

Abstract
Metal-catalyzed silylative dehydration of primary amides is an economical approach to the synthesis of nitriles. We report a copper–hydride(CuH)-catalyzed process that avoids a typically challenging 1,2-siloxane elimination step, thereby dramatically increasing the rate of the overall transformation relative to alternative metal-catalyzed systems. This new reaction proceeds at ambient temperature, tolerates a variety of metal-, acid-, or base-sensitive functional groups, and can be performed using a simple ligand, inexpensive siloxanes, and low catalyst loading.
Graphical Abstract
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.

Due to their unique reactivity and activating ability, nitriles are important functional groups in organic synthesis.1 Moreover, cyanated compounds frequently find applications in medicinal, biological, physical organic, and materials chemistry.2 Alongside substitution and rearrangement reactions that form C–CN bonds,3,4 the generation of nitriles by dehydration has been an important functional group interconversion,5,6 which is traditionally accomplished using harsh, acidic dehydrating reagents such as P4O10,5a POCl3,5b SOCl2,5c or TiCl4.5d
Recently, several research groups have reported silane-based dehydration reactions of primary amides, typically catalyzed by metals such as iron, ruthenium, or f-block elements.6 These elegant approaches feature several advantages: inexpensive hydrosilanes can be employed, only hydrogen gas and non-toxic siloxanes are produced as byproducts, and highly acidic and basic conditions can generally be avoided. Nevertheless, the reported examples require prolonged heating at refluxing temperatures and exhibit limited substrate scope, particularly if aliphatic amides are employed. It is thought that the harsh conditions are necessitated by the difficulty of a siloxane-releasing thermal syn-elimination step that is involved in each of the reaction mechanisms.6c,d,g,h,p Indeed, density functional theory (DFT) calculations suggest that this barrier is high for a typical silyl group (see Figure 1B).
Figure 1.

Overview of Amide-to-Nitrile Conversion. The computed transition state structure for the boxed step is shown in the bottom left. a Density functional theory (DFT) free energy calculations performed with 1,2-bis(dicyclohexylphosphino)ethane as the supporting ligand using M06/SDD-6-311+G(2d,p)/SMD(THF)//B3LYP/6-31G(d).
Copper–hydride(CuH)-catalyzed transformations have been extensively explored by our laboratory and others. These studies have focused mostly on stereoselective hydroamination, reduction and, more recently, hydrofunctionalization reactions.7 During our studies on imine allylation under CuH-catalysis,7j we were surprised to find that rather than undergoing the desired coupling reaction, substrates containing a primary amide functional group appeared to rapidly dehydrate to the corresponding nitriles at room temperature. Although initially we did not expect silylative dehydration under such mild conditions, we have previously reported that copper(I)-alkyl intermediates bearing a β-leaving group appear very prone to rapid elimination. For instance, a copper alkoxide is thought to be released through syn-elimination in the mechanisms of CuH-catalyzed hydroacylation using unsaturated acids,7h redox relay hydroamination,7p and reductive coupling with anhydrides.7q Suspecting by analogy that a particularly facile syn-elimination might also be involved here, we proposed the catalytic mechanism in Figure 1C.
We expected that the dehydrogenative silylation of a primary amide should first yield a monosilyl imidate. The O- and N-bound isomers are predicted by DFT to rapidly equilibrate, consistent with previous studies.6c,d,g From here, most reported methods are thought to proceed through a second silylation to produce a disilyl imidate, a species that we identified as the probable kinetic sink. When we performed DFT calculations in order to model the mechanism of our process, we found a much lower energy alternative pathway in the case of a bisphosphine-ligated copper catalyst. σ-Bond metathesis to form the disilyl imidate from the preceding intermediate is predicted to be slow, with a free energy barrier of over 30 kcal/mol; however, a nearly barrierless copper-alkoxide elimination is expected to proceed rapidly, which directly forms the desired nitrile product and LnCuOSiR3. The latter can close the mechanistic cycle through σ-bond metathesis with additional hydrosilane.8
Although concerted elimination to generate an sp-hybridized carbon center is relatively rare, we note that an analogous process, which generates a ketene, is proposed to be involved in CuH-catalyzed unsaturated acid reduction with support from both DFT calculations and multiple mechanistic experiments.7f The facile nature of this elimination step relative to the uncatalyzed elimination from the disilyl imidate may be due primarily to greatly reduced strain in the four-membered transition state. In the copper-catalyzed elimination transition state, the Cu–O and Cu–N bonds (251 pm and 199 pm) are longer on average than the Si–O and SiN bonds (187 pm and 200 pm) in the corresponding disilyl imidate elimination transition state, likely due to the larger size of Cu compared to Si. This reduced strain, combined with the thermodynamic favorability of forming a C–N triple bond, results in particularly rapid elimination.
