

Summary
Coagulation factor XIa is a candidate target for anticoagulants that better separate antithrombotic efficacy from bleeding risk. We report a co-crystal structure of the FXIa protease domain with DEF, a human monoclonal antibody that blocks FXIa function and prevents thrombosis in animal models without detectably increased bleeding. The light chain of DEF occludes the FXIa S1 subsite and active site while the heavy chain provides electrostatic interactions with the surface of FXIa. The structure accounts for the specificity of DEF for FXIa over its zymogen and related proteases, its active site-dependent binding, and its ability to inhibit substrate cleavage. The inactive FXIa protease domain used to obtain the DEF-FXIa crystal structure reversed anticoagulant activity of DEF in plasma and in vivo and the activity of a small molecule FXIa active-site inhibitor in vitro. DEF and this reversal agent for FXIa active-site inhibitors may help support clinical development of FXIa-targeting anticoagulants.
Keywords: Coagulation, factor XI, intrinsic pathway, IgG, protease-blocking antibody, protease inhibitor, active-site inhibitor, anticoagulant, crystal structure, hemostasis, thrombosis
eTOC Blurb
Inhibition of coagulation factor XI (FXI) may prevent thrombosis with less bleeding. DEF is a human monoclonal antibody that blocks the active form of FXI (FXIa). Ely et al. describe a structure that explains the anticoagulant function and specificity of DEF for FXIa, and identifies an antidote for FXIa inhibitors.
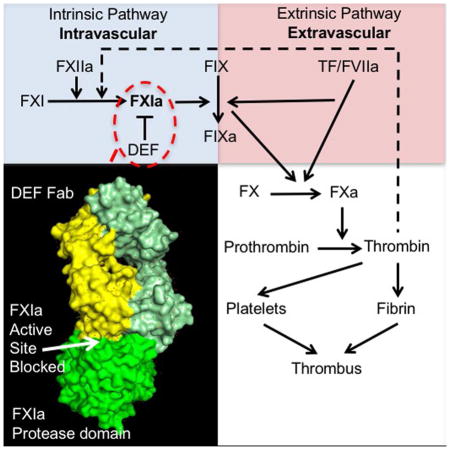
Introduction
Thrombosis, the formation of intravascular blood clots that cause myocardial infarction, stroke, pulmonary embolism, deep venous thrombosis and other disorders, remains a major cause of morbidity and mortality (Marder, 2013; Raskob et al., 2014; Wendelboe and Raskob, 2016). The coagulation protease thrombin is a key driver of clot formation, and anticoagulants that diminish thrombin generation or block its activity are used to prevent and treat thrombosis. Two separate triggers, the extrinsic and intrinsic pathways, converge on a common pathway that leads to thrombin generation. The extrinsic and common pathways are necessary for hemostasis, and all available anticoagulants target components of these pathways and decrease thrombosis but increase bleeding (De Caterina et al., 2013; Yeh et al., 2015). This trade-off limits their efficacy, safety and use in patients at increased bleeding risk. Better agents are needed, and the existence of two separate triggers for coagulation has not been exploited (Gailani et al., 2015).
The hypothesis that inhibition of the intrinsic pathway component FXI may inhibit thrombosis with less bleeding risk than current anticoagulants (Bane and Gailani, 2014; Fredenburgh et al., 2017; Muller et al., 2011) is supported by animal models (Bi et al., 1995; Bugge et al., 1996; Dewerchin et al., 2000; Erlich et al., 1999; Gruber and Hanson, 2003; Rosen et al., 1997; Sun et al., 1998; Wang et al., 1997; Wang et al., 2006), human genetics (Asakai et al., 1991; Doggen et al., 2006; Meijers et al., 2000; Peyvandi et al., 2006; Peyvandi et al., 2012; Yang et al., 2006), and a proof-of-concept trial of antisense oligonucleotides to inhibit factor XI (FXI) expression in humans (Buller et al., 2015). Toward enabling further exploration of this hypothesis, we developed DEF, a fully human, effector null IgG1 that recognizes FXIa, the activated form of FXI, and blocks its catalytic activity. DEF inhibits intrinsic pathway-triggered coagulation in human blood and thrombosis in several animal models with no discernable effect on hemostasis (David et al., 2016). DEF does not bind to FXI zymogen, other coagulation proteases or the FXIa-like protease plasma kallikrein, and small covalent modifiers of the FXIa active-site serine block DEF binding to FXIa. These observations suggest that DEF probably occupies the FXIa active site (David et al., 2016), but the structural basis for its binding properties and activities is not known.
