrocytes and platelets, and was found to play a role in vascular development113–118. Gain-of-function mutations in Piezo1 are associated with hereditary xerocytosis, an autosomal dominant hemolytic anemia due to erythrocyte dehydration and an imbalance of intracellular cation concentrations115,116. Loss of function of Piezo1 caused congenital lymphatic dysplasia in human patients, while loss of Piezo1 caused improper vessel formation in mice117,119. Piezo1 is involved in shear-induced calcium influx that mediate ATP release in red blood cells, while ATP induces nitric oxide (NO) production and relaxation of endothelial cells114. There may be a role for Piezo1 in cellular proliferation. In one study, epithelial cells had increased proliferation in response to mechanical stretching via activation of Piezo1 and subsequent calcium influx and ERK1/2 phosphorylation120. A recent study found that Piezo1 is expressed in human platelets and the Meg-01 MK cell line113. Extracellular calcium influx was induced in Meg-01 cells and platelets when exposed to fluid shear stress and when Meg-01 cells had their cell membranes mechanically deformed with a glass pipette. This influx was largely abolished by GsMTx-4, a Piezo1 inhibitor113. Interestingly, the addition of GsMTx-4 to whole blood for 30 seconds resulted in a reduction of thrombus formation under arterial flow on a collagen surface, though no difference was seen in platelet aggregation under low shear stress conditions113.112
The role of Piezo proteins in MK and proplatelet formation has not been studied yet, though it seems to be a promising mechanosensors for ECM stiffness. Piezo1 and other mechanosensitive ion channels may play a role in MK function by affecting ion currents121. One such channel is a depolarization-gated potassium-selective (Kv) channel. MKs express the α subunit (a pore forming subunit) of Kv, Kv1.3, which is encoded by the gene KCNA3122. Kv1.3 is important in early calcium influx in platelets, and Kv1.3 deficient mice had significantly higher platelet counts compared to WT, but no effects on megakaryopoeisis were seen122. Platelets and MKs also express calcium-activated potassium channels (KCa) which may help determine membrane potential and calcium influx121,123. One such channel, KCa3.1, is important in stromal cell derived factor 1 (SDF-1) mediated platelet migration124.
Bone Marrow Pathology Associated with a Fibrotic and Stiffer ECM
Given the importance of the ECM and cellular signaling evoked by it during maintenance of tissue homeostasis, it is not surprising that dysregulation of ECM components leads to tissue fibrosis that disrupts organ function and is an important cause of mortality and morbidity. Fibrosis associated with excessive ECM deposition in the BM (BMF) leads to impairment in blood cell production and sometimes extramedullary hematopoiesis (EMH). These fibers are essentially reticulin fibers composed of type III collagen that can be accompanied by type I collagen fibers. Semi-quantitative assessment of the severity of BMF is through a grading system on a scale of 0–4 for the Bauermeister system or 0–3 for the revised European Consensus system125,126. This evaluation is based on the amount of reticulin fibers, their reticulation and the presence of collagen bundles. While no collagen fibers have been observed in healthy subjects, reticulin fibers may be present at a low grade127,128. A strong increase in the quantity and reticulation of reticulin fibers beyond the normal range, associated or not with collagen bundles, is characteristic of fibrotic marrow disorders.
A relationship between increased BM stromal fibers and disease has been most extensively studied in the myeloproliferative neoplasm, primary myelofibrosis (PMF)129,130. PMF is a heterogeneous and clonal BCR-ABL1-negative hematopoietic stem cell malignancy characterized by BMF, ineffective hematopoiesis, MK proliferation and atypia, EMH, and splenomegaly. The last WHO diagnostic criteria from 2016 defined a pre-fibrotic PMF to distinguish it from other BCR-ABL-negative myeloproliferative neoplasms such as ET and polycythemia vera (PV) that may develop late secondary myelofibrosis4,131,132. BMF is also observed in other hematological malignancies, such as acute myeloid leukemia (AML), acute lymphocytic leukemia (ALL), myelodysplastic syndrome (MDS) and chronic myeloid leukemia (CML).
Nonmalignant diseases associated with BMF can be of autoimmune origin, called primary autoimmune myelofibrosis (AIMF). In these cases, patients possess autoantibodies, but do not present a well-characterized autoimmune disorder. AIMF is marked by cytopenias without splenomegaly, and high grades of BMF with lymphocytic infiltration, but without associated osteosclerosis. AIMF can also be secondary to an established disorder such as systemic lupus erythematosus (SLE)133. However, although isolated case reports are regularly presented, it is still an underdiagnosed disorder134,135.
Infectious or inflammatory diseases may also be associated with BMF. This is the case for HIV or tuberculosis infection, visceral leishmaniasis, and pulmonary hypertension. In addition, disorders associated with vitamin D deficiency are also prone to development of BMF. Furthermore, exposure to toxins or radiations may lead to the development of BMF, as well as treatment with medications such as recombinant human IL-11 (rhIL-11)136, or thrombopoietin receptor agonists137–140.
Several studies have tried to correlate the amount and type of fibrosis with disease prognosis, but the conclusions vary. The presence of reticulin fibers alone, characteristic of mild fibrosis, does not seem to correlate with disease severity or co-morbidities. On the other hand, the presence of collagen fibers, representing higher grade of BMF, correlate with abnormal blood count and severity of the underlying disorders126,130,141. Nevertheless, in both cases, provided that adequate treatment exists for the disease, reticulin fibrosis and even collagen fibrosis can decrease or resolve. This is the case for AIMF and SLE-associated myelofibrosis treated with corticoids142. Increased reticulin fibrosis due to CML is also reversible with Imatinib therapy, but not with interferon143–145. In MPN patients, ruxolitonib or interferon treatment improves aspects of the pathology in some but not all patients146,147. In cases where supportive care measures are ineffective, stem cell transplant eradicating the clone responsible for the neoplastic transformation has proven to be effective in resolving both collagen and reticulin fibrosis148,149.
