

Abstract
The acquisition of biallelic mutations in the APC gene is a rate-limiting step in the development of most colorectal cancers and occurs in the earliest lesions. APC encodes a 312-kDa protein that localizes to multiple subcellular compartments and performs diverse functions. APC participates in a cytoplasmic complex that promotes the destruction of the transcriptional licensing factor β-catenin; APC mutations that abolish this function trigger constitutive activation of the canonical WNT signaling pathway, a characteristic found in almost all colorectal cancers. By negatively regulating canonical WNT signaling, APC counteracts proliferation, promotes differentiation, facilitates apoptosis and suppresses invasion and tumor progression. APC further antagonizes canonical WNT signaling by interacting with and counteracting β-catenin in the nucleus.
APC also suppresses tumor initiation and progression in the colorectal epithelium through functions that are independent of canonical WNT signaling. APC regulates the mitotic spindle to facilitate proper chromosome segregation, localizes to the cell periphery and cell protrusions to establish cell polarity and appropriate directional migration, and inhibits DNA replication by interacting directly with DNA. Mutations in APC are often frameshifts, insertions or deletions that introduce premature stop codons and lead to the production of truncated APC proteins that lack its normal functions and possess tumorigenic properties. Therapeutic approaches in development for the treatment of APC-deficient tumors are focused on the inhibition of canonical WNT signaling, especially through targets downstream of APC in the pathway, or on the restoration of wild-type APC expression.
Keywords: APC, canonical WNT signaling, WNT-independent, colorectal cancer, therapeutics
Driving questions in colorectal cancer research
Colorectal cancer has emerged as a family of diseases that can develop along a number of different histological and molecular trajectories (Fig. 1). As the understanding of colorectal cancer at the molecular level becomes increasingly detailed, intense research interest surrounds the ability of these histological or molecular characteristics to predict prognosis. 65% of colorectal cancer patients survive 5 years or more after diagnosis, although individual outcomes are heavily dependent upon stage. Localized colorectal cancers are associated with 5-year survival of approximately 70–90% (depending on the degree of invasiveness) [1], while cancers that have spread to nearby lymph nodes are associated with only 40–50% 5-year survival. Metastatic colorectal cancers that have spread to the liver or other distant sites are associated with only 5–15% survival over a 5-year period, yet represent more than 20% of diagnoses [1]. These statistics indicate the extent to which surgical resection remains the best tool for curing colorectal cancer, while therapeutic options against cancers that have disseminated to other sites quickly become exhausted.
Figure 1. APC mutations are unequally distributed across different colorectal cancer pathways.

Molecular analysis of colorectal cancers has identified at least four subsets associated with different prognoses [66, 171]. Subsets are defined by the presence or absence of the CpG island methylator phenotype (CIMP) and the microsatellite instability phenotype (MSI), two often-linked characteristics which together with the chromosomal instability phenotype (CIN) generate mutations that drive disease progression. Most colorectal cancers are characterized by neither CIMP nor MSI (B), but follow a trajectory similar to that observed in familial adenomatous polyposis (FAP), characterized by mutations in APC and TP53 [68, 172]. CIMP in the absence of MSI is associated with poor prognosis (A), while CIMP resulting in MSI [19, 173, 174] is associated with good prognosis (C), similar to the MSI-driven cancers observed in Lynch syndrome [175, 176] (D).
The discovery and characterization of the genetic changes acquired along the malignant pathway have informed the search for novel therapeutic options. However, significant questions remain unanswered, such as whether or how colorectal tumors tend to acquire these key mutations in a particular order [2]. The field has begun to identify the patterns of mutations or aberrations in gene expression that distinguish short-term survivors from long-term survivors of advanced disease [3], or that correlate with responsiveness to certain interventions, including emerging antibody-based therapies [4]. These clinically-oriented questions may eventually be answered through a more complete understanding of how particular genetic changes translate into phenotypic changes.
The APC gene
Biallelic mutation of the APC gene occurs in 45%–80% of colorectal cancers [5–7] and is observed in the earliest detectable lesions [2]. The APC locus was originally identified based on its link to familial adenomatous polyposis coli (FAP), an inherited syndrome of cancer predisposition [8–11]. Inherited mutations in the APC gene cause affected individuals to develop hundreds to thousands of adenomatous polyps, resulting in the onset of CRC typically before the age of 40 [12]. Individuals with FAP inherit a loss-of-function mutation in a single allele of APC, followed by an additional acquired mutation in the second allele of APC in the adenomas and adenocarcinomas that develop [13–15]. Thus, the acquisition of biallelic APC mutations represents an early and rate-limiting step in all FAP-associated and most sporadic colorectal tumors.
In studies of colorectal cancer as a whole, APC mutational status does not strongly correlate with outcome [16, 17]. Nevertheless, APC mutations exhibit an interesting pattern of differential distribution in the recognized subtypes of colorectal cancer (Fig. 1). APC mutations correlate strongly with a large subset of colorectal cancers associated with intermediate prognosis [18]. On the other hand, APC mutations occur infrequently within a smaller subset derived from sessile serrated adenomas and associated with microsatellite instability and good prognosis [18]. This latter subset exhibits a relatively high proportion of activating mutations in the gene encoding β-catenin (CTNNB1) [19] that are mutually exclusive of APC mutations [20, 21]. Interestingly, CTNNB1 mutations are significantly more prevalent in small adenomas than in large adenomas or adenocarcinomas, [18], whereas APC mutations are well-represented across all stages of tumorigenesis. It has recently emerged that APC mutational status has value as a predictive marker of poor prognosis in Stage III colorectal cancers [17], raising the possibility that APC mutations not only initiate colorectal cancer development, but drive clinical phenotypes relevant to progression and metastasis as well.
Functions of the APC tumor suppressor protein
The APC gene encodes a 312-kDa protein (Fig. 2) that performs diverse cellular functions and localizes to multiple subcellular compartments. Mutations in APC are often frameshifts, insertions or deletions that introduce premature stop codons and lead to the production of a truncated APC protein. Amino acids 1000 to 1600 of APC have been identified as a mutation cluster region that represents roughly 20% of the total (2,843-amino acid) protein yet contains about 60% of all identified mutation sites [7]. The following sections detail functions of the wild-type APC protein, all of which are weakened or lost through the acquisition of pathogenic mutations. Also discussed are dominant functions of truncated APC proteins that contribute to tumorigenesis.
Figure 2. Truncating mutations disrupt key structural features in the central and C-terminal regions of the human APC protein.
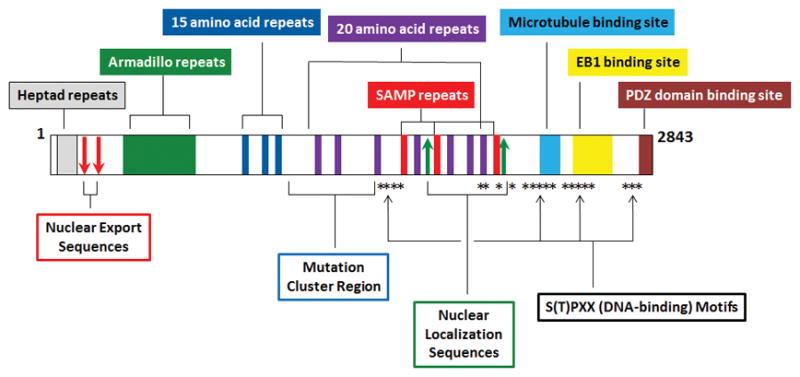
Key structural features of the human APC protein that are disrupted by most truncating mutations include its 20-amino acid repeats (purple), SAMP repeats (red), nuclear localization sequences (green arrows), microtubule binding region (light blue), EB1 binding region (yellow), DNA-interacting S(T)PXX motifs (asterisks), and C-terminal PDZ domain-binding region (brown). Truncated N-terminal APC proteins retain some ability to interact with β-catenin due to the preservation of the 15-amino acid repeats and some of the 20-amino acid repeats, while retention of the Armadillo domain preserves the ability localize to the nucleus.
Cytoplasmic APC negatively regulates canonical WNT signaling
The best-known function of APC is its ability to interact with β-catenin in the cytoplasm and promote β-catenin phosphorylation, ubiquitination and subsequent proteolytic degradation (Fig. 3) [22–25]. This function occurs within the context of a cytoplasmic complex [23, 26] that includes glycogen synthase kinase 3β (GSK-3β) [27], AXIN1 [24, 28] or AXIN2 [23, 29], and other kinases and phosphatases [30, 31]. The interaction of APC with this complex maps to a region in the center of the protein that contains three serine-alanine-methionine-proline (SAMP) repeats that mediate the binding of APC to AXIN1/AXIN2 [23], as well as three repeats of 15 amino acids each and seven repeats of 20 amino acids each. β-catenin interacts with the 15-amino acid repeats constitutively [32], and with the 20-amino acid repeats inducibly [33] following their phosphorylation by GSK-3β [34] and casein kinase I [31, 35]. Both the 15-amino acid and 20-amino acid repeats are necessary to enable APC to promote β-catenin degradation [33]; truncating mutations to APC generally disrupt these repeats either partially or completely.