We report the structure of a complex of DEF bound to the FXIa protease domain as determined by X-ray crystallography. The structure accounts for the specificity of DEF for FXIa over its relative, plasma kallikrein, and its zymogen, FXI. It also accounts for the ability of DEF to inhibit cleavage of small and large substrates and the remarkable sensitivity of DEF-FXIa binding to active-site inhibitors. Additionally, because reversal agents are desirable for clinical use of anticoagulants, we determined whether the inactive FXIa protease domain used to obtain the DEF-FXIa crystal structure might serve as a decoy to reverse inhibitor activity. We provide evidence that this protein can indeed reverse the anticoagulant activity of DEF as well as that of a small molecule FXIa active-site inhibitor. DEF and this reversal agent for DEF and other FXIa active-site inhibitors may help support the clinical development of FXIa-targeting anticoagulants.
Results
A proteolytically inactive FXIa protease domain retains high affinity for the anti-FXIa mAb DEF
FXI is a 160 kDa disulfide-linked homodimer in which each monomer consists of a ~50 kDa “disk” comprised of four apple domains and a ~30 kDa trypsin-like serine protease domain. The protease domain remains attached to the four-apple disk of each monomeric subunit via a disulfide bond after proteolytic conversion of the FXI zymogen to the active FXIa protease (Emsley et al., 2010). To define the structural basis for the activity and properties of DEF, we sought a minimum FXIa construct suitable for generating a co-crystal with DEF. We refer to amino acids in FXI in italics with both full length FXI numbering and trypsin homology (t.n.) numbering (Jin et al., 2005), e.g., the active site serine is designated Ser557 (t.n. Ser195). Antibody residues will be referred to in normal font.
As noted above, the binding and functional properties of DEF suggested that DEF binds to the active site of the FXIa protease (David et al., 2016). Accordingly, we first expressed the FXIa protease domain, Ile370 to Ala606 (t.n. Ile16 to Ala 244) without the FXI apple domains. After expression and purification, the FXIa protease domain protein, hereafter referred to as FXIaPD, was catalytically active as assessed by cleavage of the fluorogenic peptide substrate SN-59 (Figure S1). DEF potently blocked this activity in a dose-dependent manner (Figure 1A) suggesting that DEF could bind isolated FXIa protease domain.
Figure 1. Small substrate cleavage activity and DEF binding by FXIaPD.

(A) Catalytic activity of FXIa and FXIaPD was measured as SN-59 hydrolysis in the presence of DEF at the indicated concentrations. Values are the means of duplicate determinations.
(B) Biacore surface plasmon resonance sensograms and calculated affinity for DEF Fab binding to immobilized FXIa and FXIaiPD. Binding curves with kinetic curve fits overlaid for 0.5–10 nM DEF Fab binding to FXIaiPD are shown. The average calculated Kd values for two independent experiments ± standard error of the mean (SEM) are reported in the table.
To avoid autoproteolysis during purification, concentration, and crystallization, we next generated a proteolytically inert version of FXIaPD domain (denoted FXIaiPD for FXIa inactive protease domain) by converting the active site serine, Ser557 (t.n. Ser195), to alanine. FXIaiPD did not hydrolyze SN-59 (Figure S1) indicating it was indeed catalytically inactive. To assess DEF binding activity, we compared binding of DEF Fab to biotinylated FXIaiPD or biotinylated blood-derived full-length FXIa immobilized on a Biacore chip. DEF bound to FXIaiPD with an apparent affinity of 162 pM and to full-length FXIa with an apparent affinity of 59 pM under the same conditions (Figure 1B). The lower affinity of DEF for FXIaiPD than for full-length FXI might be due to effects of the Ser557Ala, other substitutions, or apple-domain deletion on the stability of the protease domain active conformation or how the protease domain is biotinylated or displayed on the chip. Regardless, taken together, our data indicate that the FXIa protease domain alone supports high-affinity DEF binding, suggesting that most if not all of the DEF epitope resides in this domain.