In the BM, the main cells responsible for the increase in ECM are fibroblasts, which are usually found in close association with collagen fibers150,151. Rather than an increase in number, it is most probably their activation or differentiation into matrix-producing cells which is responsible for augmented ECM deposition151,152. Fibroblasts respond to a number of fibrogenic factors present in the diseased marrow. Increases in cytokines such as interleukin (IL)-6, IL-12, IL-8, TNFα, IFNγ, as well as profibrogenic growth factors such as TGFβ, bFGF, VEGF and TGFβ1 have been implicated153. MKs are primarily responsible for the secretion of many of these profibrotic, angiogenic and pro-inflammatory factors stored in their alpha granules154–156. This has been well documented for PMF. In addition, non-myeloproliferative fibrosis has also been linked to MK hyperplasias as is the case for AIMF135. Interestingly, the presence of MKs in the lungs was proposed to contribute to pulmonary fibrosis and pulmonary hypertension in systemic sclerosis157. In this context, it is worth noting that pulmonary hypertension is diagnosed in some patients with PMF and other MPNs, and has been associated with pulmonary EMH and fibrosis158. Yet, the degree of abundance and role of pulmonary MKs in health and disease, MPN included, require further study. In addition to MKs (and platelets), monocytes/macrophages could be alternative sources of fibrogenic growth factors. In PMF, monocytes are activated and overexpress IL-1 and TGFβ, suggesting that they also contribute to the fibrotic process. Table 1 summarizes the above-described physiological and pathological factors of BM stiffness.
Table 1.
Physiological and pathological factors of BM fibrosis and stiffness
| BM Components | Representative References |
|---|---|
|
| |
| BONE MARROW MATRIX | 1,4 |
| Proteins: Fibrillar collagens I, II, III, V, XI ; Non fibrillar collagen IV ; Fibronectin ; Vitronectin ; Fibrinogen ; Laminin ; Fibrillin- A ; Tenascin-C ; Elastin ; Agrin ; Thrombospondin ; Matrillin-4 | |
| Proteoglycans: Heparan sulfate; Hyaluronan; Chondroitin sulfate; Dermatan sulfate; Keratin sulfate; Heparin | |
|
| |
| REMODELING FACTORS IMPACTING STIFNESS | 23, 29–31 |
|
| |
| Degradation enzymes and inhibitors MMPs ; Plasmin ; tPA; TIMPs ; PAI-1 | 4, 53, 57, 74 |
| Cross-linking enzyme: Lysyl oxidase (LOX) | |
|
| |
| FIBROGENIC SOLUBLE FACTORS | 4, 154 |
| Inflammatory cytokines: IL-1 -2, -6,-8,-12,- 13, -15; TNFa | |
| Growth factors: TGFb; bFGF; VEGF; PDGF; BMP-2, -4, -5, -6 | 4, 73, 150, 152, 156 |
|
| |
| TREATMENT-INDUCING FIBROSIS | |
| rhIL-11; TPO agonists | 136, 138–140 |
Abbreviations are as in Figure 1.
Conclusions and Future Directions
Deregulation of ECM remodeling with excess matrix deposition, increased matrix cross-linking activity and defective matrix degradation, leading to increased stiffness, are associated with bone marrow pathologies. Important efforts will be needed to identify the mechanotransduction pathways used by MKs and other BM cells to sense such changes in the ECM. This is especially important considering that pathologies associated with BMF are often linked to platelet production defects. Not only platelet production but also platelet function may be affected by modifications of the mechanics of the vascular environment; as yet, unexplored area of investigation. The main function of platelets is to ensure hemostasis by forming a plug at the site of vascular injury that seals the breach and stops blood loss159. Platelets are also involved in arterial thrombosis, which occurs in diseased vessels presenting evolved atherosclerotic plaques. Upon erosion and rupture of such a plaque, platelets accumulate and form a thrombus that can become occlusive, resulting in life-threatening ischemic pathologies such as stroke and myocardial infarction160. The stiffness of the exposed sub-endothelium in healthy vessels far differs from that found in stiffer atherosclerotic vessels. An association between arterial stiffness and platelet activation has been reported161. This link was proposed to be indirect and appears to result from the reduced ability of atherosclerotic plaques to release normal levels of nitric oxide which maintain platelets in a resting state162. How stiff surfaces exposed after atherosclerotic plaque rupture modifies platelet function remains completely unknown and represents a very attractive research area which could provide some clues on why thrombus formation can become occlusive in diseased arteries as compared to healthy vessels. Recently, it was reported that platelets sense microenvironmental mechanical properties, including substrate stiffness, which results in biological signal responses163. Evidence was provided that modifying the substrate stiffness of a fibrin surface increases platelet adhesion and spreading. Future studies are required to determine whether these observations are relevant to hemostasis and/or arterial thrombosis. They could help to identify novel mechanosensitive receptors which might represent interesting novel anti-thrombotic targets. Such a mechano-sensitive receptor, Piezo1, has recently been reported to be expressed by platelets. Piezo-1 was shown to promote Ca2+ entry and could participate in thrombus formation under arterial shear stress113. Future studies of other platelet mechanosensors could provide insights on how these cells sense both matrix stiffness and shear forces in hemostasis and arterial thrombosis.
Acknowledgments
The authors would like to thank Dr. Beatrice Hechler for reading the manuscript and for helpful discussions. KR is supported by NHLBI HL80442. OL is a recipient of an ASH MMSAP and HONORS awards. We apologize to all whose work was not cited due to limited space and limited number of citations.
