

SUMMARY
Objectives
Anti-seizure drugs are the leading therapeutic choice for treatment of epilepsy, but their efficacy is limited by pharmacoresistance and the occurrence of unwanted side effects. Here, we examined the therapeutic efficacy of KCNQ channel activation by retigabine in preventing seizures and neurocardiac dysfunction in two potassium channelopathy mouse models of epilepsy with differing severity that have been associated with increased risk of sudden unexpected death in epilepsy (SUDEP): the Kcna1−/− model of severe epilepsy and the Kcnq1A340E/A340E model of mild epilepsy.
Methods
A combination of behavioral, seizure threshold, electrophysiological, and gene expression analyses was used to determine the effects of KCNQ activation in mice.
Results
Behaviorally, Kcna1−/− mice exhibited unexpected hyperexcitability instead of the expected sedative-like response. In flurothyl-induced seizure tests, KCNQ activation decreased seizure latency by ≥50% in Kcnq1 strain mice but had no effect in the Kcna1 strain, suggesting the influence of genetic background. However, in simultaneous electroencephalography and electrocardiography recordings, KCNQ activation significantly reduced spontaneous seizure frequency in Kcna1−/− mice by ~60%. In Kcnq1A340E/A340E mice, KCNQ activation produced adverse cardiac effects including profound bradycardia and abnormal increases in heart rate variability and atrioventricular conduction blocks. Analyses of Kcnq2 and Kcnq3 mRNA levels revealed significantly elevated Kcnq2 expression in Kcna1−/− brains, suggesting drug target alterations may contribute to the altered drug responses.
Significance
This study shows that treatment strategies in channelopathy may have unexpected outcomes and that effective rebalancing of channel defects requires improved understanding of channel interactions at the circuit and tissue levels. The efficacy of KCNQ channel activation and manifestation of adverse effects were greatly affected by genetic background, potentially limiting KCNQ modulation as a way to prevent neurocardiac dysfunction in epilepsy and thereby SUDEP risk. Our data also uncovers a potential role for KCNQ2-5 channels in autonomic control of chronotropy.
Keywords: retigabine, Kcna1, Kcnq1, Kv1.1, Kv7.1
INTRODUCTION
Studies in mouse models suggest that activation of voltage-gated KCNQ (Kv7) potassium channels could be an effective pharmacological approach to prevent aberrant neurocardiac activity and thereby decrease risk for sudden unexpected death in epilepsy (SUDEP) risk. In ex vivo experiments using Kcna1 knockout (Kcna1−/−) mice, a well-characterized model of SUDEP involving brain-heart dysregulation via the parasympathetic nervous system, Kcna1−/− vagal axons exhibit abnormal spontaneous firing that is almost completely abolished by augmenting Kv7 currents with the KCNQ channel opener flupirtine.1 In Sentrin/SUMO-specific protease 2 deficient mice, which exhibit seizures and sudden death as a result of hyper-SUMOylation of multiple potassium channels, in vivo treatment with retigabine (RTG), a more potent analog of flupirtine, prevents acoustically-induced seizures and vagally-mediated, seizure-associated atrioventricular (AV) blocks.2 Thus, KCNQ openers show promise for preventing SUDEP in cases involving neurocardiac or parasympathetic mechanisms since they can not only suppress seizures, but also potentially help maintain normal vagal activity.
Here, we explored the potential of KCNQ channel activation as a pharmacological strategy to prevent seizures and neurocardiac dysfunction in two potassium channelopathy mouse models of epilepsy of differing severity that have been associated with sudden death: the Kcna1−/− model of severe epilepsy and the Kcnq1A340E/A340E model of mild epilepsy. Kcna1−/− mice lack Kv1.1 voltage-gated potassium channel α-subunits and exhibit several key features of human SUDEP including severe early-onset epilepsy characterized by frequent generalized tonic-clonic seizures (up to ~24/d); premature sudden death beginning at a young age (~P16); and seizure-evoked bradyarrhythmias that progress to cardiac arrest.3–6 Kv1.1 channel mutations have been identified as a molecular cause of epilepsy in patients and a de novo copy number variant in KCNA1 was identified in a human case of SUDEP.7,8
In contrast, Kcnq1A340E/A340E mice carry a dominant negative missense mutation (A340E) leading to loss-of-function of the Kcnq1 gene, which encodes KCNQ1 (Kv7.1) voltage-gated potassium channel α-subunits. The A340E mutation in mice causes infrequent spontaneous seizures (~0.5/d) and multiple cardiac abnormalities, including AV blocks that occur concomitantly with cortical discharges suggesting they are brain-driven.9 Although SUDEP has not been documented in Kcnq1A340E/A340E mice, the homologous A341E mutation in humans causes long QT syndrome type 1 (LQT1), which is strongly associated with sudden death due to ventricular tachyarrhythmias.9,10 KCNQ1 mutations have been identified in patients with epilepsy or a history of seizures suggesting KCNQ1 is also a human epilepsy gene.11,12 In this study, we tested the hypothesis that KCNQ channel activation with RTG normalizes neurocardiac function in Kcna1−/− and Kcnq1A340E/A340E mice leading to a reduction in seizure susceptibility and cardiac dysfunction. RTG is a potent activator of KCNQ2 (Kv7.2) and KCNQ3 (Kv7.3) voltage-gated postassium channel α-subunits but has no effect on KCNQ1.13 Our results reveal unexpected effects of genotype on the efficacy and tolerability of KCNQ activation, as well as potentially new cardiovascular roles for KCNQ2-5 channels in the autonomic control of chrontropy.
MATERIALS AND METHODS
Animals and Genotyping
Kcna1−/− knockout (KO) mice (Tac:N:NIHS-BC genetic background) carry null alleles of the Kcna1 gene resulting from targeted deletion of the open reading frame, as previously described.6 Kcnq1A340E/A340E mutant mice (C57BL/6 genetic background) carry a point mutation of the Kcnq1 gene, as previously described.14 Both strains were maintained by breeding heterozygous mice with non-sibling heterozygous mice of the same strain. Animals were housed at 22°C, fed irradiated rodent chow ad libitum (PicoLab Diet 20; LabDiet, St. Louis, MO), and submitted to a 12 h light/dark cycle. Mice of both sexes were used in experiments. All procedures were performed in accordance with the guidelines of the National Institutes of Health (NIH), as approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center-Shreveport. Genotyping was performed as described previously (see Supporting Information for details).3,9
Drug Administration
Retigabine dihydrochloride (RTG; Alomone Labs, Jerusalem, Israel) was administered at a concentration of 5, 10, or 20 mg·kg−1, as specified in the text. RTG was dissolved in 0.9% NaCl vehicle (VEH) and injected intraperitoneally (ip) at a volume of 10 mL·kg−1.