With this hypothesis in mind, a more systematic study of this reaction initially focused on the examination of a selection of common mono- and diphosphines as supporting ligands. Using each of these, we subjected primary amide 1a to typical conditions for CuH catalysis (Table 1). At ambient temperature, most ligands, in combination with Cu(OAc)2, were effective in promoting the formation of the desired nitrile product 2a. In general, the use of chelating diphosphine ligands proved to be superior to monophosphine ligands. We chose DCyPE due to its effectiveness and commercial availability.9 We note that PMHS (polymethylhydrosiloxane), a very inexpensive polysiloxane, can be used as the stoichiometric dehydration agent (Table 1, entry 8); however, for most examples we have employed monomeric DMMS (dimethylmethoxysilane) since it can be obtained in more consistent quality and more uniform composition from commercial sources.10,11,12
Table 1.
Evaluation of Conditions for Amide Dehydrationa

| |||
|---|---|---|---|
| Entry | Ligand (n) | Silane (equiv) | Yieldb (%) |
| 1 | nonec | DMMS (3) | 0 |
| 2 | PCy3 (5) | DMMS (3) | 62 |
| 3 | PPh3 (5) | DMMS (3) | 65 |
| 4 | xantphos (5) | DMMS (3) | 50 |
| 5 | BINAP (5) | DMMS (3) | 82 |
| 6 | DM-BINAP (5) | DMMS (3) | 99 |
| 7 | DCyPE (2) | DMMS (2.5) | 97 |
| 8 | DCyPE (2) | PMHS (3) | 91 |
Reaction conditions: amide (0.1 mmol), copper(II) acetate (n mol%), ligand (1.1n mol%), silane (indicated equiv), in THF (0.1 mL) at RT for 12 h. See supporting information for additional details.
The yield was assessed by GC analysis of the crude reaction mixture, using dodecane as an internal standard.
5 mol% copper(II) acetate was used, but no additional ligand was added.
When we compared this optimized procedure with representative classical and modern methods (Table 2) on four simple substrates, we found that only our system provided high yields on each of these amides. We determined that a wide range of amides could be converted efficiently to the corresponding nitrile under these optimized conditions (Table 3). Simple α-primary (2b, 2c), secondary (2a), and tertiary (2d) amides were all compatible substrates. In addition, a variety of aromatic amides were suitable substrates, regardless of whether they bore electron-withdrawing (2g), -donating (2h), or protic (2f) substituents. Moreover, several classes of heterocyclic amides, including a thiophene (2i), pyridine (2l), pyrrolidine (2j, 2k), and piperidine (2m) were successfully transformed. The mild reaction conditions were compatible with (hetero)aryl halides (2g, 2i), and acid-sensitive carbamate protecting groups like Boc (2l). Importantly, since there is no base or acid generated in the reaction, no epimerization of labile α-stereocenters is observed during the process (2k, 100% enantiospecificity).
Table 2.