References
- 1.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840:2506–2519. doi: 10.1016/j.bbagen.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leiva O, et al. The role of the extracellular matrix in primary myelofibrosis. Blood Cancer J. 2017;7:e525. doi: 10.1038/bcj.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glavey SV, et al. Proteomic characterization of human multiple myeloma bone marrow extracellular matrix. Leukemia. 2017 doi: 10.1038/leu.2017.102. [DOI] [PubMed] [Google Scholar]
- 6.Pallotta I, Lovett M, Rice W, Kaplan DL, Balduini A. Bone marrow osteoblastic niche: a new model to study physiological regulation of megakaryopoiesis. PLoS One. 2009;4:e8359. doi: 10.1371/journal.pone.0008359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steevels TA, et al. Co-expression of the collagen receptors leukocyte-associated immunoglobulin-like receptor-1 and glycoprotein VI on a subset of megakaryoblasts. Haematologica. 2010;95:2005–2012. doi: 10.3324/haematol.2010.026120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang Y, et al. Proplatelet formation is regulated by the Rho/ROCK pathway. Blood. 2007;109:4229–4236. doi: 10.1182/blood-2006-04-020024. [DOI] [PubMed] [Google Scholar]
- 9.Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 10.Malara A, et al. Brief Report: Alternative Splicing of Extra Domain A (EIIIA) of Fibronectin Plays a Tissue-Specific Role in Hematopoietic Homeostasis. Stem Cells. 2016;34:2263–2268. doi: 10.1002/stem.2381. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka R, et al. VLA-5-mediated adhesion to fibronectin accelerates hemin-stimulated erythroid differentiation of K562 cells through induction of VLA-4 expression. J Biol Chem. 2009;284:19817–19825. doi: 10.1074/jbc.M109.009860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zweegman S, Veenhof MA, Huijgens PC, Schuurhuis GJ, Drager AM. Regulation of megakaryocytopoiesis in an in vitro stroma model: preferential adhesion of megakaryocytic progenitors and subsequent inhibition of maturation. Exp Hematol. 2000;28:401–410. doi: 10.1016/s0301-472x(00)00128-4. [DOI] [PubMed] [Google Scholar]
- 13.Di Giacomo F, et al. Heparan sulfate mimetics can efficiently mobilize long-term hematopoietic stem cells. Haematologica. 2012;97:491–499. doi: 10.3324/haematol.2011.047662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khaldoyanidi S, Moll J, Karakhanova S, Herrlich P, Ponta H. Hyaluronate-enhanced hematopoiesis: two different receptors trigger the release of interleukin-1beta and interleukin-6 from bone marrow macrophages. Blood. 1999;94:940–949. [PubMed] [Google Scholar]
- 15.Matrosova VY, Orlovskaya IA, Serobyan N, Khaldoyanidi SK. Hyaluronic acid facilitates the recovery of hematopoiesis following 5-fluorouracil administration. Stem Cells. 2004;22:544–555. doi: 10.1634/stemcells.22-4-544. [DOI] [PubMed] [Google Scholar]
- 16.Goncharova V, et al. Hyaluronan expressed by the hematopoietic microenvironment is required for bone marrow hematopoiesis. J Biol Chem. 2012;287:25419–25433. doi: 10.1074/jbc.M112.376699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smaldone S, Bigarella CL, Del Solar M, Ghaffari S, Ramirez F. Fibrillin-1 microfibrils influence adult bone marrow hematopoiesis. Matrix Biol. 2016;52–54:88–94. doi: 10.1016/j.matbio.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazzon C, et al. The critical role of agrin in the hematopoietic stem cell niche. Blood. 2011;118:2733–2742. doi: 10.1182/blood-2011-01-331272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura-Ishizu A, et al. Extracellular matrix protein tenascin-C is required in the bone marrow microenvironment primed for hematopoietic regeneration. Blood. 2012;119:5429–5437. doi: 10.1182/blood-2011-11-393645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uckelmann H, et al. Extracellular matrix protein Matrilin-4 regulates stress-induced HSC proliferation via CXCR4. J Exp Med. 2016;213:1961–1971. doi: 10.1084/jem.20151713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bornstein P. Thrombospondins function as regulators of angiogenesis. J Cell Commun Signal. 2009;3:189–200. doi: 10.1007/s12079-009-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang JC, Novetsky A, Chen C, Novetsky AD. Plasma matrix metalloproteinase and tissue inhibitor of metalloproteinase in patients with agnogenic myeloid metaplasia or idiopathic primary myelofibrosis. Br J Haematol. 2002;119:709–712. doi: 10.1046/j.1365-2141.2002.03874.x. [DOI] [PubMed] [Google Scholar]
- 24.Shen Y, et al. Tissue inhibitor of metalloproteinase-3 (TIMP-3) regulates hematopoiesis and bone formation in vivo. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakajima H, et al. TIMP-3 recruits quiescent hematopoietic stem cells into active cell cycle and expands multipotent progenitor pool. Blood. 2010;116:4474–4482. doi: 10.1182/blood-2010-01-266528. [DOI] [PubMed] [Google Scholar]
- 26.Hoyer-Hansen G, Behrendt N, Ploug M, Dano K, Preissner KT. The intact urokinase receptor is required for efficient vitronectin binding: receptor cleavage prevents ligand interaction. FEBS Lett. 1997;420:79–85. doi: 10.1016/s0014-5793(97)01491-9. [DOI] [PubMed] [Google Scholar]
- 27.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Daculsi R, Bareille R, Bourget C, Amedee J. uPA and MMP-2 were involved in self-assembled network formation in a two dimensional co-culture model of bone marrow stromal cells and endothelial cells. J Cell Biochem. 2013;114:650–657. doi: 10.1002/jcb.24407. [DOI] [PubMed] [Google Scholar]
