

Abstract
The poor prognosis associated with malignant melanoma has not changed substantially over the past 30 years. Targeted molecular therapies, such as immunotherapy have shown promise, but suffer from resistance and off-target toxicities, underscoring the need for alternative therapeutic strategies that can be used in combination with existing protocols. Moreover, peptides targeting melanoma-specific markers, like the melanocortin-1 receptor (MC1-R), for imaging and therapy exhibit high renal uptake that limits clinical translation. In the current study, the application of ultrasmall fluorescent (Cy5) silica nanoparticles (C′ dots), conjugated with MC1-R targeting alpha melanocyte stimulating hormone (αMSH) peptides on the polyethylene glycol (PEG) coated surface, is examined for melanoma-selective imaging. αMSH peptide sequences, evaluated for conjugation to the PEG-Cy5-C′ dot nanoparticles, bound to MC1-R with high affinity, and targeted melanoma in syngenetic and xenografted melanoma mouse models. Results demonstrated a 10-fold improvement in MC1-R affinity over the native peptide alone following surface attachment of the optimal αMSH peptide. Systematic in vivo studies further demonstrated favorable in vivo renal clearance kinetics as well as receptor-mediated tumor cell internalization of as-developed radiolabeled particle tracers in B16F10 melanoma bearing mice. These findings highlight the ability of αMSH-PEG-Cy5-C′ dots to overcome previous hurdles that prevented clinical translation of peptide and antibody-based melanoma probes, and reveal the potential of αMSH-PEG-Cy5-C′ dots for melanoma selective imaging, image-guided surgery, and therapeutic applications.
Keywords: αMSH peptide, Ultrasmall silica nanoparticle, C′ dots, Target-or-clear, Melanoma
Graphical Abstract
INTRODUCTION
Melanoma is the deadliest form of skin cancer. Although early diagnosis and treatment results in a high percentage of cures, the survival rate significantly declines to only 15–20% for systemic melanoma disease spread.1 The mortality rate from melanoma has not changed substantially over the past 25 years. In addition, mechanisms underlying melanoma treatment resistance remain elusive. Treatment resistance is likely due to a combination of immune surveillance avoidance, multidrug resistance, and persistent melanoma stem cell populations.2–4 However, therapeutic exploitation of genetic mutations in melanoma and the discovery of targets for cancer immunotherapies have resulted in better treatment paradigms for patients.5–10 As a result of advancing such novel targeted treatment options, improved clinical responses have been achieved, although generally short-lived (6–12 months) and accompanied by immune resistance mechanisms and/or activation of alternative signaling pathways.2, 5, 11 Of critical need, therefore, are alternative systemic therapeutic options that target new melanoma-specific signaling pathway markers.
The melanoma biomarker, melanocortin-1 receptor (MC1-R), is overexpressed on the surface of human melanoma tumor cells, and serves as an attractive target for melanoma-specific imaging and the selective delivery of therapeutic agents.12–18 Its naturally occurring ligand is a 13-amino acid peptide, alpha melanocyte stimulating hormone (αMSH), which is involved in the regulation of skin pigmentation, as well as immune and anti-inflammatory responses.19 Numerous structure-activity studies have identified linear and cyclic peptide agonists and antagonists with nanomolar and subnanomolar binding affinities.20 Binding of MC1-R agonists to receptors expressed on melanoma cells results in rapid cellular internalization of the receptor-peptide complex, and this mechanism has been exploited for receptor-mediated internalization of radiolabeled, optical, and therapeutic peptide complexes.
Potent radiolabeled peptide analogs of the αMSH sequence, such as [Nle4, D-Phe7]- αMSH (NDP),21 have been extensively studied as imaging and therapeutic agents. Although early NDP radioconjugates22,23 suffered from low tumor retention and poor biodistribution properties, cyclization of the core MC1-R binding region of the αMSH sequence via metal coordination or lactam bond formation resulted in radiolabeled peptides with high tumor uptake and prolonged retention.16, 24–25 However, their high non-specific uptake in radiosensitive organs, such as the kidneys, together with limited mid-gut imaging sensitivity and low tumor-to-background ratios, have hampered clinical translation efforts.15, 26
To overcome these limitations, several strategies have been advanced, including linker optimization, peptide multimerization, and the attachment of peptides onto nanoparticle surfaces. Optimization of linkers bridging the peptide with a radiometal chelator have led to incremental improvements in tumor-to-kidney ratios, while the greatest improvements were found to occur after altering the net charge of the entire molecular complex.15–16 Further, the multimerization of αMSH peptides has resulted in constructs with improved in vitro cell binding affinities. However, these same constructs demonstrated poor in vivo pharmacokinetic properties due to increased hydrophobicity and reduced tissue penetration.27 Finally, the use of different types of nanomaterials, such as liposomes and dendrimers, conjugated with multiple copies of αMSH analogs, has generally resulted in only modest increases in in vitro MC1-R binding affinities.28 Although preliminary in vivo MC1-R targeting studies utilizing larger diameter αMSH-conjugated particles, such as polymer micelles29 (i.e., ~100 nm) and hollow gold nanospheres30 (i.e., 40 nm) have been reported, high non-specific accumulation of these products in the reticuloendothelial system (RES) has led to significantly decreased target-to-background ratios31 and/or raised concerns for off-target toxicity, thereby hindering their translational potential. As a result, successful translation of an MC1-R targeted imaging platform demonstrating favorable biological and clearance properties has not been achieved.
Herein we report on the structure-activity relationships for a small library of custom-synthesized rhenium (Re)-cyclized αMSH peptide ligands linked to positron emission tomography (PET) imaging labels conjugated to latest-generation ultrasmall “target-or-clear” silica nanoparticles (i.e., Cornell prime dots or C′ dots) with different surface chemical designs.32–33 These efforts are based on earlier work in which C′ dots were surface engineered with controllable numbers of surface functional groups.34–36 We characterize the resulting ultrasmall (diameter < 10 nm) MC1-R targeting and dual-modality C′ dot conjugates (Scheme 1) in terms of their photophysical and in vitro/in vivo biological properties in melanoma cell lines and xenograft tumor models in comparison to the native αMSH peptide and/or particle and biological controls. The coupling of different radiolabels, such as radiohalogens (i.e., iodine-125; 125I) or radiometals (i.e., zirconium-89; 89Zr), to the particle surface facilitates comparisons of the pharmacokinetics (PK), clearance, and/or tumor-targeting profiles of these radioconjugates. Results demonstrated statistically significant increases in MC1-R-specific tumor-targeted uptake and target-to-background ratios of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots over the time course of the study, as well as internalization of the targeted particle probes in tumor tissue specimens. No significant difference was found in the PK profiles measured at 24 hours post-injection (p.i.) for particles labeled with different radionuclides. We hope these findings will serve as the foundation for next-stage diagnostic and/or radiotherapeutic developments by exploiting a variety of radiometals for the image-guided treatment of human melanoma.
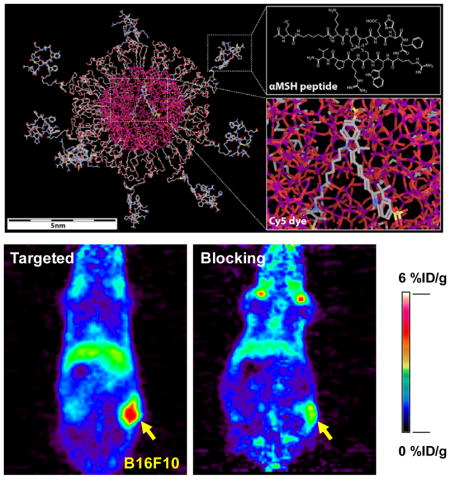
Scheme 1. Molecular rendering of αMSH-PEG-Cy5-C′ dots.

Left side shows rendering of the entire dot, with inserts on the right depicting αMSH chemical structure (top) and encapsulated Cy5 dye (bottom). Silicon, oxygen, carbon, nitrogen, sulfur and rhenium atoms are colored in purple, red, gray, blue, yellow and light green, respectively. Hydrogen atoms are not displayed in the model for better visualization.
