

Abstract
Casein kinase 1δ/ε have been identified as promising therapeutic target for oncology application, including breast and brain cancer. Here, we described our continued efforts in optimization of a lead series of purine scaffold inhibitors that led to identification of two new CK1δ/ε inhibitors 17 and 28 displaying low nanomolar values in antiproliferative assays against the human MDA-MB-231 triple negative breast cancer cell line and have physical, in vitro and in vivo pharmacokinetic properties suitable for use in proof of principle animal xenograft studies against human cancers. 2009 Elsevier Ltd. All rights reserved.
Keywords: Casein kinase 1 delta and epsilon, Kinase, Inhibitor, Structure-activity relationship, Breast cancer
Graphical abstract
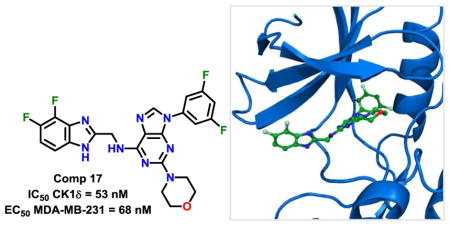
1. Introduction
The casein kinase 1 (CK1) family consists of six monomeric serine/threonine-specific protein kinases (α, δ, ε, γ1, γ2 and γ3).1 All six human CK1 isoforms are highly homologous in their active site, with the delta (CK1δ) and epsilon (CK1ε) isoforms sharing 98% sequence identity in their protein kinase domains. Elsewhere in the enzyme, such as in the C-terminal, and non-catalytic domains, significant variances exist between isoforms.2 CK1 kinases play crucial roles in regulating a variety of cellular growth and survival processes including circadian rhythm,3 membrane trafficking,4 DNA damage repair,5 cytoskeleton maintenance,6 and notably Wnt signaling.7 A recent study showed that CK1δ/CK1ε might be involved in the etiology of addictive behavior and that their inhibition prevents relapse-like alcohol drinking.8 Importantly, abnormal regulation of these two CK1 family members is implicated in human cancers and several known CK1δ and/or CK1ε substrates control tumor cell growth, apoptosis, metabolism and differentiation.9 For instance, both isoforms are overexpressed in pancreatic ductal adenocarcinoma,10 ovarian cancer11 and chronic lymphocytic leukemia.12 Interestingly, expression of constitutively active, myristoylated CK1ε in mammary epithelial cells is sufficient to drive cell transformation in vitro via stabilization of β-catenin and the activation of Wnt transcription targets.13 At the same time, forced expression of kinase defective CK1δ mutants blocks SV40-driven cellular transformation in vitro and mammary carcinogenesis in vivo.14
The Wnt/β-catenin pathway has known roles in breast cancer, where: (i) Wnt signaling can contribute to triple negative breast cancer; (ii) nuclear β-catenin connotes metastasis and poor outcome; and (iii) β-catenin contributes to mutant ErbB2-driven breast cancer.15 Importantly, gain-of-function (e.g., in β-catenin) and loss-of-function (e.g., in CK1α APC and AXIN) mutations prevalent in other cancers are not found in breast cancer. Recent discoveries from our laboratories establish casein kinase 1 delta (CK1δ) as an essential regulator of β-catenin activity that is overexpressed and amplified in human breast cancer.16
CK1δ and CK1ε are eminently tractable for small molecule targeted drug discovery.17 Among the small molecules reported to inhibit CK1δ/CK1ε are CKI-7,18 (R)-CR8 and (R)-DRF053,19 IC261,20 D4476,21 LH846,22 benzimidazole 5,23 PF480056724 and PF670462.3a, 24 Unfortunately, most of these compounds are not specific inhibitors of CK1δ/CK1ε and in some cases their pharmacological effects are now known to be (or are suspected to be) due to non-selective target engagement.20b, 25 At the same time, the contribution of CK1δ and CK1ε to human cancer is still not fully understood and the non-selective nature of previously reported CK1δ/ε inhibitors has impeded pharmacological validation of these kinases as anti-cancer targets.3a, 20b Nonetheless, available data clearly shows that targeted inhibition of CK1δ/CK1ε is a viable, highly attractive anticancer strategy yet to be fully developed.26
In research targeting the development of novel therapeutics for treatment of metastatic and resistant forms of cancer, the Roush laboratory at Scripps Florida identified highly potent and selective purine-derived dual inhibitors of CK1δ and CK1ε under the aegis of the NIH’s MLPCN program (Figure 1).25 These agents induce proliferative arrest and rapid apoptosis of CK1δ/CK1ε-expressing human luminal B, HER2+ and triple negative cancer cells ex vivo and SR-3029 induces tumor regression in orthotopic xenograft models in vivo.26 Specifically, genetic knockdown or pharmacological inhibition of CK1δ using SR-3029 blocks β-catenin nuclear localization and TCF transcriptional activity, induces rapid apoptosis and provokes tumor regression in patient derived orthotopic models of triple negative breast cancer.25–26 Mechanistic studies are consistent with the premise that CK1δ controls breast cancer tumorigenesis via its effect on β-catenin, which plays known roles as an effector of aberrant Wnt signaling in human breast cancer.26
Figure 1.
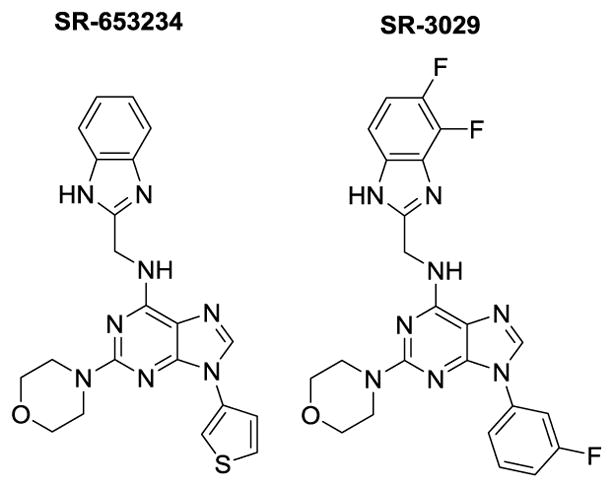
Structures of HTS hit SR-653234 and optimized probe SR-3029
Noteworthy, profiling of 442 kinases with SR-3029 (and three close analogs) confirmed their high selectivity vs. the plethora of kinases whose activity is impaired by the purportedly specific Pfizer CK1δ inhibitor (PF670462).25 The four off-target kinases that are inhibited by SR-3029 largely have no known function. High selectivity of N9-arylsubstituted purine scaffold could be explained by the unique structural features of the CK1δ/ε active site. Specifically, relatively small size of the gatekeeper residue Met82 in case of both CK1δ/ε creates large hydrophobic pocket which is occupied by N9 aryl residue of the inhibitors. This structural feature of CK1δ/ε was also encountered by Shokat and colleagues when developing analog-sensitive kinase technology.27
Herein we report structure-activity and structure-property relationship (SAR and SPR) studies that led to identification of dual selective CK1δ/ε inhibitors (including SR-3029) with physicochemical properties and in vitro and in vivo pharmacokinetic parameters suitable for use in murine xenograft studies against breast cancer.26
2. Results and Discussion
2.1. Synthetic Chemistry
The general procedure for the synthesis of purine-based CK1δ/ε inhibitors is based on a previously reported sequence 25, 28 which we improved by incorporating a more efficient method for arylation of the N9 nitrogen of dichloropurine (Scheme 1). Thus, N9 aryl purine intermediates (permutations of structure 6) were generated by copper (I) mediated coupling of commercially available dichloropurine 4 with a variety of symmetrical or unsymmetrical diaryliodonium salts 5.29 This reaction provides 6 in 65%–85% yield – a substantial improvement relative to the previously employed Chan-Lan coupling (yield 0–30%).25 The diaryliodonium salts were accessed via one-step procedures from aryl iodides 1 and arenes 2 or boronic acids 3.30 Subsequently, substituted benzimidazoles 9 and various amines were introduced at C6 and C2 of 6, respectively. This procedure provided the targeted CK1δ/ε inhibitors with excellent regioselectivity and good yields (70–90%).25 The substituted benzimidazoles (9) were accessed as previously described from o-dianilines, 7.25, 31
Scheme 1.