- 29.McWilliam NA, et al. Evidence for an active fibrinolytic system in normal human bone marrow. Br J Haematol. 1996;93:170–176. doi: 10.1046/j.1365-2141.1996.442985.x. [DOI] [PubMed] [Google Scholar]
- 30.Ibrahim AA, et al. Inhibition of plasminogen activator inhibitor type-1 activity enhances rapid and sustainable hematopoietic regeneration. Stem Cells. 2014;32:946–958. doi: 10.1002/stem.1577. [DOI] [PubMed] [Google Scholar]
- 31.Alessi MC, et al. Detection of plasminogen activator inhibitor-1 (PAI-1) mRNA in human megakaryocytes by in situ hybridization. Thromb Haemost. 1994;72:931–936. [PubMed] [Google Scholar]
- 32.Slany A, et al. Extracellular matrix remodeling by bone marrow fibroblast-like cells correlates with disease progression in multiple myeloma. J Proteome Res. 2014;13:844–854. doi: 10.1021/pr400881p. [DOI] [PubMed] [Google Scholar]
- 33.Rosc D, et al. Plasminogen activators (t-PA and u-PA) and plasminogen activators inhibitors (PAI-1 and PAI-2) in some myeloproliferative syndromes. Med Sci Monit. 2000;6:684–691. [PubMed] [Google Scholar]
- 34.Larson MK, Watson SP. Regulation of proplatelet formation and platelet release by integrin alpha IIb beta3. Blood. 2006;108:1509–1514. doi: 10.1182/blood-2005-11-011957. [DOI] [PubMed] [Google Scholar]
- 35.Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci. 2006;63:2304–2316. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Georges PC, et al. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1147–1154. doi: 10.1152/ajpgi.00032.2007. [DOI] [PubMed] [Google Scholar]
- 37.Berry MF, et al. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol. 2006;290:H2196–2203. doi: 10.1152/ajpheart.01017.2005. [DOI] [PubMed] [Google Scholar]
- 38.Jansen LE, Birch NP, Schiffman JD, Crosby AJ, Peyton SR. Mechanics of intact bone marrow. J Mech Behav Biomed Mater. 2015;50:299–307. doi: 10.1016/j.jmbbm.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Apoorva FN, et al. Lymph node stiffness-mimicking hydrogels regulate human B-cell lymphoma growth and cell surface receptor expression in a molecular subtype-specific manner. J Biomed Mater Res A. 2017 doi: 10.1002/jbm.a.36031. [DOI] [PubMed] [Google Scholar]
- 40.Muller DJ, Helenius J, Alsteens D, Dufrene YF. Force probing surfaces of living cells to molecular resolution. Nat Chem Biol. 2009;5:383–390. doi: 10.1038/nchembio.181. [DOI] [PubMed] [Google Scholar]
- 41.Asgari M, Latifi N, Heris HK, Vali H, Mongeau L. In vitro fibrillogenesis of tropocollagen type III in collagen type I affects its relative fibrillar topology and mechanics. Sci Rep. 2017;7:1392. doi: 10.1038/s41598-017-01476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keating M, Kurup A, Alvarez-Elizondo M, Levine AJ, Botvinick E. Spatial distributions of pericellular stiffness in natural extracellular matrices are dependent on cell-mediated proteolysis and contractility. Acta Biomater. 2017 doi: 10.1016/j.actbio.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolacna L, et al. Biochemical and biophysical aspects of collagen nanostructure in the extracellular matrix. Physiol Res. 2007;56(Suppl 1):S51–60. doi: 10.33549/physiolres.931302. [DOI] [PubMed] [Google Scholar]
- 44.Malara A, et al. Megakaryocytes contribute to the bone marrow-matrix environment by expressing fibronectin, type IV collagen, and laminin. Stem Cells. 2014;32:926–937. doi: 10.1002/stem.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nilsson SK, et al. Immunofluorescence characterization of key extracellular matrix proteins in murine bone marrow in situ. J Histochem Cytochem. 1998;46:371–377. doi: 10.1177/002215549804600311. [DOI] [PubMed] [Google Scholar]
- 46.Licup AJ, et al. Stress controls the mechanics of collagen networks. Proc Natl Acad Sci U S A. 2015;112:9573–9578. doi: 10.1073/pnas.1504258112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. J Biol Chem. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- 48.Iwasaki A, et al. Molecular Mechanism Responsible for Fibronectin-controlled Alterations in Matrix Stiffness in Advanced Chronic Liver Fibrogenesis. J Biol Chem. 2016;291:72–88. doi: 10.1074/jbc.M115.691519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Stefano V, et al. Major Action of Endogenous Lysyl Oxidase in Clear Cell Renal Cell Carcinoma Progression and Collagen Stiffness Revealed by Primary Cell Cultures. Am J Pathol. 2016;186:2473–2485. doi: 10.1016/j.ajpath.2016.05.019. [DOI] [PubMed] [Google Scholar]
- 51.Baker AM, Bird D, Lang G, Cox TR, Erler JT. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene. 2013;32:1863–1868. doi: 10.1038/onc.2012.202. [DOI] [PubMed] [Google Scholar]
- 52.Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nat Rev Cancer. 2012;12:540–552. doi: 10.1038/nrc3319. [DOI] [PubMed] [Google Scholar]
- 53.Eliades A, et al. Control of megakaryocyte expansion and bone marrow fibrosis by lysyl oxidase. J Biol Chem. 2011;286:27630–27638. doi: 10.1074/jbc.M111.243113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tadmor T, et al. The expression of lysyl-oxidase gene family members in myeloproliferative neoplasms. Am J Hematol. 2013;88:355–358. doi: 10.1002/ajh.23409. [DOI] [PubMed] [Google Scholar]