RESULTS AND DISCUSSION
Synthesis and screening of MC1-R targeting peptide(s)
MC1-R targeting peptide ligands used for C′ dot synthesis were developed on the basis of the ReCCMSH molecule (Ac-Re[Cys3-Cys4-Glu5-His6-d-Phe7-Arg8-Trp9-Cys10]-Arg11-Pro12-Val13-NH2; superscripts refer to amino acid number based on the native αMSH sequence), and cyclized via rhenium (Re) coordination by Cys-3, Cys-4, and Cys-10 sidechain thiols as well as the Cys-4 amide.24, 37 The initial strategy involved modification of the de-acetylated ReCCMSH molecule (Figure S1a; Peptide I) with N-succinimidyl S-acetylthiolacetate (SATA) and the Bolton-Hunter reagent, N-succinimidyl-3-(4-hydroxypheynyl) propionate (SHPP). The SATA reaction and deprotection converted a primary amine to a free thiol for thiol-ene click chemistry with a heterobifunctional maleimide containing PEG used in the C′ dot synthesis, while SHPP conjugation enabled the ReCCMSH-C′ dot to be radioiodinated. Radiochemical yields of the 131I-ReCCMSH-PEG-C’dots were poor (<2%), and an initial biodistribution study demonstrated transient tumor uptake and GI clearance (Figure S2a). A second ReCCMSH peptide analog was synthesized with a pendant tyrosine residue for direct radioiodination (Figure S1b: Peptide II). The amino terminus of the peptide was modified by SATA to facilitate C′ dot synthesis. The radiolabeling yield for the Ser-Tyr(131I)-Ser-ReCCMSH-PEG-Cy5-C′ dots was ~7%. Preliminary biodistribution results showed poor tumor uptake and evidence of in vivo deiodination (Figure S2b). The third peptide design (Figure S1c: Peptide III) contained a double aminohexanoic acid (Ahx)2-linker between an N-terminal Ac-Cys and the D-Lys-ReCCMSH peptide (Figure 1a). The D-Lys sidechain could be modified for radiolabeling via the addition of prosthetic groups (i.e., SHPP) or metal chelators. Ac-Cys-(Ahx)2-D-Lys-Re[Cys-Cys-Glu-His-dPhe-Arg-Trp-Cys]-Arg-Pro-Val-NH2 (αMSH, Figure S1c) was conjugated with maleimide-terminated heterobifunctional PEG-silane molecules via the N-terminal Cys thiol in preparation for nanoparticle PEGylation. The αMSH peptide was synthesized in its entirety using Fmoc chemistry, and high-purity batches could be produced at a large scale (Figure 1b). The IC50 value measured for the MC1-R targeting D-Lys-ReCCMSH peptide was 6.2×10−9 M (Figure 1c). A scrambled αMSH peptide (Scr-MSH: Ac-Cys-(Ahx)2-D-Lys-Re[Cys-Cys-Trp-Arg-His-dPhe-Glu-Cys]-Arg-Pro-Val-NH2, Figure S1d) was also designed as a negative control for MC1-R binding. The IC50 of the scrambled peptide was 3.7×10−6 M (data not shown). Peptide III was found to be superior to Peptides I and II in terms of synthetic yield and/or stability, and therefore was selected as the lead candidate for αMSH-PEG-C′ dot synthesis and characterization and for performing in vitro and in vivo MC1-R targeting studies.
Figure 1. Characterization of the lead αMSH targeting peptide III.

(a) Chemical structure of the αMSH peptide. (b) HPLC trace of the purified αMSH peptide sequence. (c) IC50 determination of the MC1-R targeting D-Lys-ReCCMSH peptide.
Synthesis and characterization of αMSH -PEG-Cy5-C′ dots
The αMSH-PEG-Cy5-C′ dots described in this paper were prepared using the latest synthesis protocol of Cornell dots (C dots).32 This new generation of Cornell dots is referred to as Cornell prime dots, or in short C′ dots. As compared to the original conventional Stöber process derived C dots using ethanol as the reaction medium,38–39 the synthesis of C′ dots is now conducted in water. Fast hydrolysis and condensation of silane molecules in an aqueous environment facilitates the formation of homogeneous ultra-small silica nanoparticles,32, 40–41 which is different from the process associated with larger size (>10 nm HD) silica particles.42 At the same time, silane-modified Cy5 dyes were added together with the silica source to be covalently encapsulated in the silica nanoparticles during their formation, endowing the particles with bright near-infrared fluorescence.43–45 Additionally, the aqueous environment also promotes the formation of hydrogen bonds between silica surface and peptide ligands.33 As a result, by adding monofunctional PEG-silane and αMSH modified heterobifunctional αMSH-PEG-silane into the reaction mixture, PEG-silane and and in particular αMSH-PEG-silane were quickly adsorbed by the silica nanoparticle surface, terminating the particle growth.33, 41 An overnight high temperature treatment at 80 °C was then applied to further enhance the covalent attachment of PEG-silane and αMSH-PEG-silane to the nanoparticles. The resulting αMSH-PEG-Cy5-C′ dots were finally purified by gel permeation chromatography (GPC),34, 36 and characterized by a combination of different techniques, including GPC, transmission electron microscopy (TEM), ultraviolet–visible (UV-Vis) spectroscopy, and fluorescence correlation spectroscopy (FCS).
The αMSH-PEG-Cy5-C′ dots synthesized through this approach (Scheme 1) contained Cy5 fluorescent dyes in the silica matrix. The particle surface was modified with PEG chains, ensuring their high bio-compatibility. αMSH peptides were covalently attached at the end of some of the PEG chains, enabling active tumor targeting. The GPC elugram of the resulting purified αMSH-PEG-Cy5-C′ dots showed only a single peak at around 9 min, which was well fitted by a single, symmetric Gaussian distribution function (Figure 2a), demonstrating high purity of the final particle product and narrow particle size distribution, consistent with TEM observations (inset of Figure 2a).34, 36 The average hydrodynamic diameter of the αMSH-PEG-Cy5-C′ dots was 6.2 nm, according to a fit of the fluorescence correlation spectroscopy (FCS) correlation curve (Figure 2b). In addition to particle size, the FCS fit also provides particle concentration, which in combination with optical spectroscopy can be used to estimate particle fluorescence brightness and average numbers of functional ligands on each particle.36, 43 The UV-Vis spectrum of the αMSH-PEG-Cy5-C′ dots exhibited an absorbance peak at around 650nm, consistent with the absorbance of Cy5 dye. In addition, strong absorbance in the regime of wavelength < 300 nm was observed, which was attributed to the αMSH peptides attached to the C′ dot surface (Figure 2c). As demonstrated for other ligands before, the UV-Vis spectrum of αMSH-PEG-Cy5-C′ dots was deconvoluted into the contributions from Cy5-PEG-Cy5-C′ dots and αMSH, through fitting the spectrum by a set of standard spectra of individual components (Figure 2d, e; Figure S4).36 Through dividing the ligand concentrations obtained from UV-Vis spectral analysis by the particle concentration obtained from FCS, the average numbers of Cy5 dyes and αMSH peptides per particle were estimated to be 1.5 and 6.9, respectively.
Figure 2. Characterization of αMSH-PEG-Cy5-C′ dots and NH2-αMSH-PEG-Cy5-C′ dots.

(a and b) GPC elugram (a) and FCS correlation curve with fit (b) of αMSH-PEG-Cy5-C′ dots. A TEM image of αMSH-PEG-Cy5-C′ dots is shown in the inset of (a). (c to e) Deconvolution of the UV-Vis spectrum (c) of αMSH-PEG-Cy5-C′ dots into contributions from PEG-Cy5-C′ dots (d) and αMSH (e). (f and g) GPC elugram (f) and FCS correlation curve with fit (g) of NH2-αMSH-PEG-Cy5-C′ dots. A TEM image of NH2-αMSH-PEG-Cy5-C′ dots is shown in the inset of (f). (h to j) Deconvolution of the UV-Vis spectrum (h) of NH2-αMSH-PEG-Cy5-C′ dots into contributions from PEG-Cy5-C′ dots (i) and αMSH (j).
In vitro receptor targeting and cellular uptake studies of αMSH-PEG-Cy5-C′ dots
The in vitro cell binding kinetics and targeting specificity of αMSH-PEG-Cy5-C′ dots bearing, on average, about 7 αMSH ligands per C′ dot, were evaluated by flow cytometry in murine and human MC1-R-overexpressing cell lines, B16F10 and M21, respectively, using MC1-R receptor blocking assays and particle controls (i.e., Scr-MSH-PEG-Cy5-C′ dots). Figure 3a shows the concentration-dependent cell binding and uptake of αMSH-PEG-Cy5-C′ dots in B16F10 and M21 cells, as against that found in media alone (0.5% BSA). Enhanced particle uptake in both cell lines was observed with increasing particle concentrations (i.e., 0 – 250 nM) over a 4 hour incubation period. Saturation binding was seen to occur at ~100 nM for the B16F10 cell line and 150 nM for M21 cells, which is likely related to known differences in the regulation of MC1-R expression in mouse and human cells,46 a combination of receptor density,14, 47 internalization kinetics, and receptor recycling rates. Furthermore, αMSH-PEG-C′ dot uptake in B16F10 and M21 cells using 100 nM concentrations was seen to increase as a function of incubation time by flow cytometry (Figure 3b), noting that maximum values for both cell types were found at 4–5 hours.
Figure 3. In vitro targeted uptake of αMSH-PEG-Cy5-C′ dots and competitive binding studies in MC1-R-expressing melanoma cell lines.

Cellular binding/uptake of M21 and B16F10 cells as a function of particle concentration (a) and incubation time (b) by flow cytometry. Percentage of the total events detected is displayed. Each data point represents mean±s.d. of three replicates. (c–e) Specific binding of αMSH-PEG-Cy5-C′ dots. (c, d) MC1-R blocking of M21 and B16F10 cells, respectively, using flow cytometry and anti-MC1-R antibody prior to the addition of targeted C′ dots. (e) Tumor-directed binding of αMSH-PEG-Cy5-C′ dots in B16F10 cells with one targeted particle concentration (25 nM) versus scrambled peptide-bound particle control, assayed by flow cytometry. Each bar represents mean±s.d. of three replicates. (f) Competitive MC1-R binding studies of aMSH-PEG-C’dots and Scr-αMSH-PEG-Cy5-C′ dots over a concentration range of 10−13 to 10−5 mol/L with 125I-NDP in B16/F10 melanoma cells. Each data point reflects mean±s.d. of three replicates.
To assess in vitro MC1-R binding specificity, blocking studies using an anti-MC1-R antibody were performed by using flow cytometry. As shown in Figure 3, competitive binding assays showed that uptake via MC1-R was almost completely blocked (~90%) in M21 cells (Figure 3c) and partially blocked (i.e., > 60%) in B16F10 cells (Figure 3d) using excess (×6–9) anti-MC1-R antibody prior to addition of 25 nM αMSH-PEG-Cy5-C′ dots. Differences in the degree of receptor blocking may be attributed to variations in the rates of MC1-R internalization and/or recycling rates to the cell surface in mouse and human melanoma cells. For instance, endocytosed MC1-R receptors in B16 cells do not recycle back to the cell surface,46 so that fewer receptors may be available for binding. Targeting specificity of αMSH-PEG-Cy5-C′ dots was further demonstrated in B16F10 cells by MC1-R receptor binding with 25 nM of Scr-MSH-PEG-Cy5-C′ dots under the same incubation conditions, and findings were similar to those found with anti-MC1-R antibody (i.e., >60%; Figure 3e).