Yield Comparison with Alternative Methods of Nitrile Dehydrationa
| Method |

|

|

|

|
|---|---|---|---|---|
| 2% [Cu], DMMS, rt | 97% | 85% | 80% | 93% |
| 5% [Fe], DEMS, 100 °C6g | 85% | 23% | 75% | 49% |
| 5% [Fe], DEMS, rt | <5% | <5% | 24% | <5% |
| POCl3, Et3N, rt | 98% | 27% | 38% | 42% |
| T3P, 100 °C | 86% | 48% | 13% | 68% |
| 5% TBAF, PhSiH3, 100 °C6p | 87% | 8% | 62% | 47% |
| 5% TBAF, PhSiH3, rt | 7% | <5% | 16% | 5% |
Yields were assessed by GC analysis of the crude reaction mixture, using dodecane as an internal standard. See supporting information for reaction conditions and details.
Table 3.
Examples of CuH-Catalyzed Amide Dehydrationa

|
Reaction conditions: amide (1.0 mmol), copper(II) acetate (0.02 equiv), 1,2-bis(dicyclohexylphosphino)ethane (0.022 equiv), dimethoxy(methyl)silane (3 equiv) in THF (1 mL) at RT for 12 h. Yield represents the average isolated yield of two or more replicates. The yield enclosed in parentheses indicates the isolated yield of a single run wherein polymethylhydrosiloxane (PMHS, 4 equiv) was used instead of dimethoxy(methyl)silane. See supporting information for additional details.
Toluene (1 mL) was used instead of THF.
At 50 °C instead of RT.
4 equiv silane was used.
Several experiments on more complex substrates were conducted in order to demonstrate the synthetic utility of these conditions (Table 4). The corresponding nitriles were obtained from some prototypical compounds of biological interest, such as a mimic of the eugeroic drug modafinil (2n), an NSAID (naproxen, 2o, 100% retention at the α-stereocenter), a protected sugar (2p), a beta-blocker drug (atenolol, 2q), and a drug bearing a potentially labile N-acyl indole (indomethacin, 2r). In these cases, even though many amide substrates are relatively insoluble in common organic solvents, we found that each compound slowly dissolves as the silylative process proceeds. Under nearly identical reaction conditions, we also observed the dehydration of a urea to generate common peptide coupling reagent N,N-dicyclohexylcarbodiimide (2s). We note that this transformation can be stopped at this stage since subsequent reduction of the carbodiimide group to an amidine is significantly slower. Finally, the reaction can be conducted on a preparatively useful scale with 1.0 mol % catalyst loading without need for flame-dried glassware or inert-atmosphere glovebox equipment (Figure 2).
Table 4.
Further Examples of Amide Dehydrationa

|
Reaction conditions: amide (0.5 or 1.0 mmol), copper(II) acetate (0.02 equiv), 1,2-bis(dicyclohexylphosphino)ethane (0.022 equiv), dimethoxy(methyl)silane (3 equiv) in THF (1 mL) at RT for 12 h, unless otherwise specified; see supporting information for details. Yield represents average isolated yield of two or more replicates.
5 equiv silane was used.
At 40 °C instead of RT; 1,4-dioxane (2 mL) was used instead of THF.
Yield as measured by gas chromatography, relative to an internal standard.
Figure 2.

Multigram-Scale Reaction at Low Catalyst Loading, with Inexpensive PMHS as the Silane.
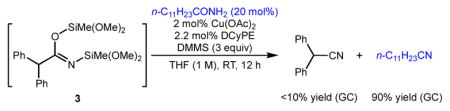
At this point, we performed some mechanistic experiments to investigate whether our original hypothesis explained the efficiency of this dehydration process. We considered the low temperature at which this reaction proceeds to be suggestive of a distinct operative mechanism compared to previously reported high-temperature procedures. More specifically, we wanted a way to directly test if we were successfully able to divert the reaction sequence away from the problematic disilyl imidate intermediate. In particular, as shown in Figure 3, our hypothesis requires that the monosilyl imidate 2 be a kinetically competent intermediate on the reaction pathway, but the disilyl imidate 3 may not be competent under the same conditions. Noting that copper(II) salts without additional ligands can catalyze the dehydrogenative silylation of acidic groups,13 but cannot catalyze the net dehydration reaction (Table 1, entry 1), we first reacted amide 1 with either a single equivalent of silane or a large excess (5 equiv) of silane, in the presence of copper(II) acetate, to generate putative intermediates 2 and 3 respectively. We then subjected the corresponding crude mixtures to the reaction conditions, including additional DCyPE-ligated catalyst. As expected, in the former case, a good yield of product was obtained; yet, only trace product could be detected in the latter case, which gave nearly quantitative (92%) recovery of starting material after protic work-up.14
Figure 3.