Behavioral Studies
Individually housed Kcna1 and Kcnq1 homozygous mutants, heterozygotes, and WT littermate controls (4–6 weeks old; n = 6 per genotype) were injected once daily for four days with vehicle or RTG according to the following injection schedule: VEH was given the first day (day 1), followed by progressively increasing RTG doses of 5 mg·kg−1 (day 2), 10 mg·kg−1 (day 3), and 20 mg·kg−1 (day 4). Injections were always performed at a similar time of day between 9:30 am to 2:00 pm. Behavioral responses to VEH or drug were video-monitored for 1 hour post-injection using digital network cameras (DCS-942L, D-Link, Fountain Valley, CA). During the first 30 min post-injection, the following behaviors were manually scored offline and quantified by an observer blinded to genotype: immobility, myoclonus, hopping, and vocalizing.
Flurothyl-Induced Seizure Susceptibility
Flurothyl-induced seizure latencies were measured using the same procedure as described previously (see Supporting Information for details).4 Forty minutes prior to seizure induction, mice (age P29–31) were administered VEH or RTG (10 mg·kg−1, ip). Mice were then placed in an air-tight Plexiglas chamber (18.4 × 15.0 × 34.5 cm) and allowed to acclimate for five minutes before exposure to the liquid convulsant flurothyl (2,2,2-trifluoroethyl ether; Sigma-Aldrich, St. Louis, MO).
Simultaneous Electroencephalography-Electrocardiography (EEG-ECG) Recordings
Mice (~4 weeks old) were anesthetized with avertin (0.02 mL·g−1, ip) and surgically implanted with bilateral silver wire EEG (temporal and frontal) and ECG (thoracic) electrodes (0.005-inch diameter) attached to a microminiature connector for recording in a tethered configuration as done previously (see Supporting Information for details).4 Avertin was prepared by mixing 250 mg 2,2,2-tribromoethanol (Sigma-Aldrich, St. Louis, MO) with 155 μL 2-methyl-2-butanol (Sigma-Aldrich, St. Louis, MO) dissolved in 19.75 mL double-distilled water heated to 40°C, followed by filter-sterilization prior to use. Simultaneous video and cortical EEG-ECG activity were recorded in freely moving mice using a digital video EEG-ECG monitoring system (Data Sciences International; St. Paul, MN). Mice were monitored by video EEG-ECG over an 8-h daytime recording period for 3 consecutive days. Vehicle was administered at the beginning of each 8-h recording session, followed by RTG 4 h later. To determine drug effects, mice were used as their own controls by comparing their responses to VEH and drug. EEG-ECG recordings were analyzed offline using Ponemah software (Data Sciences International; St. Paul, MN) to manually identify spontaneous seizures and cardiac events by an observer blinded to genotype as described in detail in the Supporting Information.
Heart Rate (HR) and Heart Rate Variability (HRV) Analyses
Ponemah software (Data Sciences International; St. Paul, MN) was used to automatically generate RR interval data for the first day of EEG-ECG recording for each animal. The RR intervals were used to calculate the average HR for each minute of recording for the 2 h immediately following VEH and RTG administration. For HR and HRV analyses, values for each animal were derived by averaging three stable 3-min ECG segments when the mouse was mostly stationary, beginning at 30-min after administration of either VEH or RTG. HRV was measured in the time domain using the standard deviation of the RR intervals (SDNN) and the root mean square of successive differences (RMSSD) as done previously.4 All cardiac parameters were measured during the light phase of the day between 10:30 a.m. and 4:30 p.m.
Quantitative PCR (qPCR) Analyses
Following isoflurane euthanasia, brains of mice (age P29–31) were quickly harvested, dissected, and collected. Total RNA was isolated using TRI-Reagent® (Zymo Research, Irvine, CA), purified using PureLink® RNA Mini Kit (Thermo Fisher, Waltham, MA), and processed for relative qPCR analyses of mRNA transcript levels using TaqMan probes (Thermo Fisher, Waltham, MA), as described in detail in the Supporting Information. Reactions were normalized to the housekeeping gene hypoxanthine phosphoribosyl-transferase 1 (Hprt1). Relative mRNA expression was calculated as normalized values by use of the 2−ΔCt formula.
Statistical Analyses
All data are expressed as means ± standard deviation. Prism 6 for Windows (GraphPad Software Inc, La Jolla, CA) was used for statistical analyses. For comparisons involving only two groups, either paired or unpaired two-tailed Student’s t tests were employed as appropriate. For comparisons involving three or more groups, one-way analysis of variance (ANOVA) or two-way repeated measures ANOVA were performed with either Tukey’s or Sidak’s multiple comparison post hoc tests.
RESULTS
RTG increases behavioral excitability in Kcna1−/− mice
Age-matched Kcna1 and Kcnq1 mutants (homozygotes and heterozygotes) and WT littermate controls were injected with either VEH or RTG (5, 10, or 20 mg·kg−1, ip) followed by video monitoring to observe behavior. KCNQ channel openers have muscle-relaxant properties in rodents,15,16 so we hypothesized that RTG would induce this type of behavior, especially at higher doses. At the highest dose tested (20 mg·kg−1), WT and heterozygous mice of both strains exhibited varying degrees of the expected muscle-relaxant behavior, which we termed immobility, which was characterized by a motionless flattened posture with thoracic and abdominal areas resting against the floor of the cage (Fig. 1A, B). However, RTG-induced immobility was much more pronounced and long lasting in mice from the Kcnq1 strain (C57BL/6) compared to the Kcna1 strain (Tac:N:NIHS-BC), even at lower doses (Fig. 1A, B).
Figure 1.
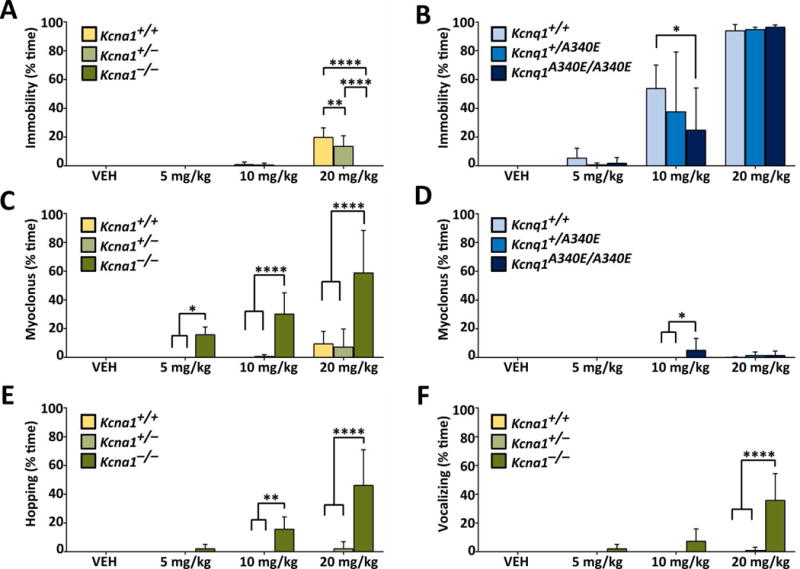
RTG increases behavioral excitability in Kcna1−/− mice. (A, B) At 20 mg·kg−1, RTG caused all mice to become immobile and lie with their abdomen and thorax against the floor of the cage, except for Kcna1−/− mice which continued to be active. (C, D) RTG induced myoclonus in Kcna1−/− mice at 5, 10, and 20 mg·kg−1 (ip). In the other genotypes, RTG induced myoclonus only sporadically at the highest doses. RTG induced hopping (E) and vocalizing (F) in Kcna1−/− mice. These two behavioral effects were not seen in the Kcnq1 mice at any dose. Two-way repeated measures ANOVA showed the following statistical results. For (A), effect of treatment: P < 0.0001, F3,45 = 61.87; genotype: P < 0.0001, F2,15 = 19.85; interaction between treatment and genotype: P < 0.0001, F6,45 = 17.10). For (B), effect of treatment: P < 0.0001, F3,30 = 101.8; genotype: P = 0.36, F2,10 = 1.118; interaction between treatment and genotype: P = 0.47, F6,30 = 0.9533). For (C), effect of treatment: P < 0.0001, F3,45 = 19.82; genotype: P < 0.0001, F2,15 = 34.09; interaction between treatment and genotype: P < 0.0001, F6,45 = 8.664). For (D), effect of treatment: P < 0.0001, F3,30 = 101.8; genotype: P = 0.36, F2,10 = 1.118; interaction between treatment and genotype: P = 0.47, F6,30 = 0.9533). For (E), effect of treatment: P < 0.0001, F3,45 = 17.81; genotype: P < 0.0001, F2,15 = 26.69; interaction between treatment and genotype: P < 0.0001, F6,45 = 15.59). For (F), effect of treatment: P < 0.0001, F3,45 = 19.86; genotype: P < 0.0001, F2,15 = 17.29; interaction between treatment and genotype: P < 0.0001, F6,45 = 18.33). *, P ≤ 0.05; **, P ≤ 0.01; ****, P ≤ 0.0001 (two-way repeated measures ANOVA, Tukey’s post hoc; n=4–6 per genotype).