These results were confirmed by competitive binding assays, which measured the IC50 of αMSH-PEG-Cy5-C′ dots for MC1-R using B16F10 melanoma cells and a radioiodinated αMSH analog 125I-(Tyr2)-[Nle4, D-Phe7]-αMSH (125I-NDP), the latter exhibiting picomolar affinity for MC1-R.21 The IC50, or concentration of nanoparticle required to inhibit 50% of a standard radiolabeled MC1-R agonist binding, was determined for both αMSH-PEG-C′ dot and Scr-MSH-PEG-C′ dot constructs over concentrations ranging from 10−13 to 10−5 mol/L (Figure 3f). The IC50 value for αMSH-PEG-Cy5-C′ dots was estimated to be 6.6×10−10 M, while that for the negative particle control (i.e., Scr-MSH-PEG-Cy5-C′ dots) was 2.3×10−8 M, suggesting that differences in MC1-R affinity between the αMSH-PEG-Cy5-C′ dots and the Scr-MSH-PEG-Cy5-C′ dots were more than two orders of magnitude.
Intracellular localization of αMSH-functionalized C′ dots in M21 and B16F10 cells was found to occur primarily via an MC1-R-driven endocytic mechanism, similar to that found for MC1-R targeting ligand–receptor complexes.48 To assess whether this process was temperature-dependent, and whether attachment of αMSH peptides to the surfaces of such ultrasmall particles promoted their accumulation in M21 and B16F10 cells, the uptake of particle-exposed cells was investigated at 3 different temperatures (i.e., 4°, 25°C and 37°C) over a 4-hour incubation period. The results, summarized in Figures 4a and 4b, demonstrated that cellular internalization of αMSH-PEG-Cy5-C′ dots progressively rose in both M21 (Figure 4a) and B16F10 (Figure 4b) cells as a function of incubation temperatures. In addition, significantly decreased particle uptake was observed at 4°C (i.e., >95%) relative to that found at 37°C, suggesting that intracellular internalization occurred primarily via MC1-R-dependent receptor-mediated endocytosis.49–50 Similar results were observed for early-generation cRGDY-functionalized C dots in ανβ3 integrin-overexpressing cell lines.51 Representative microscopic imaging of M21 cells (Figure 4c) 4 hours after particle exposure showed that internalized αMSH-PEG-Cy5-C′ dots resided in late endosomes/lysosomes, as demonstrated by co-localization with an expressed LAMP1-GFP protein that labels these compartments.
Figure 4. Temperature-dependent uptake of αMSH-PEG-Cy5-C′ dots and cellular internalization.

Specific binding and uptake of αMSH-PEG-C dots in M21(a) and B16F10 (b) cells as a function of temperature using 25nM particle concentrations and flow cytometry. Each data point represents the mean ± s.d. of 3 replicates. (c) αMSH-PEG-C′ dot (1 μM, red) colocalization with a lysosomal marker (LAMP1-GFP) after 4h incubation using M21 cells. Colocalized vesicles (yellow); Scale bar: 10 μm.
To demonstrate that no significant loss of cell viability or proliferative activity occurred due to the presence of particles, both M21 and B16F10 cells were exposed to αMSH-PEG-Cy5-C′ dots over a wide range of concentrations (0–250 nM) and incubation times (0–96 hrs) in serum-supplemented media at 37°C. In both cases, no significant loss of cell survival was observed at two post-incubation times relative to findings seen under control conditions (Figure S3a, 3b). Furthermore, time-dependent increases in the number of cells were found following multi-dose (n=4) addition of 100 nM particles to M21 and B16F10 cells, suggesting no adverse effects on cellular proliferation (Figure S3c). Taken together, these results demonstrate site-specific targeting, cellular internalization via endolysosomal pathways, and an absence of cytotoxicity of αMSH-PEG-Cy5-C′ dots in MC1-R-overexpressing cell lines.
Biodistribution and radiodosimetry of 125I-αMSH-PEG-Cy5-C′ dots
After demonstrating MC1-R specific cellular targeting in vitro, we studied the PK of radiolabeled αMSH-PEG-Cy5-C′ dots in MC1-R-overexpressing tumor models. As-synthesized αMSH-PEG-Cy5-C′ dots were initially radiolabeled with iodine-125 (125I) by reacting 125I-Bolton-Hunter reagent with primary amines associated with the αMSH-bound C′ dots, then purified using a PD-10 column to remove any free, unreacted 125I (see experimental section for more details). The in vivo biological properties of intravenously (i.v.)-injected 125I-αMSH-PEG-Cy5-C′ dots (~50 μCi, or ~1.85 MBq), including their PK, tumor-targeting efficiency, and target-to-background ratios were then evaluated over a range of post-injection time points (i.e., 1, 4, 24, 72 and 96 h) in both M21 and B16F10 xenografted mice, as shown in Figure 5. Favorable PK findings were observed for the particle tracer, including a reasonably long blood circulation half-time, dominant renal clearance, and minimal reticuloendothelial system (RES) uptake-results significantly different from those found for the native peptide radiotracer, specifically rapid renal clearance and high renal retention.15, 26
Figure 5. Biodistribution of 125I-αMSH-PEG-Cy5-C′ dots.

in (a) M21 and (c) B16F10 tumor-bearing mice (n=3) at 1, 4, 24, 72 and 96 h post-injection. Insets shows %ID/g values as ratios for tumor to organ (T/B: tumor to blood; T/M: tumor to muscle; T/L: tumor to liver). (b) and (d) are the corresponding activities of 125I-αMSH-PEG-Cy5-C′ dots in urinary specimens.
As shown in Figure 5a and Table S1, whole-blood activity concentration of 125I-αMSH-PEG-C′ dots, obtained from M21 tumor-bearing mice, was found to be ~20 %ID/g at early p.i. time points, gradually declining thereafter to ~9 and ~1 %ID/g at 24 and 96 h p.i., respectively. Dominant renal excretion was confirmed by the high activity measured in urine specimens, namely, >100 %ID/g at 1 h p.i. and >30 %ID/g at 4 h p.i. (Figure 5b). Renal uptake was only about 5 %ID/g at 4 hours p.i., followed by a decline in the activity to <1 %ID/g at 96 h p.i. Liver and spleen uptakes were also relatively low (i.e., <5 %ID/g at 24 h p.i. and ~2 %ID/g at 96 h p.i.). Maximal 125I-αMSH-PEG-Cy5-C′ dot uptake in M21 tumors was estimated to be 5–6 %ID/g. Given the clearance of 125I-αMSH-PEG-Cy5-C′ dots from the circulation and tumor retention of the particles, tumor-to-blood ratios were observed to increase from 0.2±0.1 at 1 h p.i. to 5.8±2.7 at 96 h p.i. (Figure 5a-inset, Table S2). Maximal tumor-to-muscle and tumor-to-liver ratios were found to be 8.3±3.0 and 1.8±0.6, respectively (Figure 5a-inset, Table S2). A similar PK profile was found in mice bearing MC1-R-overexpressing B16F10 xenografts, namely increased blood and urinary activity concentrations at early p.i. time points, as well as low kidney and RES uptake (Figure 5c and d, Table S3 and S4). It is important to note that B16F10 tumor-bearing mice voided before urine specimens were obtained at 1 h p.i., thereby leading to underestimates of the %ID/g for that time point (Figure 5d). While uptake of 125I-αMSH-PEG-C′ dots in B16F10 xenografts was roughly equivalent to that found in M21 xenograft models at early time points, progressively reduced uptake was measured for B16F10 tumors (from ~4.8 %ID/g down to 1.2 %ID/g) at later p.i. time points (i.e., 72 and 96 h) compared to that found for M21 tumors, presumably due to the rapid tumor doubling time (i.e., about 4 days) of the former. In both cases, over 90% of the injected 125I-αMSH-PEG-Cy5-C′ dots were renally excreted within the first 4 days p.i.
On the basis of appropriate organ mass-adjusted translation of the foregoing biodistribution data (Figure 5) to humans, time-integrated activities and human (70-kg Reference Person) normal-organ radiation absorbed dose were derived using the OLINDA computer program (yielding doses expressed in mGy/MBq 125I administered).52 The PK profiles of 125I-αMSH-PEG-C′ dots in both MC1-R-overexpressing tumor models and their normal-organ radiation doses (Table 1) were found to be nearly equivalent; the latter were also comparable to those of commonly used diagnostic radiotracers.