Figure 3. Cytoplasmic APC protein negatively regulates the canonical WNT signaling pathway.
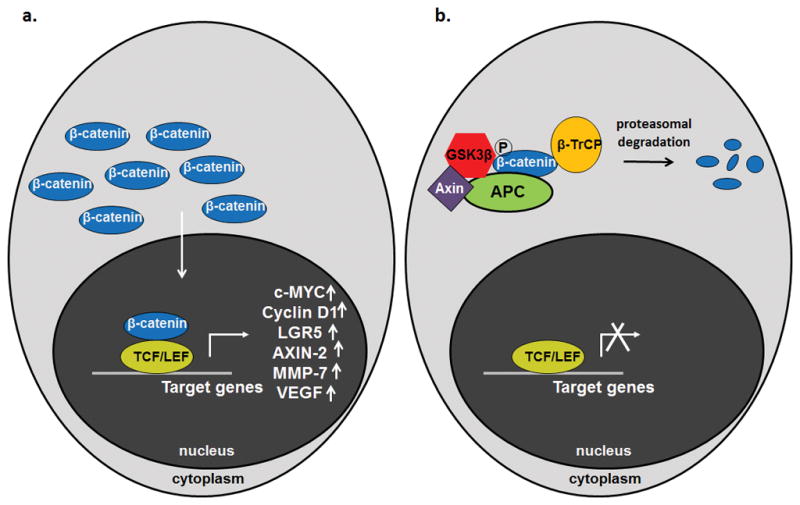
The critical difference between the active (A) and inactive (B) states of the canonical WNT signaling pathway is the accumulation of the licensing factor β-catenin, especially in the nucleus, where it binds to TCF/LEF family transcription factors to promote changes in gene transcription. The active state of the pathway (A) is characteristic of colorectal cancers and colorectal progenitor cells, as it favors proliferation and survival at the expense of differentiation and sensitivity to apoptosis. The inactive state (B) is characteristic of mature, differentiated cells of the colorectal epithelium that express APC. APC (green) is a required component of the cytoplasmic complex that limits β-catenin availability by promoting its proteolytic degradation.
The role of APC in this cytoplasmic complex negatively regulates the canonical WNT signaling pathway [26], whose ability to mediate transcriptional changes requires the licensing factor β-catenin to bind to transcription factors of the TCF/LEF family [36]. The canonical WNT signaling pathway alters the transcription of key target genes, including activation of the genes encoding c-MYC [37], Cyclin D1 [38, 39] and LGR5 [40, 41]. These and other changes in gene expression collectively drive proliferation, survival and maintenance of an undifferentiated state in progenitor cells of the colorectal epithelium [38, 42–45]. The pathway is critical to normal tissue homeostasis [46], as mature cells lining the colon and rectum must be frequently replaced as they become damaged, undergo apoptosis and are sloughed into the intestinal lumen [47]. Progenitor cells of the colorectal epithelium are characterized by activated canonical WNT signaling, while maturing cells are characterized by inactivation of the pathway and increasing expression of APC [48, 49]. Disruption of this balance initiates colorectal cancer, as the progeny of a colorectal progenitor cell lacking functional APC are unable to stop proliferating, differentiate or undergo apoptosis [48]. APC loss stabilizes β-catenin and constitutively activates the pathway even in the absence of a WNT signal [33, 50]. The importance of this particular APC function in the process of tumorigenesis is underscored by the observation that a significant percentage of the colorectal cancers with wild-type APC exhibit a point mutation in CTNNB1, the gene encoding β-catenin [20] that makes the protein resistant to degradation [21]. Together, APC and β-catenin mutations form part of a larger group of molecular changes by which approximately 93% of colorectal cancers exhibit activation of the canonical WNT signaling pathway [5].
Nuclear APC negatively regulates canonical WNT signaling
The APC protein further counteracts canonical WNT signaling by participating in at least two other mechanisms that take place in the nucleus. Nuclear import of APC is dependent upon two nuclear localization sequences (NLS) as well as a separate Armadillo domain that also promotes nuclear import [51, 52]. Once inside, nuclear APC interacts with β-catenin and facilitates its export to the cytoplasm [53–56]. This activity is dependent upon two recognized nuclear export sequences (NES) within APC [54]. The ratio of nuclear APC to cytoplasmic APC decreases as proliferation slows down and cells enter a quiescent state [57–59], and is controlled in part by phosphorylation of key residues near each APC NLS [51, 57]. The nuclear interaction between APC and β-catenin occurs in part within the chromatin fraction, in which APC promotes the removal of β-catenin from specific genomic loci (Fig. 4) [60]. Nuclear APC interacts not only with β-catenin but also with C-terminal binding protein (CtBP), a transcriptional co-repressor [61]. A detailed mechanistic study has demonstrated that APC and CtBP transiently interact with β-catenin at the WNT-activated MYC promoter and promote the removal of β-catenin from this locus, coinciding with the appearance of the more stable co-repressors TLE-1 and HDAC1 [60]. This function of chromatin-associated APC negatively regulates WNT activation of the MYC gene [60], as well as WNT activation of the AXIN2, DKK1 and SP5 genes [62]. Truncated APC proteins observed in colorectal cancer generally lack both NLS, which are located in the missing C-terminal half of the protein. However, truncated APC retains the ability to move between the nucleus and cytoplasm because of the preserved Armadillo domain [63]. It is difficult to compare the relative importance of the cytoplasmic and nuclear mechanisms by which APC negatively regulates canonical WNT signaling, as both are disrupted by pathogenic APC mutations [64]. Evidence for the importance of nuclear APC includes the observations that biallelic point mutations abolishing both NLS within APC increase proliferation and expression of canonical WNT signaling targets in the mouse intestine, and that a single mutant allele increases polyp number and size in ApcMin/+ mice [65].
Figure 4. Chromatin-associated APC negatively regulates canonical WNT activation of specific target genes by removing the licensing factor β-catenin.
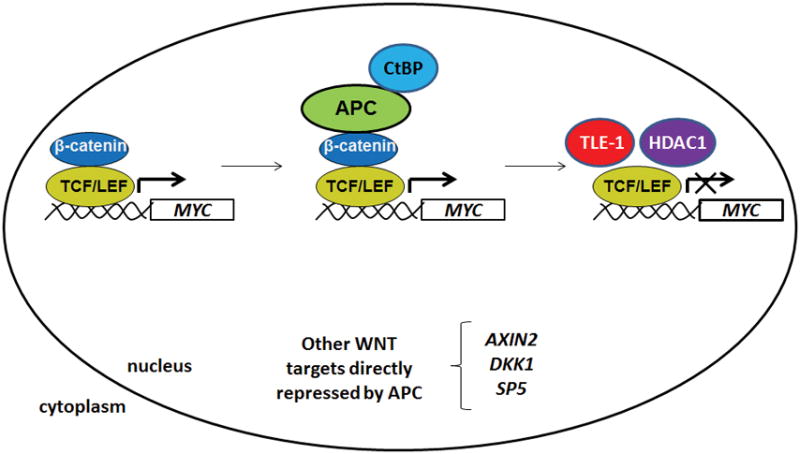
The nuclear fraction of APC further antagonizes canonical WNT signaling in colorectal cancer cells by interacting with chromatin-associated β-catenin at WNT-activated target genes such as MYC. This transient interaction leads to the removal of β-catenin, the appearance of co-repressors such as TLE-1 and HDAC1, and transcriptional repression [60, 62].
APC in mitotic spindle dynamics and genomic stability
Colorectal cancers segregate into two mutually exclusive categories based on exhibiting genomic instability at either the microsatellite level (due defects in mismatch repair) or the chromosomal level [66, 67]. APC mutations are particularly well-represented within the subset of colorectal cancers that feature widespread chromosomal damage [68], and also play an important role in this phenotype [69]. The ability of APC to interact with the microtubule network facilitates its localization to the kinetochore [69, 70], a protein structure that mediates the attachment of microtubules to sister chromatids during mitosis. Murine cells with Apc mutations exhibit chromosomal abnormalities as well as mitotic spindles characterized by numerous microtubules lacking proper connections to the kinetochore [69]. APC silencing in human colorectal cancer cells similarly decreases inter-kinetochore tension during metaphase, and results in defective progression through mitosis [71]. The APC-interacting protein EB1 collaborates with APC to regulate the mitotic spindle, and the loss of this APC-dependent mechanism facilitates errors in chromosome alignment without halting cell division [72].
