

SUMMARY
In the active HER receptor dimers, kinases play distinct roles; one is the catalytically active kinase and the other is its allosteric activator. This specialization enables signaling by the catalytically inactive HER3, which functions exclusively as an allosteric activator upon heterodimerization with other HER receptors. It is unclear whether the allosteric activation mechanism evolved before HER receptors functionally specialized. We determined the crystal structure of the kinase domain of the only EGF receptor in C. elegans, LET-23. Our structure of a non-human EGFR kinase reveals autoinhibitory features conserved in the human counterpart. Strikingly, mutations within the putative allosteric dimer interface abrogate activity of the isolated LET-23 kinase and of the full-length receptor despite these regions being only partially conserved with human EGFR. Our results indicate that ancestral EGFRs have built-in features that poise them for allosteric activation that could facilitate emergence of the catalytically dead, yet functional, orthologs.
Keywords: LET-23, EGFR, HER receptors, ERBB receptors, receptor tyrosine kinase signaling, kinase activation, asymmetric dimerization, allosteric kinase activation, kinase structure
eTOC BLURB
Liu et al. present the crystal structure of a non-human EGFR kinase, C. elegans LET-23. Their findings offer insights into the regulation of ancestral EGFRs, demonstrating that LET-23 is activated allosterically by oligomerization, similarly to human EGFR, but through a mechanism that is likely structurally distinct.
INTRODUCTION
Receptor tyrosine kinases (RTKs) play fundamental roles in cellular signaling by coupling extracellular signals to intracellular tyrosine phosphorylation (Lemmon and Schlessinger, 2010). RTK activation relies on ligand binding to the extracellular regions, which triggers conformational transitions in the receptor resulting in formation of a complex that supports catalysis of tyrosine phosphorylation. Although mechanistic details of the activation process are yet to be fully understood for each RTK, some general principles have emerged. For example, receptor oligomerization is typically a necessary step in the activation process. In addition, in most RTKs full activation requires phosphorylation of tyrosines in the activation loop, which stabilizes the active conformation of the kinase domain (Lemmon and Schlessinger, 2010).
The family of human epidermal growth factor receptor (EGFR/HER/ERBB) tyrosine kinases, which includes EGFR, HER2, HER3 and HER4, engages a quite distinct activation mechanism that so far has not been observed in other kinases. Activation of these RTKs is phosphorylation-independent and involves allosteric activation of one kinase by another through formation of an asymmetric kinase domain dimer. In this dimer, the C-lobe of one kinase domain (the “activator”) binds to the N-lobe of the other kinase domain (the “receiver”) allosterically stabilizing the active conformation of the latter (Zhang et al., 2006). The asymmetric kinase domain interface is also defined by interactions between a portion of the intracellular juxtamembrane segment of the receiver kinase and the C-lobe of the activator kinase (Jura et al., 2009a; Red Brewer et al., 2009). In this asymmetric configuration, the activator kinase does not have to be catalytically active to trigger conformational changes in the receiver kinase (Zhang et al., 2006). This is a key distinction from kinase activation achieved by trans-phosphorylation in active receptor complexes where both receptors need to be catalytically competent.
It is unknown why catalytic and non-catalytic functions of the kinase domain in active HER receptor complexes are separated. One advantage of such specialization is the ability to incorporate a catalytically inactive receptor into functional signaling complexes. The human EGFR family does have such a member, HER3, which has severely impaired catalytic activity that has not yet been linked to a biological function, and hence it is categorized as a pseudokinase (Mendrola et al., 2013; Murphy et al., 2014; Shi et al., 2010). HER3 carries mutations in two crucial catalytic residues: the glutamate in helix C, which forms a salt bridge with the catalytic lysine on the β3 strand to ensure proper ATP coordination in the active site, and the catalytic base aspartate in the conserved HRD motif. In addition, HER3 has several inactivating substitutions in the receiver interface, located in the kinase N-lobe, that prevent its allosteric activation in an asymmetric dimer. These mutations render HER3 incapable of self-activation, but do not affect its ability to interact with other HER receptors in the activator position and allosterically activate them (Jura et al., 2009b).
It is unclear if evolution of catalytically inactive HER receptors coincided with the development of the allosteric mechanism. The EGFR family is present in almost all Metazoa, including two earliest branching animal clades: ctenophores and sponges (Suga et al., 2012). HER3 orthologs on the other hand are only found in higher vertebrates, which have up to four classes of HER receptors (including EGFR, HER2, HER3 and HER4). In invertebrates, the EGFR family is much more diverse, spanning from one EGF receptor in nematodes (i.e. C. elegans) and arthropods (i.e. D. melanogaster) to up to 8 EGF receptors in plathyleminthes (i.e. M. lignano) (Barberan et al., 2016). The relationships between these receptors have thus far not been studied. Interestingly, EGF receptors that can be categorized as pseudokinases based on sequence have been found in at least two invertebrate clades, raising questions about their mechanisms of signaling (Barberan et al., 2016).
To understand the origin of the allosteric activation mechanism of the human EGF receptors, in this study we focused on an invertebrate EGFR ortholog in a nematoda clade, represented by LET-23 in C. elegans, the sole EGFR present in this species. By analyzing sequences of the EGFR kinase domains from different invertebrate and vertebrate clades, we show that LET-23 is unlike other ancestral receptors and does not share most of the canonical sequence motifs that support the allosteric activation mechanism described for human EGF receptors. However, by determining the crystal structure of the LET-23 tyrosine kinase domain (LET-23KD) in the inactive state, we show that autoinhibitory interactions within the kinase domain and the C-terminal tail are remarkably conserved between LET-23 and human EGFR. Like human EGFR, LET-23 is also inactive in solution, but its activity is significantly increased by concentrating it on the surface of lipid vesicles - implicating the involvement of intermolecular interactions in kinase activation. Although the activator and receiver interfaces are poorly conserved between LET-23 and human EGFR, we found that mutations in these interfaces blocked activation of LET-23 on lipid vesicle surfaces. Most importantly, these mutations also abrogated LET-23 signaling in cells in response to its ligand, LIN-3. In contrast to human EGFR however, signaling could not be restored through reconstitution of an asymmetric dimer. Our findings, taken together, suggest that LET-23 activation is dependent on allosteric interactions within kinase oligomers in a way that is qualitatively or quantitatively distinct from EGFR.
RESULTS
Sequence conservation of allosteric mechanism features across evolution
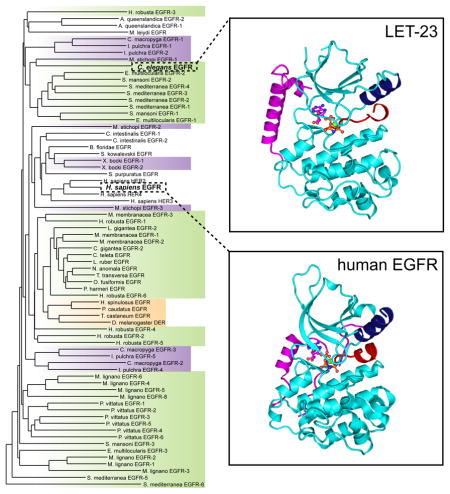
To assess the extent to which structural elements important for the allosteric mechanism are conserved across evolution, we analyzed kinase domain sequences from all fifteen metazoan clades in which EGFR family members have been found (Barberan et al., 2016). We focused on the EGFR genes assembled by Barberan and colleagues (Barberan et al., 2016) and removed genes in which the sequence of the kinase domain carried large deletions obstructing complete analysis of the kinase domain sequence. Based on these criteria, we selected 71 sequences representing EGFR kinase domains from 30 different species (Table S1) and aligned them using MUSCLE 3.7 (Edgar, 2004). We then performed phylogenetic analysis, using the phylogeny software PhyML 3.0 based on the maximum likelihood principle (Anisimova and Gascuel, 2006; Dereeper et al., 2008) (Figure 1).