In contrast to the normal sedative-like drug response in WT animals, RTG (5–20 mg·kg−1) never induced immobility in Kcna1−/− mice (Fig. 1A, B). Instead, Kcna1−/− mice exhibited unexpected behavioral responses to RTG, including full-body jerking and twitching (termed myoclonus) beginning at low doses (5 mg·kg−1), which evolved to include hopping and vocalization at higher doses (10–20 mg·kg−1; Fig. 1C, E, F). Unlike Kcna1−/− animals, Kcnq1A340E/A340E mice rarely exhibited myoclonic behavior in response to RTG (Fig. 1D), and hopping and vocalizing was never observed at any dose (data not shown).
RTG increases seizure threshold in Kcnq1 mice, but has no effect in Kcna1 mice
To determine whether RTG decreases seizure susceptibility in Kcna1 and Kcnq1 mice, animals were injected with either VEH or RTG (10 mg·kg−1) followed by exposure to the convulsant flurothyl to induce seizure activity, which manifests as initial myoclonic jerks that progress to generalized tonic-clonic seizure. In this and subsequent experiments, RTG was administered at a dose of 10 mg·kg−1 (unless otherwise stated) to avoid excessive confounding effects of behavioral toxicity such as the myoclonus, hopping, and vocalizing that occurs at higher doses in Kcna1−/− mice. Upon flurothyl exposure, VEH-injected Kcna1−/− mice exhibited latencies to myoclonic jerks and tonic-clonic seizure that were reduced by about 50% compared to their Tac:N:NIHS-BC WT littermates (Fig. 2A, C), similar to previous reports.4 However, VEH-injected Kcnq1A340E/A340E mice showed latencies that were not significantly different from their C57BL/6 WT counterparts (Fig. 2B, D), indicating the Kcnq1A340E mutation does not affect seizure threshold despite the occurrence of infrequent spontaneous seizures in this model.9 When injected with RTG, Kcna1−/− mice and Tac:N:NIHS-BC WT littermates exhibited latencies that were unchanged compared to VEH, suggesting that a 10 mg·kg−1 dose of RTG has no effect on seizure susceptibility in this mouse strain (Fig. 2A, C). Even when administered higher RTG doses of 20 mg·kg−1, Tac:N:NIHS-BC Kcna1+/+ mice still showed no changes in seizures latencies compared to VEH-treated animals (Fig. S1). In contrast, RTG (10 mg·kg−1) significantly increased the latency to tonic-clonic seizure by >50% in both C57BL/6 Kcnq1+/+ (t = 2.68, P ≤ 0.05, unpaired t-test) and Kcnq1A340E/A340E mice (t = 3.179, P ≤ 0.01, unpaired t-test; Fig. 2D), indicating a therapeutic anti-seizure response. However, RTG (10 mg·kg−1) had no significant effect on the latency to myoclonic jerks in C57BL/6 Kcnq1+/+ animals, and instead slightly shortened the latency to myoclonic jerks in Kcnq1A340E/A340E mice (t = 2.194, P ≤ 0.05, unpaired t-test; Fig. 2B).
Figure 2.
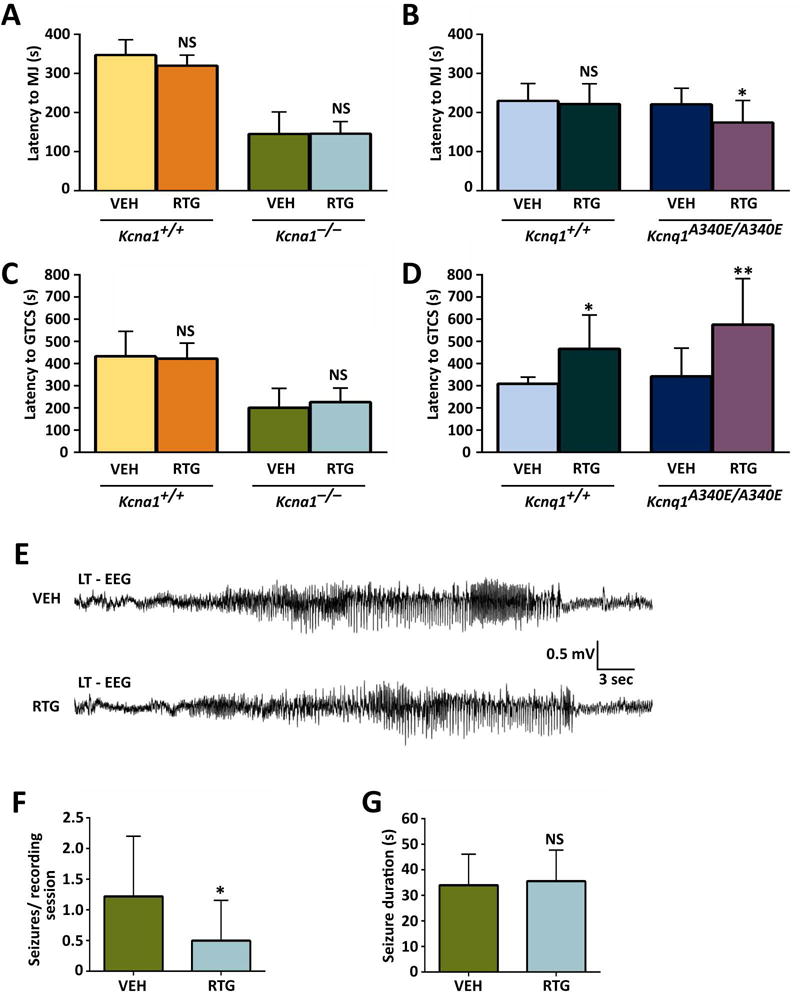
KCNQ activation increases flurothyl-induced seizure latencies in Kcnq1 mice and decreases spontaneous seizure frequency in Kcna1−/− mice. (A) Administration of RTG (10 mg·kg−1, ip) had no significant effect on latency to flurothyl-induced myoclonic jerks (MJ) in Tac:N:NIHS-BC Kcna1+/+ and Kcna1−/− mice. However, RTG decreased latency to myoclonic jerk in Kcnq1A340E/A340E mutant mice (B). (C) Administration of RTG (10 mg·kg−1, ip) had no significant effect on latency to flurothyl-induced generalized tonic-clonic seizures (GTCS) in Tac:N:NIHS-BC Kcna1+/+ and Kcna1−/− mice. However, RTG significantly increased the latency to GTCS in Kcnq1 strain (C57BL/6) mice (D). (E) EEG traces showing representative spontaneous seizure activity in Kcna1−/− mice after receiving either VEH or RTG (10 mg·kg−1). (F) Analysis of spontaneous seizure frequency in Kcna1−/− mice (n = 6) showed a significant decrease after treatment with 10 mg·kg−1 RTG. (G) Average spontaneous seizure durations were similar in Kcna1−/− mice that exhibited seizures after VEH and 10 mg·kg−1 RTG administration (n = 3). For (A-D): *, P ≤ 0.05; **, P ≤ 0.01 (two-tailed unpaired Student’s t-test; n = 6–11 per treatment). For (F) and (G): *, P ≤ 0.05 (two-tailed paired Student’s t-test). Abbreviations: LT, left temporal; VEH, vehicle; RTG, retigabine; NS, not significant compared to VEH control.