APC is modified during mitosis by phosphorylation [73], including by the spindle checkpoint kinases Bub1 and BubR1, suggesting that its contributions to mitotic spindle integrity and chromosome segregation are accompanied by a role in the spindle checkpoint [70]. Interestingly, overexpression of a truncated APC protein exerts a dominant negative effect that compromises the spindle checkpoint [74], in which defects are often linked to chromosomal instability. These functional data collectively suggest that truncating APC mutations drive genomic instability at the chromosomal level through both loss-of-function and gain-of-function mechanisms that occur independently of its role in canonical WNT signaling.
APC controls DNA replication and cell cycle progression
In addition to its role in the spindle checkpoint and mitotic progression, APC controls the cell cycle by regulating the G1/S transition. Various APC-deficient colorectal cancer cell lines stably transfected to express wild-type APC exogenously exhibit dramatic changes in doubling time and inhibition of G1/S phase progression [75]. Overexpression of RB pathway components such as Cyclin D1/CDK4, Cyclin E/CDK2, E1A, and E2F overrides cell cycle inhibition by APC, suggesting that APC re-expression restores the G1/S checkpoint [76, 77]. Negative regulation of canonical WNT signaling by APC contributes to this function by downregulating targets including Cyclin D1 itself [38, 39]. The physical interaction of APC with other proteins such as DLG may also be involved, as the C-terminal DLG-binding residues within APC are required for complete inhibition of S-phase entry in a mouse fibroblast cell model, while DLG overexpression in itself is sufficient to arrest cells at the G1/S transition [78]. The interaction of the APC C-terminus with A/T-rich DNA also blocks entry into or progression through S-phase by blocking DNA replication [79, 80]. These findings are consistent with evidence that defective G1/S-phase progression due to APC overexpression is only partially alleviated by co-transfection of a constitutively active mutant β-catenin [77]. APC thus contributes to the regulation of cell cycle progression through a combination of WNT-dependent and WNT-independent mechanisms.
Pro-apoptotic functions of APC
APC exhibits a gradient of increasing expression in the luminal direction along the colorectal crypt-villus axis [49], coinciding with mature, non-proliferative areas of the crypt where apoptotic cell death and cell shedding occur. Functional evidence linking APC to apoptosis began to accumulate with the observation that exogenously restoring APC expression in an APC-deficient colon cancer cell line triggered a 10-fold increase in the proportion of apoptotic cells [43]. In addition, overexpression of WNT-inhibitory PDZ domain-containing peptides from the Dishevelled protein induce apoptosis in an APC-dependent manner [81]. The link between APC and apoptosis is mediated at least in part by its role in canonical WNT signaling, whose targets include the BIRC5 gene that encodes the anti-apoptotic Survivin protein [82].
The ability of APC to sensitize cells to apoptosis is also partially WNT-independent [83]. Caspase family members proteolytically cleave APC in apoptotic cells [84], producing an N-terminal fragment that localizes to mitochondria and interacts with hTID-1 to promote caspase activity and cell sensitivity to apoptosis [85]. While truncated APC proteins in colorectal cancer also exhibit mitochondrial localization, they appear to exert anti-apoptotic effects instead, as their knockdown promotes apoptosis and mitochondrial membrane permeability [86]. Truncated APC proteins interact with and promote mitochondrial localization of the anti-apoptotic BCL2 protein [86]. The role of APC loss in shifting the balance between apoptosis and survival therefore is mediated by multiple mechanisms, both direct and indirect.
The role of APC in differentiation
Just as the pattern of APC expression along the colorectal crypt-villus axis indicates its role in apoptosis, the coincidence of APC expression with areas of mature, functional epithelium provided the first clue that APC affects differentiation as well. This luminal upper half of the colorectal crypt is populated with a mixture of terminally-differentiated columnar epithelial cells, goblet cells that produce mucus and neuroendocrine cells, all of which are derived from multipotent stem cells at the base of the crypt. Apc loss in the mouse small intestine disrupts commitment to all three cell fates while driving commitment to the basally-located Paneth cell lineage through a WNT-dependent mechanism [87]. Apc likely drives differentiation through suppression of canonical WNT signaling as well, as mice deficient in the canonical WNT signaling transcription factor Tcf7L2 exhibit dramatic and lethal defects in the proliferation of intestinal stem cells [88]. Consistent with these findings, canonical WNT signaling activates the expression of Lgr5, a critical marker of intestinal stem cells [89].
Interestingly, loss of Apc in the zebrafish gut produces a differentiation defect that occurs prior to nuclear β-catenin accumulation or changes in proliferation [90]. This may result from the loss of an Apc function in promoting proteasome-dependent degradation of the transcriptional co-repressor Ctbp1 [91]. Upon Apc loss, Ctbp1 suppresses the transcription of genes involved in retinoic acid biosynthesis [91], a process required for differentiation in the zebrafish gut [90, 92]. Differentiation in this model is accomplished through the ability of retinoic acid to antagonize the expression of demethylase genes, resulting in the methylation of gene promoters critical to maintaining progenitor-like phenotypes [93]. APC similarly regulates CtBP1 expression in human colon cancer cell lines [91], indicating that the zebrafish mechanism by which it drives differentiation through retinoic acid biosynthesis may be present in the human colorectal epithelium as well. Studies from the zebrafish model also indicate that impaired intestinal differentiation following Apc loss is mediated in part by a decrease in the expression of mitochondrial pyruvate carrier 1 (Mpc1), resulting in defective pyruvate metabolism that is independent of the WNT pathway and potentially linked to the Warburg effect [94]. Since differentiation and cell migration along the crypt-villus axis are coupled processes [95], APC may help link cell fate and cell migration in the colorectal epithelium.
Cytoskeletal functions link APC to adhesion, migration and cell polarity
In addition to its roles in the cytoplasm and nucleus, APC localizes to the cell border [63] and participates in at least three mechanisms regulating epithelial organization and cell migration. APC localization to the cell periphery is dependent upon the actin cytoskeleton [96] as well as the Armadillo domain within APC [97], although truncated APC proteins retaining this region still localize there with reduced efficiency [96]. The scaffolding protein IQGAP1 mediates an indirect interaction of APC with actin and also links APC with Rac1 and Cdc42, two Ras-family GTPases that regulate actin structures [98]. APC also regulates the activity of these GTPases to influence actin organization [99]. APC also interacts with actin filaments and stress fibers and has the ability to bundle actin filaments in vitro [100], raising the possibility that it connects and coordinates crosstalk between the actin and microtubule cytoskeletons. APC collaborates with the formin mDia1 to nucleate actin filament formation [101], and this function is required for directed cell migration and focal adhesion turnover in cultured cells [102].
APC interacts with both β-catenin and plakoglobin (γ-catenin) at cell-cell junctions [103], which function to connect adjacent epithelial cells, organize them into layers and promote cell polarity. β-catenin and plakoglobin physically connect cytoskeletal components including actin and intermediate filaments with transmembrane adhesion molecules such as cadherins. APC loss promotes a decrease in cell-cell and cell-matrix adhesion by altering the subcellular distribution of E-cadherin [104, 105]. This consequence of APC loss occurs at the protein level, independently of canonical WNT signaling [104]. Truncated APC proteins contribute in a dominant negative manner to this loss of migration directionality [106]. APC is furthermore linked to cell polarity through interactions with DLG [107] and hScrib [108], the respective human homologues of the Drosophila proteins Discs Large and Scribble. DLG interacts with PDZ domain-binding residues at the C-terminus of APC, and the two proteins form a complex with hScrib at lateral regions of cell-cell contact in canine kidney epithelial cells [108].
Finally, APC localizes to leading edges of migrating cells in a microtubule-dependent manner [109]. This function of APC maps not to its N-terminal Armadillo domain, but to C-terminal regions of APC that directly contact the microtubule network [110]. Specifically, amino acids 2200–2400 are enriched for basic residues that facilitate microtubule binding and that promote polymerization [110] and bundling [111, 112] of microtubules in vitro. The APC C-terminal region also binds the microtubule-interacting protein EB1, which specifically interacts with and localizes APC to microtubule plus ends [113]. These microtubule-related functions of APC are proposed to drive the formation of membrane protrusions and influence the balance between adhesion and motility [114]. The well-characterized decrease in cell migration that follows APC loss in vivo [45] may be explained at least in part by the decreased formation of membrane protrusions and alterations in microtubule stability that follow APC loss in cultured cells [115]. APC also functions in mouse fibroblasts as an RNA-binding protein that localizes key RNA molecules to cell protrusions with consequences for cell migration [116]. In dying cells, APC may be required to target microtubules to the appropriate side of the cell, promoting proper extrusion from the apical rather than the basal side of the epithelium [117]. Collectively, these studies of APC at the cell periphery argue for roles in linking the actin cytoskeleton with the microtubule network and in the establishment of cell polarity and suppression of invasive behaviors [118].