Crystal structure of the DEF Fab bound to the human FXIa protease domain
To aid crystallization, asparagines in two predicted N-linked glycosylation sites (Asn432 and Asn473 (t.n. Asn73 and Asn113)) were replaced by glutamine to prevent glycosylation. The aglycosylated FXIaiPD was mixed with DEF Fab protein and the resulting complex was purified by size exclusion chromatography before setting up crystallization trials. Crystals of the FXIaiPD:DEF Fab complex grew in space group I422 and diffracted X-rays to a resolution of 1.8 Å (Table 1). The structure (PDB code 6AOD) revealed that DEF Fab recognizes FXIa using contacts centered on the FXIa active site made predominantly by light chain variable loops CDR L1 and CDR L3 (Figure 2A–C and Table 2) and augmented by contacts from heavy chain loops CDR H1 and CDR H3 (Table 2). The resulting buried surface area between FXIa and the DEF Fab light and heavy chains is 976.4 Å2 and 276.6 Å2, respectively.
Table 1.
DEF Fab-FXIa crystallographic data collection.
Crystallographic data collection and refinement statistics
| FXIaiPD-DEF FAB (native) | FXIaiPD-DEF FAB (derivative) | |
|---|---|---|
| Data Collection | ||
| Space group | I422 | I422 |
| Cell dimensions | ||
| a/b/c (Å) | 136.12/136.12/175.95 | 136.61/136.61/176.31 |
| α/β/γ(°) | 90.0/90.0/90.0 | 90.0/90.0/90.0 |
| Resolution (Å) | 64.94 – 1.80 (1.86 – 1.80) | 107.99 – 2.50 (2.59 – 2.50) |
| Rmerge (%) | 5.4 | 3.4 |
| I/σI | 12.99 (0.83) | 25.77 (3.30) |
| Correlation Coefficient | 0.99 (0.28) | 0.99 (0.92) |
| Completeness (%) | 100 (98.0) | 100 (100) |
| Redundancy | 5.7 (5.4) | 15.9 (14.6) |
| Unique reflections | 75924 (7371) | 28898 (2817) |
| Wilson B-factor | 33.99 | 47.30 |
| Refinement | ||
| Rwork/Rfree (%) | 18.2/20.7 | |
| No. of chains in AU | 3 | |
| No. of protein atoms | 5089 | |
| No. of ligand atoms | 102 | |
| No. of water atoms | 560 | |
| RMSD bond lengths (Å) | 0.025 | |
| RMSD angles (°) | 2.05 | |
| Ramachandran best/disallowed regions (%) | 97.37/0.15 | |
Figure 2. Structure of the DEF-FXIa catalytic domain complex.

(A) Cartoon and surface representation of the DEF-FXIa catalytic domain complex. DEF Heavy chain (cyan) and Light chain (marine) are indicated. Inset shows CDR interactions with FXIa (light orange). Catalytic triad residues are red. (B) Cutaway view showing DEF light chain engagement with the active site. Black shows the DEF light chain interior. DEF light chain loops are shown and labeled. Select DEF residues are indicated.
(C) Details of the DEF-FXIa interaction. FXIa residues are indicated by italics.
(D) Orientation of DEF CDR L1 loop residues relative to the FXIa catalytic triad. The structure of trypsin (PDB code 1PQA, light yellow) was superimposed on that of FXIaiPD (light orange) to predict the location of the oxygen nucleophile in the active site serine of FXIa (an alanine in FXIaiPD, introduced to prevent autolysis during protein concentration). The distance between this location and Gly28 in DEF CDR L1 is shown.
Table 2. FXIa-DEF Fab interface contacts.
DEF residues within 5Å of FXIa are shown. FXIa amino acids are indicated by full-length human protein numbering and standard (chymo)trypsin homology numbering. Electrostatic, hydrogen bonding, and cation-p interactions are highlighted in red. FXIa catalytic triad residues His413 and Asp462 (t.n. His57 and Asp102) and Asp551 (t.n. Asp189), the keystone for the S1 subsite, are in italics.