- 55.Malara A, et al. Extracellular matrix structure and nano-mechanics determine megakaryocyte function. Blood. 2011;118:4449–4453. doi: 10.1182/blood-2011-04-345876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fox NE, Kaushansky K. Engagement of integrin alpha4beta1 enhances thrombopoietin-induced megakaryopoiesis. Exp Hematol. 2005;33:94–99. doi: 10.1016/j.exphem.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 57.Abbonante V, et al. A new path to platelet production through matrix sensing. Haematologica. 2017 doi: 10.3324/haematol.2016.161562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang F, Jia Y, Cohen I. Fibronectin- and protein kinase C-mediated activation of ERK/MAPK are essential for proplateletlike formation. Blood. 2002;99:3579–3584. doi: 10.1182/blood.v99.10.3579. [DOI] [PubMed] [Google Scholar]
- 59.Matsunaga T, et al. Potentiated activation of VLA-4 and VLA-5 accelerates proplatelet-like formation. Ann Hematol. 2012;91:1633–1643. doi: 10.1007/s00277-012-1498-y. [DOI] [PubMed] [Google Scholar]
- 60.Schick PK, et al. Integrins involved in the adhesion of megakaryocytes to fibronectin and fibrinogen. Blood. 1998;92:2650–2656. [PubMed] [Google Scholar]
- 61.Malara A, et al. Megakaryocyte-matrix interaction within bone marrow: new roles for fibronectin and factor XIII-A. Blood. 2011;117:2476–2483. doi: 10.1182/blood-2010-06-288795. [DOI] [PubMed] [Google Scholar]
- 62.Choi JS, Harley BA. Marrow-inspired matrix cues rapidly affect early fate decisions of hematopoietic stem and progenitor cells. Sci Adv. 2017;3:e1600455. doi: 10.1126/sciadv.1600455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leven RM, Tablin F. Extracellular matrix stimulation of guinea pig megakaryocyte proplatelet formation in vitro is mediated through the vitronectin receptor. Exp Hematol. 1992;20:1316–1322. [PubMed] [Google Scholar]
- 64.Majka M, et al. In vitro expansion of human megakaryocytes as a tool for studying megakaryocytic development and function. Platelets. 2001;12:325–332. doi: 10.1080/09537100120068152. [DOI] [PubMed] [Google Scholar]
- 65.Wohn KD, et al. The role of plasminogen activator inhibitor-1 as inhibitor of platelet and megakaryoblastic cell adhesion. Br J Haematol. 1999;104:901–908. doi: 10.1046/j.1365-2141.1999.01242.x. [DOI] [PubMed] [Google Scholar]
- 66.Kyriakides TR, et al. Megakaryocytes require thrombospondin-2 for normal platelet formation and function. Blood. 2003;101:3915–3923. doi: 10.1182/blood.V101.10.3915. [DOI] [PubMed] [Google Scholar]
- 67.Tajika K, Ikebuchi K, Dan K, Asano S. A role of GAGs in ECM on morphogenesis of megakaryocytes. Br J Haematol. 1996;94:34–39. doi: 10.1046/j.1365-2141.1996.d01-1781.x. [DOI] [PubMed] [Google Scholar]
- 68.Petrey AC, Obery DR, Kessler SP, Flamion B, de la Motte CA. Hyaluronan Depolymerization by Megakaryocyte Hyaluronidase-2 Is Required for Thrombopoiesis. Am J Pathol. 2016;186:2390–2403. doi: 10.1016/j.ajpath.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Currao M, et al. Hyaluronan based hydrogels provide an improved model to study megakaryocyte-matrix interactions. Exp Cell Res. 2016;346:1–8. doi: 10.1016/j.yexcr.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kashiwakura I, et al. The effects of glycosaminoglycans on thrombopoietin-induced megakaryocytopoiesis. Haematologica. 2006;91:445–451. [PubMed] [Google Scholar]
- 71.Lucero HA, et al. Lysyl oxidase oxidizes cell membrane proteins and enhances the chemotactic response of vascular smooth muscle cells. J Biol Chem. 2008;283:24103–24117. doi: 10.1074/jbc.M709897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsuura S, et al. Lysyl oxidase is associated with increased thrombosis and platelet reactivity. Blood. 2016;127:1493–1501. doi: 10.1182/blood-2015-02-629667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bock O, et al. Bone morphogenetic proteins are overexpressed in the bone marrow of primary myelofibrosis and are apparently induced by fibrogenic cytokines. Am J Pathol. 2008;172:951–960. doi: 10.2353/ajpath.2008.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Papadantonakis N, Matsuura S, Ravid K. Megakaryocyte pathology and bone marrow fibrosis: the lysyl oxidase connection. Blood. 2012;120:1774–1781. doi: 10.1182/blood-2012-02-402594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eliades A, et al. Megakaryocyte polyploidy is inhibited by lysyl oxidase propeptide. Cell Cycle. 2013;12:1242–1250. doi: 10.4161/cc.24312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aguilar A, et al. Importance of environmental stiffness for megakaryocyte differentiation and proplatelet formation. Blood. 2016;128:2022–2032. doi: 10.1182/blood-2016-02-699959. [DOI] [PubMed] [Google Scholar]
- 77.Shin JW, Swift J, Spinler KR, Discher DE. Myosin-II inhibition and soft 2D matrix maximize multinucleation and cellular projections typical of platelet-producing megakaryocytes. Proc Natl Acad Sci U S A. 2011;108:11458–11463. doi: 10.1073/pnas.1017474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Buduo CA, et al. Programmable 3D silk bone marrow niche for platelet generation ex vivo and modeling of megakaryopoiesis pathologies. Blood. 2015;125:2254–2264. doi: 10.1182/blood-2014-08-595561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 80.Lo CM, Wang HB, Dembo M, Wang YL. Cell movement is guided by the rigidity of the substrate. Biophys J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chitteti BR, Kacena MA, Voytik-Harbin SL, Srour EF. Modulation of hematopoietic progenitor cell fate in vitro by varying collagen oligomer matrix stiffness in the presence or absence of osteoblasts. J Immunol Methods. 2015;425:108–113. doi: 10.1016/j.jim.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 82.Lazar-Karsten P, et al. The influence of extracellular matrix proteins and mesenchymal stem cells on erythropoietic cell maturation. Vox Sang. 2011;101:65–76. doi: 10.1111/j.1423-0410.2010.01453.x. [DOI] [PubMed] [Google Scholar]