Figure 1. Phylogenetic tree of EGFR kinase domain protein sequences in the animal kingdom.

The tree was assembled from 71 sequences representing EGFR kinase domains in 30 different species aligned using MUSCLE 3.7 (Edgar, 2004). The tree was built using the phylogeny software PhyML 3.0 based on the maximum likelihood principle (Anisimova and Gascuel, 2006; Dereeper et al., 2008). Degrees of protein sequence conservation in the regions corresponding to the allosteric activator interface (Activator), receiver interface (Receiver), and the juxtamembrane latch (JM latch) are indicated by different colors. EGF receptors predicted to be inactive are marked by boxes. See also Tables S1 and S2.
We focused on several structural elements that have been demonstrated as essential for the allosteric activation mechanism described for human EGF receptors. The first element, the activator interface, is defined as a network of hydrophobic residues at the bottom of the kinase C-lobe (Zhang et al., 2006). We considered this interface to be conserved (green “Activator” label in Figure 1) when all residues were either unchanged or were replaced by residues of similar chemical character (Table S2). We considered this interface partially conserved (grey “Activator” label in Figure 1) when there was only one substitution in the interface that significantly altered chemical properties, such as substitution of a hydrophobic residue with a charged residue. In all other instances, we considered this interface as not conserved. We observed that the activator interface in all analyzed EGFR kinases was 56% absolutely conserved and 21% partially conserved, demonstrating significant similarity of this interface across evolution. In all analyzed metazoan clades: Xenacoelomorpha, Ecdysoa, Spiralia, Deuterostomia, Porifera and Ctenophora, we could find at least one EGFR gene that fell into one of the two categories: conserved or partially conserved.
The analysis of the receiver interface located in the kinase domain N-lobe was less straightforward, and this interface displayed much higher sequence variation. Nevertheless, many substitutions preserved the chemical character of the residue found in the equivalent position in human EGFR. As a result, this interface seems to be at least partially conserved in all analyzed species, and highly conserved (blue “Receiver” label in Figure 1) in 18% of species (Table S2).
We next examined the juxtamembrane latch, a segment preceding the N-terminal end of the kinase domain. This feature plays an essential role in the activation of human EGF receptors by binding to the C-terminal lobe of the dimerization partner to stabilize the asymmetric kinase domain dimer (Jura et al., 2009a; Littlefield et al., 2014; Red Brewer et al., 2009). Using criteria analogous to those applied to the sequence analysis of the activator and receiver interfaces, we established that the juxtamembrane latch is significantly less well conserved, with only 19% of the analyzed EGFR genes showing full conservation (magenta “JM latch” label in Figure 1) and 13% partial conservation (grey “JM latch” label in Figure 1). This feature was present in the majority of clades belonging to Deuterostomia, Ecdysoa and Spiralia. The corresponding sequence was either poorly or not at all conserved in the analyzed species from the Xenacoelomorpha, Porifera and Ctenophora clades.
Given that the role of activation loop tyrosine phosphorylation in the regulation of human EGFR still remains unclear, it was interesting to observe that this residue is highly conserved throughout evolution (in 65% of all sequences (46 of 71 sequences)) (Figure 1). The activation loop tyrosine was present in the same position as in human EGFR in 41 sequences (yellow “Y” label in Figure 1), and displaced by only a few residues in the remaining 5 sequences (grey “Y” label in Figure 1). In some organisms, such as M. lignano 1, M. lignano 2, M. lignano 8 or M. stichopi 1, the activation loop tyrosine was present even in the absence of the structural elements defined as hallmarks of allosteric EGFR activation (the activator interface and the juxtamembrane latch). However, in the majority of sequences the presence of the activation loop tyrosine was concomitant with the conservation of these structural elements. This likely indicates an essential role of the activation loop tyrosine in regulating EGFR signaling in these organisms.
In light of the degree of evolutionary conservation of structural elements involved in the allosteric activation of human EGF receptors, we hypothesized that the unique activation mechanism described for the human receptors might also operate outside vertebrates. Similar composition of the EGFR family in H. sapiens and in invertebrate species such as S. mediterranea, namely the presence of multiple EGF receptors and members predicted to be catalytically inactive (Table S2), suggests that the allosteric mechanism may mediate receptor cross-activation in these species as well. In an effort to understand whether evolution of the allosteric mechanism preceded expansion of the EGFR family to include multiple members (and the emergence of pseudokinase receptors), we chose to further investigate the single EGF receptor in C. elegans, LET-23. As shown in Figure 1 (and Table S3), LET-23 lacks most of the hallmarks of allosteric EGFR regulation. It carries several substitutions at hydrophobic residues in the activator interface (such as V924E, M921Q and I917Q in human EGFR numbering) that are predicted to significantly destabilize this interface (Zhang et al., 2006). LET-23 also has no identifiable juxtamembrane latch sequence and lacks both the conserved receiver interface and the activation loop tyrosine, yet motifs important for catalysis (DFG and HRD motifs) are conserved (Figure 1). Accordingly, LET-23 represents a good EGFR model system for an active kinase in which an alternative mode of regulation might be anticipated.
Structural analysis of the C. elegans EGF receptor, LET-23
To initiate studies of LET-23 regulation, we determined the crystal structure of its kinase domain flanked by a 33 residue-long portion of the C-terminal tail (residues 866 – 1191). To our knowledge, this is the first structure determined for a non-human EGFR family kinase domain. Crystals grown in the presence of Mg2+ ions and the nonhydrolyzable ATP analogue, AMP-PNP, diffracted to 2.4 Å resolution (Table 1). The asymmetric unit contains one copy of the LET-23 kinase domain bound to a single AMPPNP and Mg2+ ion and adopts a conformation frequently referred to as the Src/CDK-like conformation observed in the inactive structures of the human EGFR kinase (Figure 2A), as well as the inactive HER3 pseudokinase and the inactive HER4 kinase (Figure S1). In this conformation, helix C is swung away from the active site, disrupting a catalytically crucial salt bridge between the catalytic lysine (K919 in LET-23) and a glutamate on helix C (E934 in LET-23) (Figure 2B). The activation loop (residues 1028 – 1048) in LET-23 is largely disordered with the exception of the N-terminal fragment encompassing the DFG motif (residues 1028 – 1035). The ordered region of the activation loop forms a short helical turn observed in many other kinases adopting the Src/CDK-like inactive conformation, including human EGFR kinase (Huse and Kuriyan, 2002; Wood et al., 2004) (Figure 2A). In these structures, the central glutamate in helix C forms an ionic interaction with an arginine or lysine residue in the N-terminal portion of the activation loop. In LET-23, this interaction is seemingly conserved, however, the activation loop lysine (K1033) interacts with a more peripheral glutamate residue in helix C (E930) (Figure 2B). The presence of an AMP-PNP molecule and one Mg2+ ion is not an uncommon feature of kinases crystallized in the Src/CDK-like inactive conformation, and has previously been proposed to represent an intermediate step during catalysis in active kinases (Shan et al., 2009). Overall, the LET-23 kinase domain demonstrates remarkable sequence and structural conservation of autoinhibitory features between human and worm EGFRs.