Hydrogen Bond and Electrostatic Interactions
| DEF Light Chain | DEF Heavy Chain | FXIa | Trypsin Numbering | Interaction |
|---|---|---|---|---|
| Arg24 | Tyr416 | 59A | VdW | |
| Met456 | 96 | VdW | ||
| Ala457 | 97 | VdW | ||
| Ala25 | Tyr416 | 59A | H bond | |
| Ser26 | His413 | 57 | VdW | |
| Tyr416 | 59A | VdW | ||
| Gln27 | His413 | 57 | VdW | |
| Tyr416 | 59A | VdW | ||
| Tyr454 | 94 | VdW | ||
| Glu458 | 98 | VdW | ||
| Ser459 | 99 | H bond | ||
| Asp462 | 102 | H bond | ||
| Ser576 | 214 | H bond | ||
| Trp577 | 215 | VdW | ||
| Gly28 | His413 | 57 | VdW | |
| Lys554 | 192 | VdW | ||
| Ile29 | Lys554 | 192 | VdW | |
| Arg30 | Leu506 | 146 | VdW | |
| Asp551 | 189 | salt bridge | ||
| Ala552 | 190 | VdW | ||
| Cys553 | 191 | VdW | ||
| Lys554 | 192 | VdW | ||
| Tyr575 | 213 | VdW | ||
| Ser576 | 214 | VdW | ||
| Trp577 | 215 | VdW | ||
| Gly578 | 216 | VdW | ||
| Glu579 | 217 | VdW | ||
| Gly580 | 218 | H bond | ||
| Cys581 | 219 | VdW | ||
| Gly588 | 226 | VdW | ||
| Val589 | 227 | VdW | ||
| Asn31 | His534 | 174 | VdW | |
| Trp577 | 215 | VdW | ||
| Gly578 | 216 | VdW | ||
| Glu579 | 217 | VdW | ||
| Gly580 | 218 | VdW | ||
| Asp 32 | Leu506 | 146 | VdW | |
| Arg507 | 147 | salt bridge | ||
| Lys554 | 192 | salt bridge | ||
| Arg46 | Arg507 | 147 | VdW | |
| Ser52 | Glu579 | 217 | VdW | |
| Gly66 | His534 | 174 | VdW | |
| Ser67 | Ala457 | 97 | H bond | |
| Glu458 | 98 | VdW | ||
| Ser459 | 99 | VdW | ||
| Gly68 | Met456 | 96 | VdW | |
| Ala457 | 97 | VdW | ||
| Glu458 | 98 | VdW | ||
| Ser459 | 99 | VdW | ||
| Thr69 | Tyr416 | 59A | VdW | |
| Tyr454 | 94 | VdW | ||
| Met 456 | 96 | H bond | ||
| Ala457 | 97 | VdW | ||
| Glu458 | 98 | VdW | ||
| His91 | Arg507 | 147 | VdW | |
| Lys554 | 192 | H bond | ||
| Asp92 | Tyr503 | 143 | H bond | |
| Tyr554 | 192 | VdW | ||
| Ile93 | Tyr503 | 143 | H bond | |
| Asp508 | 148 | VdW | ||
| Ile510 | 151 | VdW | ||
| Lys554 | 192 | VdW | ||
| Gly555 | 193 | VdW | ||
| Tyr94 | Arg395 | 37D | Cation-p | |
| His396 | 38 | H bond | ||
| Leu397 | 39 | VdW | ||
| Tyr503 | 143 | VdW | ||
| Ile510 | 151 | VdW | ||
| Tyr33 | Lys505 | 145 | H bond | |
| Arg507 | 147 | H bond | ||
| Asp508 | 148 | VdW | ||
| Asp551 | 189 | H bond | ||
| His35 | Arg507 | 147 | VdW | |
| Trp50 | Asp508 | 148 | VdW | |
| Lys509 | 149 | VdW | ||
| Asp52 | Lys505 | 145 | H bond | |
| Glu55 | Arg504 | 144 | salt bridge | |
| Asp57 | Lys509 | 149 | VdW | |
| Thr58 | Lys509 | 149 | VdW | |
| Asn59 | Lys509 | 149 | VdW | |
| Leu99 | Arg507 | 147 | H bond | |
| Ala100 | Arg507 | 147 | VdW | |
| Ser101 | Leu506 | 146 | VdW | |
| Arg507 | 147 | VdW |
FXIa-DEF Fab interface contacts. DEF residues within 5 Å of FXIa are shown. FXIa amino acids are indicated by full-length human protein numbering and standard (chymo)trypsin homology numbering. Electrostatic, hydrogen bonding, and cation-π interactions are listed. FXIa catalytic triad residues His413 and Asp462 (t.n. His57 and Asp102) and Asp551 (t.n. Asp189), the keystone for the S1 subsite, are in italics. VdW, van der Waals.