RTG decreases spontaneous seizure frequency in Kcna1−/− mice
To determine whether short-term administration of RTG decreases the frequency and/or duration of spontaneous seizures in Kcna1−/− and Kcnq1A340E/A340E mice, video EEG-ECG recordings were performed over three consecutive days. In Kcna1−/− mice, RTG (10 mg·kg−1) significantly reduced the number of spontaneous seizures per 4-h recording session from 1.2 ± 1.0 seizures with VEH to 0.5 ± 0.7 seizures following treatment (t = 2.6, P < 0.05, paired t-test), without modifying seizure durations (Fig. 2E–G). Although spontaneous seizures have been reported in Kcnq1A340E/A340E mice, they are relatively rare, occurring only about once every 1–2 d.9 Consistent with these previous studies, no EEG seizures were observed during the 3-day recording period in Kcnq1A340E/A340E mice; therefore, modification of spontaneous seizure phenotypes could not be readily assessed in this strain. In addition, interictal spikes did not occur at a high enough frequency in our cohort to allow utility as a marker of spontaneous epileptiform activity.
RTG increases HRV in Kcnq1A340E/A340E mice
To determine whether KCNQ activation normalizes heart function in Kcna1 and Kcnq1 mice, various cardiac measures were analyzed using video EEG-ECG recordings: HR, HRV, and skipped heart beats (i.e., AV conduction blocks). At baseline with VEH, mice exhibited strain-dependent differences in cardiac function. Mice from the Kcna1 strain exhibited faster HR with less HRV than mice from the Kcnq1 strain (Fig. 3A–D and Table 1). In response to 10 mg·kg−1 RTG injection, WT and mutant mice of both strains exhibited trends towards a transient decrease in HR. However, the degree and duration of this HR reduction varied by strain and genotype (Fig. 3A, B, E, F). Whereas Tac:N:NIHS-BC WT mice from the Kcna1 strain exhibited relatively mild (~15%) and short-lived (~15 min) HR reductions (Fig. 3E), C57BL/6 WT mice from the Kcnq1 strain exhibited more profound (~40%) and long-lasting (~30 min) HR reductions (Fig. 3F). RTG responses differed in mutant mice of both strains: Kcna1−/− mice exhibited ~5% reductions in HR that lasted about 5 min (Fig. 3E) compared to Kcnq1A340E/A340E mice, which showed HR reductions of ~45% that lasted up to ~90 min (Fig. 3F).
Figure 3.
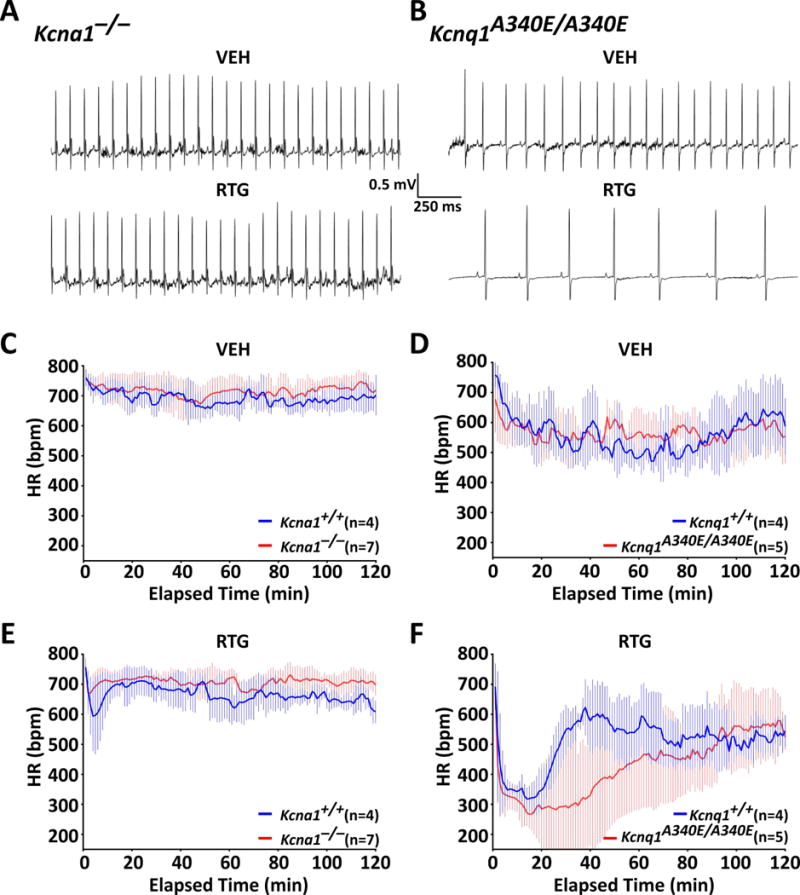
KCNQ activation transiently decreases heart rate in Kcna1 and Kcnq1 mice. Representative 2-s ECG traces taken ~30 min after injection of either VEH or RTG showing HR effects in Kcna1−/− (A) and Kcnq1A340E/A340E mice (B). Plots showing HR responses for the 2 h immediately following VEH (C, D) and 10 mg·kg−1 RTG injections (E, F) in Kcna1 and Kcnq1 mice. Baseline HRs were not significantly different between WTs and mutants of either strain. Following RTG, HR transiently decreased from baseline rates by approximately 50–100 bpm and 250–300 bpm in Kcna1 (Tac:N:NIHS-BC) and Kcnq1 (C57BL/6) strain mice, respectively. At 38 min after RTG administration, HR was significantly different (P ≤ 0.05) between C57BL/6 Kcnq1+/+ and Kcnq1A340E/A340E mice (effect of time: P < 0.0001, F119,833 = 6.664; genotype: P = 0.21, F1,7 = 1.935; interaction between time and genotype: P < 0.0001, F119,883 = 2.634; two-way repeated measures ANOVA, Sidak’s post hoc). HR did not differ significantly between RTG-treated Tac:N:NIHS-BC Kcna1+/+ and Kcna1−/− mice.