Table 1.
Crystallographic data collection and refinement statistics
| Data Collection | |
|---|---|
| Beamline | ALS 8.3.1 |
| Wavelength (Å) | 1.11 |
| Space group | P 21 21 21 |
| Cell dimensions | |
| a, b, c (Å) | 48.1, 78.0, 106.2 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution (Å) | 63 – 2.4 (2.45 – 2.4)a |
| Rmergeb (%) | 10.3 (89.0) |
| <I>/<σ>c | 10.8 (1.1) |
| Completeness (%) | 98.8 (98.5) |
| Refinement | |
| No. of reflections | 16,260 |
| Rworkc (%) | 21.5 |
| Rfreed (%) | 24.7 |
| No. of atoms | |
| Protein | 2441 |
| Ligand/ion | 32 |
| Water | 11 |
| RMSD | |
| Bond lengths (Å) | 0.002 |
| Bond angle (°) | 0.45 |
| Ramachandran plot (%) | |
| Favored | 97.0 |
| Allowed | 3.0 |
| Outliers | 0.0 |
Values in parentheses are for the highest resolution shell.
Rmerge= Σhkl Σj | I j − 〈 I 〉 |/Σhkl ΣIj, where j is the jth measurement and <I> is the weighted mean of I.
<I>/<σ> is the mean intensity divided by the mean error.
Rwork= Σhkl || Fo | − k | Fc ||/Σhkl | Fo |, where Fo and Fc are the observed and calculated structure factor amplitudes, and k is a weighting factor.
Rfree is the same as Rwork calculated on 5% of the reflections.
Figure 2. Crystal structure of the LET-23 kinase domain.

(A) The structure of the inactive C. elegans LET-23 kinase domain in complex with AMP-PNP compared to the structures of the inactive human EGFR V924R kinase in complex with AMP-PNP (PDB ID: 5CNO) and active human EGFR kinase in complex with an ATP analog (PDB ID: 2GS6). (B) Detailed view of structural elements in the N-lobe of the LET-23 kinase domain overlaid on the structure of the inactive human EGFR V924R kinase domain (PDB ID: 5CNO) highlighting features of the C helix interaction with the activation loop. (C) Comparison of C-tail binding modes in inactive human EGFR V924R kinase domains (PDB ID: 5CNO) (left panel), active human EGFR kinase domain (PDB ID: 2GS6), and inactive LET-23 kinase domain. (D) Detailed view of interactions between the C-tail and the hinge region in the crystal structures of LET-23 and inactive human EGFR V924R kinase domains (PDB ID: 5CNO). (E) The left panel shows two LET-23 kinases interacting via the C-terminal tail/C-lobe interface observed in the crystal lattice. The two right panels highlight interactions in the C-lobe interface in LET-23 (middle panel) and EGFR (right panel) with the C-tail in LET-23 or Mig6 in EGFR (PDB ID: 2RFE). See also Figure S1.
Another structural feature conserved between human HER receptors and LET-23 is the presence of an intramolecular interaction between the back of the kinase domain and the beginning of the receptor’s C-terminal tail (C-tail) (Figure 2C). Similar interactions have been observed in both active and inactive structures of EGFR kinase and in active structure of HER2 (Aertgeerts et al., 2011; Jura et al., 2009a; Stamos et al., 2002). In the inactive state in EGFR, these C-tail-mediated interactions (Figure 2C, left panel) have been implicated as important for negative regulation of kinase activity through restriction of the conformational dynamics of the hinge region, and through direct overlap between the C-tail binding to the region in the C-lobe that binds the juxtamembrane segment in the active state of the receptor (Jura et al., 2009a; Kovacs et al., 2015). The latter is accomplished by a 180° turn of the C-tail at the hinge region that directs the tail back towards the C-lobe where the juxtamembrane segment binding site is located.
The functional importance of the C-tail binding observed in the active structures of EGFR and HER2 kinase domains (Aertgeerts et al., 2011; Jura et al., 2009a; Kovacs et al., 2015; Stamos et al., 2002; Wood et al., 2004) is less understood. In these structures the trajectory of the C-tail is shifted to the right of the hinge region and continues towards the N-lobe, leaving the juxtamembrane segment binding site exposed (Figure 2C, middle panel). In the majority of the active structures, the portion of the C-tail in the proximity of the hinge region also tends to be poorly resolved. Somewhat surprisingly, because the LET-23 kinase is in the inactive conformation, the binding of the C-tail in LET-23 is more reminiscent of how the C-tail interacts with the EGFR kinase domain in the active conformation (Stamos et al., 2002) (Figure 2C, right panel). In both activated EGFR and inactive LET-23, the C-tails extend towards the N-lobe, and then take a similar turn underneath the β2-β3 loop, continuing towards the N-lobe region defined previously as the AP-2 pocket. The exposure of the juxtamembrane segment binding site in the inactive LET-23 could indicate that this regulatory mechanism is lacking in the worm receptor, which is further supported by the lack of conservation between LET-23 and EGFR juxtamembrane segment sequences (Figure 1, Table S2). Despite the difference in C-tail orientations, the unifying feature of the inactive EGFR and LET-23 is the presence of a network of interactions between the C-tail and the kinase hinge region, which are predicted to be inhibitory and are lacking in the active EGFR structures (Figure 2D).
One possibility for the observed differences in the C-tail orientation is the effect of crystal packing in the structure of LET-23 in which the C-tail of one kinase engages the C-lobe of another kinase in the crystal lattice (Figure 2E – left panel). What is intriguing about this crystal packing is that the C-tail interaction with the activator interface in LET-23 mimics how the negative regulator MIG6 binds to EGFR (Zhang et al., 2007) (Figure 2E). This interaction mode has also been observed for a more distal portion of the C-tail of EGFR in a recent inactive structure of the EGFR kinase (Kovacs et al., 2015). Binding of both MIG6 and the C-tail to the activator interface in EGFR has been suggested to restrict EGFR activity by competing with the formation of the active asymmetric dimer. The MIG6-like motif is also present in the ACK1 kinase, another protein interaction partner of EGFR, predicted to bind to EGFR’s activator interface (Shen et al., 2007). The observation that LET-23 engages the activator interface to mediate protein-protein interactions in the crystal lattice may point to the importance of this binding site in LET-23 for signaling.