APC counteracts invasion, cancer progression and metastasis through targets of canonical WNT signaling
The previous section discussed non-transcriptional roles of APC at the cell periphery, which explain at least in part the phenotype of defective intestinal epithelial cell migration observed along crypt-villus axes in both Apc-deficient [119] and Apc-overexpressing [120] mice. Additional evidence has linked APC loss to cell motility and invasive phenotypes through its role in the regulation of canonical WNT signaling. At first glance, this appears to conflict with the common view that APC mutations initiate colorectal tumorigenesis and drive adenoma formation, while other subsequent hits drive invasion and metastasis. This point of view is supported by the findings that APC mutations occur early in colorectal tumorigenesis [6] and are typically concordant both between paired primary and metastatic tumors [121] and between metastatic and non-metastatic intratumoral lineages [122]. On the other hand, histological analysis of β-catenin staining in primary colorectal tumors indicates that most tumors exhibit a predominantly peripheral rather than nuclear distribution of β-catenin, coinciding with a more differentiated epithelial growth phenotype, while nuclear β-catenin staining tends to characterize a smaller population of poorly-differentiated cells at the invasive front that lack membrane-associated E-cadherin staining [123]. Two representative cases (Figs. 5A and 5B) depict a higher proportion of tumor cells at the invasive edge exhibiting nuclear β-catenin staining relative to their counterparts in the tumor center. This evidence has led to the hypothesis that the tumor microenvironment at these sites drives loss of differentiation and epithelial-to-mesenchymal transition to promote invasion in a manner dependent on nuclear β-catenin [123].
Figure 5. Colorectal tumor cells at the invasive front exhibit higher levels of nuclear β-catenin than cells at the tumor center.

Stage II colorectal cancer cases were selected from The Ohio State University College of Medicine, Department of Pathology archives and stained immunohistochemically to characterize β-catenin localization. Two representative cases are shown in Figures 5A and 5B and include cells in the tumor center and at the invasive tumor edge at 10x, 20x and 40x magnification. Cases were selected representatives of a common pattern in which a higher proportion of tumor cells at the invasive edge exhibit nuclear β-catenin staining relative to their counterparts in the tumor center. These observations are consistent with published reports of variations in β-catenin staining across tumor sub-compartments as well as with evidence that some canonical WNT targets are differentially activated at the invasive tumor edge.
This finding raises the question of which targets of canonical WNT signaling might mediate cell migration, invasion and metastatic dissemination. Activation of canonical WNT signaling downregulates expression of E-cadherin, a key driver of epithelial cell identity that counteracts both invasion and metastasis [124]. The extent to which this regulation occurs directly through WNT transcriptional activity [125] or indirectly through WNT targeting of the transcription factor Slug (SNAI2) [126] is not clear. Other targets of canonical WNT signaling that facilitate invasive and/or metastatic phenotypes include extracellular matrix components such as fibronectin (FN1) [127] and LAMC2 [128], matrix-remodeling enzymes such as matrilysin (MMP7) [129, 130], MMP14 [131] and ADAM10 [132], and cell adhesion receptors such as uPAR [133], CD44 [134], Nr-CAM [135] and L1-CAM [136]. Relevant targets also include ligands and receptor tyrosine kinases of the EphB/EphrinB pathway [137] and fascin (FSCN1), which induces the formation of filopodia [138]. Some of these targets are more highly expressed at the invasive front relative to the well-differentiated central region of colorectal tumors [139], consistent with the proposed role of elevated nuclear β-catenin at these sites driving invasion. CD44 is a particularly interesting target, as the variant isoform CD44v6 marks cancer stem cells with metastatic potential within primary colorectal tumors, exhibits higher expression in mesenchymal-like cells at the invasive front, and enhances both cell migration and metastatic capability [140]. CD44v6 expression remains elevated in metastases [140] despite the fact that these lesions are generally characterized by a predominantly differentiated epithelial histology and cell peripheral β-catenin staining pattern similar to their primary tumor counterparts [123]. Overall, APC is linked to the suppression of colorectal cancer progression and metastasis not only through its role in maintaining chromosomal stability, but also through both WNT-dependent and WNT-independent contributions to cell migration and invasion.
Targeted treatment of APC-deficient colorectal cancers
Re-establishing Apc expression can restore normal development and differentiation of the intestinal crypt epithelium in mice with Apc-deficient intestinal adenomas and adenocarcinomas [46]. This is consistent with studies in human colon cancer cell lines in which re-expression of APC modifies cancer-related characteristics such as proliferation [75], adhesion [104] and sensitivity to apoptosis [43]. These reports on re-establishing APC function as a therapeutic strategy, and have led to the exploration of several different methodologies for restoring APC expression. Tumors with nonsense mutations in APC respond to aminoglycoside and macrolide agents that promote readthrough of premature stop codons [141]. These treatments partially restore APC expression in an APC-deficient human colon cancer cell line sufficiently to reduce tumor size in a mouse xenograft model [141]. Treatment with the macrolide compound tylosin reduces the size and number of intestinal tumors and extends the lifespan of ApcMin/+ mice possessing a nonsense mutation at codon 850 of the Apc gene [141]. In breast and lung cancer cell lines with hypermethylation of the APC promoter, APC expression has been successfully restored by treatment with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine [142]. Finally, herpesvirus particles tested as vehicles for delivery of wild-type APC to APC-deficient colon cancer cell lines cause a reduction in both their proliferation and migration in culture [143]. In spite of the demonstrated benefits of reversing the effects of APC loss in multiple model systems, clinical application of this strategy is not considered technically feasible and has not been explored outside of preliminary macrolide studies in patients with FAP.
Research strategies to improve the treatment of APC-deficient colorectal cancers have also focused on the development of agents targeting the canonical WNT signaling pathway, as several lines of evidence point to the pathway’s activation as a key tumorigenic consequence of APC loss [20, 21, 46, 144–146] and a driver of key cancer stem cell lineages within colorectal tumors [147]. In spite of significant efforts to develop inhibitors of canonical WNT signaling for cancer treatment, clinically relevant inhibitors do not yet exist as of 2017. One speculated reason has been the lack of convenient enzymatic targets in canonical WNT signaling relative to other developmental signaling pathways [148], as well as the challenge of targeting components of the pathway downstream of APC in colorectal cancers harboring APC mutations [149]. Another proposed obstacle has been the possibility that the synthetic TOPFLASH reporter system used for candidate discovery fails to recapitulate the endogenous complexity of WNT activation and inactivation sufficiently to facilitate identification of clinically relevant targets [150]. Finally, even an effective WNT inhibitor may be accompanied by significant safety concerns due to the critical role of canonical WNT signaling in normal processes such as somatic stem cell maintenance [148].
Many of the more promising inhibitors of canonical WNT signaling target early steps of the pathway that lie upstream of APC, and therefore are less effective in the absence of functional APC [148]. Nevertheless, evidence suggests that APC-deficient cells rely on endogenous WNT ligands to fine-tune the signaling pathway to optimal levels [151], and inhibitors of Porcupine O-Acyltransferase (required for WNT ligand palmitoylation and secretion) [152] effectively reduce the ability of colon cancer cell lines to proliferate and form tumors in xenograft mouse models [153]. The Porcupine inhibitors LGK974 [154] and ETC-159 [155] are currently in clinical trials for treatment of a subset of colorectal cancers harboring BRAF mutations as well as for other solid tumors. Other antagonists of WNT signal transduction currently in clinical trials include inhibitors of the WNT receptor Frizzled, such as the antibody vantictumab [156] and the fusion protein ipafricept [157].
Potential therapeutic targets in the canonical WNT signaling pathway include other components of the cytoplasmic complex through which APC negatively regulates β-catenin stability. β-catenin degradation can be promoted therapeutically by activators of casein kinase that can enhance β-catenin phosphorylation [158], and by inhibitors of tankyrase enzymes that modulate the stability of the key scaffolding component AXIN [159]. The potential efficacy of these strategies in the treatment of APC-deficient colorectal cancers remains a matter of some debate [160], and tankyrase inhibition in particular has been shown to partially reverse the effects of APC loss on β-catenin levels in cell culture models [161]. Other mechanisms negatively regulate β-catenin levels independently of canonical WNT signaling, including activation of the Vitamin D signaling pathway [162–164].