- 83.Choi JS, Harley BA. The combined influence of substrate elasticity and ligand density on the viability and biophysical properties of hematopoietic stem and progenitor cells. Biomaterials. 2012;33:4460–4468. doi: 10.1016/j.biomaterials.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 84.Oswald J, et al. Gene-expression profiling of CD34+ hematopoietic cells expanded in a collagen I matrix. Stem Cells. 2006;24:494–500. doi: 10.1634/stemcells.2005-0276. [DOI] [PubMed] [Google Scholar]
- 85.Sugahara H, et al. Induction of programmed cell death in human hematopoietic cell lines by fibronectin via its interaction with very late antigen 5. J Exp Med. 1994;179:1757–1766. doi: 10.1084/jem.179.6.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hamamura K, et al. A critical role of VLA-4 in erythropoiesis in vivo. Blood. 1996;87:2513–2517. [PubMed] [Google Scholar]
- 87.Hurley RW, McCarthy JB, Verfaillie CM. Direct adhesion to bone marrow stroma via fibronectin receptors inhibits hematopoietic progenitor proliferation. J Clin Invest. 1995;96:511–519. doi: 10.1172/JCI118063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vuillet-Gaugler MH, et al. Loss of attachment to fibronectin with terminal human erythroid differentiation. Blood. 1990;75:865–873. [PubMed] [Google Scholar]
- 89.Simon Davis DA, Parish CR. Heparan sulfate: a ubiquitous glycosaminoglycan with multiple roles in immunity. Front Immunol. 2013;4:470. doi: 10.3389/fimmu.2013.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Coombe DR, Watt SM, Parish CR. Mac-1 (CD11b/CD18) and CD45 mediate the adhesion of hematopoietic progenitor cells to stromal cell elements via recognition of stromal heparan sulfate. Blood. 1994;84:739–752. [PubMed] [Google Scholar]
- 91.Thon JN, et al. Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol. 2010;191:861–874. doi: 10.1083/jcb.201006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mossuz P, Schweitzer A, Molla A, Berthier R. Expression and function of receptors for extracellular matrix molecules in the differentiation of human megakaryocytes in vitro. Br J Haematol. 1997;98:819–827. doi: 10.1046/j.1365-2141.1997.3013118.x. [DOI] [PubMed] [Google Scholar]
- 93.Sun Z, Guo SS, Fassler R. Integrin-mediated mechanotransduction. J Cell Biol. 2016;215:445–456. doi: 10.1083/jcb.201609037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sabri S, et al. Differential regulation of actin stress fiber assembly and proplatelet formation by alpha2beta1 integrin and GPVI in human megakaryocytes. Blood. 2004;104:3117–3125. doi: 10.1182/blood-2003-12-4398. [DOI] [PubMed] [Google Scholar]
- 95.Chen Z, et al. The May-Hegglin anomaly gene MYH9 is a negative regulator of platelet biogenesis modulated by the Rho-ROCK pathway. Blood. 2007;110:171–179. doi: 10.1182/blood-2007-02-071589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pecci A, et al. Megakaryocytes of patients with MYH9-related thrombocytopenia present an altered proplatelet formation. Thromb Haemost. 2009;102:90–96. doi: 10.1160/TH09-01-0068. [DOI] [PubMed] [Google Scholar]
- 97.Pecci A, et al. Mutations responsible for MYH9-related thrombocytopenia impair SDF-1-driven migration of megakaryoblastic cells. Thromb Haemost. 2011;106:693–704. doi: 10.1160/TH11-02-0126. [DOI] [PubMed] [Google Scholar]
- 98.Di Buduo CA, et al. The importance of calcium in the regulation of megakaryocyte function. Haematologica. 2014;99:769–778. doi: 10.3324/haematol.2013.096859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Di Buduo CA, Balduini A, Moccia F. Pathophysiological Significance of Store-Operated Calcium Entry in Megakaryocyte Function: Opening New Paths for Understanding the Role of Calcium in Thrombopoiesis. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17122055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 101.Parekh AB. Store-operated CRAC channels: function in health and disease. Nat Rev Drug Discov. 2010;9:399–410. doi: 10.1038/nrd3136. [DOI] [PubMed] [Google Scholar]
- 102.Mountford JC, Melford SK, Bunce CM, Gibbins J, Watson SP. Collagen or collagen-related peptide cause (Ca2+)i elevation and increased tyrosine phosphorylation in human megakaryocytes. Thromb Haemost. 1999;82:1153–1159. [PubMed] [Google Scholar]
- 103.Nangalia J, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–2405. doi: 10.1056/NEJMoa1312542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pietra D, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia. 2016;30:431–438. doi: 10.1038/leu.2015.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thodeti CK, et al. TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ Res. 2009;104:1123–1130. doi: 10.1161/CIRCRESAHA.108.192930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bagriantsev SN, Gracheva EO, Gallagher PG. Piezo proteins: regulators of mechanosensation and other cellular processes. J Biol Chem. 2014;289:31673–31681. doi: 10.1074/jbc.R114.612697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Coste B, et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature. 2012;483:176–181. doi: 10.1038/nature10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Coste B, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55–60. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chesler AT, et al. The Role of PIEZO2 in Human Mechanosensation. N Engl J Med. 2016;375:1355–1364. doi: 10.1056/NEJMoa1602812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Coste B, et al. Gain-of-function mutations in the mechanically activated ion channel PIEZO2 cause a subtype of Distal Arthrogryposis. Proc Natl Acad Sci U S A. 2013;110:4667–4672. doi: 10.1073/pnas.1221400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McMillin MJ, et al. Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am J Hum Genet. 2014;94:734–744. doi: 10.1016/j.ajhg.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rocio Servin-Vences M, Moroni M, Lewin GR, Poole K. Direct measurement of TRPV4 and PIEZO1 activity reveals multiple mechanotransduction pathways in chondrocytes. Elife. 2017;6 doi: 10.7554/eLife.21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ilkan Z, et al. Evidence for shear-mediated Ca2+ entry through mechanosensitive cation channels in human platelets and a megakaryocytic cell line. J Biol Chem. 2017;292:9204–9217. doi: 10.1074/jbc.M116.766196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cinar E, et al. Piezo1 regulates mechanotransductive release of ATP from human RBCs. Proc Natl Acad Sci U S A. 2015;112:11783–11788. doi: 10.1073/pnas.1507309112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zarychanski R, et al. Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood. 2012;120:1908–1915. doi: 10.1182/blood-2012-04-422253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Albuisson J, et al. Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat Commun. 2013;4:1884. doi: 10.1038/ncomms2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ranade SS, et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci U S A. 2014;111:10347–10352. doi: 10.1073/pnas.1409233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li J, et al. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515:279–282. doi: 10.1038/nature13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lukacs V, et al. Impaired PIEZO1 function in patients with a novel autosomal recessive congenital lymphatic dysplasia. Nat Commun. 2015;6:8329. doi: 10.1038/ncomms9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gudipaty SA, et al. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature. 2017;543:118–121. doi: 10.1038/nature21407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mahaut-Smith MP. The unique contribution of ion channels to platelet and megakaryocyte function. J Thromb Haemost. 2012;10:1722–1732. doi: 10.1111/j.1538-7836.2012.04837.x. [DOI] [PubMed] [Google Scholar]
- 122.McCloskey C, et al. Kv1.3 is the exclusive voltage-gated K+ channel of platelets and megakaryocytes: roles in membrane potential, Ca2+ signalling and platelet count. J Physiol. 2010;588:1399–1406. doi: 10.1113/jphysiol.2010.188136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mahaut-Smith MP. Calcium-activated potassium channels in human platelets. J Physiol. 1995;484(Pt 1):15–24. doi: 10.1113/jphysiol.1995.sp020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schmidt EM, et al. Ion channels in the regulation of platelet migration. Biochem Biophys Res Commun. 2011;415:54–60. doi: 10.1016/j.bbrc.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 125.Gianelli U, et al. The European Consensus on grading of bone marrow fibrosis allows a better prognostication of patients with primary myelofibrosis. Mod Pathol. 2012;25:1193–1202. doi: 10.1038/modpathol.2012.87. [DOI] [PubMed] [Google Scholar]
- 126.Thiele J, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90:1128–1132. [PubMed] [Google Scholar]
- 127.Bauermeister DE. Quantitation of bone marrow reticulin--a normal range. Am J Clin Pathol. 1971;56:24–31. doi: 10.1093/ajcp/56.1.24. [DOI] [PubMed] [Google Scholar]
- 128.Beckman EN, Brown AW., Jr Normal reticulin level in iliac bone marrow. Arch Pathol Lab Med. 1990;114:1241–1243. [PubMed] [Google Scholar]
- 129.Kuter DJ, Bain B, Mufti G, Bagg A, Hasserjian RP. Bone marrow fibrosis: pathophysiology and clinical significance of increased bone marrow stromal fibres. Br J Haematol. 2007;139:351–362. doi: 10.1111/j.1365-2141.2007.06807.x. [DOI] [PubMed] [Google Scholar]
- 130.Bain BJ, Clark DM, Lampert IA, Wilkins BS. Bone Marrow Pathology. 2. Blackwell Science Ltd; 2001. [Google Scholar]
- 131.Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22:14–22. doi: 10.1038/sj.leu.2404955. [DOI] [PubMed] [Google Scholar]
- 132.Tefferi A, et al. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT) Blood. 2006;108:1497–1503. doi: 10.1182/blood-2006-03-009746. [DOI] [PubMed] [Google Scholar]
- 133.Chalayer E, et al. Bone Marrow Involvement in Systemic Lupus Erythematosus. QJM. 2017 doi: 10.1093/qjmed/hcx102. [DOI] [PubMed] [Google Scholar]
- 134.Piatek CI, et al. Autoimmune Myelofibrosis: Clinical Features, Course, and Outcome. Acta Haematol. 2017;138:129–137. doi: 10.1159/000479103. [DOI] [PubMed] [Google Scholar]
- 135.Vergara-Lluri ME, et al. Autoimmune myelofibrosis: an update on morphologic features in 29 cases and review of the literature. Hum Pathol. 2014;45:2183–2191. doi: 10.1016/j.humpath.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 136.Orazi A, et al. Effects of recombinant human interleukin-11 (Neumega rhIL-11 growth factor) on megakaryocytopoiesis in human bone marrow. Exp Hematol. 1996;24:1289–1297. [PubMed] [Google Scholar]
- 137.Kuter DJ, et al. Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood. 2009;114:3748–3756. doi: 10.1182/blood-2009-05-224766. [DOI] [PubMed] [Google Scholar]