Investigation of the Role of Mono- and Disilyl Imidate Intermediates.
In summary, we have identified a function-group-tolerant copper-catalyzed method for the synthesis of nitriles, which circumvents the uncatalyzed thermal elimination step required by typical metal-catalyzed processes. Although further studies are underway to elucidate the fundamental metal-ligand interactions responsible for facilitating this low-barrier elimination pathway, we believe that this type of general process may serve as a platform for the design of other useful, non-reductive CuH-catalyzed reactions.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health (GM058160, GM122483, GM058160-17S1). The content of this communication solely reflects the research and opinion of the authors and does not necessarily represent the official views of the NIH. R.Y.L thanks MIT for Presidential Graduate Fellowships and Bristol–Myers Squibb for a Fellowship in Synthetic Organic Chemistry. M.B. thanks Sam-sung Group for a Samsung Scholarship and MIT for support through the Undergraduate Research Opportunities Program. We are grateful to Drs. Andy Thomas and Christine Nguyen for advice on the preparation of this manuscript.
Footnotes
Notes
The authors declare no competing financial interests.
The Supporting Information is available free of charge on the ACS Publications website.
Full procedures, computational details, characterization for known and new compounds (PDF).
References
- 1.(a) Subramanian LR. Nitriles. In: Trost BM, Lautens M, editors. Science of Synthesis. Vol. 19. Thieme; Stuttgart: 2011. p. 79. [Google Scholar]; (b) Katritzky AR, Taylor RJK, editors. Comprehensive Organic Functional Group Transformations II. 2. Vol. 2 Elsevier; Amsterdam: 2004. [Google Scholar]
- 2.(a) Acton QA. Nitriles—Advances in Research and Application, 2013 Edition. ScholarlyEditions; Atlanta: 2013. [Google Scholar]; (b) Pollak P, Romeder G, Hagedorn F, Gelbke H-P. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley; Weinheim: 2012. Nitriles. [Google Scholar]; (c) Fleming FF, Yao L, Ravikumar PC, Funk L, Shook BC. J Med Chem. 2010;53:7902. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fleming FF, Wang Q. Chem Rev. 2003;103:2035. doi: 10.1021/cr020045d. [DOI] [PubMed] [Google Scholar]; (e) Kleeman A, Engels J, Kutscher B, Reichert D. Pharmaceutical Substances: Syntheses, Patents and Applications. 4. Thieme; Stuttgart: 2001. [Google Scholar]; (f) Miller JS, Manson JL. Acc Chem Res. 2001;34:563. doi: 10.1021/ar0000354. [DOI] [PubMed] [Google Scholar]; (g) Gregory RJH. Chem Rev. 1999;99:3649. doi: 10.1021/cr9902906. [DOI] [PubMed] [Google Scholar]; (h) Rappoport Z, editor. The Chemistry of the Cyano Group. Wiley; Weinheim: 1971. [Google Scholar]
- 3.For a recent, comprehensive overview of nitrile synthesis, see: Larock RC, Yao T. Comprehensive Organic Transformations. 3. Wiley; 2018. Formation of Nitriles, Carboxylic Acids, and Derivatives by Oxidation, Substitution, and Addition.