Other promising preclinical studies have characterized small molecules designed to disrupt the nuclear interaction between β-catenin and TCF/LEF family transcription factors [165] or between β-catenin and the transcriptional coactivator CBP [166]. Disruptors of the β-catenin and CBP interaction include PRI-724 (also known as ICG-001), currently in clinical trials for newly-diagnosed metastatic colorectal cancer [150]. Small molecule antagonists of the β-catenin / TCF complex include PKF115-584 and CGP049090, which appear to function by binding directly β-catenin [165] but are not currently in clinical trials.
The Traf2 and Nck-interacting kinase (TNIK) is a required collaborator of β-catenin and TCF7L2 in nuclear transcription factor complexes [167], and is under investigation as a potential therapeutic target for APC-deficient colorectal cancers [149, 168, 169]. A recently-discovered TNIK inhibitor, NCB-0846, has not yet been studied in a clinical context [169]. Other more targeted strategies have been designed to manipulate a single gene activated by canonical WNT signaling, particularly the MYC proto-oncogene [170]. The specificity of these strategies may help mitigate the anticipated shortcoming of more general WNT antagonists in that the canonical WNT signaling pathway functions to maintain the somatic stem cells responsible for normal homeostasis of the colorectal epithelium and other tissues [88, 148]. Toxicity to normal cells is therefore a significant concern as these therapeutic candidates for APC-deficient colorectal cancer continue to progress into clinical studies.
Summary
In conclusion, the APC tumor suppressor protein performs multiple functions that contribute to its role in preventing colorectal tumorigenesis. It is difficult to assign relative importance to individual functions, other than to point out that APC contributions to relevant phenotypes such as cytoskeletal regulation, cell cycle progression, chromosomal stability, sensitivity to apoptosis, differentiation and invasion/metastasis are mediated by multiple mechanisms, both WNT-mediated and WNT-independent. Dominant negative effects of truncated APC proteins further counteract wild-type APC functions by promoting migration, chromosomal instability and evasion of apoptosis. Therapeutic strategies to restore functions of the APC tumor suppressor protein are not yet fully developed but are progressing along several different lines of investigation, particularly in the form of inhibitors of the canonical WNT signaling pathway.
Materials and Methods
Immunohistochemical analysis
Stage II colorectal cancer tumor samples were previously formalin-fixed, embedded in paraffin and prepared as 4 μm sections by the Department of Pathology at The Ohio State University College of Medicine. Antigen retrieval was performed with a high pH EDTA solution (Agilent Technologies, Santa Clara, CA) before immunohistochemical staining with an anti-human β-catenin antibody (Agilent Technologies, clone b-catenin-1; 1:400 dilution). Detection was performed with the Novolink polymer detection system (Leica Biosystems, Buffalo Grove, IL) and an Autostainer Link 48 (Agilent Technologies). Image capture was performed at 10x, 20x and 40x magnification.
Acknowledgments
Funding: NIH Awards R01CA063507 (J.G.), UL1TR001070 (J.G.), F31CA174260 (W.H.), HHMI MED into GRAD (W.H.), and Pelotonia Fellowship Program (W.H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding institutions or the National Institutes of Health.
The authors thank Debbie Knight, Research Associate in the Department of Pathology at The Ohio State University Wexner Medical Center, for performance of key immunohistochemical staining experiments, image generation, and figure design and construction. This is a post-peer- review, pre-copyedit version of an article published in Cancer and Metastasis Reviews. The final authenticated version is available online at: http://dx.doi.org/10.1007/s10555-017-9725-6.
Footnotes
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
References
- 1.Howlader NNA, Krapcho M, Miller D, Bishop K, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA, editors. SEER Cancer Statistics Review, 1975–2013. National Cancer Institute; Bethesda, MD: 2016. [Google Scholar]
- 2.Jen J, et al. Molecular determinants of dysplasia in colorectal lesions. Cancer Res. 1994;54(21):5523–6. [PubMed] [Google Scholar]
- 3.Stein U, et al. MACC1, a newly identified key regulator of HGF-MET signaling, predicts colon cancer metastasis. Nat Med. 2009;15(1):59–67. doi: 10.1038/nm.1889. [DOI] [PubMed] [Google Scholar]
- 4.Linardou H, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: A systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9(10):962–72. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 5.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powell SM, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359(6392):235–7. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 7.Miyoshi Y, et al. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum Mol Genet. 1992;1(4):229–33. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 8.Groden J, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66(3):589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 9.Nishisho I, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253(5020):665–9. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 10.Joslyn G, et al. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991;66(3):601–13. doi: 10.1016/0092-8674(81)90022-2. [DOI] [PubMed] [Google Scholar]
- 11.Kinzler KW, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253(5020):661–5. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 12.Giardiello F. Gastrointestinal polyposis syndromes and hereditary nonpolyposis colorectal cancer. In: AKR, editor. Gastrointestinal Cancers: Biology, Diagnosis, and Therapy. Lippincott-Raven; Philadelphia: 1995. pp. 367–377. [Google Scholar]
- 13.Ichii S, et al. Detailed analysis of genetic alterations in colorectal tumors from patients with and without familial adenomatous polyposis (FAP) Oncogene. 1993;8(9):2399–405. [PubMed] [Google Scholar]
- 14.Levy DB, et al. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. 1994;54(22):5953–8. [PubMed] [Google Scholar]
- 15.Luongo C, et al. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994;54(22):5947–52. [PubMed] [Google Scholar]
- 16.Conlin A, et al. The prognostic significance of K-ras, p53, and APC mutations in colorectal carcinoma. Gut. 2005;54(9):1283–6. doi: 10.1136/gut.2005.066514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Broek E, et al. Genomic profiling of stage II and III colon cancers reveals APC mutations to be associated with survival in stage III colon cancer patients. Oncotarget. 2016;7(45):73876–73887. doi: 10.18632/oncotarget.12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samowitz WS, et al. β-Catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res. 1999;59(7):1442–4. [PubMed] [Google Scholar]
- 19.Mirabelli-Primdahl L, et al. Beta-catenin mutations are specific for colorectal carcinomas with microsatellite instability but occur in endometrial carcinomas irrespective of mutator pathway. Cancer Res. 1999;59(14):3346–51. [PubMed] [Google Scholar]
- 20.Sparks AB, et al. Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58(6):1130–4. [PubMed] [Google Scholar]
- 21.Morin PJ, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275(5307):1787–90. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 22.Orford K, et al. Serine phosphorylation-regulated ubiquitination and degradation of beta-catenin. J Biol Chem. 1997;272(40):24735–8. doi: 10.1074/jbc.272.40.24735. [DOI] [PubMed] [Google Scholar]
- 23.Behrens J, et al. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998;280(5363):596–9. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- 24.Sakanaka C, Weiss JB, Williams LT. Bridging of beta-catenin and glycogen synthase kinase-3beta by axin and inhibition of beta-catenin-mediated transcription. Proc Natl Acad Sci U S A. 1998;95(6):3020–3. doi: 10.1073/pnas.95.6.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aberle H, et al. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16(13):3797–804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munemitsu S, et al. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci U S A. 1995;92(7):3046–50. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yost C, et al. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10(12):1443–54. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 28.Ikeda S, et al. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17(5):1371–84. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto H, et al. Axil, a member of the Axin family, interacts with both glycogen synthase kinase 3beta and beta-catenin and inhibits axis formation of Xenopus embryos. Mol Cell Biol. 1998;18(5):2867–75. doi: 10.1128/mcb.18.5.2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seeling JM, et al. Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science. 1999;283(5410):2089–91. doi: 10.1126/science.283.5410.2089. [DOI] [PubMed] [Google Scholar]
- 31.Gao ZH, et al. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc Natl Acad Sci U S A. 2002;99(3):1182–7. doi: 10.1073/pnas.032468199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science. 1993;262(5140):1734–7. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- 33.Rubinfeld B, et al. Loss of beta-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res. 1997;57(20):4624–30. [PubMed] [Google Scholar]