General strategy for the synthesis of N9-arylsubstituted CK1δ/ε inhibitors.
2.2. Structure-activity relationship (SAR) studies
We synthesized analogs in an iterative fashion using the chemistry outlined in Scheme 1, refining our compound design on the basis of biochemical and biological data obtained from prior rounds of tested inhibitors. Compounds with low nanomolar biochemical affinity (IC50 < 100 nM, 20 μM ATP concentration) were assessed in cell proliferation assays vs. MDA-MB-231 breast cancer cell lines using 3-day MTT and longer term clonogenicity assays (see Experimental section for details).25
Our SAR optimization efforts commenced with the synthesis of agents with various N9 alkyl and aryl substitution (R1) (Table 1). Alkyl substitution of the purine N9 position (11, 12) resulted in the complete loss of CK1δ/ε activity. Consequently, subsequent SAR efforts focused on aryl R1 substituents. Since we looked at only one example of a N9-difluorophenyl substitution in our original publication,25 other mono- and disubstituted fluorophenyl analogs were explored. Notably, all the analogs with a N9 4-fluorophenyl substitution resulted in an approximately 10 fold loss of activity against CK1δ compared to SR-3029 (14, 15, 19, Table 1). Other difluorophenyl analogs 2,3-difluorophenyl 16, 3,5-difluorophenyl 17 and 2,5-difluorophenyl 18 showed similar activity to SR-3029 with 18 having a slightly better biochemical profile.
Table 1.
SAR data for CK1δ/ε inhibitors with alkyl and aryl substituents at N9 (R1)
| ||||
|---|---|---|---|---|
| № | R1 | CK1δ IC50 (nM)[a] | CK1ε IC50 (nM)[a] | MDA-MB-231 EC50 (nM)[b] |
| 11 | -Me | > 10000 | > 10000 | - [c] |
| 12 | -Et | 4010 | 6560 | - [c] |
| SR-3029 (13) |
|
44 | 260 | 26 |
| 14 |
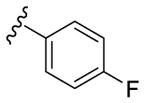
|
535 | 395 | - [c] |
| 15 |
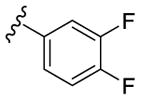
|
4520 | 990 | - [c] |
| 16 |
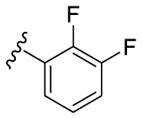
|
48 | 80 | 101 |
| 17 |
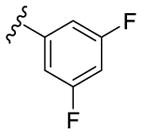
|
53 | 145 | 68 |
| 18 |
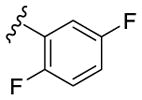
|
10 | 16 | 38 |
| 19 |
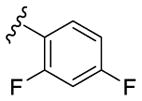
|
525 | 250 | - [c] |
| 20 |
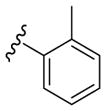
|
110 | 135 | 6280 |
| 21 |
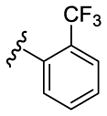
|
230 | 175 | 9790 |
Biochemical assay.
Cellular proliferation assay.
Not determined.
Introduction of other substituents at the ortho position of the N9 aromatic moiety (R1), such as in inhibitors 20 and 21, resulted in a modest reduction of biochemical activity (2–4 fold) but substantial loss of cell-based activity (EC50 = 6–10 μM, Table 1).
While performing the initial rounds of SAR optimization, we noticed a moderate correlation between the calculated logD (pH = 7.4) values of tested analogs and their antiproliferative properties (Figure 2, library of 224 inhibitors from Bibian et al25 and related patent32). In particular, the majority of poorly active analogs (EC50 > 750 nM) were more lipophilic (logD > 4) than those agents with significant activity. The logD distribution of compounds with EC50 < 750 nM and compounds with EC50 > 750 nM also suggests that partition coefficients peak around 2.5 for active and 3.8 for inactive analogs (Figures 2A, B).
Figure 2.

Correlation between calculated logD (pH 7.4) and antiproliferative properties (cell based assay, EC50, nM) of CK1δ/ε inhibitors available library (total 224 inhibitors including compounds from Bibian et al25 and related patent31). A: probability of logD distribution for active compounds (EC50 < 750 nM, n = 52). B: probability of logD distribution for inactive compounds (EC50 > 750 nM, n = 172).
In an effort to lower the lipophilicity of the CK1δ/ε inhibitors, we synthesized and tested a series of N9 heteroaryl-substituted analogs (Table 2). Although N9 thiophene substitution resulted in inhibitors with low nanomolar values in both biochemical and cell based assays (22 and 23), an unsubstituted thiophene is generally considered to be a metabolic liability.33 To avoid this potential problem, we selected alternate groups that could be used at the purine N9 position (R1) to lower lipophilicity while, hopefully, maintaining CK1δ/ε inhibitory activity. Replacement of the thiophene ring by a variety of pyridyl groups retained the logD values in the targeted range (≤ 3.5), however all these compounds were less potent against CK1δ/ε (IC50 > 143) compared to either 23 or SR-3029.
Table 2.
SAR and clogD data for CK1δ/ε inhibitors with heteroaryl substituents at N9 (R1)
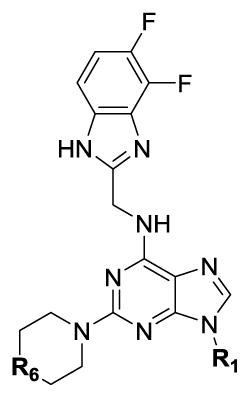
| ||||||
|---|---|---|---|---|---|---|
| № | R1 | R6 | CK1δ IC50 (nM)[a] | CK1ε IC50 (nM)[a] | MDA-MB-231EC50 (nM)[b] | LogD [c] |
| 22 |
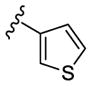
|
N-Me | 47 | 125 | 75 | 2.6 |
| 23 |
|
O | 54 | 105 | 43 | 3.0 |
| 24 |
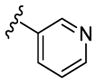
|
N-Me | 700 | 1145 | - [d] | 1.8 |
| 25 |
|
O | 335 | 500 | - [d] | 2.8 |
| 26 |
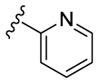
|
N-Me | 260 | 860 | - [d] | 2.4 |
| 27 |
|
O | 440 | 1905 | - [d] | 3.3 |
| 28 |
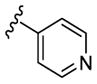
|
N-Me | 175 | 315 | 28 | 3.0 |
| 29 |
|
O | 260 | 385 | - [d] | 3.3 |
| 30 |
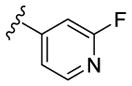
|
N-Me | 295 | 660 | - [d] | 3.0 |
| 31 |
|
O | 145 | 245 | 8 | 3.4 |
| 32 |
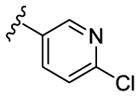
|
N-Me | 1120 | 860 | - [d] | 2.7 |
| 33 |
|
O | > 9900 | 1195 | - [d] | 3.1 |
| 34 |
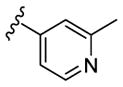
|
N-Me | 295 | 385 | - [d] | 3.2 |
| 35 |
|
O | 1195 | 245 | - [d] | 3.5 |
Biochemical assay.
Cellular proliferation assay.
Calculated with Pipeline Pilot workflow application (Accelrys) at pH 7.4.