- 4.For selected modern methods of preparing nitriles, see, along with ref. 6, the following: Wang D, Zhu N, Chen P, Lin Z, Liu G. J Am Chem Soc. 2017;139:15632. doi: 10.1021/jacs.7b09802.McManus JB, Nicewicz DA. J Am Chem Soc. 2017;139:2880. doi: 10.1021/jacs.6b12708.Shi S, Szostak M. Org Lett. 2017;19:3095. doi: 10.1021/acs.orglett.7b01199.Zhang W, Wang F, McCann SD, Wang D, Chen P, Stahl SS, Liu G. Science. 2016;353:1014. doi: 10.1126/science.aaf7783.Fang X, Yu P, Morandi B. Science. 2016;351:832. doi: 10.1126/science.aae0427.Wu Q, Luo Y, Lei A, You J. J Am Chem Soc. 2016;138:2885. doi: 10.1021/jacs.5b10945.Ping Y, Ding Q, Peng Y. ACS Catal. 2016;6:5989.Cohen DT, Buchwald SL. Org Lett. 2015;17:202. doi: 10.1021/ol5032359.Jagadeesh RV, Junge H, Beller M. Nat Commun. 2014;5:4123. doi: 10.1038/ncomms5123.Yu L, Li H, Zhang X, Ye J, Liu J, Xu Q, Lautens M. Org Lett. 2014;16:1346. doi: 10.1021/ol500075h.Lambert KM, Bobbitt JM, Eldirany SA, Wiberg KB, Bailey WF. Org Lett. 2014;16:6484. doi: 10.1021/ol503345h.Shen T, Wang T, Qin C, Jiao N. Angew Chem Int Ed. 2013;52:6677. doi: 10.1002/anie.201300193.Yin W, Wang C, Huang Y. Org Lett. 2013;15:1850. doi: 10.1021/ol400459y.Dornan LM, Cao Q, Flanagan JCA, Crawford JJ, Cook MJ, Muldoon MJ. Chem Commun. 2013;49:6030. doi: 10.1039/c3cc42231c.Shimojo H, Moriyama K, Togo H. Synthesis. 2013;45:2155.Enthaler S, Weidauer M, Schroeder F. Tetrahedron Lett. 2012;53:882.Laulhé S, Gori SS, Nantz MH. J Org Chem. 2012;77:9334. doi: 10.1021/jo301133y.Anbarasan P, Schareina T, Beller M. Chem Soc Rev. 2011;40:5049. doi: 10.1039/c1cs15004a.Liskey CW, Liao X, Hartwig JF. J Am Chem Soc. 2010;132:11389. doi: 10.1021/ja104442v.Veisi H. Synthesis. 2010:2631.Augustine JK, Atta RN, Ramappa BK, Boodappa C. Synlett. 2009:3378.Nicolaou KC, Mathison CJN. Angew Chem Int Ed. 2005;44:5992. doi: 10.1002/anie.200501853.Czekelius C, Carreira EM. Angew Chem Int Ed. 2005;44:612. doi: 10.1002/anie.200461879.Nielsen MA, Nielsen MK, Pittelkow T. Org Proc Res Dev. 2004;8:1059.Zanon J, Klapars A, Buchwald SL. J Am Chem Soc. 2003;125:2890. doi: 10.1021/ja0299708.Ellis GP, Romney-Alexander TM. Chem Rev. 1987;87:779.