- 34.Rubinfeld B, et al. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272(5264):1023–6. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 35.Liu C, et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108(6):837–47. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 36.Behrens J, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382(6592):638–42. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 37.He TC, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 38.Shtutman M, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96(10):5522–7. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto Y, et al. Overexpression of orphan G-protein-coupled receptor, Gpr49, in human hepatocellular carcinomas with beta-catenin mutations. Hepatology. 2003;37(3):528–33. doi: 10.1053/jhep.2003.50029. [DOI] [PubMed] [Google Scholar]
- 41.Van der Flier LG, et al. The intestinal Wnt/TCF signature. Gastroenterology. 2007;132(2):628–32. doi: 10.1053/j.gastro.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 42.Fevr T, et al. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol. 2007;27(21):7551–9. doi: 10.1128/MCB.01034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morin PJ, Vogelstein B, Kinzler KW. Apoptosis and APC in colorectal tumorigenesis. Proc Natl Acad Sci U S A. 1996;93(15):7950–4. doi: 10.1073/pnas.93.15.7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandra SH, et al. A common role for various human truncated adenomatous polyposis coli isoforms in the control of beta-catenin activity and cell proliferation. PLoS One. 2012;7(4):e34479. doi: 10.1371/journal.pone.0034479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sansom OJ, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18(12):1385–90. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dow LE, et al. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell. 2015;161(7):1539–52. doi: 10.1016/j.cell.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strater J, et al. In situ detection of enterocytic apoptosis in normal colonic mucosa and in familial adenomatous polyposis. Gut. 1995;37(6):819–25. doi: 10.1136/gut.37.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van de Wetering M, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111(2):241–50. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 49.Senda T, et al. Adenomatous polyposis coli (APC) plays multiple roles in the intestinal and colorectal epithelia. Med Mol Morphol. 2007;40(2):68–81. doi: 10.1007/s00795-006-0352-5. [DOI] [PubMed] [Google Scholar]
- 50.Korinek V, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275(5307):1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 51.Zhang F, White RL, Neufeld KL. Phosphorylation near nuclear localization signal regulates nuclear import of adenomatous polyposis coli protein. Proc Natl Acad Sci U S A. 2000;97(23):12577–82. doi: 10.1073/pnas.230435597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galea MA, Eleftheriou A, Henderson BR. ARM domain-dependent nuclear import of adenomatous polyposis coli protein is stimulated by the B56 alpha subunit of protein phosphatase 2A. J Biol Chem. 2001;276(49):45833–9. doi: 10.1074/jbc.M107149200. [DOI] [PubMed] [Google Scholar]
- 53.Henderson BR. Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat Cell Biol. 2000;2(9):653–60. doi: 10.1038/35023605. [DOI] [PubMed] [Google Scholar]
- 54.Neufeld KL, et al. APC-mediated downregulation of beta-catenin activity involves nuclear sequestration and nuclear export. EMBO Rep. 2000;1(6):519–23. doi: 10.1093/embo-reports/kvd117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosin-Arbesfeld R, Townsley F, Bienz M. The APC tumour suppressor has a nuclear export function. Nature. 2000;406(6799):1009–12. doi: 10.1038/35023016. [DOI] [PubMed] [Google Scholar]
- 56.Rosin-Arbesfeld R, et al. Nuclear export of the APC tumour suppressor controls beta-catenin function in transcription. EMBO J. 2003;22(5):1101–13. doi: 10.1093/emboj/cdg105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang F, White RL, Neufeld KL. Cell density and phosphorylation control the subcellular localization of adenomatous polyposis coli protein. Mol Cell Biol. 2001;21(23):8143–56. doi: 10.1128/MCB.21.23.8143-8156.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fagman H, et al. Nuclear accumulation of full-length and truncated adenomatous polyposis coli protein in tumor cells depends on proliferation. Oncogene. 2003;22(38):6013–22. doi: 10.1038/sj.onc.1206731. [DOI] [PubMed] [Google Scholar]
- 59.Davies JR, et al. Potential link between the NIMA mitotic kinase and nuclear membrane fission during mitotic exit in Aspergillus nidulans. Eukaryot Cell. 2004;3(6):1433–44. doi: 10.1128/EC.3.6.1433-1444.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sierra J, et al. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 2006;20(5):586–600. doi: 10.1101/gad.1385806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hamada F, Bienz M. The APC tumor suppressor binds to C-terminal binding protein to divert nuclear beta-catenin from TCF. Dev Cell. 2004;7(5):677–85. doi: 10.1016/j.devcel.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 62.Choi SH, et al. α-Catenin interacts with APC to regulate β-catenin proteolysis and transcriptional repression of Wnt target genes. Genes Dev. 2013;27(22):2473–88. doi: 10.1101/gad.229062.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson CB, Neufeld KL, White RL. Subcellular distribution of Wnt pathway proteins in normal and neoplastic colon. Proc Natl Acad Sci U S A. 2002;99(13):8683–8. doi: 10.1073/pnas.122235399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kouzmenko AP, et al. Truncation mutations abolish chromatin-associated activities of adenomatous polyposis coli. Oncogene. 2008;27(36):4888–99. doi: 10.1038/onc.2008.127. [DOI] [PubMed] [Google Scholar]
- 65.Zeineldin M, et al. A knock-in mouse model reveals roles for nuclear Apc in cell proliferation, Wnt signal inhibition and tumor suppression. Oncogene. 2012;31(19):2423–37. doi: 10.1038/onc.2011.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng YW, et al. CpG island methylator phenotype associates with low-degree chromosomal abnormalities in colorectal cancer. Clin Cancer Res. 2008;14(19):6005–13. doi: 10.1158/1078-0432.CCR-08-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goel A, et al. The CpG island methylator phenotype and chromosomal instability are inversely correlated in sporadic colorectal cancer. Gastroenterology. 2007;132(1):127–38. doi: 10.1053/j.gastro.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 68.Samowitz WS, et al. APC mutations and other genetic and epigenetic changes in colon cancer. Mol Cancer Res. 2007;5(2):165–70. doi: 10.1158/1541-7786.MCR-06-0398. [DOI] [PubMed] [Google Scholar]
- 69.Fodde R, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3(4):433–8. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 70.Kaplan KB, et al. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol. 2001;3(4):429–32. doi: 10.1038/35070123. [DOI] [PubMed] [Google Scholar]
- 71.Dikovskaya D, et al. Loss of APC induces polyploidy as a result of a combination of defects in mitosis and apoptosis. J Cell Biol. 2007;176(2):183–95. doi: 10.1083/jcb.200610099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Green RA, Wollman R, Kaplan KB. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol Biol Cell. 2005;16(10):4609–22. doi: 10.1091/mbc.E05-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trzepacz C, et al. Phosphorylation of the tumor suppressor adenomatous polyposis coli (APC) by the cyclin-dependent kinase p34. J Biol Chem. 1997;272(35):21681–4. doi: 10.1074/jbc.272.35.21681. [DOI] [PubMed] [Google Scholar]
- 74.Green RA, Kaplan KB. Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J Cell Biol. 2003;163(5):949–61. doi: 10.1083/jcb.200307070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Groden J, et al. Response of colon cancer cell lines to the introduction of APC, a colon-specific tumor suppressor gene. Cancer Res. 1995;55(7):1531–9. [PubMed] [Google Scholar]
- 76.Baeg GH, et al. The tumour suppressor gene product APC blocks cell cycle progression from G0/G1 to S phase. EMBO J. 1995;14(22):5618–25. doi: 10.1002/j.1460-2075.1995.tb00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heinen CD, et al. The APC tumor suppressor controls entry into S-phase through its ability to regulate the cyclin D/RB pathway. Gastroenterology. 2002;123(3):751–63. doi: 10.1053/gast.2002.35382. [DOI] [PubMed] [Google Scholar]
- 78.Ishidate T, et al. The APC-hDLG complex negatively regulates cell cycle progression from the G0/G1 to S phase. Oncogene. 2000;19(3):365–72. doi: 10.1038/sj.onc.1203309. [DOI] [PubMed] [Google Scholar]
- 79.Qian J, et al. The APC tumor suppressor inhibits DNA replication by directly binding to DNA via its carboxyl terminus. Gastroenterology. 2008;135(1):152–62. doi: 10.1053/j.gastro.2008.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brocardo MG, Borowiec JA, Henderson BR. Adenomatous polyposis coli protein regulates the cellular response to DNA replication stress. Int J Biochem Cell Biol. 2011;43(9):1354–64. doi: 10.1016/j.biocel.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 81.Zhang Y, et al. Inhibition of Wnt signaling by Dishevelled PDZ peptides. Nat Chem Biol. 2009;5(4):217–9. doi: 10.1038/nchembio.152. [DOI] [PubMed] [Google Scholar]