Not determined
Contemporaneously, we sought to vary the substitution on the benzimidazole unit (R5). The data presented in Table 3 are for one series in which the purine N9 substituent was held constant as m-pyridyl. Additionally three different amines (N-methylpiperazine, morpholine and piperazine; Table 3) were utilized at purine C2 throughout this compound set. Compounds 36, 37 and 38, which have an unsubstituted benzimidazole ring, showed decreased CK1δ/ε inhibition (IC50 vs CK1δ = 824 – 1566 nM) compared to other analogs in this set (Table 3). Substitution of the benzimidazole with either 5-chloro (39–41) or 5-methoxy (45–47) groups led to a 2–3 fold increase in CK1δ/ε inhibition (IC50 vs CK1δ = 336 – 624 nM) relative to the unsubstituted benzimidazole, derivatives 36–38. Further improvement of CK1δ/ε inhibitor activity was achieved through the incorporation of two fluorine atoms into the 5 and 6 positions of the benzimidazole ring (48, 49). Finally, the most active compounds (IC50 vs CK1δ < 100 nM) in this series were obtained through the introduction of a 5,6-dichloro substitution pattern (42–44) into the benzimidazole unit. These three analogs were additionally tested in a cell based assay vs. MDA-MB-231 breast cancer cell lines, where they showed excellent antiproliferative properties (EC50 < 6 nM). Interestingly, compound 42 is almost 100-fold more potent in the cell-based assay than in in vitro tests against purified CK1δ. This observation suggests that 42 may possibly be engaging additional kinases that contribute to cell-based activity of this compound. This assumption, however, remains to be validated experimentally.
Table 3.
SAR data for CK1δ/ε inhibitors with a substituted benzimidazole unit (R5)
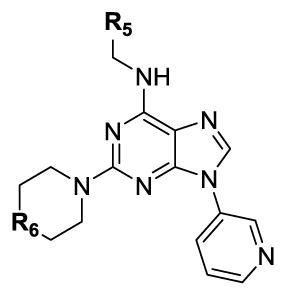
| |||||
|---|---|---|---|---|---|
| № | R5 | R6 | CK1δ IC50 (nM)[a] | CK1ε IC50 (nM)[a] | MDA-MB-231EC50 (nM)[b] |
| 36 |
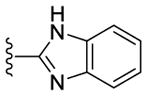
|
N-Me | 1565 | 2955 | - [c] |
| 37 |
|
O | 1155 | 2095 | - [c] |
| 38 |
|
NH | 825 | 1840 | - [c] |
| 39 |
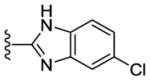
|
N-Me | 335 | 630 | - [c] |
| 40 |
|
O | 520 | 715 | - [c] |
| 41 |
|
NH | 430 | 915 | - [c] |
| 42 |
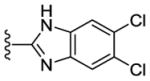
|
N-Me | 100 | 320 | < 1 |
| 43 |
|
O | 50 | 105 | 4 |
| 44 |
|
NH | 80 | 255 | 6 |
| 45 |
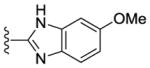
|
N-Me | 610 | 335 | - [c] |
| 46 |
|
O | 520 | 975 | - [c] |
| 47 |
|
NH | 625 | 1765 | - [c] |
| 48 |
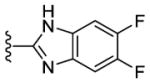
|
N-Me | 200 | 660 | 1040 |
| 49 |
|
O | 175 | 340 | 60 |
Biochemical assay.
Cellular proliferation assay.
Not determined.
Lastly, inclusion of morpholine or piperazine units at C2 of the purine scaffold generally conferred greater CK1δ/ε inhibition activity than compounds with N-methylpiperazine units at this position.
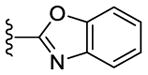
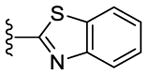
In addition to SAR studies performed on the benzimidazole ring itself, we also examined the possibility of using benzimidazole isosteres or replacing the benzimidazoles with different heterocycles (Table 4). The benzimidazole unit (R5) was replaced with structurally similar benzoxazole (50) and benzothiazole (51) units.
Table 4.
SAR and apparent cell permeability data for CK1δ/ε inhibitors with bioisosters or heterocyclic replacements of the benzimidazole unit (R5)
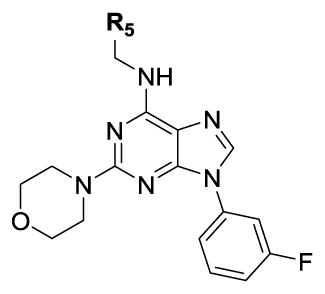
| |||||
|---|---|---|---|---|---|
| № | R5 | CK1δ IC50 (nM)[a] | CK1ε IC50 (nM)[a] | MDA-MB-231EC50 (nM)[b] | PAMPA[c] Papp·10−6 |
| 13 |
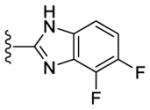
|
44 | 260 | 26 | 0.4 |
| 50 |
|
42 | 150 | > 10000 | < 0.1 |
| 51 |
|
9 | 115 | > 10000 | < 0.1 |
| 52 |
|
215 | 280 | 1400 | < 0.1 |
| 53 |
|
> 10000 | > 10000 | - [d] | < 0.1 |
| 54 |
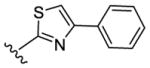
|
> 10000 | > 10000 | - [d] | < 0.1 |
| 55 |
|
245 | 380 | 1850 | 0.3 |
| 56 |
|
50 | 54 | 7160 | 7.1 |
| 57 |
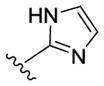
|
180 | 205 | > 10000 | 0.1 |
| 58 |
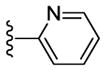
|
85 | - [d] | > 10000 | 6.8 |
| 59 |
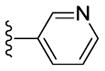
|
98 | - [d] | > 10000 | - [d] |
| 60 |
|
80 | - [d] | 6460 | - [d] |
| 61 |
|
37 | - [d] | > 10000 | - [d] |
| 62 |
|
89 | - [d] | > 10000 | < 0.1 |
Biochemical assay.
Cellular proliferation assay.
Permeability, Parallel Artificial Membrane Permeability Assay (PAMPA), see experimental section for details
Not determined
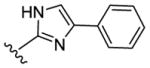
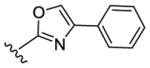
Despite evincing excellent biochemical activity (51, IC50 vs CK1δ = 9 nM), these two compounds were inactive in the cell based assay (EC50 > 10 μM). Other inhibitors with benzimidazole replacements, such as phenyl-substituted imidazole (52), oxazole (53) and thiazole (54), were synthesized and tested in biological assays. Of these three compounds, 52 showed good activity against CK1δ/ε (IC50 vs CK1δ = 214 nM). Unfortunately 52 displayed markedly diminished potency in cell-based assays against MDA-MB-231 cancer cells (EC50 = 1400 nM). Replacing the benzimidazole with 1-methyl-4-(pyridin-3-yl)piperazine (55) or 1-methyl-4-phenylpiperazine (56) led to improvement in biochemical activity (IC50 of 56 vs CK1δ/ε = 50 nM).
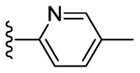
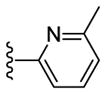
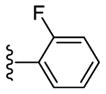
Sadly, these compounds also displayed a significant disparity between biochemical and cell-based potency (EC50 of 56 vs. MDA-MB-231 = 7160 nM). Replacement of the benzimidazole unit with an imidazole decreased CK1δ/ε inhibition activity significantly (57). Finally, eschewing the benzimidazole in favor of a 2-pyridyl (58), 3-pyridyl (59), 4-methyl-2-pyridyl (60), 3-methyl-2-pyridyl (61) or 2-flurophenyl (62) groups resulted in a series of highly potent CK1δ/ε inhibitors, but which were inactive against MDA-MB-231 breast cancer cell lines (EC50 > 10 μM).