- 5.(a) Reisner DB, Horning EC. Org Synth. 1963:144. Coll. IV. [Google Scholar]; (b) Rickborn B, Jensen FR. J Org Chem. 1962;27:4608. [Google Scholar]; (c) Krynitsky JA, Carhart HW. Org Synth. 1963:436. Coll. IV. [Google Scholar]; (d) Lehnert W. Tetrahedron Lett. 1971;19:1501. [Google Scholar]
- 6.For examples of metal-catalyzed dehydrative synthesis of nitriles from amides, see: Xue B, Sun H, Wang Y, Zheng T, Li X, Fuhr O, Fenske D. Catal Commun. 2016;86:148.Elangovan S, Duque SQ, Dorcet V, Roisnel T, Norel L, Darcel C, Sortais JB. Organometallics. 2015;34:4521.Enthaler S, Inoue S. Chem Asian J. 2012;7:169. doi: 10.1002/asia.201100493.Enthaler S. Chem Eur J. 2011;17:9316. doi: 10.1002/chem.201101478.Enthaler S, Weidauer M. Catal Lett. 2011;141:1079.Sueoka S, Mitsudome T, Mizugaki T, Jitsukawa K, Kaneda K. Chem Commun. 2010;46:8243. doi: 10.1039/c0cc02412k.Zhou S, Addis D, Das S, Junge K, Beller M. Chem Commun. 2009:4883. doi: 10.1039/b910145d.Hanada S, Motoyama Y, Nagashima H. Eur J Org Chem. 2008:4097. doi: 10.1021/jo070591c.Campbell JA, McDougald G, McNab H, Rees LVC, Tyas RG. Synthesis. 2007:3179.Furuya Y, Ishihara K, Yamamoto H. Bull Chem Soc Jpn. 2007;80:400.Maffioli SI, Marzorati E, Marazzi A. Org Lett. 2005;7:5237. doi: 10.1021/ol052100l.Ishihara K, Furuya Y, Yamamoto H. Angew Chem Int Ed. 2002;41:2983. doi: 10.1002/1521-3773(20020816)41:16<2983::AID-ANIE2983>3.0.CO;2-X.Watanabe Y, Okuida F, Tsuji Y. J Mol Catat. 1990;58:87.Blum J, Fisher A, Greener E. Tetrahedron. 1973;29:1073.Blum J, Fisher A. Tetrahedron Lett. 1970;11:1963.This elegant example using catalytic fluoride is also highly relevant: Zhou S, Junge K, Addis D, Das S, Beller M. Org Lett. 2009;11:2461. doi: 10.1021/ol900716q.
- 7.For reviews on CuH catalysis, see: Pirnot MT, Wang YM, Buchwald SL. Angew Chem, Int Ed. 2016;55:48. doi: 10.1002/anie.201507594.Jordan AJ, Lalic G, Sadighi JP. Chem Rev. 2016;116:8318. doi: 10.1021/acs.chemrev.6b00366.Rendler S, Oestreich M. Angew Chem Int Ed. 2007;46:498. doi: 10.1002/anie.200602668.Lipshutz BH. In: Modern Organocopper Chemistry. Krause N, editor. Wiley-VCH; Weinheim: 2002. pp. 167–187.Deutsch C, Krause N, Lipshutz BH. Chem Rev. 2008;108:2916. doi: 10.1021/cr0684321. For selected examples of recently discovered CuH-catalyzed transformations, see: Zhou Y, Bandar JS, Liu RY, Buchwald SL. J Am Chem Soc. 2018;140:606. doi: 10.1021/jacs.7b12260.Lee J, Torker S, Hoveyda AH. Angew Chem, Int Ed. 2017;56:821. doi: 10.1002/anie.201611444.Zhou Y, Bandar JS, Buchwald SL. J Am Chem Soc. 2017;139:8126. doi: 10.1021/jacs.7b04937.Friis SD, Pirnot MT, Dupuis LN, Buchwald SL. Angew Chem Int Ed. 2017;56:7242. doi: 10.1002/anie.201703400.Liu RY, Yang Y, Buchwald SL. Angew Chem Int Ed. 2016;55:14077. doi: 10.1002/anie.201608446.Han JT, Jang WJ, Kim N, Yun J. J Am Chem Soc. 2016;138:15146. doi: 10.1021/jacs.6b11229.Xi Y, Butcher TW, Zhang J, Hartwig JF. Angew Chem, Int Ed. 2016;55:77. doi: 10.1002/anie.201509235.Xi Y, Hartwig JF. J Am Chem Soc. 2016;138:6703. doi: 10.1021/jacs.6b02478.Yang Y, Perry IB, Lu G, Liu P, Buchwald SL. Science. 2016;353:144. doi: 10.1126/science.aaf7720.Wang YM, Buchwald SL. J Am Chem Soc. 2016;138:5024. doi: 10.1021/jacs.6b02527.Zhu S, Niljianskul N, Buchwald SL. Nat Chem. 2016;8:144. doi: 10.1038/nchem.2418.Bandar JS, Ascic E, Buchwald SL. J Am Chem Soc. 2016;138:5821. doi: 10.1021/jacs.6b03086.Yang Y, Shi SL, Liu P, Buchwald SL. Science. 2015;349:62. doi: 10.1126/science.aab3753.Zhu S, Niljianskul N, Buchwald SL. J Am Chem Soc. 2013;135:15746. doi: 10.1021/ja4092819.Miki Y, Hirano K, Satoh T, Miura M. Angew Chem Int Ed. 2013;52:10830. doi: 10.1002/anie.201304365.