DEF Fab light chain interactions from CDR L1, CDR L3, and the 63-74 loop (LC63-74) fill the substrate-binding pocket (Figure 2B–D). CDR L1 forms the heart of the FXIa recognition element by using three contiguous residues to interact with the FXIa S1 subsite and catalytic triad. L1 Arg30 occupies the S1 subsite, which recognizes P1 basic residues of substrates for FXIa and other trypsin-like proteases. It is coordinated by FXIa Asp551 (t.n. Asp 189) sidechain and the Gly580 (t.n. Gly 218) backbone. Gly28 covers the FXIa catalytic triad residues His413 and Ala557 (t.n. His57 and Ser195; recall that Ala557 is a serine in the active protease). Gln27 donates a hydrogen bond to a carboxylate oxygen in the remaining FXIa catalytic triad residue, Asp462 (t.n. Asp102) (Polgar, 2005) (Figure 2B–D; Table 2); this might reduce the ability of Asp462 to function as a proton acceptor during catalysis. The LC63-74 loop buttresses the CDR L1 conformation through hydrophobic interactions of LC Phe71 with CDR L1 Ile29 and Leu33. The extensive contacts made by CDR L1 and the 63-74 loop are flanked by an additional set made by CDR L3 that center around a cation-π interaction between the guanidino group in FXIa residue Arg395 (t.n. 37D) and CDR L3 Tyr94 and hydrophobic interactions made by Ile93. Together, these light chain interactions completely cover and occlude the active site of FXIa in a manner that would prevent access of peptide and protein substrates (Figure 2B–D). This recognition mode explains the ability of DEF to block cleavage of both small substrates and protein substrates by FXIa and hence its anticoagulant activity.
Co-crystal structure predicts and accounts for DEF specificity and binding properties
The proximity of CDR L1 to the FXIa active site is consistent with the observation that FXIa binding of Fab C24, which has an identical light chain to DEF, loses binding affinity for FXIa modified by phenylmethylsulfonyl fluoride (PMSF) (David et al., 2016), which adds only a phenylmethyl group to the active-site serine. Treatment of FXIa with D-phenylalanyl-L-propyl-L-arginyl chloromethyl ketone (PPACK), which occupies the S1 subsite of trypsin-like serine proteases and also covalently modifies the active-site serine like PMSF, caused the same loss of binding with DEF (Figure 3A, B). In the DEF-FXIaiPD complex, the DEF CDR L1 is in close proximity to the FXIa active-site Ser557 (t.n. Ser195, an alanine here to remove protease function). Superimposition of published structures of serine protease domains bound to PPACK (Thrombin, PDB 1Z8I) or PMSF (Trypsin, PDB 1PQA) on the DEF-FXIaiPD complex predicts that both inhibitors sterically block CDR L1 loop interactions with FXIa (Figure 3C, D), consistent with their inhibiting DEF binding.
Figure 3. Structure predicts sensitivity of DEF binding to modification of the FXIa active site and specificity for FXIa over plasma kallikrein.

(A, B) Effect of PPACK on C24 Fab binding to FXIa was evaluated by surface plasmon resonance. For each experiment, an equivalent amount of FXIa (A) without and (B) with PPACK prior treatment was captured on two different Biacore chip flow cells. C24 Fab passed over for 3 minutes to allow binding and allowed to dissociate for a further 20 minutes.
(C, D) Structure comparison of the DEF-FXIaiPD complex with the (C) Thrombin-PPACK (1Z8I) and (D) Trypsin-PMSF (1PQA) complexes. DEF light chain is marine and shown as cartoon and surface. FXIa is light orange with the catalytic residues show as red sticks and labeled. Thrombin and Trypsin are yellow and white, respectively. PPACK and PMSF are shown as violet and magenta spheres, respectively. DEF light chain elements are labeled.
(E) DEF-FXIa/kallikrein structure comparison. DEF LC63-74 residues (space filling, marine) likely to clash with plasma kallikrein (PK) (light green, PDB: 4OGY). FXIa is light orange with amino acid labels in italics. Kallikrein amino acids are underlined.