Table 1. Radiodosimetry of 125I-αMSH-PEG-Cy5-C′ dots.
in a 70-kg standard human based on the pharmacokinetics in M21 and B16F10 tumor-bearing mice and the OLINDA dosimetry program.
| Tissue | Absorbed Dose (mGy/MBq) | |
|---|---|---|
| Based on M21 biodistribution data | Based on B16F10 biodistribution data | |
| Adrenals | 0.204 | 0.206 |
| Brain | 0.140 | 0.168 |
| Breasts | 0.129 | 0.147 |
| Gallbladder Wall | 0.223 | 0.224 |
| Lower Large Intestine Wall | 0.276 | 0.263 |
| Small Intestine | 0.243 | 0.248 |
| Stomach Wall | 0.270 | 0.247 |
| Upper Large Intestine Wall | 0.249 | 0.240 |
| Heart Wall | 0.206 | 0.206 |
| Kidneys | 0.324 | 0.242 |
| Liver | 0.335 | 0.251 |
| Lungs | 0.249 | 0.192 |
| Muscle | 0.179 | 0.169 |
| Ovaries | 0.204 | 0.226 |
| Pancreas | 0.215 | 0.220 |
| Red Marrow | 0.212 | 0.193 |
| Bone | 0.354 | 0.308 |
| Skin | 0.115 | 0.128 |
| Spleen | 0.262 | 0.195 |
| Testes | 0.152 | 0.178 |
| Thymus | 0.164 | 0.178 |
| Thyroid | 0.161 | 0.179 |
| Urinary Bladder Wall | 0.281 | 0.359 |
| Uterus | 0.206 | 0.243 |
| Total Body | 0.192 | 0.194 |
| Effective Dose | 0.222 | 0.223 |
In vivo tumor-targeted PET imaging of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots
αMSH MSH-PEG-Cy5-C′ dots in B16F10 xenografted mice, in vivo PET/CT imaging was initially performed after i.v.-injection of αMSH-PEG-Cy5-C′ dots conjugated with Bolton-Hunter reagent and radiolabeled with the positron-emission radioisotope, iodine-124 (124I, t1/2=4.2 d). Enhanced tumor uptake was observed in this MC1-R-targeted cohort relative to that found in a second targeted cohort blocked with NDP. It should be noted that mice were not pretreated with KI to prevent free radioiodine uptake. Activity in the thyroid gland and stomach was felt to be a consequence of free iodine-124 associated with the specific radioiodination conditions needed to achieve high specific activity 124I-αMSH-PEG-C′ dot preparations, along with excess non-specifically bound iodine (Figure S5).
To address issues related to free iodine, zirconium-89 (89Zr, t1/2=3.3 d) with a lower mean β+ energy (396 keV vs. 820 keV) and a comparable decay half-life (3.3 d vs. 4.2 d) to those of 124I was selected as radioisotope tracer replacing iodine-124. In order to label αMSH-PEG-Cy5-C′ dots with zirconium-89, the surface of αMSH-PEG-Cy5-C′ dots were first functionalized with amine groups, using a recently developed post-PEGylation surface modification by insertion (PPSMI) approach,36 which were subsequently reacted with radiometal chelator p-SCN-Bn-deferoxamine (DFO-NCS). Sandwiching the amine-functionalization step between PEGylation and purification steps in the C′ dot synthesis allowed to maintain the high-quality of the C′ dot synthesis. For PPSMI, amine-functionalized silane molecules were inserted in-between the chains of the C′ dot PEG layer and covalently attached to the remaining silanol groups on the silica surface underneath. The resulting NH2-αMSH-PEG-Cy5-C′ dots enabled further modification with amine-reactive functional ligands, e.g., DFO-NCS chelator molecules for zirconium-89. The GPC elugram of the purified NH2-αMSH-PEG-Cy5-C′ dots exhibited only one peak, well fitted by a single, symmetric Gaussian distribution (Figure 2f). This indicated high purity and a narrow particle size distribution, further confirmed by TEM (inset in Figure 2f). The fit of the corresponding particle FCS correlation curve suggested an average hydrodynamic diameter of NH2-αMSH-PEG-Cy5-C′ dots of 6.2nm (Figure 2g). The average numbers of Cy5 and αMSH molecules per C′ dot were estimated by FCS and UV-Vis spectroscopic analysis to be 1.5 and 8.0, respectively (Figure 2h–i). The NH2-αMSH-PEG-Cy5-C′ dots were subsequently mixed with DFO-NCS in PBS, allowing the isothiocyanate-functionalized DFO molecules to diffuse into the PEG layer of NH2-αMSH-PEG-Cy5-C′ dots and covalently attach to the amine groups under the PEG layer. As synthesized DFO-αMSH-PEG-Cy5-C′ dots could then be labeled with 89Zr through DFO chelation (see Experimental Section for more details). Unlike 124I, which is prone to dehalogenation after cellular uptake, 89Zr has been reported to stably residualized within cells after its internalization,53 thereby enabling accurate quantitation of in vivo cellular uptake. A flow cytometry study of NH2-αMSH-PEG-Cy5-C′ dots in B16F10 cells was performed to confirm that specific targeting is not substantially influenced by introducing amines to the αMSH-PEG-Cy5-C′ dots (as shown in Figure S6).
Prior to the PET imaging study, the biodistribution pattern of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots in B16F10 tumor-bearing mice was performed and compared with that of 125I-αMSH-PEG-Cy5-C′ dots in the same tumor model. As shown in Figure 6a, both radiotracers showed nearly equivalent biodistribution profiles, noting ~5 %ID/g uptake in B16F10 tumor at 24 h p.i., a result that likely reflects limitations in overall tumor uptake imposed by the relatively low number of MC1-R receptors expressed by this tumor as opposed to other melanoma markers (i.e., integrins).14, 47 Less than 5 %ID/g accumulation of both radio-tracers were observed in liver, spleen and kidney. Figure 6b presents H&E staining, Cy5 fluorescence microscopy, and quantitative autoradiography of a representative ex vivo B16F10 tumor tissue specimen demonstrating excellent colocalization of optical and radiotracer signal across the specimen.
Figure 6. Biodistribution and ex vivo histological studies.

(a) Comparison of the biodistribution profiles in B16F10 tumor-bearing mice 24 h p.i. of 89Zr-DFO-αMSH-PEG-C′ dot (n=5) or 125I-αMSH-PEG-C′ dot (n=3); (b) H&E staining (left, scale bar: 1mm), Cy5-fluorescence microscopy (middle), and autoradiography (right) images of tumor tissue specimen harvested from B16F10 tumor-bearing mouse 24 h p.i. of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots.
After i.v. injection of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots into B16F10 tumor-bearing mice, in vivo PET imaging (Figure 7) showed significant bladder activity 2 h p.i. High 89Zr-DFO-αMSH-PEG-C′ dot uptake found in cardiac tissue (10.6±1.9 %ID/g) clearly indicated significant blood pool activity. Time-activity curves, shown in Figure 8a, demonstrate the clearance of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots from the blood, with activity concentrations of ~3 and ~1 %ID/g at 24 and 72 h post-injection, respectively. The clearance of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots by the RES (e.g., liver) was estimated to be 5.0±0.6 %ID/g at 2 h p.i., with a nearly 50% reduction (i.e., 2.3±0.2 %ID/g) after 3 days. Splenic uptake was found to be only half of that found in liver over the course of 3 days. Muscle uptake was low, no greater than1 %ID/g. B16F10 tumor uptake peaked at 24 h post-injection at 5.5±0.9 %ID/g (Figure 7a and 8a), and retention of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots was seen over this 3-day time period (Figure 7a). As expected, mice injected with 89Zr-DFO-αMSH-PEG-Cy5-C′ dots showed minimal bone and joint uptake, indicating high in vivo radiostability (Figure 7a) (free 89Zr4+ is an osteophilic cation, which could be readily accreted into bone mineral54).
Figure 7. Serial PET imaging of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots.

at 2, 24, 48 and 72 h post-injection of the MC1-R-targeting particle tracer into two cohorts of B16F10 xenografted mice: (a) targeted group and (b) NDP blocking group. For the blocking study, each mouse was co-injected with 89Zr-DFO-αMSH-PEG-Cy5-C′ dots and 200 μg of NDP. N=3 for each group.
Figure 8. Quantitative region-of-interest studies in B16F10 xenografted mice.

Time activity curves of (a) targeted group (n=3), and (b) blocking group (n=3). (c) Tumor uptake comparison between targeted and blocking groups (n=3). (d–f) Tumor-to-blood, tumor-to-muscle and tumor-to-liver ratios at 2, 24, 48 and 72 h post-injection. For blocking study, the mice were co-injected with 89Zr-DFO-αMSH-PEG-Cy5-C′ dots and 200 μg of NDP. *P<0.05, **P<0.005.
To further demonstrate in vivo target specificity of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots, a blocking study was performed in a separate cohort of B16F10 tumor-bearing mice co-injected with 89Zr-DFO-αMSH-PEG-Cy5-C′ dots and 200 μg of NDP inhibitor. Results showed similar particle distributions in major organs, such as bladder, heart, liver and muscle, but with a nearly 50% reduction in tumor uptake (mean: 2–3 %ID/g), as shown in Figures 7b and 8b,c. For the targeted group, the highest tumor-to-blood and tumor-to-muscle ratios were estimated to be 5.8±0.7 and 8.4±1.1 at 72 h p.i., respectively, which are ~2-fold greater than the corresponding ratios obtained for the blocked group (tumor-to-blood: 3.0±0.6; tumor-to-muscle: 3.5±1.0, Figures 8d,e). Finally, on the basis of high tumor uptake and low RES accumulation, we observed tumor-to-liver ratios of about 2 in the targeted group (Figure 8f). These findings successfully demonstrated in vivo targeting specificity and concomitant renal clearance for 89Zr-DFO-αMSH-PEG-Cy5-C′ dots in MC1-R overexpressing xenograft models.