- 8.For early examples establishing the utility of hydrosilanes in converting Cu–OR complexes to CuH complexes, see: Lee DW, Yun J. Tetrahedron Lett. 2005;46:2037.Lipshutz BH, Servesko JM, Petersen TB, Papa PP, Lover AA. Org Lett. 2004;6:1273. doi: 10.1021/ol0400185.Chiu P, Li Z, Fung KCM. Tetrahedron Lett. 2003;44:455.Moritani Y, Appella DH, Jurkauskas V, Buchwald SL. J Am Chem Soc. 2000;122:6967.Lipshutz BH, Chrisman W, Noson K, Papa P, Scalfani JA, Vivian RW, Keith JM. Tetrahedron. 2000;56:2779.Mori A, Fujita A, Kajiro H, Nishihara Y, Hiyama T. Tetrahedron. 1999;55:4573.Appella DH, Moritani Y, Shintani R, Ferreira EM, Buchwald SL. J Am Chem Soc. 1999;121:9473.Lipshutz BH, Kieth J, Papa P, Vivian R. Tetrahedron Lett. 1998;39:4627.
- 9.For copper-catalyzed transformations for which DCyPE is an effective ligand, see ref. 7j, and: Gribble MWG, Jr, Pirnot MT, Bandar JS, Liu RY, Buchwald SL. J Am Chem Soc. 2017;139:2192. doi: 10.1021/jacs.6b13029.Wakamatsu T, Nagao K, Ohmiya H, Sawamura M. Organometallics. 2016;35:1354.Van Hoveln R, Hudson BM, Wedler HB, Bates DM, Le Gros G, Tantillo DJ, Schomaker JM. J Am Chem Soc. 2015;137:5346. doi: 10.1021/ja511236d.Grigg RD, Van Hovelin R, Schomaker JM. J Am Chem Soc. 2012;134:16131. doi: 10.1021/ja306446m.
- 10.Commercial PMHS can be obtained in highly variable quality, in several different chain lengths, and with a choice of terminal groups. We evaluated several batches of PMHS from different vendors and of different ages, and the model reaction was successful in all cases. However, the yields can vary by up to 20% between batches without obvious indication of cause, unless a larger excess (4 equiv) of PMHS is employed. In the interest of maximum reproducibility, we chose to use DMMS as the silane in the substrate scope studies. The use of PMHS is demonstrated on several representative substrates in Table 2, and on large scale in Figure 2.
- 11.For this study, we obtained DMMS from TCI America, and the material was stored without further purification in the refrigerator at 0 °C for up to 4 months after opening. 1H NMR analysis indicated no detectable decomposition, including methanol formation by hydrolysis. Even if slight decomposition should occur, minimal detrimental effect is expected since the addition of 0.1 equiv of either water or MeOH did not significantly alter the yield of the model reaction. Furthermore, several batches of DMMS of different age and storage condition were employed with minimal variation in yield.
- 12.For detailed information about the safe handling of DMMS, please see the supporting information.
- 13.Alcoholysis and aminolysis of Si–H bonds is known to be catalyzed by a variety of metals and metal salts; for instance, see: Lukevics E, Dzintara M. J Organomet Chem. 1985;295:265. and references therein.
-
14.To confirm that the failure of the second experiment is due to the conversion of 1 to an inactive species, rather than, for instance, decomposition of the catalyst in the presence of some detrimental contaminant, we added a small amount of a second primary amide to 3and conducted the CuH-catalyzed dehydration reaction. The unsilylated amide was fully converted to the corresponding, while 3 was not converted to the corresponding nitrile:
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.