Table 1.
Effects of RTG (10 mg·kg−1) on HRV in Kcna1 and Kcnq1 mice at 30-min post-injection.
| Gene | Genotype | n | Condition | SDNN (ms) | RMSSD (ms) | HR (bpm) |
|---|---|---|---|---|---|---|
| Kcna1 | +/+ | 4 | VEH | 2.9 ± 1.3 | 1.6 ± 0.6 | 697 ± 25 |
| RTG | 2.6 ± 1.5 | 1.4 ± 0.7 | 682 ± 47 | |||
| −/− | 7 | VEH | 3.6 ± 1.9 | 3.0 ± 2.7 | 700 ± 56 | |
| RTG | 3.6 ± 1.2 | 3.0 ± 2.0 | 711 ± 21 | |||
| Kcnq1 | +/+ | 4 | VEH | 11.1 ± 1.7 | 5.9 ± 0.4 | 549 ± 55 |
| RTG | 9.8 ± 2.8 | 6.3 ± 3.0 | 593 ± 50 | |||
| A340E/A340E | 5 | VEH | 11.0 ± 3.5 | 7.2 ± 3.5 | 555 ± 34 | |
| RTG | 47.2 ± 26.0* | 57.8 ± 36.8* | 340 ± 190* |
, P < 0.05 (two-tailed paired Student’s t-test). Abbreviations: HR, heart rate; RMSSD, root mean square of successive beat-to-beat differences; RTG, retigabine; SDNN, standard deviation of the beat-to-beat intervals; VEH, vehicle.
To assess RTG-induced modification of autonomic control of the heart, HRV was analyzed at 30-min post-injection using the following time domain measures: the standard deviation of the beat-to-beat intervals (SDNN), an index of the total autonomic variability; and the root mean square of the successive beat-to-beat differences (RMSSD), an index of parasympathetic tone. Following VEH injection, mice from the Kcnq1 strain exhibited 2- to 4-fold higher SDNN and RMSSD values relative to Kcna1 strain mice, suggesting higher parasympathetic tone at baseline (Table 1). Mutant mice from both strains exhibited HRV values that were approximately similar to their WT counterparts (Table 1). However, baseline RMSSD was nearly twice as high in Kcna1−/− mice compared to their Tac:N:NIHS-BC WT controls, consistent with a previous study showing elevated HRV measures of parasympathetic tone in these animals.4 Upon 10 mg·kg−1 RTG administration, SDNN (t = 3.488, P = 0.025, paired t-test) and RMSSD (t = 3.373, P = 0.028, paired t-test) significantly increased by 4-fold and 8-fold, respectively, in Kcnq1A340E/A340E mice compared to VEH (Table 1), suggesting dysregulation of autonomic control of the heart in response to the drug. In contrast, Kcna1−/− mice showed no change in HRV following RTG injection (Table 1).
Next, we examined whether acute RTG administration affects the frequency of interictal skipped heart beats, which we use as a biomarker of abnormal cardiac function indicative of potential autonomic dysregulation. Previous studies have shown that Kcna1−/− mice exhibit an increased incidence of skipped heart beats due to vagally-driven AV conduction blocks.3 Similarly, Kcnq1A340E/A340E mice have also been reported to have increased susceptibility to AV blocks in ECG recordings.9 Acute RTG (10 mg·kg−1) treatment did not significantly alter the frequency of skipped beats in Kcna1−/− and Tac:N:NIHS-BC Kcna1+/+ mice (Fig. 4A, B). However, this dose of RTG induced a dramatic increase in the rate of skipped beats in mice of the Kcnq1 strain, especially Kcnq1A340E/A340E mice which exhibited an increase of ~200–800 per h (equivalent to a ≥ 20-fold increase; t = 2.891, P = 0.045, paired t-test), suggesting drastic augmentation of parasympathetic tone (Fig. 4C, D).
Figure 4.
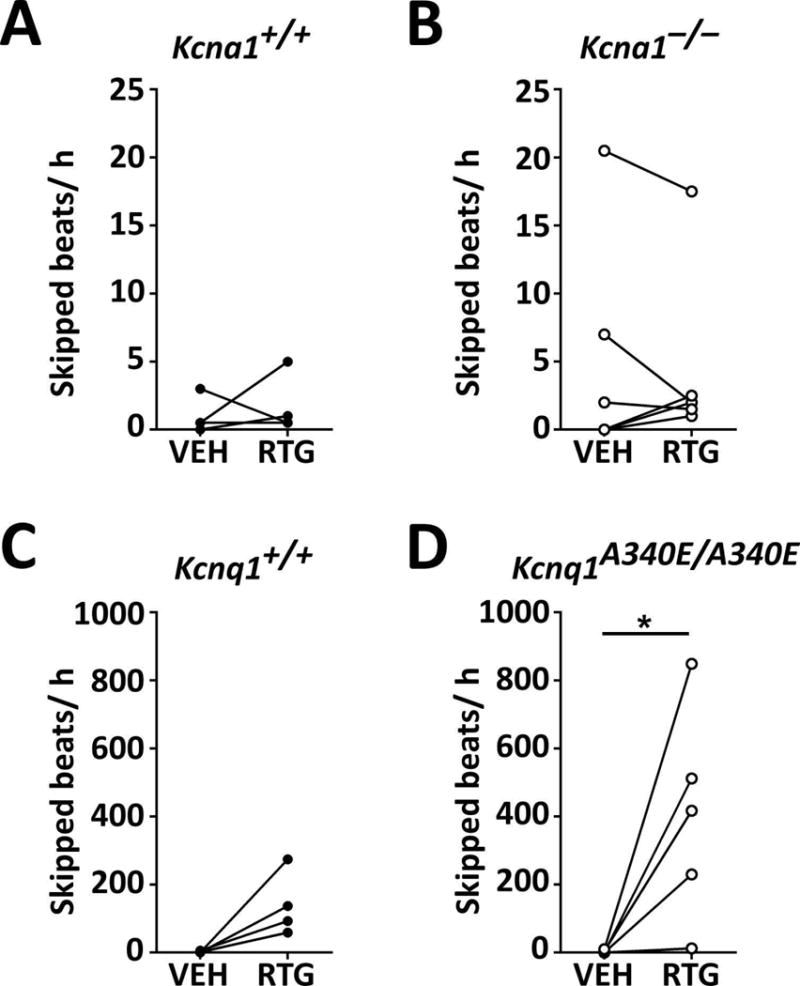
KCNQ activation increases the frequency of skipped heart beats in Kcna1A340E/A340E mice. RTG treatment did not significantly alter the frequency of skipped beats (i.e., AV conduction blocks) in Tac:N:NIHS-BC Kcna1+/+ (A) and Kcna1−/− (B) mice. However, RTG increased the rate of skipped beats in Kcnq1 mice (C; P = 0.06, two-tailed paired Student’s t-test), especially in Kcnq1A340E/A340E animals which exhibited a ≥20-fold increase (D). *, P < 0.05 (two-tailed paired Student’s t-test.)