Catalytic activity of the LET-23 kinase domain in solution and associated with lipid vesicles
The isolated human EGFR kinase domain is monomeric in solution, and its catalytic activity has been shown to be intrinsically autoinhibited (Shan et al., 2012; Zhang et al., 2006). We measured the activity of the LET-23 kinase domain in solution toward a generic substrate peptide, poly-4Glu:Tyr, and found that its basal activity at 1 μM is also almost undetectable – even lower than the activity of the human EGFR kinase domain at the same concentration (Figure 3A). The activity of the human EGFR kinase is substantially stimulated through dimerization, and this can be mimicked in vitro by concentrating the histidine-tagged kinase on the surface of small unilamellar vesicles that contain lipids with a nickel-nitrilotriacetate salt head group (DOGS-NTA-Ni) (Zhang et al., 2006). Because the LET-23 kinase domain (LET-23KD), like human EGFR, purifies as a monomeric kinase, we anticipated that its activity might also be stimulated by concentration on vesicle surfaces where intramolecular interactions between protein monomers leading to allosteric activation can occur. Indeed, wild-type LET-23KD attached to DOGS-NTA-Ni containing vesicles displayed a significantly increased catalytic rate relative to that observed for the kinase domain free in solution (Figure 3A). Increasing the density of DOGS-NTA-Ni lipids in the vesicles from 0.5 to 5.0 mole percent, thus increasing the effective local concentration of LET-23KD from 0.4 mM to 4 mM at the vesicle surface, further elevated the activity (Figure 3B). This result indicates that, as with human EGFR, intermolecular interactions are important for kinase activation in LET-23. In many tyrosine kinases, concentration-dependent activation is a result of trans-phosphorylation of the activation loop on a tyrosine (Huse and Kuriyan, 2002). LET-23 has no such tyrosine phosphorylation site in its activation loop (Figure 3C), suggesting that the observed concentration-dependent increase in kinase activity likely arises through the allosteric effects of intermolecular interactions within kinase oligomers stabilized on the vesicle surfaces.
Figure 3. Catalytic activity of the LET-23 kinase domain in solution and associated with lipid vesicles.

(A) Specific activity of the LET-23 and human EGFR kinase domain, each at 1 μM, in solution (light grey) and linked to vesicles (dark grey) containing 5 mole percent DOGS-NTA-Ni. (B) Concentration-dependent activation of the LET-23 kinase domain upon association with lipid vesicles. The specific activities of the kinase domain in solution or on vesicles containing increasing mole percentages of DOGS-NTA-Ni are shown. In all measurements, the ratio of protein to total lipids in bulk solution was fixed at 1 μM and 12.5 μM, respectively. Data are represented as mean ± S.D. (C) Alignment of the activation loop sequences of LET-23 and the human EGFR kinase domain.
Model of the altered asymmetric dimer interface in LET-23
Residues of the activator C-lobe and the receiver N-lobe comprising the asymmetric kinase dimerization interface described for human EGFR (and responsible for its allosteric activation) are not strictly conserved in LET-23. Despite a lack of sequence identity, the majority of residues forming the C-lobe activator interface are still hydrophobic in nature in the LET-23 kinase domain, with the exception of three residues in human EGFR (I917, M921 and V956) that are replaced in LET-23 by polar (Q1114 and Q1118) or charged (E1153) residues (Figure 4A, top panel). Analogous mutations in human EGFR have been shown to impair kinase activation and receptor signaling significantly (Zhang et al., 2006). Notably, the residue equivalent to V924 in human EGFR, the mutation of which to a charged residue most potently disrupts the activator interface, is also a hydrophobic residue in LET-23 (L1121) (Figure 2E). While the activator interface in LET-23 is still partially conserved with human EGFR, the amino acid composition of the LET-23 N-lobe receiver interface is substantially different (Figure 4B, top panel). Since the architecture of the receiver interface in our structure of the LET23 kinase domain is in the autoinhibited state (Figure 2A, B), it is difficult to predict with confidence whether the LET-23 N-lobe architecture in the active state could function as a receiver interface to support asymmetric dimerization as is observed in the EGFR activation mechanism.
Figure 4. Structural analysis of the LET-23 asymmetric dimer model.

(A) Upper panel - sequence alignment of LET-23 and human EGFR kinase domain within the region containing residues involved in the activator interface. Residues in the activator interface are denoted by rectangles, which are blue if the residues are depicted on cartoon images in lower panels. Lower panels – detailed view of the asymmetric dimer interface in the human EGFR kinase dimer (PDB ID: 2GS6) (left panels), and in the homology model of the LET-23 asymmetric dimer (right panels). The key interfacial residues on the activator interface are shown in stick and dot representation. Residue E1153 in the LET-23 C-lobe was included in the original structure, but removed during generation of the homology model, likely due to unfavorable interactions with the LET-23 N-lobe in the asymmetric dimer model. (B) Upper panel - sequence alignment of LET-23 and human EGFR kinase domain within the region containing residues involved in the receiver interface. Residues in the activator interface are denoted by ovals, which are dark orange if the residues are depicted on cartoon images in lower panels. Lower panels – detailed view of the receiver interface of the active human EGFR kinase dimer (PDB ID: 2GS6) (left panels), and in the homology model of the LET-23 asymmetric dimer (right panels). The key interfacial residues on the activator interface are shown in stick and dot representation. The electrostatic surface potentials shown for the activator and receiver interfaces were calculated using APBS (Baker et al., 2001).
To better understand the capacity of the LET-23 kinase domain N-lobe to function as a receiver interface during allosteric activation, we used SWISS-MODEL Workspace (Arnold et al., 2006; Bordoli et al., 2009) to generate a homology model of the LET-23 kinase domain in the active conformation engaged in an EGFR-like dimer. To generate the model, we used the LET-23 kinase domain sequence and the model of the EGFR asymmetric dimer based on the crystal structure of the active state of the human EGFR kinase (PDB ID: 2GS6) as a structural template. In the LET-23 asymmetric dimer model, the receiver interface is now engaged at the dimer interface in an active conformation with comparable calculated buried surface areas (1979 Å2 for LET-23 and 2052 Å2 for EGFR) (Figure 4B, bottom panel). In both activator and receiver interfaces, the electrostatic surface of LET-23 overall is less hydrophobic than in EGFR, with the N-lobe receiver interface of LET-23 being most polar (Figure 4). These differences are predicted to destabilize asymmetric dimerization between LET-23 kinase domains in an EGFR-like manner, which relies largely on hydrophobic interactions. This weakened asymmetric interface may also account for the lack of dimer formation through the N-lobe/C-lobe interface in the LET-23 kinase domain crystal lattices typically observed for structures of catalytically active human EGF receptor kinases (EGFR, HER2 and HER4) (Aertgeerts et al., 2011; Qiu et al., 2008; Stamos et al., 2002).
Role of the asymmetric dimer interface in activation of LET-23KD
Our data show that activity of LET-23KD can be induced by concentration on lipid vesicle surfaces, pointing to oligomerization as a potential activation mechanism (Figure 3B). To test whether the increase in LET-23KD activity observed when concentrated on vesicle surfaces depends on dimerization through an asymmetric interface analogous to the mechanism employed by human EGFR, we generated LET-23KD variants with mutations equivalent to those that disrupt the asymmetric interface in human EGFR (Zhang et al., 2006). To mimic the commonly used V924R mutation in human EGFR, which blocks asymmetric dimerization by mutating a central residue in the hydrophobic activator interface (Figure 2E, middle panel), we generated the L1121R C-lobe mutant of LET-23KD. To mimic the I682Q mutation in the N-lobe of the human EGFR, which disrupts receiver function (Zhang et al., 2006), we generated L877Q LET-23KD. Interestingly, both of these mutations abolished vesicle-dependent elevation of LET-23 kinase activity to the same extent as mutating the crucial catalytic base aspartate in the LET-23 HRD motif to asparagine (D1010N) (Figure 5A).