CONCLUSION
To address technical challenges associated with translation of potent αMSH analogs, a small library of custom-synthesized rhenium (Re)-cyclized αMSH peptide ligands were sythesized and adapted for attachment to ultrasmall fluorescent silica nanoparticles. The resulting MC1-R particle probes were shown to have subnanomolar receptor affinity and rapid receptor-mediated internalization in melanoma cells. In vivo, radiolabeled αMSH-PEG-Cy5-C′ dots displayed favorable pharmacokinetics and dosimetry, highlighted by their accumulation and retention in MC1-R-expressing tumors, efficient renal excretion, and low RES uptake. Ex vivo, tumor penetration and distribution of αMSH-PEG-Cy5-C′ dots were visualized by Cy5 fluorescence imaging and autoradiography. Site-specific uptake in B16F10 xenografts was assessed by blocking with excess NDP peptide using PET imaging and 89Zr- and 124I-labeled αMSH-PEG-Cy5-C′ dots. These results highlight the promising clinical potential of αMSH-PEG-Cy5-C′ dots as dual-modality particle probes for the image-guided surgical treatment and targeted radiotherapy of melanoma by labeling αMSH-PEG-Cy5-C′ dots with alpha and beta emitting radionuclides.
MATERIALS AND METHODS
General Synthesis procedures of Peptides
Three MC1-R targeting ReCCMSH peptide analogs (Figure S1) were synthesized using standard fluorenylmethyloxycarbonyl (Fmoc) solid-phase peptide chemistry on a multiple peptide synthesizer (Model 396, AAPPTEC or Tetras, Advanced ChemTech).37 4-Methyltrityl (Mmt) protection groups were selectively removed from the cysteines involved in peptide cyclization by treatment with 1% trifluoroacetic acid (TFA) and 5% triisopropylsilane (TIS) in dichloromethane (DCM). Rhenium (Re) cyclization was achieved directly on solid phase by overnight reaction with 4 molar excess of ReOCl3(PPh2)3 in N-methyl-2-pyrrolidone (NMP). Cleavage and sidechain deprotection were achieved by treating the peptidyl-resin with 85% TFA and non-reducing scavengers (phenol/water/TIS, 5% each). The peptides were purified and analyzed on a Beckman Coulter High Performance Liquid Chromatography (HPLC) system coupled with an LCQ FLEET Ion Trap Mass spectrometer (MS; Thermo Fisher Scientific). The final products were recovered by lyophilization and stored at −20˚C.
Synthesis of αMSH peptide I
Peptide I (NH2-Re[Cyc-Cys-Glu-His-dPhe-Arg-Trp-Cys]-Arg-Pro-Val-NH2 (Figure S1a) was synthesized, as described above. The purified peptide was subjected to HPLC MS analysis; calculated and experimental mass-to-charge (m/z) ratios were 1632 and 1633, respectively. Peptide I was modified at both termini, first with N-succinimidyl S-acetylthiolacetate (SATA) reagent, to convert one amine to a sulfhydryl; this was followed by a second amine modification to introduce a N-succinimidyl-3-(4-hydroxyphenyl) propionate (SHPP; Bolton-Hunter reagent) group for radioiodination. Peptide I was reacted with SATA reagent at a 9:1 SATA-to-peptide molar ratio in phosphate buffered saline (PBS) pH 7.4 at 25˚C for 30 min. The reaction was quenched by the addition of 1 mM ethanolamine and purified over a size exclusion column. The acetylthioacetate-modified peptide I was reacted with SHPP at a SHPP-to-peptide ratio of 10:1 in 200 mM borate buffer (pH 9.0) on ice for 3 h. Modified peptides were purified from unreacted SHPP by size exclusion chromatography. The sulfhydryl acetyl protection group was removed with 0.5 M hydroxylamine and 25 mM EDTA in PBS (pH 7.4) for 2 h at 25˚C, and purified on a G25 size exclusion column to yield the final HPP-Peptide I-thiolacetate.
Synthesis of αMSH peptide II
Peptide II (NH2-Ser-Tyr-Ser-Re[Cys-Cys-Glu-His-dPhe-Arg-Trp-Cys]-Arg-Pro-Val-NH2 (Figure S1b) was synthesized using standard solid-phase Fmoc peptide synthesis, and modified with SATA reagent for the introduction of a terminal sulfhydryl as described above. The purified peptide was subjected to HPLC MS analysis; m/z, calculated 1972, experimental 1972.
Synthesis of αMSH peptide III
Peptide III (Ac-Cys-(Ahx)2-dLys-Re[Cys-Cys-Glu-His-dPhe-Arg-Trp-Cys]-Arg-Pro-Val-NH2 (αMSH, Figure S1c) was synthesized with a N-acetylated-cysteine, a double aminohexanoic acid (Ahx2) aliphatic linker and a dLys using standard solid-phase Fmoc peptide chemistry, as described above. The purified peptide was subjected to HPLC MS analysis; m/z, calculated 2131, experimental 2131.
Synthesis of scrambled (Scr) MSH peptide
The Scr-MSH peptide control, Ac-Cys-(Ahx)2-D-Lys-Re[Cys-Cys-Trp-Arg-His-dPhe-Glu-Cys]-Arg-Pro-Val-NH2 was synthesized using standard solid phase peptide synthesis chemistry. It contained the same N-terminal Ac-Cys, (Ahx)2 linker and D-Lys as αMSH peptide III. Only the Re-cyclized MC1-R binding sequence was scrambled. The purified peptide was subjected to HPLC MS analysis; m/z, calculated 2131, experimental 2131.
Synthesis of αMSH-PEG-Cy5-C′ dots and NH2-αMSH-PEG-Cy5-C′ dots
The αMSH-PEG-Cy5-C′ dots were synthesized based on a previously published protocol.32 More specifically, NHS ester/maleimido heterobifunctional polyethylene glycol (PEG) with molar mass around 860 (12 ethylene glycol units) was first conjugated with (3-aminopropyl)triethoxysilane at room temperature under nitrogen through amine-NHS ester reaction, forming mal-PEG-silane. αMSH was then added into the mixture at room temperature under nitrogen to further conjugate mal-PEG-silane with αMSH through thiol-ene reaction, forming αMSH-PEG-silane. At the same time, Cy5-silane conjugate was prepared by conjugating maleimido functionalized Cy5 dyes with (3-mercaptopropyl)trimethoxysilane at room temperature under nitrogen. In the next step, tetramethyl orthosilicate (TMOS) and Cy5-silane were added to an aqueous solution with ammonium hydroxide adjusting the pH around 8.5 at room temperature under vigorous stirring. One day later, αMSH-PEG-silane and monofunctional PEG-silane with molar mass around 500 (6 to 9 ethylene glycol units) were added into the reaction at room temperature under vigorous stirring. The reaction mixture was further left at room temperature under vigorous stirring overnight, followed by an overnight high temperature treatment at 80 °C without stirring. After cooling the reaction to room temperature, the resulting αMSH-PEG-Cy5-C′ dots were dialyzed in DI water, purified using GPC, filtered by sterile syringe filters and finally stored at 4 °C. The full name of the αMSH-PEG-Cy5-C′ dots using a comprehensive nomenclature introduced in a recent publication to differentiate between different C′ dot chemistries is CD(Cy5)-PEG12-mal-thiol-αMSH_PEG6. 36
The synthesis of NH2-αMSH-PEG-Cy5-C′ dots followed a similar protocol as described above, in which a PPSMI approach was employed to functionalize αMSH-PEG-Cy5-C′ dots with amine groups.36 More specifically, after the reaction temperature of the post-PEGylation heat treatment was reduced from 80 °C to room temperature, (3-aminopropyl)trimethoxysilane was added at room temperature under vigorous stirring. The reaction solution was left at room temperature under vigorous stirring overnight to insert the small amine-silane molecules in-between the PEG layer of αMSH-PEG-Cy5-C′ dots and to covalently attach them to the silanol groups of the underlying silica particle surface. The resulting NH2-αMSH-PEG-Cy5-C′ dots were dialyzed in DI water, purified using GPC, filtered by sterile syringe filters and finally stored at 4 °C. The full name of the NH2-αMSH-PEG-Cy5-C′ dots using the comprehensive nomenclature described above is CD(Cy5)-PEG12-mal-thiol-αMSH_PEG6_NH2. 36
GPC purification and characterization of the particles was conducted using a biologic LP system (Bio-Rad) equipped with a 275nm UV detector and a chromatography column packed with Superdex 200 resin from GE healthcare. TEM images were taken on an FEI T12 Spirit TEM operated at an acceleration voltage of 120kV. UV-Vis absorbance spectra were measured on a Varian Cary 5000 spectrophotometer. FCS measurements were conducted using a home-built FCS setup with a 633nm solid state laser as the excitation source.
Cells and cell culture
Human melanoma M21 was obtained from D.A. Cheresh (University of California San Diego, San Diego, California, USA). Cells were maintained in RPMI 1640 media/10% FBS, and 2 mM l-glutamine, penicillin, and streptomycin (Core Media Preparation Facility, Memorial Sloan Kettering Cancer Center). B16F10 cells were obtained from American Type Culture Collection (ATCC). Cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and Penicillin Streptomycin.
In vitro MC1-R-overexpressing cell binding studies
To assay particle binding in MC1-R-overexpressing cell lines, 24-well plates were coated with 10 μg/ml collagen type I (BD Biosciences) in PBS, incubated at 37°C for 30 minutes, and washed once with PBS. M21 and B16F10 cells (3.0×105 – 4.0×105 cells/well) were grown to confluency. Differential binding of αMSH-PEG-Cy5-C′ dots to M21 or B16F10 cells was evaluated over a range of incubation times (0–5 hours) and particle concentrations (10–300 nM). After incubation, cells were washed with RPMI 1640 media/0.5% BSA, detached using 0.25% trypsin/EDTA, pelleted in a microcentrifuge tube (5 minutes at 153 g, 25°C), re-suspended in BD FACSFlow solution (BD Biosciences), and analyzed by flow cytometry in the Cy-5 channel to determine the percentage of particle-bound probe (FACSCalibur, Becton Dickinson, Mountain View, CA). Blocking studies were performed after incubation of αMSH-PEG-Cy5-C′ dots with M21 cells (or B16F10) in the presence of Anti-MC1 Receptor antibody (EPR6530, ab125031, http://www.abcam.com/mc1-receptor-antibody-epr6530-ab125031.html), and analyzed by flow cytometry. Binding studies were also conducted in B16F10 cells using a particle control (i.e., Scr-MSH-PEG-Cy5-C′ dots, 25 nM).