Kcnq2 mRNA levels are increased in Kcna1−/− mouse brain
To test whether the observed differential drug effects were associated with strain- or genotype-based differences in the expression of the main molecular targets of RTG, qPCR was performed to measure the mRNA levels of Kcnq2 and Kcnq3 in the hippocampi and cortices of Kcna1 and Kcnq1 mice (n=5–6/genotype per brain region). Regional qPCR revealed that the levels of Kcnq2 mRNA, but not Kcnq3, were significantly higher in both the hippocampus (F = 4.815, P = 0.012, one-way ANOVA) and cortex (F = 4.275, P = 0.017, one-way ANOVA) of Kcna1−/− mice compared to Kcnq1 mice (Fig. 5).
Figure 5.
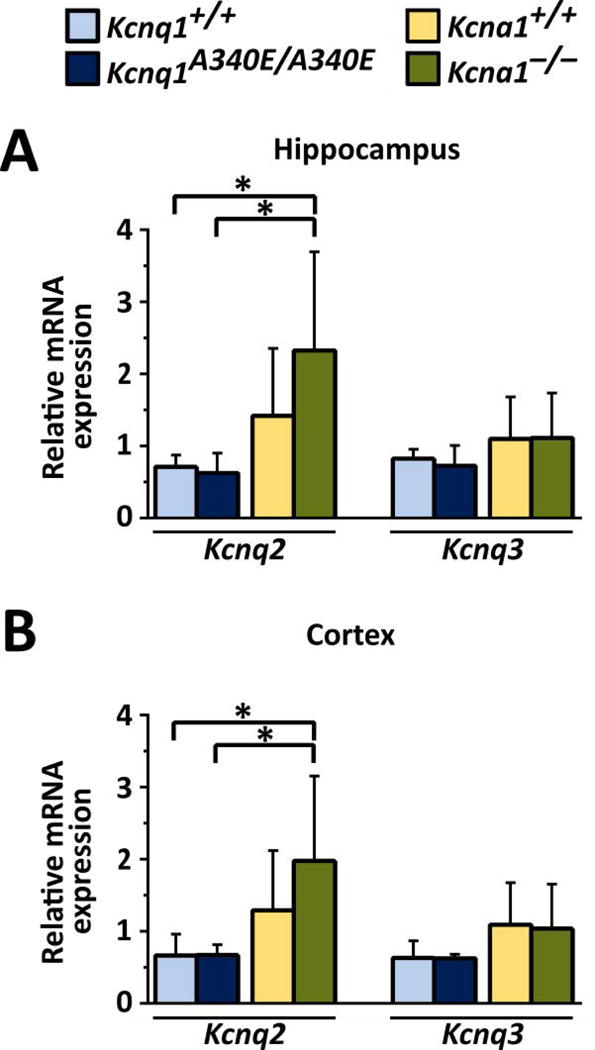
Kcnq2 mRNA levels are increased in Kcna1−/− mouse brain. qPCR analyses of relative Kcnq2 and Kcnq3 mRNA expression revealed Kcnq2 mRNA levels in hippocampus (A) and cortex (B) were significantly increased in Kcna1−/− mice compared to Kcnq1 strain mice. *, P ≤ 0.05 (one-way ANOVA, Tukey’s post hoc; n=5–6/genotype per brain region).
DISCUSSION
In this study, the efficacy of the KCNQ channel opener RTG and its potential for severe adverse behavioral or cardiac effects was profoundly affected by genetic background, suggesting KCNQ activation may not be an optimal strategy to combat SUDEP. RTG treatment has been used as an effective adjunctive therapy for the management of drug-resistant partial-onset seizures, but its production was recently discontinued due to limited usage and continued decline in patient initiation. However, RTG is simply one drug that targets KCNQ channels and many other KCNQ activators are currently under development for controlling disorders of neuronal hyperexcitability.17,18 Another factor in the demise of RTG as a clinical pharmaceutical was likely the occurrence of unwanted side effects in a subset of patients, as is seen for most anti-seizure drugs. The most common adverse events associated with RTG in patients include central nervous system effects, increased risk of urinary retention, and altered skin and retinal pigmentation.19,20 In rodents, the main side-effect of RTG is a transient reduction in locomotor activity described as flat body posture and some muscle relaxation16, as was observed in WT and Kcnq1A340E/A340E mice in this study. We also observed an additional and previously unreported response to KCNQ activation that was specific to mice with the Kcna1−/− mutation, namely behavioral hyperexcitability that was characterized by myoclonus, hopping, and vocalization. These behaviors may be the rodent correlate of the tremors that occur in 9% of patients at higher doses of RTG (900 mg·d−1).19 In one case report, a patient overdosed on the KCNQ opener flupirtine, leading to intermittent myoclonus and active and resting tremor of the extremities reminiscent of the behavior seen in Kcna1−/− mice.21 Although the EEGs in Kcna1−/− mice showed some abnormal waveforms following RTG administration, these were more consistent with movement artifacts due to hopping rather than increased cortical excitability (Fig. S2). The hyperexcitable behavioral response to RTG in Kcna1−/− mice provides one possible pharmocogenetic explanation for the adverse behavioral effects of RTG in some patients and suggests that future KCNQ activator drugs should be evaluated for potential behavioral hyperexcitability side effects.
KCNQ activation also produced novel genotype- and strain-dependent effects on HR suggesting KCNQ2-5 channels play a role in autonomic control of chronotropy, which varies based on genetic background. Whereas RTG had no significant cardiac effects in Kcna1 mice, Kcnq1 mice exhibited transient HR decreases that were most pronounced in Kcnq1A340E/A340E mutants, which exhibited a prolonged fall in HR lasting ≥60 min coupled with 4- to 8-fold increases in HRV. Cardiovascular roles for KCNQ2-5 channels have been described in visceral sensory neurons and the vasculature, but less is known about their potential roles in regulating cardiac function.22 Heteromeric KCNQ2/3 channels are expressed in superior cervical ganglion (SCG) neurons where they regulate release of noradrenaline (NA) onto the heart.23 Unlike KCNQ1, KCNQ2-5 channels are not expressed at significant levels in the heart24; however, their potential compensatory expression in the absence of functional KCNQ1 cannot be ruled out. Previously, in a co-culture preparation of rat SCG sympathetic neurons and mouse cardiomyocytes, RTG abolished the increase in cardiomyocyte constriction rate caused by nicotine-induced stimulation of SCG neurons; however, RTG application to cardiomyocytes cultured alone had no effect.25 These results suggest that RTG induces up-regulation of KCNQ2/3-mediated M-current preventing NA release from SCG cells, which could lead to a bradycardic effect due to sympathetic inhibition consistent with our observations.