Figure 5. Role of the asymmetric dimer interface in activation of the LET-23 kinase domain.

(A) Specific activity of the wild-type and mutant LET-23 kinase constructs at 1 μM, linked to vesicles containing 5 mole percent DOGS-NTA-Ni. (B) Specific activity measurements for the wild-type, L877Q, and L1121R constructs alone collected at 2 μM protein concentrations. In the L877Q/L1121R condition, each protein was present in the reaction mixture at 1 μM (2 μM total concentration). Data were collected on vesicles containing 5 mole percent DOGS-NTA-Ni. (C) Western blot analysis of LIN-3EGF-induced phosphorylation of full-length wild type of mutant LET-23 constructs upon transient transfection in COS-7 cells and immunoprecipitation with the anti-FLAG antibody. LIN-3EGF corresponds to the purified EGF domain from LIN-3, as previously described (Freed et al., 2015). (D) Specific activity of the wild-type and mutant LET-23 kinase constructs linked to vesicles containing 5 mole percent DOGS-NTA-Ni. (E) Hypothesized EGFR-like asymmetric dimer pairings between the wild type and mutant LET-23 kinases mixed at equimolar concentrations. Green check marks indicate activity, black crosses indicate no activity. Specific activity data are represented as mean ± S.D.
In the case of human EGFR, the I682Q and V924R variants are individually inactive, but can form an active heterodimer in which I682Q EGFR plays the activator role and V924R EGFR plays the receiver role to recover ~50% of the activity observed in wild-type EGFR alone (Zhang et al., 2006). We would expect that the activity of LET-23 could similarly be recovered on vesicles if asymmetric dimerization through the interface supporting EGFR activation was essential for activity. However, mixing the L877Q LET-23KD with its activator-impaired counterpart (the L1121R mutant) does not reconstitute activity to the same extent as has been observed for EGFR (Zhang et al., 2006) (Figure 5B). The most plausible explanation for these results is that the dimerization interface required for allosteric activation of the LET-23 kinase domain differs from that seen in human EGFR, and is perhaps not asymmetric in a way that allows for the complementation of interfaces observed in the human receptor. Alternatively, interactions in the LET23 allosteric interface may be significantly weaker than observed in human EGFR and therefore more difficult to complement at the concentrations of the isolated kinase domain achievable in our experiments.
The extracellular region of LET-23 has been shown to dimerize constitutively, however kinase activation is ligand-dependent implying that receptor activation upon extracellular ligand binding is driven primarily by conformational changes propagated within a pre-existing full-length receptor dimer (Freed et al., 2015). We considered the possibility that the weak complementation observed during concentration of the L877Q and L1121R mutants of isolated LET-23 kinase domains on vesicles (Figure 5B) is due to the lack of other receptor domains essential for activation and to the non-physiological properties of the reconstituted membrane system that may have prevented conformational transitions necessary for effective LET-23 activation. To address these limitations, we introduced the L1121R and L877Q mutations into full-length LET-23 and transiently expressed the individual variants either alone or together in COS7 cells. The LET-23 ligand, LIN-3, promotes robust activation of the wild-type receptor as assessed by anti-phosphotyrosine immunoblotting of immunoprecipitated receptor (Figure 5C). Both L1121R and L877Q mutations abolish ligand-dependent LET-23 activation as expected. However, consistent with the in vitro kinase assay data, no rescue of activity was detected when the L1121R and L877Q mutants were co-expressed together, further indicating an inherent difference in the mechanism of activation between LET-23 and human EGFR (Figure 5C).
Dimer complementation through asymmetric interactions with wild-type LET-23KD
One possible explanation for why we do not observe complementation in either the in vitro kinase assay or the cell-based assay for the L877Q and L1121R-mutated LET-23KD is that these residues participate in the formation of additional, and possibly structurally distinct, oligomeric assemblies that contribute to the regulation of activity in LET-23. To investigate this further, we examined the ability of the L877Q-mutated (receiver-impaired), L1121R-mutated (activator-impaired), and D1010N-mutated (kinase-dead) variants to 14 form active complexes with wild-type LET-23KD on the surface of lipid vesicles (Figure 5D). As shown in Figure 5D, neither the addition of the LET-23 N-lobe variant (L877Q) nor the kinase-dead variant (D1010N) had any influence on the activity of wild-type LET-23KD. Surprisingly, an equimolar quantity of the C-lobe mutant (L1121R) increased the kinase activity nearly 1.5-fold over 1 μM wild-type LET-23KD alone suggesting that the L1121R mutant engages the wild-type LET-23 kinase domain in a catalytically productive manner.
This partial rescue was puzzling and at present its functional significance is unclear, but we considered this result under the assumption that the activation mechanism of LET-23 might follow the EGFR-like activation mechanism (Figure 5E). In the context of an EGFRlike asymmetric dimer of LET-23, the presence of the L1121R mutation in the center of the activator kinase C-lobe (Figure 2E, right panel) introduces an electrostatically unfavorable basic arginine residue into the dimerization interface and is unlikely to interact with an unaltered receiver kinase N-lobe of wild-type or L1121R mutant of the LET23 kinase domain. In the opposite configuration, the unaltered C-lobe activator interface of wild-type LET-23 kinase would presumably be able to engage both the unaltered N-lobe of either the wild-type or L1121R variant of LET-23 in the receiver kinase position. Preference for the formation of the L1121Rreceiver/wild-typeactivator dimer pairings would likely be able to contribute to elevated activity as is observed in the presence of an increased total protein concentration (2 μM contributed by 1 μM wild-type and 1 μM L1121R LET-23KD).
Given the unique properties of the N- and C-lobe dimerization interfaces in the LET-23 kinase, we would not expect the same distribution of catalytically productive dimers in the wild-type/L877Q mutant mixture in the context of the EGFR-like asymmetric dimer (Figure 5E). In contrast to human EGFR in which the equivalent I682Q mutation alters a very hydrophobic interface in the N-lobe to block the ability of the kinase domain to engage in a dimerization interface as a receiver kinase, the wild-type LET-23 N-lobe is already more charged than in human EGFR kinase (Figure 4B). It is possible then that the L877Q variant of LET-23 can still engage an activator kinase in a non-active, but stable dimer. Introduction of the polar residue (L877Q) in the receiver interface might in fact improve the binding between these two interfaces given that the LET-23 C-lobe activator interface also contains at least three additional polar and charged residues (Q1114, Q1118, and E1153) (Figure 4A). The differences in the chemical nature of amino acids making up each interface may result in a failure to promote structural changes that underlie stabilization of the active conformation of the receiver kinase necessary for catalysis. If this is the case, then the preferential formation of L877Qreceiver/wild-typeactivator complexes may become trapped in a catalytically “disengaged” asymmetric dimer that would not contribute to an increase in kinase activity in the presence of increased total protein (Figure 5E), as is observed in our results (Figure 5D). Such “disengaged” asymmetric dimers have been previously seen in crystals structures of the catalytically dead human kinase, K721M EGFR (Red Brewer et al., 2009), and HER4 kinase bound to an inhibitor (Wood et al., 2008). Similarly, adding the catalytically-dead D1010N LET-23 variant would result in the formation of 50% of the complexes becoming trapped in catalytically inactive dimers in which the wild-type kinase in the activator position dimerizes with the D1010N mutant functioning as a receiver (Figure 5E). Thus no increase in activity would be expected when this variant is added, as is also the case in our experiments (Figure 5D).