Cellular internalization and microscopic imaging
M21 melanoma cells expressing LAMP1-GFP were generated by retroviral transduction of a pRetro-Lamp1-GFP construct and selection in puromycin (2μg/ml). Cells were then cultured overnight on glass-bottom 35mm dishes (MatTek, Ashland, MA), incubated with 1.5μM aMSH-PEG-C′ dots for 6 hours, and imaged by widefield microscopy, using a Nikon Ti-E inverted microscope and a CoolSnap HQ2 CCD camera (Photometrics, Tucson, AZ). Images were processed with Elements software (Nikon, Melville, NY) and ImageJ software.
Cell Viability and Proliferation
Viability and proliferation of M21 and B16F10 cells were monitored under control conditions (10% FBS) or after exposure to particles at 37°C. Cells were split (4–5×104 cells/well) in a 24-well plate coated with collagen, as described above. Media was changed to RPMI, supplemented with 10% FBS, 24 h later, and different concentrations of αMSH-PEG-C′ dots were added (25–250 nM) for 24–48 hours at 37ºC. The percent (%) cell viability was determined 24 or 48 h post-incubation after trypsinization using a Vi-Cell series viability analyzer (Beckman Coulter, Inc, Fullerton, CA). To assess proliferative activity, particle-exposed cells were counted every 24 h over a 96-h time interval.
Competitive binding assay (IC50)
The IC50 (concentration of peptide required to inhibit 50% of radioligand binding) was determined for αMSH-PEG-Cy5-C′ dots and Scr-MSH-PEG-Cy5-C′ dots in competitive binding assays with 125I-(Tyr2)-NDP, a radioiodinated αMSH analog with picomolar affinity for the MC1-R receptor.55 Murine melanoma B16F10 cells were plated at a density of 0.2 million/well in 24-well tissue culture plates and incubated overnight. Individual wells were incubated at 25°C for 3 h with approximately 50,000 cpm (62,480 dpm) 125I-(Tyr2)-NDP in 0.3 mL binding medium (DMEM with 25 mM N-(2-hydroxyethyl)-piperazine-N’-(2-ethanesulfonic acid) (HEPES), pH 7.4, 0.02% BSA, and 0.3 mM 1,10-phenanthroline) with concentrations of the αMSH-PEG-Cy5-C′ dots and Scr-MSH-PEG-Cy5-C′ dots ranging from 10−13 to 10−5 mol/L. The binding media was aspirated after incubation. Cells were rinsed with 0.5 mL of ice-cold pH 7.1, 0.02% BSA/0.01 M PBS twice and lysed with 0.5 mL of 1 M NaOH for 10 min. The radioactivity in the cells was collected and measured in a γ-counter. The IC50 values of αMSH-PEG-Cy5-C′ dots and Scr-MSH-PEG-Cy5-C′ dots were calculated with GraphPad Prism version 4.0.
Animal models and tumor inoculation
All animal experiments were done in accordance with protocols approved by the Institutional Animal Care and Use Committee of Memorial Sloan-Kettering Cancer Center and followed NIH guidelines for animal welfare. M21 and B16F10 xenografts were generated by co-injecting equal volumes of cells (~5×106 cells/100 μL) and Matrigel subcutaneously into the hind legs of female athymic nu/nu mice (6–8 weeks old, Taconic Farms Inc.). Average tumor volumes of 200 mm3 were used for all studies.
125I radiolabeling of αMSH-PEG- and Scr-MSH-PEG-Cy5-C′ dots for cell binding and biodistribution studies
1.8 mCi of 125I Bolton Hunter reagent (PerkinElmer) was evaporated to dryness in a glass vial with N2 gas, followed by the addition of 100 μL of DMSO, 15 μL of DIEA and an equal molar concentration of αMSH-PEG-Cy5-C′ dots or Scr-MSH-PEG-Cy5-C′ dots (15μM, <10 MSH). The mixture was kept at room temperature for 30 minutes before it was loaded into a pre-conditioned PD10 column and eluted with 0.5 mL of saline. The eluent was collected for biodistribution studies in B16/F10 and M21 melanoma-bearing mice. The yield was approximately 7%, and product specific activity was 3100 Ci/mol. 125I-αMSH-PEG-C′ dots (~50 μCi, or ~1.85 MBq) were i.v.-injected in both tumor models.
124I radiolabeling of αMSH-PEG-C’dots for a pilot PET/CT imaging
Ten times molar excess of N-succinimidyl-3-(4-hydroxyphenyl) propionate (Bolton-Hunter Reagent; SHPP) was added to αMSH-PEG-Cy5-C′ dots (15 μM) in 0.2M Borax (pH 8.7), vortexed, and allowed to proceed at room temperature (RT) shielded from light for 3 hours. The mixture was dialyzed in triplicate using a 10,000 molecular weight cut-off membrane (Thermo Scientific) against phosphate-buffered saline (PBS, 0.1 M sodium phosphate, 150mM sodium chloride, pH 7.2). Equal molar amounts of 3-(4-hydroxyphenyl) propionate (HPP)-αMSH-PEG-C′ dots and 124I were then added into iodogen-coated (20 μg) vials containing PBS; the total volume was 100 μL. After vortexing, the reaction was allowed to proceed at RT for 20 minutes. The radioiodinated product was purified four times with a Zeba spin column (7K). The yield was 25%, and product specific activity was 5.5×106 Ci/mol. The radiochemical purity was greater than 92% using ITLC. For the mobile phase, MeOH: Ethyl Acetate (1:1) was used; Rf values for the particle tracer (124I-αMSH-PEG-C′ dots) and free radioiodine (124I−) were 0 and 0.7–0.8, respectively.
89Zr-oxalate production
89Zr was produced at Memorial Sloan Kettering Cancer Center on a TR19/9 cyclotron (Ebco Industries Inc.) via the 89Y(p,n)89Zr reaction, and purified to yield 89Zr with a specific activity of 5.28–13.43 mCi/μg (470–1195 Ci/mmol) of zirconium.56 Activity measurements were performed using a CRC-15R Dose Calibrator (Capintec). For the quantification of activities, experimental samples were counted on an Automatic Wizard2 γ-Counter (PerkinElmer). All in vivo experiments were performed according to protocols approved by the Memorial Sloan Kettering Institutional Animal Care and Use Committee. A purity of greater than 95% was confirmed using radio-TLC for all of the 89Zr-labeled DFO-αMSH-PEG-Cy5-C′ dots.
Synthesis of DFO-αMSH-PEG-Cy5-C′ dots and 89Zr radiolabeling
The conjugation of DFO to NH2-αMSH-PEG-Cy5-C′ dots was achieved by following a previously reported protocol with slight modifications.57 NH2-αMSH-PEG-Cy5-C′ dots (200 μL, 15 μM) in DI water were first added to 400 μL PBS (pH 7.4), followed by adding DFO-NCS (30 nmol in DMSO). The final pH was adjusted to 8–9. The reaction was kept at RT with agitation (640 rpm) for 2 h. About 2–3 DFO per αMSH-PEG-C′ dot was estimated. To remove the un-reacted DFO-NCS, as-synthesized DFO-αMSH-PEG-Cy5-C′ dots were then purified by passing through a PD-10 column using PBS as the mobile phase.
To label DFO-αMSH-PEG-Cy5-C′ dots with 89Zr, 0.75 nmol of DFO-αMSH-PEG-Cy5-C′ dots were mixed with 1 mCi of 89Zr-oxalate in HEPES buffer (pH 8) at 37 ºC for 60 min; final labeling pH was kept as 7–7.5. An EDTA challenge process was introduced to remove any unconjugated 89Zr. The final 89Zr labeling yield was on the order of 70 to 80% (n>5). As-synthesized 89Zr-DFO-αMSH-PEG-Cy5-C′ dots were then purified using a PD-10 column. The final radiochemical purity was estimated to be greater than 99% by ITLC.
Ex vivo biodistribution and in vivo tumor-targeting PET imaging studies
For biodistribution studies, B16F10 tumor-bearing mice (n=5) were i.v.-injected with 100–150 μCi (3.7–5.55 MBq) 89Zr-DFO-αMSH-PEG-Cy5-C′ dots, and major organs/tissues harvested and weighed 24 h post-injection. Accumulated activity was counted using an Automatic Wizard2 γ-Counter (PerkinElmer), and presented as %ID/g (mean ± s.d.).
For PET imaging studies, B16F10 tumor-bearing mice (n=3) were i.v. injected with 200–300 μCi (7.4–11.1 MBq) 89Zr-DFO-αMSH-PEG-Cy5-C′ dots and serially imaged in a small-animal PET scanner (Focus 120 microPET; Concorde Microsystems) at 2, 24, 48, and 72 h post-injection. An energy window of 350−700 keV and a coincidence timing window of 6 ns were used. Data were sorted into 2D histograms by Fourier rebinning, and transverse images were reconstructed by filtered back-projection into a 128 × 128 × 63 (0.72 × 0.72 × 1.3 mm3) matrix. The PET imaging data was normalized to correct for non-uniformity of response, dead-time count losses, positron branching ratio, and physical decay to the time of injection; no attenuation, scatter, or partial-volume averaging corrections were applied. The counting rates in the reconstructed images were converted to activity concentrations (percentage injected dose per gram of tissue, %ID/g) by use of a system calibration factor derived from the imaging of a mouse-sized water-equivalent phantom containing 89Zr. Region-of-interest (ROI) analyses of the PET data were performed using IRW software. Blocking studies were performed by co-injecting mice (n=3) with similar amounts of 89Zr-DFO-αMSH-PEG-Cy5-C′ dots and 200 mg NDP.