We tested a subset of eleven compounds for their ability to permeate cell membranes (PAMPA assay, see Experimental section for details). The majority of the analogs tested possessed low permeability of Papp ≤ 2.5 · 10−6 cm/s (Table 4), suggesting that this property could be a significant contributor to the disconnect between biochemical and cell-based activity for this analogs series.
We completed this series of SAR studies of the purine core by varying the substitution at the C2 position (Table 5). Analogs 63 and 64 retained good potency but lost antiproliferative activity. Next, the morpholine unit of SR-3029 was exchanged for a thiomorpholine (65) and its oxidized variant (66), which led to a 2–3 fold decrease in CK1δ/ε biochemical and cell based activities. We also introduced a number of different piperidines at the R4 position to obtain compounds 67, 68, 73, 75 and 76. Each of these agents displayed moderate biochemical activity against CK1δ/ε. The best compound in this series was 68, which evinced an IC50 of 69 nM against CK1δ and 133 nM in the MDA-MB-321 cellular assay. Extending the morpholine unit by two or three carbons from the purine core resulted in compounds 70 and 71, both of which were are approximately 20 fold less active than SR-3029. Finally, tetrahydropyran derivative 69 and butyl analog 72 did not show any improvement in activity (69, IC50 vs CK1δ = 321 nM; 72, IC50 vs CK1δ = 1031 nM).
Table 5.
SAR data for CK1δ/ε inhibitors with other amine substituents at purine position C2 (R4)
| ||||
|---|---|---|---|---|
| № | R4 | CK1δ IC50 (nM)[a] | CK1ε IC50 (nM)[a] | MDA-MB-231EC50 (nM)[b] |
| 13 |
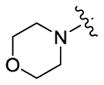
|
44 | 260 | 26 |
| 63 |
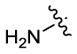
|
55 | 170 | 580 |
| 64 |
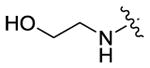
|
76 | 205 | 185 |
| 65 |
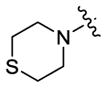
|
185 | 310 | 130 |
| 66 |
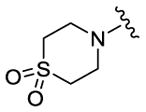
|
175 | 370 | 145 |
| 67 |
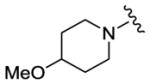
|
230 | 205 | 190 |
| 68 |
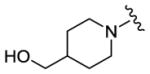
|
69 | 50 | 130 |
| 69 |
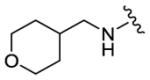
|
320 | 375 | 1290 |
| 70 |
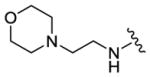
|
830 | 1230 | - [c] |
| 71 |
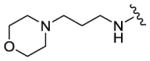
|
915 | 1310 | - [c] |
| 72 |
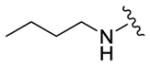
|
1030 | 2010 | - [c] |
| 73 |
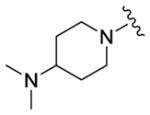
|
1180 | 1200 | 625 |
| 74 |
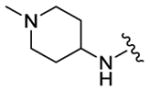
|
160 | 305 | 175 |
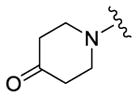
| 75 |
|
150 | 265 | 390 |
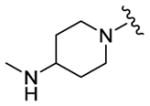
| 76 |
|
620 | 655 | 195 |
Biochemical assay.
Cellular proliferation assay.
Not determined.
2.3. Physicochemical and ADME Properties
In addition to testing inhibitory activity against CK1δ/ε and antiproliferative activity against MDA-MB-231 breast cancer cell lines, standard physicochemical properties including logP, logD7.4, and TPSA (total polar surface area) were assessed for all newly synthesized analogs. Compounds were prioritized on the basis of both their biological data and physicochemical properties, and high priority agents were assessed for in vitro drug metabolism and pharmacokinetics (DMPK, including aqueous solubility, microsomal stability and CYP450 inhibition; Table 6, PAMPA data for selected compounds are in Table 4).
Table 6.
ADME properties of selected CK1δ/ε inhibitors
| № | clogD pH 7.4[a] | Solubility (μM)[b] | Microsome stability, min (H/M/R)[c] | CYP % inhibition[d] | |||
|---|---|---|---|---|---|---|---|
| 1A2 | 2C9 | 2D6 | 3A4 | ||||
| 13 | 3.6 | 0.1 | 18/5/17 | 12 | 62 | −3 | 37 |
| 12 | 2.7 | - [e] | 16/3/5 | 40 | 49 | 14 | 76 |
| 16 | 3.7 | 0.4 | 17/5/10 | 23 | 89 | 47 | 72 |
| 17 | 3.7 | 0.1 | 26/5/27 | −48 | 42 | −2 | 38 |
| 18 | 4.4 | 0.3 | 12/3/6 | −41 | 73 | 8 | 64 |
| 22 | 2.6 | - [e] | 13/2/4 | 46 | 41 | 47 | 34 |
| 23 | 3.0 | - [e] | 15/2/6 | 91 | 84 | 53 | 62 |
| 25 | 2.8 | 0.1 | 25/5/9 | 96 | 95 | 67 | 86 |
| 28 | 3.0 | 28 | 36/4/11 | −1 | 21 | 47 | 44 |
| 29 | 3.3 | 0.4 | 20/5/9 | 61 | 53 | 50 | 74 |
| 31 | 3.4 | 0.1 | 32/12/9 | −9 | 31 | 10 | 18 |
| 40 | 3.1 | - [e] | 9/3/10 | 97 | 96 | 79 | 86 |
| 42 | 2.6 | 1.6 | 5/1/4 | 30 | 44 | 67 | 60 |
| 43 | 3.9 | - [e] | 55/16/26 | 90 | 91 | 80 | 88 |
| 44 | 2.3 | 2.8 | 19/5/18 | -[e] | -[e] | -[e] | -[e] |
| 49 | 2.9 | 1.1 | 22/7/9 | 91 | 92 | 62 | 85 |
| 50 | 3.5 | 0.1 | 16/3/- [e] | 59 | 61 | 31 | −38 |
| 51 | 4.4 | 0.1 | 30/9/- [e] | 66 | 52 | 42 | −12 |
| 58 | 3.1 | - [e] | - [e] | 68 | 31 | 1 | −17 |
| 59 | 3.4 | - [e] | - [e] | 74 | 74 | 18 | 74 |
| 61 | 3.4 | - [e] | 6/2/n.d. | 37 | 35 | −12 | −31 |
| 62 | 4.3 | - [e] | 15/6/- [e] | 85 | 67 | 38 | −51 |
| sunitinib | 46/13/30 | ||||||
Calculated with Pipeline Pilot workflow application (Accelrys) at pH 7.4.
Solubility in pH 7.4 phosphate buffered saline.
Microsome stability using human, mouse and rat liver microsomes, with sunitinib as the reference, half life in 1 mg/ml hepatic microsomes.
Cytochrome P450 inhibition assay.
Not determined.
Kinetic aqueous solubility in PBS buffer at pH 7.4 was determined using an HPLC-based protocol and reported as the average of two measurements (see Experimental Section for details). Apparent cell permeability was determined using the standard parallel artificial membrane permeability assay (PAMPA)34 using propanolol and ranitidine as controls and analyzed by HPLC-MS/MS. A large set of the most promising CK1δ/ε inhibitors was additionally tested for hepatic microsomal stability (human, mouse and rat) and cytochrome P450 inhibition as described previously.25 Briefly, the compounds were incubated together with hepatic microsomes and NADPH was used to initiate enzymatic oxidation. Acetonitrile was then added to quench the reaction at different time points and processed samples were analyzed using LC-MS/MS. Cytochrome P450 inhibition (CYP1A2, CYP2C9, CYP2D6, and CYP3A4) was evaluated in human liver microsomes using four selective marker substrates in the presence or absence of 10 μM test compound and reported as percentage inhibition (see Experimental section for details).