Although RTG has been shown to have broad spectrum anti-seizure activity, our results show that its efficacy depends on the mouse strain and testing paradigm in which it is evaluated. RTG normally exhibits potent anti-seizure effects, especially when administered prior to seizure induction, in a variety of in vitro and in vivo rodent models of epilepsy.16,26 However, to our knowledge, this is the first study to examine the anti-seizure efficacy of RTG in the flurothyl seizure model in mice. We found that while RTG could effectively increase flurothyl-induced seizure latency similar to other chemically-induced seizure models, this anti-seizure effect was limited to mice from the Kcnq1 strain, suggesting an influence of genetic background of the mouse strain. Interestingly, a 10 mg·kg−1 dose of RTG still significantly reduced the occurrence of spontaneous EEG seizures in Kcna1−/− mice despite having no effect on flurothyl seizure latency. A similar dissociation has been observed in flurothyl-kindled DBA/2J mice. Following exposure to eight flurothyl-induced seizures, DBA/2J mice exhibit spontaneous seizures without a significant change in flurothyl seizure threshold from pre-epileptic levels.27 The generalized running bouncing seizures that are evoked by prolonged flurothyl exposure involve activation of brainstem centers, whereas the spontaneous seizures in Kcna1−/− mice show evidence of limbic origin.27–29 Therefore, RTG could have contrasting effects on flurothyl-induced and spontaneous seizures due to differences in the neuronal networks underlying onset or propagation of the two types of seizures.
Quantitative PCR analyses revealed high Kcnq2 transcript levels in the hippocampus and cortex of Kcna1−/− mice that could be partly due to a neuronal homeostatic control response that is the product of the expression and functional complementarity between Kv1.1 and KCNQ2 channels. Kv1.1 and KCNQ2 channels share widespread expression throughout many common brain areas including cortex, hippocampus, thalamus, and cerebellum.30,31 At the compartmental level, Kv1.1 and KCNQ2 channels show strong expression in axons, including at the AIS.32,33 Prior studies of axonal and nerve excitability in animal models have uncovered a potential functional synergy between Kv1.1 and KCNQ2 channels. Peripheral nerves from Kcna1−/− mice exhibit abnormal nerve activity that is amplified by non-specific KCNQ blockade with TEA and ameliorated by KCNQ activation with flupirtine.1,34 More recently, functional cooperativity between Kv1.1 and KCNQ2 channels was demonstrated in the AISs of the avian cochlear nucleus, which, following deprivation of afferent inputs by removing the cochlea, switch their dominant Kv channel composition from Kv1.1 to KCNQ2 to maintain homeostasis of auditory circuits.38 The ability of Kv1.1 and KCNQ2 channels to partially compensate for one another is facilitated by their similar electrophysiological properties, including similar low-voltage activation characteristics and the ability to directly affect action potential threshold according to their different activation kinetics.35,36 In patients, gene mutations in either Kcna1 or Kcnq2 lead to similar functional consequences including epilepsy or peripheral nerve hyperexcitability.8,37 In addition to potential homeostatic compensation, Kcnq2 transcripts may also be elevated in Kcna1−/− mice partly due to genetic background of the mouse strain since the Kcna1 strain exhibits slightly elevated basal levels of Kcnq2/3 expression. Epilepsy in Kcna1−/− mice could also play a role, leading to seizure-induced increases in the transcription of Kcnq2, which could alter the effects of RTG in accordance with the target hypothesis of pharmacoresistance, which states that epilepsy-related changes in the properties of drug targets themselves may result in altered drug sensitivity.38 Since expression changes at the mRNA level do not always correlate with alterations in protein abundance, future studies will be needed to address whether the changes in Kcnq2 mRNA expression correlate with changes in KCNQ2 protein.
In summary, from a clinical perspective, this study demonstrates the principle that treatment strategies in channelopathy may have unexpected outcomes. Our findings show that activation of KCNQ channels does not prevent neurocardiac dysfunction, at least in the mouse models tested. Furthermore, KCNQ activation led to unexpected adverse strain- and genotype-dependent effects in mice that suggest this class of drugs may be contraindicated in patients with Kcna1 or Kcnq1 mutations. The differential effects of KCNQ activation may be partly due to underlying differences in Kcnq2/3 channel expression, which could alter drug target levels. Our findings emphasize the importance of an individual’s genomic profile in determining drug responsiveness, toxicity, and sensitivity, providing a clear example of pharmacogenetic interactions in epilepsy which could contribute to pharmacoresistance. To achieve effective therapeutic rebalancing of channel defects will require improved understanding of channel interactions at the circuit and tissue levels.
Supplementary Material
KEY POINTS.
Effects of KCNQ channel activation in Kcna1 and Kcnq1 mutant mice were evaluated by behavior, electrophysiology, and gene expression
KCNQ channel activation caused adverse behavioral and cardiac responses and variable anti-seizure effects depending on genotype
Levels of Kcnq2 mRNA were increased in Kcna1−/− mice, possibly contributing to their abnormal drug responses
Pharmacogenetic interactions may limit KCNQ modulation as a strategy to prevent deleterious neurocardiac dysfunction in epilepsy
Treatment strategies in channelopathy may have unexpected outcomes highlighting the complexity of rebalancing channel defects
Acknowledgments
This work was supported by Citizens United for Research in Epilepsy (grant number 249950); the National Institutes of Health (grant numbers R00HL107641, R01NS100954, R01NS099188); a predoctoral fellowship from the Epilepsy Foundation (grant number 367385); and a Louisiana State University Health Sciences Center Ike Muslow Predoctoral Fellowship.