Digital Autoradiography
10 μm B16F10 tumor tissue sections were exposed to phosphor-imaging plates (Fujifilm BAS-MS2325, Fuji Photo Film, Japan) at −20°C. Following exposure, the phosphor-imaging plate was read at a resolution of 25 μm using a Typhoon 7000 IP (GE Life Sciences) plate reader to produce digital autoradiograms. Images were saved as both TIFF and GEL formats for future analysis and processing with ImageJ software (https://imagej.nih.gov/ij/).
Statistics
All comparisons were based on statistical significance from pairwise t-tests from an analysis of variance model based on three replicates.
Supplementary Material
Acknowledgments
Funding Sources
This study was funded by grants from the National Institutes of Health (1R01CA161280-01A1 to M.B. and U.W.; 1U54 CA199081-01 to M.B. and U.W.); and Sloan Kettering Institute (Core Grant P30 CA008748CCSG). University of Missouri Peptide Synthesis Core and Harry S Truman VA Hospital Biomolecular Imaging Center and Research Animal Core.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
ASSOCIATED CONTENT
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org. Sequences of four custom-synthesized αMSH peptides for particle attachment. Biodistribution profile of 131I-αMSH-PEG-Cy5-C′ dots (Version I, FCS size: 6.3 nm, number of Cy5/C′ dot: 2.3), synthesized using Peptide I. Biodistribution profile of 131I-αMSH-PEG-Cy5-C′ dots (Version II, FCS size: 4.8 nm, number of Cy5/C′ dot: 2.6), synthesized using Peptide II. Viability and proliferation of M21 cells and B16F10 cells as a function of particle concentration and incubation time. Deconvolution of the UV-Vis spectrum of αMSH from the spectrum of αMSH-PEG-Cy5-C′ dots. PET/CT maximum intensity projection (MIP) images at 2 hours after i.v.-injection of 124I-αMSH-PEG-Cy5-C′ dots in B16F10 tumor-bearing mice. Competitive MC1-R receptor binding with NH2-αMSH-PEG-Cy5-C′ dots (10 nM) and NDP blocking peptide (100 mg, or 50 mg/mL) in B16F10 cells. Tables of biodistribution data from M21 (and B16F10) tumor-bearing mice injected with 125I-αMSH-PEG-Cy5-C′ dots. Tables of tumor-to-background ratios (blood, muscle, and liver) from M21 (and B16F10) tumor-bearing mice injected with 125I-αMSH-PEG-Cy5-C′ dots.
References
- 1.Society, A. C. Cancer Facts & Figures 2016. American Cancer Society; 2017. [Google Scholar]
- 2.Chen KG, Valencia JC, Gillet JP, Hearing VJ, Gottesman MM. Involvement of Abc Transporters in Melanogenesis and the Development of Multidrug Resistance of Melanoma. Pigment Cell Melanoma Res. 2009;22:740–749. doi: 10.1111/j.1755-148X.2009.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M, Hoth M, Bastian BC, Vogt T, Herlyn M. Overcoming Intrinsic Multidrug Resistance in Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling Jarid1b(High) Cells. Cancer Cell. 2013;23:811–825. doi: 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabatino M, Stroncek DF, Klein H, Marincola FM, Wang E. Stem Cells in Melanoma Development. Cancer Lett. 2009;279:119–125. doi: 10.1016/j.canlet.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salama AK. Evolving Pharmacotherapies for the Treatment of Metastatic Melanoma. Clin Med Insights Oncol. 2013;7:137–149. doi: 10.4137/CMO.S9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain JF, Testori A, Grob JJ, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Jr, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen TT, Humphrey R, Hoos A, Wolchok JD. Ipilimumab Plus Dacarbazine for Previously Untreated Metastatic Melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 7.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, Activity, and Immune Correlates of Anti-Pd-1 Antibody in Cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Dronca R, Zarour H, Joseph RW, Boasberg P, Chmielowski B, Mateus C, Postow MA, Gergich K, Elassaiss-Schaap J, Li XN, Iannone R, Ebbinghaus SW, Kang SP, Daud A. Anti-Programmed-Death-Receptor-1 Treatment with Pembrolizumab in Ipilimumab-Refractory Advanced Melanoma: A Randomised Dose-Comparison Cohort of a Phase 1 Trial. Lancet. 2014;384:1109–1117. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 10.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab Plus Ipilimumab in Advanced Melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sullivan RJ, Flaherty KT. Resistance to Braf-Targeted Therapy in Melanoma. Eur J Cancer. 2013;49:1297–1304. doi: 10.1016/j.ejca.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 12.Tatro JB, Reichlin S. Specific Receptors for Alpha-Melanocyte-Stimulating Hormone Are Widely Distributed in Tissues of Rodents. Endocrinology. 1987;121:1900–1907. doi: 10.1210/endo-121-5-1900. [DOI] [PubMed] [Google Scholar]
- 13.Siegrist W, Solca F, Stutz S, Giuffre L, Carrel S, Girard J, Eberle AN. Characterization of Receptors for Alpha-Melanocyte-Stimulating Hormone on Human Melanoma Cells. Cancer Res. 1989;49:6352–6358. [PubMed] [Google Scholar]
- 14.Miao Y, Whitener D, Feng W, Owen NK, Chen J, Quinn TP. Evaluation of the Human Melanoma Targeting Properties of Radiolabeled Alpha-Melanocyte Stimulating Hormone Peptide Analogues. Bioconjug Chem. 2003;14:1177–1184. doi: 10.1021/bc034069i. [DOI] [PubMed] [Google Scholar]
- 15.Miao Y, Quinn TP. Alpha-Melanocyte Stimulating Hormone Peptide-Targeted Melanoma Imaging. Front Biosci. 2007;12:4514–4524. doi: 10.2741/2406. [DOI] [PubMed] [Google Scholar]
- 16.Raposinho PD, Correia JD, Oliveira MC, Santos I. Melanocortin-1 Receptor-Targeting with Radiolabeled Cyclic Alpha-Melanocyte-Stimulating Hormone Analogs for Melanoma Imaging. Biopolymers. 2010;94:820–829. doi: 10.1002/bip.21490. [DOI] [PubMed] [Google Scholar]
- 17.Quinn T, Zhang X, Miao Y. Targeted Melanoma Imaging and Therapy with Radiolabeled Alpha-Melanocyte Stimulating Hormone Peptide Analogues. G Ital Dermatol Venereol. 2010;145:245–258. [PMC free article] [PubMed] [Google Scholar]
- 18.Eberle AN, Rout B, Qi MB, Bigliardi PL. Synthetic Peptide Drugs for Targeting Skin Cancer: Malignant Melanoma and Melanotic Lesions. Curr Med Chem. 2017;24:1797–1826. doi: 10.2174/0929867324666170605105942. [DOI] [PubMed] [Google Scholar]
- 19.Nasti TH, Timares L. Mc1r, Eumelanin and Pheomelanin: Their Role in Determining the Susceptibility to Skin Cancer. Photochem Photobiol. 2015;91:188–200. doi: 10.1111/php.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hruby VJ. Designing Peptide Receptor Agonists and Antagonists. Nat Rev Drug Discov. 2002;1:847–858. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 21.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-Phenylalanine-Alpha-Melanocyte-Stimulating Hormone: A Highly Potent Alpha-Melanotropin with Ultralong Biological Activity. Proc Natl Acad Sci U S A. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garg PK, Alston KL, Welsh PC, Zalutsky MR. Enhanced Binding and Inertness to Dehalogenation of Alpha-Melanotropic Peptides Labeled Using N-Succinimidyl 3-Iodobenzoate. Bioconjug Chem. 1996;7:233–239. doi: 10.1021/bc960001+. [DOI] [PubMed] [Google Scholar]
- 23.Giblin MF, Jurisson SS, Quinn TP. Synthesis and Characterization of Rhenium-Complexed Alpha-Melanotropin Analogs. Bioconjug Chem. 1997;8:347–353. doi: 10.1021/bc9700291. [DOI] [PubMed] [Google Scholar]
- 24.Giblin MF, Wang N, Hoffman TJ, Jurisson SS, Quinn TP. Design and Characterization of Alpha-Melanotropin Peptide Analogs Cyclized through Rhenium and Technetium Metal Coordination. Proc Natl Acad Sci U S A. 1998;95:12814–12818. doi: 10.1073/pnas.95.22.12814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo H, Gallazzi F, Miao Y. Gallium-67-Labeled Lactam Bridge-Cyclized Alpha-Msh Peptides with Enhanced Melanoma Uptake and Reduced Renal Uptake. Bioconjug Chem. 2012;23:1341–1348. doi: 10.1021/bc300191z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao Y, Fisher DR, Quinn TP. Reducing Renal Uptake of 90y- and 177lu-Labeled Alpha-Melanocyte Stimulating Hormone Peptide Analogues. Nucl Med Biol. 2006;33:723–733. doi: 10.1016/j.nucmedbio.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 27.Bapst JP, Froidevaux S, Calame M, Tanner H, Eberle AN. Dimeric Dota-Alpha-Melanocyte-Stimulating Hormone Analogs: Synthesis and in Vivo Characteristics of Radiopeptides with High in Vitro Activity. J Recept Signal Transduct Res. 2007;27:383–409. doi: 10.1080/10799890701723528. [DOI] [PubMed] [Google Scholar]