- 82.Zhang T, et al. Evidence that APC regulates survivin expression: A possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res. 2001;61(24):8664–7. [PubMed] [Google Scholar]
- 83.Steigerwald K, et al. The APC tumor suppressor promotes transcription-independent apoptosis in vitro. Mol Cancer Res. 2005;3(2):78–89. doi: 10.1158/1541-7786.MCR-03-0189. [DOI] [PubMed] [Google Scholar]
- 84.Qian J, et al. Caspase cleavage of the APC tumor suppressor and release of an amino-terminal domain is required for the transcription-independent function of APC in apoptosis. Oncogene. 2007;26(33):4872–6. doi: 10.1038/sj.onc.1210265. [DOI] [PubMed] [Google Scholar]
- 85.Qian J, et al. The mitochondrial protein hTID-1 partners with the caspase-cleaved adenomatous polyposis cell tumor suppressor to facilitate apoptosis. Gastroenterology. 2010;138(4):1418–28. doi: 10.1053/j.gastro.2009.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brocardo M, et al. Mitochondrial targeting of adenomatous polyposis coli protein is stimulated by truncating cancer mutations: Regulation of Bcl-2 and implications for cell survival. J Biol Chem. 2008;283(9):5950–9. doi: 10.1074/jbc.M708775200. [DOI] [PubMed] [Google Scholar]
- 87.Andreu P, et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development. 2005;132(6):1443–51. doi: 10.1242/dev.01700. [DOI] [PubMed] [Google Scholar]
- 88.Korinek V, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19(4):379–83. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- 89.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449(7165):1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 90.Nadauld LD, et al. Adenomatous polyposis coli control of retinoic acid biosynthesis is critical for zebrafish intestinal development and differentiation. J Biol Chem. 2004;279(49):51581–9. doi: 10.1074/jbc.M408830200. [DOI] [PubMed] [Google Scholar]
- 91.Nadauld LD, et al. Adenomatous polyposis coli control of C-terminal binding protein-1 stability regulates expression of intestinal retinol dehydrogenases. J Biol Chem. 2006;281(49):37828–35. doi: 10.1074/jbc.M602119200. [DOI] [PubMed] [Google Scholar]
- 92.Nadauld LD, et al. The zebrafish retinol dehydrogenase, rdh1l, is essential for intestinal development and is regulated by the tumor suppressor adenomatous polyposis coli. J Biol Chem. 2005;280(34):30490–5. doi: 10.1074/jbc.M504973200. [DOI] [PubMed] [Google Scholar]
- 93.Jette C, et al. The tumor suppressor adenomatous polyposis coli and caudal related homeodomain protein regulate expression of retinol dehydrogenase L. J Biol Chem. 2004;279(33):34397–405. doi: 10.1074/jbc.M314021200. [DOI] [PubMed] [Google Scholar]
- 94.Sandoval IT, et al. A metabolic switch controls intestinal differentiation downstream of Adenomatous polyposis coli (APC) Elife. 2017:6. doi: 10.7554/eLife.22706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cheng H, Leblond CP. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am J Anat. 1974;141(4):537–61. doi: 10.1002/aja.1001410407. [DOI] [PubMed] [Google Scholar]
- 96.Rosin-Arbesfeld R, Ihrke G, Bienz M. Actin-dependent membrane association of the APC tumour suppressor in polarized mammalian epithelial cells. EMBO J. 2001;20(21):5929–39. doi: 10.1093/emboj/20.21.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kawasaki Y, et al. Asef, a link between the tumor suppressor APC and G-protein signaling. Science. 2000;289(5482):1194–7. doi: 10.1126/science.289.5482.1194. [DOI] [PubMed] [Google Scholar]
- 98.Watanabe T, et al. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. 2004;7(6):871–83. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 99.Sudhaharan T, et al. Rho GTPase Cdc42 is a direct interacting partner of adenomatous polyposis coli protein and can alter its cellular localization. PLoS One. 2011;6(2):e16603. doi: 10.1371/journal.pone.0016603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moseley JB, et al. Regulated binding of adenomatous polyposis coli protein to actin. J Biol Chem. 2007;282(17):12661–8. doi: 10.1074/jbc.M610615200. [DOI] [PubMed] [Google Scholar]
- 101.Okada K, et al. Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J Cell Biol. 2010;189(7):1087–96. doi: 10.1083/jcb.201001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Juanes MA, et al. Adenomatous polyposis coli nucleates actin assembly to drive cell migration and microtubule-induced focal adhesion turnover. J Cell Biol. 2017;216(9):2859–2875. doi: 10.1083/jcb.201702007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aberle H, Schwartz H, Kemler R. Cadherin-catenin complex: Protein interactions and their implications for cadherin function. J Cell Biochem. 1996;61(4):514–23. doi: 10.1002/(SICI)1097-4644(19960616)61:4%3C514::AID-JCB4%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 104.Faux MC, et al. Restoration of full-length adenomatous polyposis coli (APC) protein in a colon cancer cell line enhances cell adhesion. J Cell Sci. 2004;117(Pt 3):427–39. doi: 10.1242/jcs.00862. [DOI] [PubMed] [Google Scholar]
- 105.Hulsken J, Birchmeier W, Behrens J. E-cadherin and APC compete for the interaction with beta-catenin and the cytoskeleton. J Cell Biol. 1994;127(6 Pt 2):2061–9. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nelson SA, et al. Tumorigenic fragments of APC cause dominant defects in directional cell migration in multiple model systems. Dis Model Mech. 2012;5(6):940–7. doi: 10.1242/dmm.008607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Matsumine A, et al. Binding of APC to the human homolog of the Drosophila discs large tumor suppressor protein. Science. 1996;272(5264):1020–3. doi: 10.1126/science.272.5264.1020. [DOI] [PubMed] [Google Scholar]
- 108.Takizawa S, et al. Human Scribble, a novel tumor suppressor identified as a target of high-risk HPV E6 for ubiquitin-mediated degradation, interacts with adenomatous polyposis coli. Genes Cells. 2006;11(4):453–64. doi: 10.1111/j.1365-2443.2006.00954.x. [DOI] [PubMed] [Google Scholar]
- 109.Nathke IS, et al. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol. 1996;134(1):165–79. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Munemitsu S, et al. The APC gene product associates with microtubules in vivo and promotes their assembly in vitro. Cancer Res. 1994;54(14):3676–81. [PubMed] [Google Scholar]
- 111.Zumbrunn J, et al. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr Biol. 2001;11(1):44–9. doi: 10.1016/s0960-9822(01)00002-1. [DOI] [PubMed] [Google Scholar]
- 112.Mogensen MM, et al. The adenomatous polyposis coli protein unambiguously localizes to microtubule plus ends and is involved in establishing parallel arrays of microtubule bundles in highly polarized epithelial cells. J Cell Biol. 2002;157(6):1041–8. doi: 10.1083/jcb.200203001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Su LK, et al. APC binds to the novel protein EB1. Cancer Res. 1995;55(14):2972–7. [PubMed] [Google Scholar]
- 114.Iizuka-Kogo A, Shimomura A, Senda T. Colocalization of APC and DLG at the tips of cellular protrusions in cultured epithelial cells and its dependency on cytoskeletons. Histochem Cell Biol. 2005;123(1):67–73. doi: 10.1007/s00418-004-0729-2. [DOI] [PubMed] [Google Scholar]
- 115.Kroboth K, et al. Lack of adenomatous polyposis coli protein correlates with a decrease in cell migration and overall changes in microtubule stability. Mol Biol Cell. 2007;18(3):910–8. doi: 10.1091/mbc.E06-03-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mili S, Moissoglu K, Macara IG. Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature. 2008;453(7191):115–9. doi: 10.1038/nature06888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marshall TW, et al. The tumor suppressor adenomatous polyposis coli controls the direction in which a cell extrudes from an epithelium. Mol Biol Cell. 2011;22(21):3962–70. doi: 10.1091/mbc.E11-05-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bellis J, et al. The tumor suppressor Apc controls planar cell polarities central to gut homeostasis. J Cell Biol. 2012;198(3):331–41. doi: 10.1083/jcb.201204086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mahmoud NN, et al. Apc gene mutation is associated with a dominant-negative effect upon intestinal cell migration. Cancer Res. 1997;57(22):5045–50. [PubMed] [Google Scholar]
- 120.Wong MH, et al. Forced expression of the tumor suppressor adenomatosis polyposis coli protein induces disordered cell migration in the intestinal epithelium. Proc Natl Acad Sci U S A. 1996;93(18):9588–93. doi: 10.1073/pnas.93.18.9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim KP, et al. Paired Primary and Metastatic Tumor Analysis of Somatic Mutations in Synchronous and Metachronous Colorectal Cancer. Cancer Res Treat. 2017;49(1):161–167. doi: 10.4143/crt.2015.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Leung ML, et al. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 2017;27(8):1287–1299. doi: 10.1101/gr.209973.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brabletz T, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A. 2001;98(18):10356–61. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bracke ME, Van Roy FM, Mareel MM. The E-cadherin/catenin complex in invasion and metastasis. Curr Top Microbiol Immunol. 1996;213( Pt 1):123–61. doi: 10.1007/978-3-642-61107-0_9. [DOI] [PubMed] [Google Scholar]