This interpretation of the effects of mutations in the N- and C-lobe on LET-23 activation, under the assumption that it proceeds through an EGFR-like mechanism, is based on the idea that the LET-23 interface mutants are more likely to become trapped in non-functional asymmetric dimers than functional dimers. This reasoning offers an explanation for how LET-23 could still use the asymmetric dimerization activation mechanism observed in human EGFR, but its activity would not be rescued through complementation between the N- and C-lobe mutants. However, the chemical properties distinguishing the N- and C-lobe interfaces in LET-23 from human EGFR are predicted to significantly reduce the affinity for such interactions and, alternatively, may promote the formation of completely different oligomerization interfaces in LET-23.
DISCUSSION
The unique kinase activation mechanism seen in the vertebrate EGF receptor family incorporates both catalytic and non-catalytic functions of each kinase domain in a dimer, and allows signaling by the HER3 pseudokinase receptor in complexes with other HER family members. The presence of seemingly catalytically inactive EGFR variants in some invertebrate species (Figure 1), suggests that their phosphorylation and signaling might be also dependent on the ability to allosterically activate their catalytically active family members. At present, the evolutionary roots of the allosteric mechanism for EGFR activation remain unknown. Through analysis of EGFR kinase domain sequences across the animal kingdom, we observe significant conservation of the structural features important for allosteric activation, suggesting that this mechanism might control EGFR activation in both vertebrate and invertebrate species. Among all interfaces critical for human EGFR activation by asymmetric dimerization, the allosteric activator interface is the most conserved. In addition to playing a critical role in catalytic activation, this interface in human EGFR engages in a number of regulatory interactions either intra-molecularly with the C-terminal tail of the receptor (Kovacs et al., 2015) or inter-molecularly with a negative feedback inhibitor of HER receptors, MIG6 (Zhang et al., 2007), as well as other signaling molecules such as ACK1 protein kinase (Shen et al., 2007; Zhang et al., 2007). It is possible that high conservation of the activator interface reflects its broader functional range, and that this interface evolved early to mediate protein–protein interactions between EGF receptors and their binding partners.
Our analysis of the only EGF receptor in C. elegans, LET-23, demonstrates that its kinase domain adopts several characteristic autoinhibitory features seen in the human EGFR kinase. Despite their large evolutionary distance, both kinases adopt almost identical inactive conformations and use intermolecular interactions between the kinase domain and the proximal region of the C-tail for stabilization of the inactive state. Interestingly, the binding mode between the C-tail and the kinase domain differ, which might coincide with the evolution of new regulatory elements for the control of receptor activation, such as the juxtamembrane segment, in higher order vertebrates. The C-tail-mediated interactions in the human EGFR kinase directly compete with the binding of the juxtamembrane segment, suggesting that these interactions co-evolved to regulate the activation of human EGFR (Jura et al., 2009a). In the absence of a conserved juxtamembrane segment in LET-23, the C-tail interactions with the kinase domain are subject to fewer structural constraints, thus allowing the proximal segment of the C-tail to play new functions in the activation of LET-23. One such function is hinted at by the observation that in our crystal structure the C-tail binding results in the occlusion of its activator interface in a MIG6-like fashion (Figure 2E). While this may be an artifact of crystallization, it suggests that the LET-23 C-lobe may participate in protein-protein interactions, which is an important characteristic of human EGFRs. Binding of the C-tail to this interface would then provide a point of regulation for such binding events in LET-23.
The inactive conformation of the LET-23 kinase domain appears to be very stable. All catalytically active human EGF receptor kinases (EGFR, HER2 and HER4) almost exclusively adopt an active conformation in crystal lattices in the absence of mutations or conformationally selective inhibitors due to the high protein concentrations in crystals promoting asymmetric kinase oligomerization (Aertgeerts et al., 2011; Qiu et al., 2008; Stamos et al., 2002). In contrast, the wild-type LET-23 kinase domain crystallizes in the inactive conformation and does not form the same asymmetric kinase domain interactions in the crystal lattice. Nonetheless, our data show that, like human EGFR, LET-23 undergoes oligomerization-dependent activation. In the absence of an activation loop tyrosine in LET-23, its activation seems to be independent of kinase autophosphorylation, pointing to an allosteric mechanism. Moreover, mutational analysis suggests that asymmetric kinase domain interfaces analogous to the allosteric activator and receiver sites in human EGFR are important for LET-23 activation. The main difference observed in our studies is that activator and receiver interface mutations can complement one another in the human receptor, but not to a detectable extent in the worm counterpart. While this finding could mean that the activation mechanism of LET-23 is quite different from that of human EGFR despite relying on similar interfaces, it is also possible that the failure to complement is a result of a significant divergence in the active dimer interfaces between LET-23 and EGFR, which could result in unspecific effects of the studied interface mutations, a scenario we discuss in Figure 5E. It is also possible that the failure to complement might simply be due to the fact that the asymmetric dimer of the kinase domain has evolved to be much weaker in LET-23 than in human EGFR and may incorporate additional inhibitory interfaces that raise the energetic barrier for allosteric activation.
The structural and functional differences described in this study may have evolved to accommodate the LET-23 extracellular region, which exists as a constitutive dimer. The pressure to maintain low kinase activity in the absence of ligand-dependent activation would be more stringent for a constitutively dimeric receptor such as LET-23, and would mandate the preservation of highly efficient intracellular autoinhibitory interactions that are only released after ligand binding. In such preformed dimers, low affinity interactions between the activator and receiver kinase would be offset by proximity to provide an additional level of control over spontaneous receptor activation in the absence of a ligand. These details will surely be informed by future structural characterizations of activated LET-23 kinase domain complexes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead contact, Natalia Jura (natalia.jura@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cloning
The gene encoding the kinase domain (KD, residues 866-1191) of LET-23 (isoform a) was inserted into pFastBac HTA (ThermoFisher) vector engineered to yield a fusion protein bearing an N-terminal His10-tag, followed by a Tobacco Etch Virus (TEV) protease recognition site and a three-residue linker (A-M-G).
After many failed attempts to clone the original full-length LET-23 sequence into a mammalian expression vector, we synthesized the codon-optimized gene using GenScript. The optimized full-length LET-23 cDNAs were inserted into pcDNA4/TO vector (ThermoFisher) with a 3X-FLAG tag at the C-terminus.
Mutations were introduced into constructs of LET-23 kinase domain or full-length receptor by Quikchange mutagenesis (Stratagene) and verified by DNA sequencing (Elim Biopharmaceuticals, Inc).