The calculated distribution coefficients (logD7.4) was in the acceptable range (1 < log D < 4) for the majority of new analogs, with the exception of 18, 51 and 62 (Table 6). This represents an improvement over the initial set of CK1δ/ε inhibitors,25 as the average logD value of our compound collection was reduced by approximately 1 order of magnitude. Despite improvements in lipophilicity, the majority of the compounds that were advanced to in vitro DMPK assessment possessed poor aqueous solubility (less than 0.5 μM). Fortunately, during our iterative analog synthesis efforts, we observed that aqueous solubility could be improved by appending a piperazine unit to the scaffold in lieu of a morpholine (i.e., 28, Table 6). Importantly, agent potency was not affected greatly by this substitution (though morpholine analogs are slightly more potent than their N-methylpiperazine counterparts).
Hepatic microsomal stability was considered to be an important factor when selecting candidates for in vivo testing. We sought compounds with half-lives resembling the FDA-approved small molecule tyrosine kinase inhibitor sunitinib, which was used as positive control (t1/2 = 46, 13 and 30 min in human, mouse and rat microsomes, respectively). Among the compounds advanced to metabolic stability studies, 43, 28, 31 and 51 emerged as the most stable candidates (Table 6). Agent stability was marginally redued or stayed comparable relative to reference compound SR-3029 for the thiophene (22, 23), benzylpyridine (61) and 2,3-difluorophenyl analogs 16 and 18.
Finally, CYP inhibition for a set of the most active compounds was assessed. It is well-known that 3-substituted pyridine-containing compounds can act as CYP3A4 inhibitors.35 Indeed, N9 m-pyridine-containing analogs 25, 40, 43 and 49 were the most potent inhibitors of all four cytochrome P450s tested (1A2, 2C9, 2D6, and 3A4, Table 6). However, it has also been reported that CYP3A4 inhibition can be decreased through the introduction of ortho-halogen or alkyl substitution into the pyridine.35 On this basis, we synthesized a series of 2-fluoro (30, 31), 2-chloro (32, 33) and 2-methyl (34, 35) substituted pyridines, which displayed significantly improved CYP inhibition profile (31, < 30% inhibition against all four tested CYP substrates at 10 μM, Table 6). However, these modifications led to reduced CK1δ/ε potency in some cases (32, 33, 35).
Based on the combination of biological activity and ADME properties, we selected 17 and 28 to advance into in vivo PK testing. While analog 28 is the most soluble compound in this series (28 μM, selected for PO administration), both compounds 17 and 28 possess acceptable stability in human, mouse and rat liver microsomes and a favorable CYP inhibition profile (Table 6) when compared to SR-3029 (13).
2.4. Molecular Modelling
Docking studies were performed using the co-crystal structure of CK1δ and PF670462 (PDB ID:3UYT)36 to predict the binding modes of designed and synthesized inhibitors. The original co-crystal structure was refined using the Protein Preparation Wizard37 implemented in the Maestro 11.1 (Schrödinger Release 2017-2) interface, and invalid atom types were corrected using this same wizard. A receptor grid was generated from the refined structure using default values. The docked models for PF670462 were in good agreement with the reported crystal structures coordinates (see Supporting information for details). Designed inhibitors were docked into the grid using Glide 7.438 in standard precision (SP) mode, without any constraints. The proposed binding pose of 17 within the CK1δ active site is shown in Figure 3A, B.
Figure 3.

A, docking pose of 17 (green) in the CK1δ active site (surface representation); B, docking pose 17 (green) in the CK1δ active site (mesh representation). C, docking pose of 28 (grey) in the active site of CK1δ. Hydrogen bond interactions are shown in red.
As revealed by docking studies, the key contacts between 17 and the binding pocket of CK1δ are two hydrogen bonding interactions between the purine scaffold and Leu85 in the hinge region (Figure 3B). The benzimidazole moiety projects into the solvent-exposed area and also makes additional contacts with the same residue Leu85, (Figure 3A and B), while the N9 m-fluorophenyl ring occupies a relatively large hydrophobic pocket created by the gatekeeper residue Met82. An image of 28 docked in CK1δ active site is presented in Figure 3C. The binding mode of this agent closely mirrors the pose adopted by 17 (Figure 3B F 3C). Specifically, 28 is positioned in a manner analogous to 17 so as to maintain two key interactions with the hinge region (Leu85) of CK1δ. Additionally, as predicted by Glide, the N9 para-pyridyl ring of 28 projects into the kinase hydrophobic pocket and picks up additional stabilizing interactions with Tyr56 (Figure 3C). Despite this prediction, we observed a 4 fold decrease in CK1δ inhibition activity for 28 (Table 2). We hypothesize that reduced inhibitory activity compared to 17 could be the result of an undesirable repulsive interaction between gatekeeper Met80 and the pyridyl unit of 28.
2.5. Kinase selectivity
In order to address the question of selectivity, kinase binding was performed using compound 17 (at 10 μM concentration) against a panel of 97 targets distributed across the kinome (DiscoverX scanEDGE Kinase Assay Panel). Analog 17 was exceptionally selective under the assay conditions, with only CK1δ being inhibited >90% of the 97 kinases tested (Figure 4, i.e., 5.2% of active kinase remaining at 10 μM). The only other off-target kinase, FLT3 (17% of active kinase remaining at 10 μM), was previously shown by us not to be responsible for the potent antiproliferative effects demonstrated by the purine-based CK1δ/ε inhibitors.25 The 95 remaining kinases in this assay showed greater than 35% control activity at 10 μM (see Supporting information for details).
Figure 4.
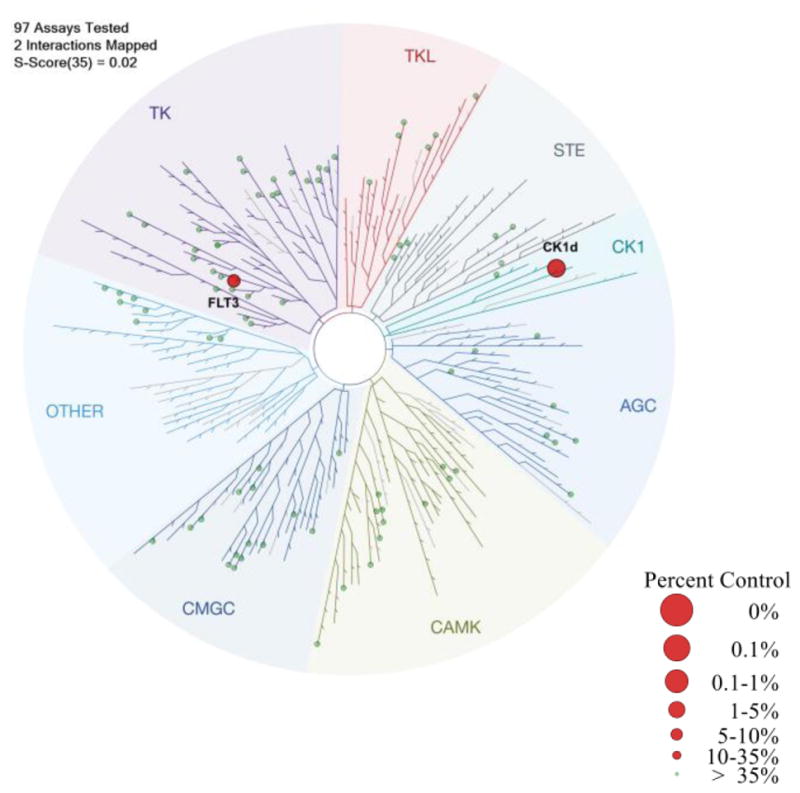
Dendrogram presentation of results of DiscoverRX scanEDGE kinase binding selectivity analysis of compound 17. Data are presented for all kinases that have <35% control activity at 10 μM (% control is the percentage of kinase remaining bound to the bead-bound active-site ligand in the presence of the inhibitor), represented as red circles on kinome tree and >35% control activity 10 μM, represented as green circles.