ABBREVIATIONS
- 4-AP
4-aminopyridine
- AIS
axon initial segment
- ANOVA
analysis of variance
- ASD
anti-seizure drug
- AV
atrioventricular
- BG/Th
basal ganglia/thalamus
- BS
brain stem
- Cbl
cerebellum
- Ctx
cortex
- ECG
electrocardiography
- EEG
electroencephalography
- Hipp
hippocampus
- HR
heart rate
- HRV
heart rate variability
- ip
intraperitoneal
- iv
intravenous
- KO
knockout
- LQT
long QT syndrome
- NA
noradrenaline
- qPCR
quantitative polymerase chain reaction
- RMSSD
root mean square of successive differences
- RTG
retigabine
- SCG
superior cervical ganglion
- SDNN
standard deviation of the RR intervals
- SUDEP
sudden unexpected death in epilepsy
- TEA
tetraethylammonium
- VEH
vehicle
- WT
wildtype
Footnotes
DISCLOSURE OF CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Glasscock E, Qian J, Kole MJ, et al. Transcompartmental reversal of single fibre hyperexcitability in juxtaparanodal Kv1.1-deficient vagus nerve axons by activation of nodal KCNQ channels. J Physiol. 2012;590:3913–26. doi: 10.1113/jphysiol.2012.235606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qi Y, Wang J, Bomben VC, et al. Hyper-SUMOylation of the Kv7 Potassium Channel Diminishes the M-Current Leading to Seizures and Sudden Death. Neuron. 2014;83:1159–71. doi: 10.1016/j.neuron.2014.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glasscock E, Yoo JW, Chen TT, et al. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–75. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mishra V, Karumuri BK, Gautier NM, et al. Scn2a deletion improves survival and brain-heart dynamics in the Kcna1-null mouse model of sudden unexpected death in epilepsy (SUDEP) Hum Mol Genet. 2017;26:2091–103. doi: 10.1093/hmg/ddx104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore BM, Jerry Jou C, Tatalovic M, et al. The Kv1.1 null mouse, a model of sudden unexpected death in epilepsy (SUDEP) Epilepsia. 2014;55:1808–16. doi: 10.1111/epi.12793. [DOI] [PubMed] [Google Scholar]
- 6.Smart SL, Lopantsev V, Zhang CL, et al. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–19. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- 7.Klassen TL, Bomben VC, Patel A, et al. High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia. 2014;55:e6–12. doi: 10.1111/epi.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuberi SM, Eunson LH, Spauschus A, et al. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. 1999;122:817–25. doi: 10.1093/brain/122.5.817. [DOI] [PubMed] [Google Scholar]
- 9.Goldman AM, Glasscock E, Yoo J, et al. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 11.Anderson JH, Bos JM, Cascino GD, et al. Prevalence and spectrum of electroencephalogram-identified epileptiform activity among patients with long QT syndrome. Heart Rhythm. 2014;11:53–7. doi: 10.1016/j.hrthm.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 12.Auerbach DS, McNitt S, Gross RA, et al. Genetic biomarkers for the risk of seizures in long QT syndrome. Neurology. 2016;87:1660–8. doi: 10.1212/WNL.0000000000003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gunthorpe MJ, Large CH, Sankar R. The mechanism of action of retigabine (ezogabine), a first-in-class K(+) channel opener for the treatment of epilepsy. Epilepsia. 2012;53:412–24. doi: 10.1111/j.1528-1167.2011.03365.x. [DOI] [PubMed] [Google Scholar]
- 14.Casimiro MC, Knollmann BC, Yamoah EN, et al. Targeted point mutagenesis of mouse Kcnq1: phenotypic analysis of mice with point mutations that cause Romano-Ward syndrome in humans. Genomics. 2004;84:555–64. doi: 10.1016/j.ygeno.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Friedel HA, Fitton A. Flupirtine. A review of its pharmacological properties, and therapeutic efficacy in pain states. Drugs. 1993;45:548–69. doi: 10.2165/00003495-199345040-00007. [DOI] [PubMed] [Google Scholar]
- 16.Rostock A, Tober C, Rundfeldt C, et al. D-23129: a new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res. 1996;23:211–23. doi: 10.1016/0920-1211(95)00101-8. [DOI] [PubMed] [Google Scholar]
- 17.Kalappa BI, Soh H, Duignan KM, et al. Potent KCNQ2/3-specific channel activator suppresses in vivo epileptic activity and prevents the development of tinnitus. J Neurosci. 2015;35:8829–42. doi: 10.1523/JNEUROSCI.5176-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar M, Reed N, Liu R, et al. Synthesis and Evaluation of Potent KCNQ2/3-Specific Channel Activators. Mol Pharmacol. 2016;89:667–77. doi: 10.1124/mol.115.103200. [DOI] [PubMed] [Google Scholar]
- 19.Brodie MJ, Lerche H, Gil-Nagel A, et al. Efficacy and safety of adjunctive ezogabine (retigabine) in refractory partial epilepsy. Neurology. 2010;75:1817–24. doi: 10.1212/WNL.0b013e3181fd6170. [DOI] [PubMed] [Google Scholar]
- 20.French JA, Abou-Khalil BW, Leroy RF, et al. Randomized, double-blind, placebo-controlled trial of ezogabine (retigabine) in partial epilepsy. Neurology. 2011;76:1555–63. doi: 10.1212/WNL.0b013e3182194bd3. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann O, Gommert LR, Egert M. Paradoxical cerebral cortical hyperexcitability following flupirtine overdose. J Toxicol Clin Toxicol. 2004;42:913–6. doi: 10.1081/clt-200035096. [DOI] [PubMed] [Google Scholar]
- 22.Mackie AR, Brueggemann LI, Henderson KK, et al. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325:475–83. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernandez CC, Zaika O, Tolstykh GP, et al. Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol. 2008;586:1811–21. doi: 10.1113/jphysiol.2007.148304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–95. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaika O, Zhang J, Shapiro MS. Functional role of M-type (KCNQ) K+ channels in adrenergic control of cardiomyocyte contraction rate by sympathetic neurons. J Physiol. 2011;589:2559–68. doi: 10.1113/jphysiol.2010.204768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kay HY, Greene DL, Kang S, et al. M-current preservation contributes to anticonvulsant effects of valproic acid. J Clin Invest. 2015;125:3904–14. doi: 10.1172/JCI79727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kadiyala SB, Ferland RJ. Dissociation of spontaneous seizures and brainstem seizure thresholds in mice exposed to eight flurothyl-induced generalized seizures. Epilepsia Open. 2017;2:48–58. doi: 10.1002/epi4.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gautier NM, Glasscock E. Spontaneous seizures in Kcna1-null mice lacking voltage-gated Kv1.1 channels activate Fos expression in select limbic circuits. J Neurochem. 2015;135:157–64. doi: 10.1111/jnc.13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samoriski GM, Applegate CD. Repeated generalized seizures induce time-dependent changes in the behavioral seizure response independent of continued seizure induction. J Neurosci. 1997;17:5581–90. doi: 10.1523/JNEUROSCI.17-14-05581.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper EC, Harrington E, Jan YN, et al. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci. 2001;21:9529–40. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Kunkel DD, Schwartzkroin PA, et al. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci. 1994;14:4588–99. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg EM, Clark BD, Zagha E, et al. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58:387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan Z, Kao T, Horvath Z, et al. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou L, Messing A, Chiu SY. Determinants of excitability at transition zones in Kv1.1-deficient myelinated nerves. J Neurosci. 1999;19:5768–81. doi: 10.1523/JNEUROSCI.19-14-05768.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuba H, Yamada R, Ishiguro G, et al. Redistribution of Kv1 and Kv7 enhances neuronal excitability during structural axon initial segment plasticity. Nat Commun. 2015;6:8815. doi: 10.1038/ncomms9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnston J, Forsythe ID, Kopp-Scheinpflug C. Going native: voltage-gated potassium channels controlling neuronal excitability. J Physiol. 2010;588:3187–200. doi: 10.1113/jphysiol.2010.191973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dedek K, Kunath B, Kananura C, et al. Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci U S A. 2001;98:12272–7. doi: 10.1073/pnas.211431298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain. 2006;129:18–35. doi: 10.1093/brain/awh682. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.