E. coli strains
TOP10 (ThermoFisher) and DH10Bac (Invitrogen) E. coli cells were used for cloning and bacmid generation for insect cell expression, respectively. Transformed cells were cultured in Luria Broth (LB) media at 37°C supplemented with gentamicin (7 μg/ml), tetracycline (10 μg/ml), and kanamycin (50 μg/ml) during bacmid generation.
Insect cell culture
sf9 insect cells were cultured in ESF921 media (Expression Systems) at 26°C for up to 20 passages.
COS-7 cell culture
COS-7 cells were cultured in DMEM media (Sigma) supplemented with fetal bovine serum (10%) and antibiotics (penicillin/streptomycin) at 37°C and 5% CO2.
METHOD DETAIL
Protein Expression and Purification
Protein was expressed using the Bac-to-Bac system (Invitrogen) in Sf9 cells cultured at 26°C. Briefly, P1 virus was first generated by plating 2 ml of sf9 cells at 1.0 million cells/mL density on the surface of a tissue culture flask with 25 cm2 of surface area. Cells were allowed to adhere for at least 45 min, after which the media was replaced with 2 ml of fresh media. The cells were then treated with a 2 ml transfection mix containing 15 μl of purified bacmid DNA (200 – 3000 ng/μl) and 15 μl of cellfectin II transfection reagent (Invitrogen). After 6 hours of incubation, the transfection mix on the cells was replaced with 6 ml of fresh media. Following incubation for 5 – 7 days, the P1 virus was harvested by centrifugation at 1000xg for 5 minutes at room temperature to remove cell debris, and the supernatant collected and filtered through a 0.22 μm syringe filter. To generate P2 virus, P1 virus was diluted 50-fold into an sf9 cell culture at a density of 2.0 million cells/mL and cultured for 3 days with shaking in non-baffled flasks at 120 rpm. For protein expression, supernatant from the P2 virus culture was collected by centrifugation and supernatant diluted 40-fold into 1L of Sf9 cells at a density of 2.0 million cells/ml. After 2.5 days, cell pellets were harvested by centrifugation at 3500xg at 4°C for 20 minutes and flash frozen in liquid nitrogen prior to storage at −80°C until purification.
An Sf9 cell pellet (~30 g) from 4 liters of culture was thawed on ice and homogenized with 3 passages on an EmulsiFlex-C5 (Avestin) in 100 ml of lysis buffer [20 mM HEPES, pH 7.0, 500 mM NaCl, 10 mM imidazole, 5 mM MgCl2, 1 mg DNase I, 0.1% Triton X-100, and 5 mM β-mercaptoethanol (βME)]. After centrifugation of the lysate, the supernatant was filtered with a 5 μm PVDF membrane (EMD Millipore) and affinity purified at 4°C using a 1 ml HisTrap FF Crude column (GE) at a flow rate of 1 ml/min. Following loading, the column was washed with 50 ml buffer A (20 mM HEPES, pH 7.0, 500 mM NaCl, 10 mM imidazole, and 3 mM BME), and eluted with a linear gradient (40 ml) from buffer A to 100% buffer B (20 mM HEPES, pH 7.0, 20 mM NaCl, 250 mM imidazole, and 3 mM βME). Fractions containing pure LET-23KD eluted between 40 – 100% buffer B. For biochemical assays, pooled protein was dialyzed overnight at 4°C against 2 L of buffer C (20 mM HEPES, pH 7.0, 2 mM βME), and dilute sample purified by multiple rounds of size exclusion chromatography at 4°C using the Superdex 200 10/300 GL column (GE Healthcare) equilibrated in buffer D (20 mM HEPES, pH 7.0, 100 mM NaCl, 2 mM DTT). For protein used in crystallization trials, the His10 tag was removed using TEV protease by dialysis overnight at 4°C against 2 liters of buffer C with TEV protease in the sample bag at a molar ratio of TEV:LET-23 = 1:25. Precipitant from the digest reaction was removed by centrifugation, and the resulting solution flowed through a refreshed 1 ml HisTrap FF Crude column to trap uncleaved protein. The flow-through containing cleaved LET-23KD was collected, quickly concentrated in a 30 KDa cutoff concentrator (Amicon) at 4°C, and purified by size exclusion chromatography at 4°C in buffer D. Purified protein was concentrated and aliquots flash frozen in liquid nitrogen stored at −80°C.
Crystallization, Data Collection, Structure Determination
Crystallization screening was conducted with purified LET-23KD in buffer D and preincubated with AMP-PNP and MgCl2 added to final concentrations of 1 mM and 2 mM, respectively. Initial crystallization conditions were identified in the JCSG Core I screen (Qiagen). Following optimization, well-diffracting crystals were grown using the hanging-drop vapor diffusion method and harvested from drops consisting of 1 μl of protein (10 mg/ml) and 1 μl of reservoir solution (20% PEG5000 MME, 200 mM NaCl, and 100 mM HEPES, pH 7.5) equilibrated against 1 ml of reservoir solution at 20°C. Crystals were cryo-protected by briefly soaking in a solution containing 30% PEG5000 MME, 200 mM NaCl, and 100 mM HEPES, pH 7.5 prior to flash-freezing in liquid nitrogen.
Diffraction data were collected at the Advanced Light Source beamline 8.3.1 (ALS, Berkeley, CA) at 100K and recorded on an ADSC Quantum 315r CCD detector. Data were processed using HKL2000 (Otwinowski and Minor, 1997). Crystallographic data processing and refinement statistics are reported in Table 1. The structure of LET-23 bound to AMP-PNP was determined by molecular replacement using Phaser (Collaborative Computational Project, 1994) and the human EGFR kinase domain (PDB ID: 2GS6) as a search model, with residues differing between the human EGFR kinase domain and LET-23 replaced by alanine. Coot was used for graphical modeling (Emsley and Cowtan, 2004) following iterative rounds of refinement conducted in Refmac5 (Vagin et al., 2004) and PHENIX (Adams et al., 2010) and translation-liberation-screw (TLS) refinement (Painter and Merritt, 2006) to improve model quality. Figures were prepared using PyMOL (Schrodinger, LLC.). Homology models of the active conformation of the LET-23 kinase domain were generated using SWISS-MODEL Workspace (Bordoli et al., 2009), the sequence of LET-23 kinase domain, and the structure of the active conformation of human EGFR kinase domain (PDB ID: 2GS6) as the structural template.
In vitro Kinase Assay
Kinase activity was measured using a continuous enzyme-coupled reaction system performed at 30°C as previously described (Zhang et al., 2006). Briefly, the reaction buffer contained 20 mM Tris (pH 7.5), 10 mM MgCl2, and 1 mM ATP. Poly-4Glu:Tyr peptide (Sigma-Aldrich) was used as the phosphorylation substrate at a concentration of 1 mg/ml. Small unilamellar vesicles (SUV) containing DOPC and DOGS-NTA-Ni lipids (Avanti Polar Lipids) in a buffer containing 20 mM Tris (pH 7.5) were produced by extrusion through a membrane containing 100 nm pores (Whatman) using a mini-Extruder apparatus. The total lipid concentration of 10X SUV stocks was fixed at 2 mg/ml with Ni-NTA-DGS lipid concentration varied from 1 to 5 mole percent.