2.6. In Vivo Pharmacokinetics
The pharmacokinetic properties of 17 and 28 were assessed in male C57Bl6 mice following IP administration at 20 mg/kg (17) and IV and PO administration at 1 mg/kg and 10 mg/kg, respectively (28, Table 7, route of administration was selected based on solubility data, Table 6). Compound 17 showed good exposure after IP administration (Figure 5). The plasma concentration was maintained above 1 μM for more than 3 hours and above 100 nM for about 7 hours (EC50 MDA-MB-231 = 68 nM), albeit a short half-life (Figure 5, Table 7). At the same time, 28 exhibited a modest in vivo half-life (1.3 h) and a high volume of distribution (9.1 L/kg) following intravenous administration. The apparent oral bioavailability of 28 after PO dosing was approximately 16% (Table 7), marginally better than SR-3029 (13), despite improvement in aqueous solubility.
Table 7.
Mouse PK data for selected CK1δ/ε inhibitors 28 (PO dosing), 17 (IP dosing) and SR-3029 (13) (PO dosing)
| № | Route [a] | Dose (mg/kg) | Cmax (μM) | Cl (ml/min/kg) | AUC (μM·h) | t1/2 (h) | % F |
|---|---|---|---|---|---|---|---|
| 13 | PO | 3 | 1.1 | 49 | 2.4 | 0.9 | 13 |
| 28 | PO | 10 | 0.7 | -[c] | 0.32 | 1.3 | 16 |
| 17 | IP | 20 | 7.1 | 43[b] | 15.6 | 1.0 | - |
10/10/80 DMSO/Tween/water;
assumes 100% absorption;
Not determined
Figure 5.
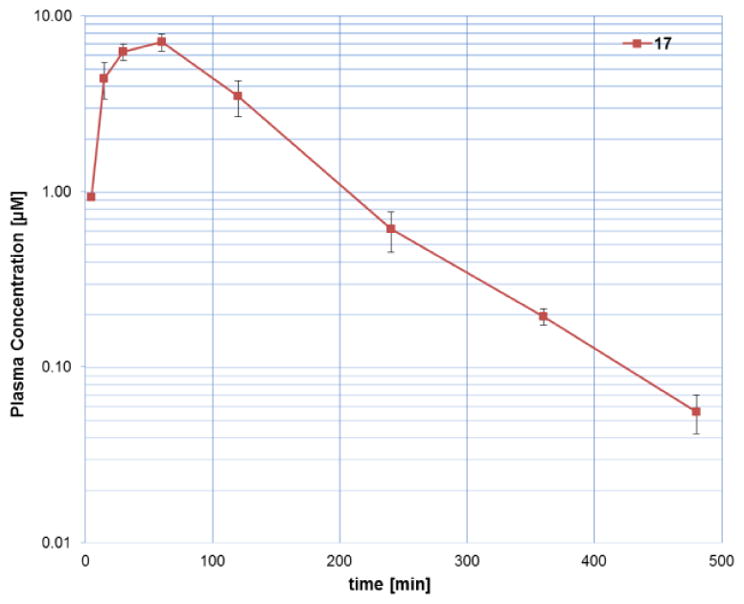
Plasma concentration versus time profile for 17 (points are the average of 3 mice) following IP administration at dose of 20 mg/kg using 10/10/80 DMSO/Tween/water.
3. Conclusions
We have developed a series of potent and selective purine-based CK1δ/ε inhibitors with excellent antiproliferative activities. A set of the most active compounds was also subjected to extensive physicochemical testing for solubility, permeability, and microsomal stability in an effort to predict their in vivo profile. These efforts led to the identification of 17 and 28 that has physical, in vitro and in vivo PK properties suitable for use in xenograph studies of human cancer. Such studies in mice are planned and will be reported in due course.
4. Experimental
4.1. Material and Methods
All reagents were purchased from commercial suppliers and were used without further purification. Dichloromethane, diethyl ether, N,N-dimethylformamide and tetrahydrofuran were dried by being passed through a column of desiccant (activated A-1 alumina). Triethylamine and diisopropyl amine was purified by distillation from calcium hydride. Reactions were either monitored by thin layer chromatography or analytical LC-MS. Thin layer chromatography was performed on Kieselgel 60 F254 glass plates pre-coated with a 0.25 mm thickness of silica gel. TLC plates were visualized with UV light and/or by staining with ninhydrin solution. Normal phase column chromatography was performed on a Biotage Isolera automated flash system. Compounds were loaded onto pre-filled cartridges filled with KP-Sil 50 μm irregular silica. For microwave reactions, a Biotage Initiator Microwave system was used. Some of the final products were isolated by reverse-phase HPLC using Shimadzu Prep LC system with photodiode array detector, Waters SunFire C18 OBD Prep Column, 100Å, 10 μm, 30 mm × 250 mm. Compounds were eluted using a gradient elution of 90/10 to 0/100 A/B over 10 min at a flow rate of 50.0 mL/min, where solvent A was water (+0.1 % TFA) and solvent B was acetonitrile/methanol (1:1).
The structures of all compounds were verified via 1H NMR, 13C NMR, 19F NMR and HPLC/HRMS. The purity of isolated products was determined using an LC-MS instrument (Agilent 1260 Infinity series LC with 500 Ion Trap MS) equipped with Kinetex® 5 μm EVO C18 100 Å LC Column 100 × 4.6 mm (Phenomenex) column. Elution was performed using the following conditions: 2% (v/v) acetonitrile (+0.1% FA) in 98% (v/v) H2O (+0.1% FA), ramped to 98% acetonitrile over 8 min, and holding at 98% acetonitrile for 1 min with a flow rate of 1.75 mL/min; UV absorption was detected from 200 to 950 nm using a diode array detector. The purity of each compound was ≥95% based on this analysis.
NMR spectra were recorded at ambient temperature on a 400 or 700 MHz Bruker NMR spectrometer in DMSO-d6. All 1H NMR data are reported in parts per million (ppm) downfield of TMS and were measured relative to the signals for dimethyl sulfoxide (2.50 ppm). All 13C NMR spectra are reported in ppm relative to the signals for dimethyl sulfoxide (39.5 ppm) with 1H decoupled observation. 19F NMR experiments were performed with 1H decoupling. Data for 1H NMR are reported as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), integration, and coupling constant (Hz), whereas 13C NMR analyses were obtained at 101 or 176 MHz and reported in terms of chemical shift. NMR data was analyzed and processed by using MestReNova software. High-resolution mass spectra were recorded on a spectrometer (ESI-TOF) at the University of Illinois Urbana-Champaign Mass Spectrometry Laboratory.
The synthesis and analysis of 2,6-dichloro-9-aryl-9H-purines 6 and benzimidazoles 9 were previously described.25, 28–29 Diaryliodonium salts 5 were prepared according to the reported procedures.30 Compounds 11–76 were newly synthesized and described in the Supporting Information.