- 138.Bussel JB, et al. Repeated short-term use of eltrombopag in patients with chronic immune thrombocytopenia (ITP) Br J Haematol. 2013;160:538–546. doi: 10.1111/bjh.12169. [DOI] [PubMed] [Google Scholar]
- 139.Saleh MN, et al. Safety and efficacy of eltrombopag for treatment of chronic immune thrombocytopenia: results of the long-term, open-label EXTEND study. Blood. 2013;121:537–545. doi: 10.1182/blood-2012-04-425512. [DOI] [PubMed] [Google Scholar]
- 140.Ghanima W, et al. Bone marrow fibrosis in 66 patients with immune thrombocytopenia treated with thrombopoietin-receptor agonists: a single-center, long-term follow-up. Haematologica. 2014;99:937–944. doi: 10.3324/haematol.2013.098921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rizvi H, et al. United Kingdom immune thrombocytopenia registry: retrospective evaluation of bone marrow fibrosis in adult patients with primary immune thrombocytopenia and correlation with clinical findings. Br J Haematol. 2015;169:590–594. doi: 10.1111/bjh.13330. [DOI] [PubMed] [Google Scholar]
- 142.Pullarkat V, Bass RD, Gong JZ, Feinstein DI, Brynes RK. Primary autoimmune myelofibrosis: definition of a distinct clinicopathologic syndrome. Am J Hematol. 2003;72:8–12. doi: 10.1002/ajh.10258. [DOI] [PubMed] [Google Scholar]
- 143.Beham-Schmid C, et al. Treatment of chronic myelogenous leukemia with the tyrosine kinase inhibitor STI571 results in marked regression of bone marrow fibrosis. Blood. 2002;99:381–383. doi: 10.1182/blood.v99.1.381. [DOI] [PubMed] [Google Scholar]
- 144.Narang NC, Rusia U, Sikka M, Kotru M. Morphological Changes in Bone Marrow Post Imatinib Therapy in Chronic Phase CML: A Follow up Study on Sequential Bone Marrow Aspirates and Biopsies. J Clin Diagn Res. 2017;11:EC25–EC29. doi: 10.7860/JCDR/2017/25173.9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hasserjian RP, et al. ST1571 (imatinib mesylate) reduces bone marrow cellularity and normalizes morphologic features irrespective of cytogenetic response. Am J Clin Pathol. 2002;117:360–367. doi: 10.1309/NR81-VCU0-CKW1-4HT9. [DOI] [PubMed] [Google Scholar]
- 146.Tefferi A, Pardanani A. Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol. 2015;1:97–105. doi: 10.1001/jamaoncol.2015.89. [DOI] [PubMed] [Google Scholar]
- 147.Harrison CN, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701–1707. doi: 10.1038/leu.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kroger N, et al. Rapid regression of bone marrow fibrosis after dose-reduced allogeneic stem cell transplantation in patients with primary myelofibrosis. Exp Hematol. 2007;35:1719–1722. doi: 10.1016/j.exphem.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 149.Domm J, et al. Unrelated stem cell transplant for infantile idiopathic myelofibrosis. Pediatr Blood Cancer. 2009;52:893–895. doi: 10.1002/pbc.21910. [DOI] [PubMed] [Google Scholar]
- 150.Bock O, et al. Aberrant expression of platelet-derived growth factor (PDGF) and PDGF receptor-alpha is associated with advanced bone marrow fibrosis in idiopathic myelofibrosis. Haematologica. 2005;90:133–134. [PubMed] [Google Scholar]
- 151.Brouty-Boye D. Developmental biology of fibroblasts and neoplastic disease. Prog Mol Subcell Biol. 2005;40:55–77. doi: 10.1007/3-540-27671-8_3. [DOI] [PubMed] [Google Scholar]
- 152.Shehata M, et al. TGF-beta1 induces bone marrow reticulin fibrosis in hairy cell leukemia. J Clin Invest. 2004;113:676–685. doi: 10.1172/JCI19540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013;24:133–145. doi: 10.1016/j.cytogfr.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 154.Castro-Malaspina H, Rabellino EM, Yen A, Nachman RL, Moore MA. Human megakaryocyte stimulation of proliferation of bone marrow fibroblasts. Blood. 1981;57:781–787. [PubMed] [Google Scholar]
- 155.Ciurea SO, et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood. 2007;110:986–993. doi: 10.1182/blood-2006-12-064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Le Bousse-Kerdiles MC, et al. Differential expression of transforming growth factor-beta, basic fibroblast growth factor, and their receptors in CD34+ hematopoietic progenitor cells from patients with myelofibrosis and myeloid metaplasia. Blood. 1996;88:4534–4546. [PubMed] [Google Scholar]
- 157.Thachil J. The lung megakaryocytes and pulmonary fibrosis in systemic sclerosis. Med Hypotheses. 2009;72:291–293. doi: 10.1016/j.mehy.2008.09.045. [DOI] [PubMed] [Google Scholar]
- 158.Mathew R, Huang J, Wu JM, Fallon JT, Gewitz MH. Hematological disorders and pulmonary hypertension. World J Cardiol. 2016;8:703–718. doi: 10.4330/wjc.v8.i12.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93:327–358. doi: 10.1152/physrev.00016.2011. [DOI] [PubMed] [Google Scholar]
- 160.Kaplan ZS, Jackson SP. The role of platelets in atherothrombosis. Hematology Am Soc Hematol Educ Program. 2011;2011:51–61. doi: 10.1182/asheducation-2011.1.51. [DOI] [PubMed] [Google Scholar]
- 161.Yamasaki F, et al. Association between arterial stiffness and platelet activation. J Hum Hypertens. 2005;19:527–533. doi: 10.1038/sj.jhh.1001861. [DOI] [PubMed] [Google Scholar]
- 162.Wilkinson IB, et al. Oral vitamin C reduces arterial stiffness and platelet aggregation in humans. J Cardiovasc Pharmacol. 1999;34:690–693. doi: 10.1097/00005344-199911000-00010. [DOI] [PubMed] [Google Scholar]
- 163.Qiu Y, et al. Platelet mechanosensing of substrate stiffness during clot formation mediates adhesion, spreading, and activation. Proc Natl Acad Sci U S A. 2014;111:14430–14435. doi: 10.1073/pnas.1322917111. [DOI] [PMC free article] [PubMed] [Google Scholar]