- 125.Jamora C, et al. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature. 2003;422(6929):317–22. doi: 10.1038/nature01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Conacci-Sorrell M, et al. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J Cell Biol. 2003;163(4):847–57. doi: 10.1083/jcb.200308162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gradl D, Kuhl M, Wedlich D. The Wnt/Wg signal transducer beta-catenin controls fibronectin expression. Mol Cell Biol. 1999;19(8):5576–87. doi: 10.1128/mcb.19.8.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hlubek F, et al. Expression of the invasion factor laminin gamma2 in colorectal carcinomas is regulated by beta-catenin. Cancer Res. 2001;61(22):8089–93. [PubMed] [Google Scholar]
- 129.Crawford HC, et al. The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene. 1999;18(18):2883–91. doi: 10.1038/sj.onc.1202627. [DOI] [PubMed] [Google Scholar]
- 130.Brabletz T, et al. beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol. 1999;155(4):1033–8. doi: 10.1016/s0002-9440(10)65204-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Takahashi M, et al. Identification of membrane-type matrix metalloproteinase-1 as a target of the beta-catenin/Tcf4 complex in human colorectal cancers. Oncogene. 2002;21(38):5861–7. doi: 10.1038/sj.onc.1205755. [DOI] [PubMed] [Google Scholar]
- 132.Gavert N, et al. Expression of L1-CAM and ADAM10 in human colon cancer cells induces metastasis. Cancer Res. 2007;67(16):7703–12. doi: 10.1158/0008-5472.CAN-07-0991. [DOI] [PubMed] [Google Scholar]
- 133.Mann B, et al. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci U S A. 1999;96(4):1603–8. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wielenga VJ, et al. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol. 1999;154(2):515–23. doi: 10.1016/S0002-9440(10)65297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Conacci-Sorrell ME, et al. Nr-CAM is a target gene of the beta-catenin/LEF-1 pathway in melanoma and colon cancer and its expression enhances motility and confers tumorigenesis. Genes Dev. 2002;16(16):2058–72. doi: 10.1101/gad.227502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gavert N, et al. L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol. 2005;168(4):633–42. doi: 10.1083/jcb.200408051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Batlle E, et al. β-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111(2):251–63. doi: 10.1016/s0092-8674(02)01015-2. [DOI] [PubMed] [Google Scholar]
- 138.Vignjevic D, et al. Fascin, a novel target of beta-catenin-TCF signaling, is expressed at the invasive front of human colon cancer. Cancer Res. 2007;67(14):6844–53. doi: 10.1158/0008-5472.CAN-07-0929. [DOI] [PubMed] [Google Scholar]
- 139.Hlubek F, et al. Heterogeneous expression of Wnt/beta-catenin target genes within colorectal cancer. Int J Cancer. 2007;121(9):1941–8. doi: 10.1002/ijc.22916. [DOI] [PubMed] [Google Scholar]
- 140.Todaro M, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. 2014;14(3):342–56. doi: 10.1016/j.stem.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 141.Zilberberg A, Lahav L, Rosin-Arbesfeld R. Restoration of APC gene function in colorectal cancer cells by aminoglycoside- and macrolide-induced read-through of premature termination codons. Gut. 2010;59(4):496–507. doi: 10.1136/gut.2008.169805. [DOI] [PubMed] [Google Scholar]
- 142.Virmani AK, et al. Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin Cancer Res. 2001;7(7):1998–2004. [PubMed] [Google Scholar]
- 143.Macnab SA, et al. Herpesvirus saimiri-mediated delivery of the adenomatous polyposis coli tumour suppressor gene reduces proliferation of colorectal cancer cells. Int J Oncol. 2011;39(5):1173–81. doi: 10.3892/ijo.2011.1130. [DOI] [PubMed] [Google Scholar]
- 144.Sansom OJ, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446(7136):676–9. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 145.Reed KR, et al. B-catenin deficiency, but not Myc deletion, suppresses the immediate phenotypes of APC loss in the liver. Proc Natl Acad Sci U S A. 2008;105(48):18919–23. doi: 10.1073/pnas.0805778105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wilkins JA, Sansom OJ. C-Myc is a critical mediator of the phenotypes of Apc loss in the intestine. Cancer Res. 2008;68(13):4963–6. doi: 10.1158/0008-5472.CAN-07-5558. [DOI] [PubMed] [Google Scholar]
- 147.Schepers AG, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337(6095):730–5. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 148.Garber K. Drugging the Wnt pathway: Problems and progress. J Natl Cancer Inst. 2009;101(8):548–50. doi: 10.1093/jnci/djp084. [DOI] [PubMed] [Google Scholar]
- 149.Shitashige M, et al. Traf2- and Nck-interacting kinase is essential for Wnt signaling and colorectal cancer growth. Cancer Res. 2010;70(12):5024–33. doi: 10.1158/0008-5472.CAN-10-0306. [DOI] [PubMed] [Google Scholar]
- 150.Lu B, et al. Wnt Drug Discovery: Weaving Through the Screens, Patents and Clinical Trials. Cancers (Basel) 2016;8(9) doi: 10.3390/cancers8090082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vincan E, Barker N. The upstream components of the Wnt signalling pathway in the dynamic EMT and MET associated with colorectal cancer progression. Clin Exp Metastasis. 2008;25(6):657–63. doi: 10.1007/s10585-008-9156-4. [DOI] [PubMed] [Google Scholar]
- 152.Chen B, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5(2):100–7. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Voloshanenko O, et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun. 2013;4:2610. doi: 10.1038/ncomms3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu J, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110(50):20224–9. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Madan B, et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene. 2016;35(17):2197–207. doi: 10.1038/onc.2015.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gurney A, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109(29):11717–22. doi: 10.1073/pnas.1120068109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Fischer MM, et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci Adv. 2017;3(6):e1700090. doi: 10.1126/sciadv.1700090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Thorne CA, et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat Chem Biol. 2010;6(11):829–36. doi: 10.1038/nchembio.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Huang SM, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461(7264):614–20. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 160.Waaler J, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012;72(11):2822–32. doi: 10.1158/0008-5472.CAN-11-3336. [DOI] [PubMed] [Google Scholar]
- 161.Lau T, et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013;73(10):3132–44. doi: 10.1158/0008-5472.CAN-12-4562. [DOI] [PubMed] [Google Scholar]
- 162.Larriba MJ, et al. Vitamin D receptor deficiency enhances Wnt/beta-catenin signaling and tumor burden in colon cancer. PLoS One. 2011;6(8):e23524. doi: 10.1371/journal.pone.0023524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Palmer HG, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154(2):369–87. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shah S, et al. The molecular basis of vitamin D receptor and beta-catenin crossregulation. Mol Cell. 2006;21(6):799–809. doi: 10.1016/j.molcel.2006.01.037. [DOI] [PubMed] [Google Scholar]
- 165.Lepourcelet M, et al. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004;5(1):91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 166.Emami KH, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] Proc Natl Acad Sci U S A. 2004;101(34):12682–7. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mahmoudi T, et al. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 2009;28(21):3329–40. doi: 10.1038/emboj.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shitashige M, et al. Regulation of Wnt signaling by the nuclear pore complex. Gastroenterology. 2008;134(7):1961–71. 1971e1–4. doi: 10.1053/j.gastro.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 169.Masuda M, et al. TNIK inhibition abrogates colorectal cancer stemness. Nat Commun. 2016;7:12586. doi: 10.1038/ncomms12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Morton JP, Myant KB, Sansom OJ. A FAK-PI-3K-mTOR axis is required for Wnt-Myc driven intestinal regeneration and tumorigenesis. Cell Cycle. 2011;10(2):173–5. doi: 10.4161/cc.10.2.14350. [DOI] [PubMed] [Google Scholar]
- 171.Issa JP. Colon cancer: It’s CIN or CIMP. Clin Cancer Res. 2008;14(19):5939–40. doi: 10.1158/1078-0432.CCR-08-1596. [DOI] [PubMed] [Google Scholar]
- 172.Shen L, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 2007;104(47):18654–9. doi: 10.1073/pnas.0704652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Akiyama Y, et al. Transforming growth factor beta type II receptor gene mutations in adenomas from hereditary nonpolyposis colorectal cancer. Gastroenterology. 1997;112(1):33–9. doi: 10.1016/s0016-5085(97)70216-6. [DOI] [PubMed] [Google Scholar]
- 174.Calin GA, et al. Genetic progression in microsatellite instability high (MSI-H) colon cancers correlates with clinico-pathological parameters: A study of the TGRbetaRII, BAX, hMSH3, hMSH6, IGFIIR and BLM genes. Int J Cancer. 2000;89(3):230–5. [PubMed] [Google Scholar]
- 175.Peltomaki P. Lynch syndrome genes. Fam Cancer. 2005;4(3):227–32. doi: 10.1007/s10689-004-7993-0. [DOI] [PubMed] [Google Scholar]
- 176.Deng G, et al. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10(1 Pt 1):191–5. doi: 10.1158/1078-0432.ccr-1118-3. [DOI] [PubMed] [Google Scholar]