Cell-based Analysis of LET-23 Phosphorylation
Wild-type or mutant full-length LET-23 was transfected into COS-7 cells using Lipofectamine™ 3000 Reagent (ThermoFisher). Following a 16 hour expression, the cells were serum-starved for 6 hours in unsupplemented Opti-MEM media (ThermoFisher), washed twice with PBS on ice, then stimulated with 10 μM LIN-3EGF for 10 minutes on ice in PBS buffer containing 0.5% (w/v) BSA. LIN-3EGF corresponds to the purified EGF domain of LIN-3, which was prepared as described previously (Freed et al., 2015). Following stimulation, the cells were washed with ice-cold PBS buffer and lysed in buffer containing 25mM Tris, pH 7.5, 150 mM NaCl, 1% Nonidet P40 and phosphatase and protease 30 inhibitors. The lysates were passaged through a 27-gauge needle 5 times, incubated on ice for 30 minutes, spun down, and supernatants collected. The lysate supernatants (500 μl) were incubated with 2 μl of anti-FLAG antibody (Sigma-Aldrich) for 1 hour at 4°C with rocking, followed by incubation with 1:1 prewashed protein-A Sepharose 4B slurry (40 μL; Life Technologies) for an additional 1 hour at 4°C. After 3 washes, the protein was eluted from the resin with 2X SDS Gel Loading Buffer and analyzed by Western Blotting. The anti-pY20 antibody (1:500; Santa Cruz Biotechnology, Cat# sc-508) was used to detect tyrosine phosphorylation.
Data and Software Availability
Atomic coordinates and structure factors for the reported crystal structure have been deposited in the PDB under the ID code 5WNO.
Supplementary Material
HIGHLIGHTS.
Autoinhibitory features of EGFR kinases are conserved across evolution
Structure of the C. elegans LET-23 kinase, a non-human EGFR ortholog
Activation of the LET-23 kinase is oligomerization-dependent
Mutation of N- and C-lobe interfaces inhibits LET-23 kinase activation
Acknowledgments
We thank D. Ma and K. Ashrafi for providing C. elegans cDNA libraries, and P. Sternberg for full-length LET-23 cDNA. We thank the staff at ALS 8.3.1 beamline for assisting with X-ray diffraction experiments. We also thank C. Agnew for critical reading of the manuscript and members of the Jura lab for insightful discussions. This work was supported by grants from the National Institute of General Medical Sciences to N.J. (R01 GM109176), National Cancer Institute to M.A.L. (R01 CA198164), Susan G. Komen Foundation Training Grant to N.J. (CCR14299947), and National Cancer Institute to T.M.T. (F32 CA216928).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization: L.L. and N.J.; Investigation: L.L., T.M.T., D.E.F., N.F., K.M. and N.J.; Writing – Original Draft: L.L., T.M.T, M.A.L, and N.J.; Writing – Review & Editing: T.M.T and N.J.; Funding Acquisition: T.M.T. M.A.L, and N.J.; Supervision: N.J.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aertgeerts K, Skene R, Yano J, Sang BC, Zou H, Snell G, Jennings A, Iwamoto K, Habuka N, Hirokawa A, et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem. 2011;286:18756–18765. doi: 10.1074/jbc.M110.206193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Systematic biology. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberan S, Martin-Duran JM, Cebria F. Evolution of the EGFR pathway in Metazoa and its diversification in the planarian Schmidtea mediterranea. Scientific reports. 2016;6:28071. doi: 10.1038/srep28071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nature protocols. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Freed DM, Alvarado D, Lemmon MA. Ligand regulation of a constitutively dimeric EGF receptor. Nature communications. 2015;6:7380. doi: 10.1038/ncomms8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009a;137:1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci U S A. 2009b;106:21608–21613. doi: 10.1073/pnas.0912101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs E, Das R, Wang Q, Collier TS, Cantor A, Huang Y, Wong K, Mirza A, Barros T, Grob P, et al. Analysis of the Role of the C-Terminal Tail in the Regulation of the Epidermal Growth Factor Receptor. Mol Cell Biol. 2015;35:3083–3102. doi: 10.1128/MCB.00248-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlefield P, Liu L, Mysore V, Shan Y, Shaw DE, Jura N. Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci Signal. 2014;7:ra114. doi: 10.1126/scisignal.2005786. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Mendrola JM, Shi F, Park JH, Lemmon MA. Receptor tyrosine kinases with intracellular pseudokinase domains. Biochem Soc Trans. 2013;41:1029–1036. doi: 10.1042/BST20130104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Zhang Q, Young SN, Reese ML, Bailey FP, Eyers PA, Ungureanu D, Hammaren H, Silvennoinen O, Varghese LN, et al. A robust methodology to subclassify pseudokinases based on their nucleotide-binding properties. Biochem J. 2014;457:323–334. doi: 10.1042/BJ20131174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Vol. 276. Elsevier; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Qiu C, Tarrant MK, Choi SH, Sathyamurthy A, Bose R, Banjade S, Pal A, Bornmann WG, Lemmon MA, Cole PA, et al. Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure. 2008;16:460–467. doi: 10.1016/j.str.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, Carpenter G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell. 2009;34:641–651. doi: 10.1016/j.molcel.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger (LLC.) The PyMOL Molecular Graphics System. Version 1.8. [Google Scholar]
- Shan Y, Eastwood MP, Zhang X, Kim ET, Arkhipov A, Dror RO, Jumper J, Kuriyan J, Shaw DE. Oncogenic mutations counteract intrinsic disorder in the EGFR kinase and promote receptor dimerization. Cell. 2012;149:860–870. doi: 10.1016/j.cell.2012.02.063. [DOI] [PubMed] [Google Scholar]
- Shan Y, Seeliger MA, Eastwood MP, Frank F, Xu H, Jensen MO, Dror RO, Kuriyan J, Shaw DE. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc Natl Acad Sci U S A. 2009;106:139–144. doi: 10.1073/pnas.0811223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen F, Lin Q, Gu Y, Childress C, Yang W. Activated Cdc42-associated kinase 1 is a component of EGF receptor signaling complex and regulates EGF receptor degradation. Mol Biol Cell. 2007;18:732–742. doi: 10.1091/mbc.E06-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A. 2010;107:7692–7697. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- Suga H, Dacre M, de Mendoza A, Shalchian-Tabrizi K, Manning G, Ruiz-Trillo I. Genomic survey of premetazoans shows deep conservation of cytoplasmic tyrosine kinases and multiple radiations of receptor tyrosine kinases. Sci Signal. 2012;5:ra35. doi: 10.1126/scisignal.2002733. [DOI] [PubMed] [Google Scholar]
- Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- Wood ER, Shewchuk LM, Ellis B, Brignola P, Brashear RL, Caferro TR, Dickerson SH, Dickson HD, Donaldson KH, Gaul M, et al. 6-Ethynylthieno[3,2-d]-and 6-ethynylthieno[2,3-d]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc Natl Acad Sci U S A. 2008;105:2773–2778. doi: 10.1073/pnas.0708281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450:741–744. doi: 10.1038/nature05998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.