The extensive and close interactions of the DEF light chain with the active site of FXIa also appear to explain the inability of DEF to bind or inhibit plasma kallikrein (David et al., 2016), the protease most highly related to FXIa (65% identity and 79% similarity in the protease domain). Such specificity is potentially desirable given the role of plasma kallikrein in complement activation and bradykinin generation. Superimposition of the structure of the plasma kallikrein protease domain on the DEF-FXIaiPD structure (Figure 3E) shows that DEF LC 63-74 loop residues Ser67 and Thr69 clash with the PK loop leading to the catalytic triad Asp438 (t.n. Asp102) and that the junction formed by DEF LC 63-74 loop Gly66 and CDR L1 loop Asn31 clashes with the PK loop containing Tyr555. These clashes both impinge on the key CDR L1 loop and likely contribute to the lack of DEF binding to plasma kallikrein.
Despite occupancy of the FXIa active site by DEF CDR L1, DEF was not cleaved by FXIa (Figure S2). In addition to a possible contribution from altered catalytic triad function associated with the Gln27-Asp462 (t.n. Asp102) interaction described above, the orientation of DEF CDR L1 in the FXIa substrate binding cleft and its distance from the active-site serine hydroxyl likely explain this lack of cleavage (Figure 2D). The side chain of Arg30 in CDR L1 does occupy the S1 subsite of FXIa like the P1 Arg of a typical FXIa or trypsin substrate, but the orientation of CDR L1 peptide chain with respect to the FXIa S1 subsite and active-site serine is carboxyl to amino — opposite that of a typical substrate. Additionally, the peptide bond in DEF CDR L1 that is closest to the active site serine is that between Gly28 and Ile29, and the carbonyl carbon in this bond is 5.5 Å away from the predicted location of the active site oxygen atom that attacks substrates (Figure 2D).
The DEF-FXIaiPD complex also yielded insight into the basis for increased affinity observed with optimization of CDR2 in the DEF heavy chain (David et al., 2016). CDR H2 positions negatively charged residues opposite a positively charged pocket of FXIa (Figure 4A). Although these DEF sites do not directly contact FXIa, being ~6 Å away from the nearest residues, they do coordinate several water molecules shared between FXIa and DEF and appear to contribute to antibody potency, as substitutions that sequentially introduced these negatively charged residues during affinity maturation of antibody D4 (David et al., 2016) increased antibody potency for inhibition of FXIa catalytic activity (Figure 4B).
Figure 4. Electrostatic interactions enhance DEF affinity for FXIa.

(A) DEF-FXIaiPD complex showing the FXIa electrostatic surface potential (PyMol version 1.3). Inset shows heavy chain CDR2 positions where incorporation of acidic residues increases binding affinity. FXIa residues are indicated by italics. (B) Inhibition of fluorogenic substrate cleavage by FXIa. IC50 values for anti-FXIa antibodies with CDRs identical except for the indicated substitutions of charged amino acids (red).
Divalent anion binding sites
The structure also revealed a set of four anions bound to DEF that, on the basis of anomalous signal and presence of ~ 2M sulfate ions in the crystallization buffer, we assigned as sulfate ions (Figure 5). There may be partial occupancy by phosphate at these sites, as this anion was present during purification and in the crystallization conditions.
Figure 5. DEF anion binding sites.

Electron density maps showing 2Fo-Fc (1σ) (slate) and anomalous difference densities (5σ) (violet) for the anion binding sites. Inset shows details of the hydrogen bond and electrostatic interactions surrounding the R507 network.
Two of the anions, designated SO4−2-I and SO4−2-II, are buried within the DEF structure and are part of an extensive network of interactions that includes FXIa residue Arg507 (t.n. Arg147)(Figure 5). SO4−2-I resides between the light and heavy chains and is coordinated by interactions with Tyr36, Arg46, and His91 from the light chain and Arg507 from FXIa. The Arg507 guanidino group is further coordinated by light chain Asp32 and SO4−2-II, which is in turn coordinated by heavy chain His35.