4.2. General procedure for preparation of compounds 11–76
A 5 mL Biotage microwave vial was charged with 2,6-dichloropurine-9-(aryl/alkyl)-purine (1 eq, 0.35 mmol), 6, 2-(aminomethyl)-benzimidazole, 9, (or the benzylamine corresponding to R5CH2NH2 in Scheme 1) (1.1 eq 0.39 mmol), freshly distilled N,N-diisopropylethylamine (5 eq, 1.77 mmol, 0.31 mL), and isopropanol (1.4 mL. 0.25M). The vial was sealed with a microwave cap, and then the reaction mixture was heated to 90 °C for 30 minutes in microwave reactor. The completion of the reaction was confirmed by LC-MS and resulting product 10 was collected by filtration. In cases where the product was soluble in isopropanol, the reaction mixture was concentrated and used in the next step without further purification. A large excess (>30 eq) of amine corresponding to R4 (Scheme 1, N-methyl piperazine, morpholine, etc) was then added to the vial containing the concentrated crude mixture or collected solid. The vial was resealed and the reaction was heated to 130 °C for 30 minutes in the microwave unit (monitored by LC-MS). The cooled reaction mixture was concentrated on a rotary evaporator, then the crude product was purified by flash chromatography using 10 g Biotage column (gradient, 0%–10% MeOH in DCM over 25 column volumes). Collected fractions were washed with solution of saturated NH4Cl to remove remaining amine (monitored by TLC using ninhydrin stain). In some cases (as specified), reverse-phase HPLC was used after the normal phase column chromatography to obtain product with purity of >95%.
4.3. Biochemical Assays
CK1δ inhibitor IC50 values were measured by using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay. Briefly, final assay concentrations for CK1δ (Signal Chem), Ulight peptide substrate (ULight-Topo-Ila(Thr1342) peptide, Perkin Elmer) and ATP were 2 nM, 200 nM and 20 μM respectively. The reaction was performed at room temperature in a 10 μL final volume (384-well low volume plate, Greiner) containing: 50 mM Hepes, pH 7.5, 5 mM MgCl2, 0.1 mg/ml bovine serum albumin, 1 mM dl-dithiothreitol, 0.01% Triton X-100 and 1% DMSO (Sigma-Aldrich). After 10 min, the reaction was terminated by addition of 10 μL of 4 nM Eu-anti-p-Topo-Ila (Cat:TRF-0218, PerkinElmer) in Lance Detection Buffer (Cat: CR97-100, PerkinElmer). The fluorescent signal was detected using an EnVision plate reader (PerkinElmer). 10 point dose-response curves with 3–10 fold dilutions starting from 10 μM for each compound was generated in duplicate and data fit to a four parameter logistic.
4.4. Cell Culture and Proliferation Assays of CK1δ/ε Inhibitors
Human MDA-MB-231 breast cancer cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, LifeTechnologies) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C, 5% CO2. To evaluate the anti-proliferative activity of newly synthesized CK1δ/ε inhibitors against MDA-MB-231 breast cancer cells, cells were plated into a 384-well plate. 10 point dose-response curves with 3–10 fold dilutions starting from 10 μM for each compound was generated in duplicate including DMSO control. Cell proliferation was measured 72 h after compound treatment using CellTiter-Glo (Promega) according to the manufacturer’s instructions. EC50 values were determined by nonlinear regression and a four-parameter algorithm (GraphPad Prism 5).
4.5. Methods for In Vitro and In Vivo PK Studies
4.5.1. Solubility
Compounds from 10 mM DMSO stock solutions were introduced to pre-warmed pH 7.4 phosphate buffered saline in a 96-well plate. The final DMSO concentration was 1% and the plate was maintained at 37°C for 24 hours on an orbital shaker. The samples were centrifuged through a Millipore Multiscreen Solvinter 0.45 micron low binding PTFE hydrophilic filter plate and analyzed by HPLC with peak area compared to standards of known concentration.
4.5.2. Permeability
An assessment of permeability was done using a commercial PAMPA (Parallel Artificial Membrane Permeability Assay) kit from BD Biosciences (Cat# 353015).34 Compound (5μM) was added to 300 μl PBS in the bottom donor plate and 200 μl of blank PBS was added to the top receiver plate. The plates were incubated in an orbital shaker temperature at 37°C for 5 h, aliquots were taken from the donor and receiver plates and the concentration of drug was determined. Compound permeability was calculated using the equation
where Papp is expressed in units of cm/s, CA(t) is drug concentration in the acceptor at time t, VD is donor well volume, VA is acceptor well volume, A is the area of the filter (0.3 cm2), t is time in seconds.
4.5.3. Hepatic microsomal stability
Microsome stability was evaluated by incubating 1 μM compound with 1 mg/ml hepatic microsomes (human, rat, or mouse) in 100 mM potassium phosphate buffer, pH 7.4. The reactions were held at 37° C with continuous shaking. The reaction was initiated by adding NADPH, 1 mM final concentration. The final incubation volume was 300 μl and 40 μl aliquots were removed at 0, 5, 10, 20, 40, and 60 min. The removed aliquot was added to 160 μl acetonitrile to stop the reaction and precipitate the protein. NADPH dependence of the reaction was evaluated in parallel incubations without NADPH. At the end of the assay, the samples are centrifuged through a 0.45 micron filter plate (Millipore Solventer low binding hydrophilic plates, cat# MSRLN0450) and analyzed by LC-MS/MS. The data was log transformed and results are reported as half-life.
4.5.4. P450 inhibition
Cytochrome P450 inhibition was evaluated in human liver microsomes using four selective marker substrates (CYP1A2, phenaceten demethylation to acetaminophen; CYP2C9, tolbutamide hydroxylation to hydroxytolbutamide; CYP2D6, bufuralol hydroxylation to 4′-Hydroxybufuralol; and CYP3A4, midazolam hydroxylation to 1′-hydroxymidazolam) in the presence or absence of 10 μM test compound. The reaction is initiated by the addition of 1 mM NADPH and stopped after ten min by the addition of 2-times volume of acetonitrile containing dextrorphan as an internal standard. The concentration of each marker substrate is approximately its Km.39 Furafylline, sulfaphenazole, quinidine, and ketoconazole were included to each run to validate that the assay could identify selective inhibitors of each isoform.
4.5.5. Pharmacokinetics
All procedures described are covered under existing protocols and have been approved by the Scripps Florida IACUC to be conducted in the Scripps vivarium, which is fully AAALAC accredited. Pharmacokinetics were determined in n=3 male C57Bl/6 mice. Compounds were dosed as indicated in the text via intravenous injection via tail vein or oral gavage. 25 μL of blood was collected via a small nick in the tail using heparin coated hematocrit capillary tubes which were sealed with wax and kept on ice until plasma was generated by centrifugation using a refrigerated centrifuge equipped with a hematocrit rotor. Dose levels are provided in the text. Time points for determination of pharmacokinetic parameters were 5m, 15m, 30m, 1h, 2h, 4h, 6h, and 8h. Plasma concentrations were determined via LC-MS/MS using a nine point standard curve between 0.4 and 2000 ng/ml prepared in mouse plasma. Pharmacokinetic analysis was done with WinNonLin, Pharsight inc. using a noncompartimental model.
4.6. LogD calculations
cLogD values were calculated with Pipeline Pilot workflow application (Accelrys) at pH 7.4 as previously described.40
Supplementary Material
Acknowledgments
We thank Benjamin Huffman and Alexander Burns for contributions to the synthesis of several CK1δ/ε inhibitors in this work. We also thank the Scripps Florida NMR facility and Xiangming Kong for assistance. This work was supported in part by NIH NCI grant R01CA175094 (W.R.R., and D.R.D.) and NIH NCI NRSA postdoctoral fellowship F32CA200105 (A.M.)