- 28.Lin CH, Al-Suwayeh SA, Hung CF, Chen CC, Fang JY. Camptothecin-Loaded Liposomes with Alpha-Melanocyte-Stimulating Hormone Enhance Cytotoxicity toward and Cellular Uptake by Melanomas: An Application of Nanomedicine on Natural Product. J Tradit Complement Med. 2013;3:102–109. doi: 10.4103/2225-4110.110423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barkey NM, Tafreshi NK, Josan JS, De Silva CR, Sill KN, Hruby VJ, Gillies RJ, Morse DL, Vagner J. Development of Melanoma-Targeted Polymer Micelles by Conjugation of a Melanocortin 1 Receptor (Mc1r) Specific Ligand. J Med Chem. 2011;54:8078–8084. doi: 10.1021/jm201226w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu W, Xiong C, Zhang R, Shi L, Huang M, Zhang G, Song S, Huang Q, Liu G, Li C. Receptor-Mediated Transcytosis: A Mechanism for Active Extravascular Transport of Nanoparticles in Solid Tumors. J Control Release. 2012;161:959–966. doi: 10.1016/j.jconrel.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu W, Xiong C, Zhang G, Huang Q, Zhang R, Zhang JZ, Li C. Targeted Photothermal Ablation of Murine Melanomas with Melanocyte-Stimulating Hormone Analog-Conjugated Hollow Gold Nanospheres. Clin Cancer Res. 2009;15:876–886. doi: 10.1158/1078-0432.CCR-08-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma K, Mendoza C, Hanson M, Werner-Zwanziger U, Zwanziger J, Wiesner U. Control of Ultrasmall Sub-10 Nm Ligand-Functionalized Fluorescent Core-Shell Silica Nanoparticle Growth in Water. Chem Mater. 2015;27:4119–4133. [Google Scholar]
- 33.Ma K, Zhang DH, Cong Y, Wiesner U. Elucidating the Mechanism of Silica Nanoparticle Pegylation Processes Using Fluorescence Correlation Spectroscopies. Chem Mater. 2016;28:1537–1545. [Google Scholar]
- 34.Chen F, Ma K, Benezra M, Zhang L, Cheal SM, Phillips E, Yoo B, Pauliah M, Overholtzer M, Zanzonico P, Sequeira S, Gonen M, Quinn T, Wiesner U, Bradbury MS. Cancer-Targeting Ultrasmall Silica Nanoparticles for Clinical Translation: Physicochemical Structure and Biological Property Correlations. Chem Mater. 2017 doi: 10.1021/acs.chemmater.1027b03033. [DOI] [PMC free article] [PubMed]
- 35.Piao Y, Burns A, Kim J, Wiesner U, Hyeon T. Designed Fabrication of Silica-Based Nanostructured Particle Systems for Nanomedicine Applications. Adv Funct Mater. 2008;18:3745–3758. [Google Scholar]
- 36.Ma K, Wiesner U. Modular and Orthogonal Post-Pegylation Surface Modifications by Insertion Enabling Penta-Functional Ultrasmall Organic-Silica Hybrid Nanoparticles. Chem Mater. 2017;29:6840–6855. [Google Scholar]
- 37.Miao Y, Benwell K, Quinn TP. 99mtc- and 111in-Labeled Alpha-Melanocyte-Stimulating Hormone Peptides as Imaging Probes for Primary and Pulmonary Metastatic Melanoma Detection. J Nucl Med. 2007;48:73–80. [PubMed] [Google Scholar]
- 38.Ow H, Larson DR, Srivastava M, Baird BA, Webb WW, Wiesner U. Bright and Stable Core-Shell Fluorescent Silica Nanoparticles. Nano Lett. 2005;5:113–117. doi: 10.1021/nl0482478. [DOI] [PubMed] [Google Scholar]
- 39.Burns A, Ow H, Wiesner U. Fluorescent Core-Shell Silica Nanoparticles: Towards “Lab on a Particle” Architectures for Nanobiotechnology. Chem Soc Rev. 2006;35:1028–1042. doi: 10.1039/b600562b. [DOI] [PubMed] [Google Scholar]
- 40.Ma K, Sai H, Wiesner U. Ultrasmall Sub-10 Nm near-Infrared Fluorescent Mesoporous Silica Nanoparticles. J Am Chem Soc. 2012;134:13180–13183. doi: 10.1021/ja3049783. [DOI] [PubMed] [Google Scholar]
- 41.Ma K, Werner-Zwanziger U, Zwanziger J, Wiesner U. Controlling Growth of Ultrasmall Sub-10 Nm Fluorescent Mesoporous Silica Nanoparticles. Chem Mater. 2013;25:677–691. [Google Scholar]
- 42.Yang P, Gai S, Lin J. Functionalized Mesoporous Silica Materials for Controlled Drug Delivery. Chem Soc Rev. 2012;41:3679–3698. doi: 10.1039/c2cs15308d. [DOI] [PubMed] [Google Scholar]
- 43.Larson DR, Ow H, Vishwasrao HD, Heikal AA, Wiesner U, Webb WW. Silica Nanoparticle Architecture Determines Radiative Properties of Encapsulated Fluorophores. Chem Mater. 2008;20:2677–2684. [Google Scholar]
- 44.Herz E, Burns A, Bonner D, Wiesner U. Large Stokes-Shift Fluorescent Silica Nanoparticles with Enhanced Emission over Free Dye for Single Excitation Multiplexing. Macromol Rap Commun. 2009;30:1907–1910. doi: 10.1002/marc.200900389. [DOI] [PubMed] [Google Scholar]
- 45.Herz E, Ow H, Bonner D, Burns A, Wiesner U. Dye Structure-Optical Property Correlations in near-Infrared Fluorescent Core-Shell Silica Nanoparticles. J Mater Chem. 2009;19:6341–6347. [Google Scholar]
- 46.Rosenkranz AA, Slastnikova TA, Durymanov MO, Sobolev AS. Malignant Melanoma and Melanocortin 1 Receptor. Biochemistry (Mosc) 2013;78:1228–1237. doi: 10.1134/S0006297913110035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang J, Guo H, Miao Y. Technetium-99m-Labeled Arg-Gly-Asp-Conjugated Alpha-Melanocyte Stimulating Hormone Hybrid Peptides for Human Melanoma Imaging. Nucl Med Biol. 2010;37:873–883. doi: 10.1016/j.nucmedbio.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong W, Minchin RF. Binding and Internalization of the Melanocyte Stimulating Hormone Receptor Ligand [Nle4, D-Phe7] Alpha-Msh in B16 Melanoma Cells. Int J Biochem Cell Biol. 1996;28:1223–1232. doi: 10.1016/s1357-2725(96)00074-x. [DOI] [PubMed] [Google Scholar]
- 49.Chithrani BD, Chan WC. Elucidating the Mechanism of Cellular Uptake and Removal of Protein-Coated Gold Nanoparticles of Different Sizes and Shapes. Nano Lett. 2007;7:1542–1550. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 50.Kam NW, Liu Z, Dai H. Carbon Nanotubes as Intracellular Transporters for Proteins and DNA: An Investigation of the Uptake Mechanism and Pathway. Angew Chem Int Ed Engl. 2006;45:577–581. doi: 10.1002/anie.200503389. [DOI] [PubMed] [Google Scholar]
- 51.Benezra M, Phillips E, Overholtzer M, Zanzonico PB, Tuominen E, Wiesner U, Bradbury MS. Ultrasmall Integrin-Targeted Silica Nanoparticles Modulate Signaling Events and Cellular Processes in a Concentration-Dependent Manner. Small. 2015;11:1721–1732. doi: 10.1002/smll.201402331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stabin MG, Sparks RB, Crowe E. Olinda/Exm: The Second-Generation Personal Computer Software for Internal Dose Assessment in Nuclear Medicine. J Nucl Med. 2005;46:1023–1027. [PubMed] [Google Scholar]
- 53.Lee FT, Scott AM. Immuno-Pet for Tumor Targeting. J Nucl Med. 2003;44:1282–1283. [PubMed] [Google Scholar]
- 54.Abou DS, Ku T, Smith-Jones PM. In Vivo Biodistribution and Accumulation of 89zr in Mice. Nucl Med Biol. 2011;38:675–681. doi: 10.1016/j.nucmedbio.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei L, Zhang X, Gallazzi F, Miao Y, Jin X, Brechbiel MW, Xu H, Clifford T, Welch MJ, Lewis JS, Quinn TP. Melanoma Imaging Using (111)in-, (86)Y- and (68)Ga-Labeled Chx-a″-Re(Arg11)Ccmsh. Nucl Med Biol. 2009;36:345–354. doi: 10.1016/j.nucmedbio.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holland JP, Sheh Y, Lewis JS. Standardized Methods for the Production of High Specific-Activity Zirconium-89. Nucl Med Biol. 2009;36:729–739. doi: 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen F, Ma K, Zhang L, Madajewski B, Zanzonico P, Sequeira S, Gonen M, Wiesner U, Bradbury MS. Target-or-Clear Zirconium-89 Labeled Silica Nanoparticles for Enhanced Cancer-Directed Uptake in Melanoma: A Comparison of Radiolabeling Strategies. Chem Mater. 2017;29:8269–8281. doi: 10.1021/acs.chemmater.7b02567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.