A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cheong JK, Virshup DM. The International Journal of Biochemistry & Cell Biology. 2011;43:465. doi: 10.1016/j.biocel.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Fish KJ, Cegielska A, Getman ME, et al. J Biol Chem. 1995;270:14875. doi: 10.1074/jbc.270.25.14875. [DOI] [PubMed] [Google Scholar]
- 3.(a) Badura L, Swanson T, Adamowicz W, et al. J Pharmacol Exp Ther. 2007;322:730. doi: 10.1124/jpet.107.122846. [DOI] [PubMed] [Google Scholar]; (b) Gallego M, Virshup DM. Nat Rev Mol Cell Biol. 2007;8:139. doi: 10.1038/nrm2106. [DOI] [PubMed] [Google Scholar]
- 4.Pooler AM, Usardi A, Evans CJ, et al. Neurobiol Aging. 2012;33:431 e27. doi: 10.1016/j.neurobiolaging.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Hoekstra MF, Liskay RM, Ou AC, et al. Science. 1991;253:1031. doi: 10.1126/science.1887218. [DOI] [PubMed] [Google Scholar]
- 6.Li G, Yin H, Kuret J. J Biol Chem. 2004;279:15938. doi: 10.1074/jbc.M314116200. [DOI] [PubMed] [Google Scholar]
- 7.(a) Price MA. Genes Dev. 2006;20:399. doi: 10.1101/gad.1394306. [DOI] [PubMed] [Google Scholar]; (b) Peters JM, McKay RM, McKay JP, et al. Nature. 1999;401:345. doi: 10.1038/43830. [DOI] [PubMed] [Google Scholar]
- 8.Perreau-Lenz S, Vengeliene V, Noori HR, et al. Neuropsychopharmacology. 2012;37:2121. doi: 10.1038/npp.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knippschild U, Wolff S, Giamas G, et al. Onkologie. 2005;28:508. doi: 10.1159/000087137. [DOI] [PubMed] [Google Scholar]
- 10.Brockschmidt C, Hirner H, Huber N, et al. Gut. 2008;57:799. doi: 10.1136/gut.2007.123695. [DOI] [PubMed] [Google Scholar]
- 11.Kaucka M, Plevova K, Pavlova S, et al. Cancer Res. 2013;73:1491. doi: 10.1158/0008-5472.CAN-12-1752. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez N, Yang J, Hasselblatt K, et al. EMBO Mol Med. 2012;4:952. doi: 10.1002/emmm.201101094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SY, Dunn IF, Firestein R, et al. PLoS One. 2010;5:e8979. doi: 10.1371/journal.pone.0008979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirner H, Gunes C, Bischof J, et al. PLoS One. 2012;7:e29709. doi: 10.1371/journal.pone.0029709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Dey N, Barwick BG, Moreno CS, et al. BMC Cancer. 2013;13:537. doi: 10.1186/1471-2407-13-537. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Khramtsov AI, Khramtsova GF, Tretiakova M, et al. Am J Pathol. 2010;176:2911. doi: 10.2353/ajpath.2010.091125. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Khalil S, Tan GA, Giri DD, et al. PLoS One. 2012;7:e33421. doi: 10.1371/journal.pone.0033421. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schade B, Lesurf R, Sanguin-Gendreau V, et al. Cancer Res. 2013;73:4474. doi: 10.1158/0008-5472.CAN-12-3925. [DOI] [PubMed] [Google Scholar]
- 16.Rosenberg LH, Lafitte M, Quereda V, et al. Sci Transl Med. 2015;7:318ra202. doi: 10.1126/scitranslmed.aac8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knippschild U, Krüger M, Richter J, et al. Frontiers in Oncology. 2014:4. doi: 10.3389/fonc.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chijiwa T, Hagiwara M, Hidaka H. J Biol Chem. 1989;264:4924. [PubMed] [Google Scholar]
- 19.(a) Bettayeb K, Oumata N, Echalier A, et al. Oncogene. 2008;27:5797. doi: 10.1038/onc.2008.191. [DOI] [PubMed] [Google Scholar]; (b) Oumata N, Bettayeb K, Ferandin Y, et al. J Med Chem. 2008;51:5229. doi: 10.1021/jm800109e. [DOI] [PubMed] [Google Scholar]
- 20.(a) Behrend L, Milne DM, Stoter M, et al. Oncogene. 2000;19:5303. doi: 10.1038/sj.onc.1203939. [DOI] [PubMed] [Google Scholar]; (b) Cheong JK, Nguyen TH, Wang H, et al. Oncogene. 2011;30:2558. doi: 10.1038/onc.2010.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Rena G, Bain J, Elliott M, et al. EMBO Rep. 2004;5:60. doi: 10.1038/sj.embor.7400048. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) MacLaine NJ, Oster B, Bundgaard B, et al. J Biol Chem. 2008;283:28563. doi: 10.1074/jbc.M804433200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JW, Hirota T, Peters EC, et al. Angew Chem Int Ed Engl. 2011;50:10608. doi: 10.1002/anie.201103915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bischof J, Leban J, Zaja M, et al. Amino Acids. 2012;43:1577. doi: 10.1007/s00726-012-1234-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walton KM, Fisher K, Rubitski D, et al. J Pharmacol Exp Ther. 2009;330:430. doi: 10.1124/jpet.109.151415. [DOI] [PubMed] [Google Scholar]
- 25.Bibian M, Rahaim RJ, Choi JY, et al. Bioorg Med Chem Lett. 2013;23:4374. doi: 10.1016/j.bmcl.2013.05.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenberg LH, Lafitte M, Quereda V, et al. Sci Transl Med. 2015:7. doi: 10.1126/scitranslmed.aac8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang C, Lopez MS, Dar AC, et al. ACS Chem Biol. 2013;8:1931. doi: 10.1021/cb400376p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding S, Gray NS, Ding Q, et al. Tetrahedron Lett. 2001;42:8751. [Google Scholar]
- 29.Niu HY, Xia C, Qu GR, et al. Org Biomol Chem. 2011;9:5039. doi: 10.1039/c1ob05333g. [DOI] [PubMed] [Google Scholar]
- 30.(a) Bielawski M, Aili D, Olofsson B. J Org Chem. 2008;73:4602. doi: 10.1021/jo8004974. [DOI] [PubMed] [Google Scholar]; (b) Bielawski M, Malmgren J, Pardo LM, et al. ChemistryOpen. 2014;3:19. doi: 10.1002/open.201300042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Casagrande M, Barteselli A, Basilico N, et al. Bioorg Med Chem. 2012;20:5965. doi: 10.1016/j.bmc.2012.07.040. [DOI] [PubMed] [Google Scholar]
- 32.Roush WR, Rahaim R, Bibian M. Wee1 degradation inhibitors. Google Patents. 2016 [Google Scholar]
- 33.Gramec D, Masic LP, Dolenc MS. Chem Res Toxicol. 2014;27:1344. doi: 10.1021/tx500134g. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Murawski A, Patel K, et al. Pharm Res. 2008;25:1511. doi: 10.1007/s11095-007-9517-8. [DOI] [PubMed] [Google Scholar]
- 35.Zlokarnik G, Grootenhuis PDJ, Watson JB. Drug Discov Today. 2005;10:1443. doi: 10.1016/S1359-6446(05)03580-4. [DOI] [PubMed] [Google Scholar]
- 36.Long AM, Zhao H, Huang X. J Med Chem. 2012;55:10307. doi: 10.1021/jm301336n. [DOI] [PubMed] [Google Scholar]
- 37.Sastry GM, Adzhigirey M, Day T, et al. J Comput Aided Mol Des. 2013;27:221. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 38.(a) Friesner RA, Banks JL, Murphy RB, et al. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]; (b) Friesner RA, Murphy RB, Repasky MP, et al. J Med Chem. 2006;49:6177. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]; (c) Halgren TA, Murphy RB, Friesner RA, et al. J Med Chem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 39.Testino SA, Jr, Patonay G. J Pharm Biomed Anal. 2003;30:1459. doi: 10.1016/s0731-7085(02)00480-6. [DOI] [PubMed] [Google Scholar]
- 40.Csizmadia F, Tsantili-Kakoulidou A, Panderi I, et al. J Pharm Sci. 1997;86:865. doi: 10.1021/js960177k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.