Use of FXIaiPD as a decoy to reverse effects of DEF on FXIa enzymatic activity
Given the ability of FXIaiPD to bind DEF, we next explored the potential of using this molecule to sequester DEF and other inhibitors of the FXIa active site to reverse their anticoagulant effect. Given its bivalency, 10 nM DEF would provide 20 nM of FXIa/FXIaiPD binding sites, and FXIa was present at only 0.7 nM in this assay. We chose 100 nM FXaiPD to rapidly saturate DEF and prevent its interaction with FXIa. Under conditions in which DEF at 10 nM completely inhibited cleavage of the fluorogenic substrate SN-59 by FXIa, inclusion of FXIaiPD at 100 nM with FXIa at 0.7 nM prevented such inhibition (Figure 6A). FXIaiPD had no effect on FXIa activity in the absence of DEF. These results are consistent with FXIaiPD blocking the ability of DEF to bind to and inhibit the FXIa active site.
Figure 6. FXIaiPD prevents inhibition of FXIa catalytic activity by DEF and related anti-FXIa mAbs.

(A) Initial reaction rates from a FXIa-mediated SN-59 hydrolysis assay in presence of increasing concentrations of FXIaiPD. FXIa and FXIaiPD were mixed before adding 10 nM DEF IgG, followed by SN-59 peptide to start the reaction. Data shown are representative of two independent experiments with similar results.
(B) Initial reaction rates plotted for FXIa assay in which FXIaiPD was mixed with the indicated concentration of DEF or related anti-FXIa mAbs of different potency, all at a in a 10 to 1 molar ratio, prior to addition of FXIa and SN-59 peptide to start reaction. Data shown are mean of duplicate determinations. This experiment was repeated two times with similar results.
(C) Initial reaction rates plotted for FXIa assay, where decoy is pre-mixed in at varying ratios with the small molecule FXIa active site inhibitor SM1 (at 500 and 200 nM), prior to addition of FXIa and SN-59 peptide to start the reaction. Data shown are mean +/− SEM (n= 3).
We next investigated the ability of FXIaiPD to prevent the actions of other FXIa active site blockers in the same assay. Addition of FXIaiPD at a 10:1 molar ratio blocked the inhibitory activity of DEF-related anti-FXIa mAbs of varying potency (David et al., 2016) (Figure 6B). Furthermore, FXIaiPD reversed the effect of SM1, a small molecule active-site inhibitor of FXIa with an IC50 for inhibition of FXIa small substrate cleavage of 200 nM (Figure 6C). Thus, FXIaiPD can prevent the ability of antibody-based inhibitors and a small molecule active site inhibitor to block small substrate cleavage by FXIa.
FXIaiPD blocks the ability of DEF to inhibit FXIIa-driven thrombin generation in human plasma
An effective agent for reversal of anticoagulation will have to function in plasma. Addition of FXIIa, the triggering protease for the intrinsic pathway, to human plasma initiates FXIa-dependent thrombin generation that is blocked by DEF (David et al., 2016). FXIaiPD reversed the inhibitory effects of DEF in this assay. Specifically, addition of FXIaiPD to human plasma at 12.5 μg/ml or above completely reversed the effect of 16 μg/ml DEF on lag to onset and peak thrombin generation (Figure 7). FXIaiPD at 12.5 μg/ml and DEF at 16 μg/ml corresponds to an ~2-fold molar ratio of FXIaiPD to DEF binding sites considering that one molecule of DEF IgG1 contains two FXIaiPD binding sites. Inclusion of FXIaiPD with DEF at a 1:1 molar ratio had a partial effect (Figure 7).
Figure 7. FXIaiPD blocks inhibition of intrinsic pathway-triggered thrombin generation by DEF in human plasma.

(A) FXIIa-induced thrombin activity in human plasma as a function of time was determined in the presence of DEF at 16 μg/ml and the indicated concentration of FXIaiPD. A representative set of curves is shown.
(B, C) Quantitation of thrombin peak activity (B) and lag time (C) plotted as a function of FXIaiPD concentration. Data shown are mean +/− SEM (n=3).
FXIaiPD does not bind the FXIa substrate FIX or alter thrombin generation in human plasma in the absence of DEF
To be useful as a reversal agent for DEF and other FXIa inhibitors in vivo, FXIaiPD should be unable to sequester natural FXIa substrates and inhibitors. Coagulation factor IX is the key FXI substrate that links the intrinsic pathway to the coagulation cascade. The A3 domain of FXIa appears to be required for FXIa to recognize and activate FIX (Emsley et al., 2010; Gailani et al., 2014) and is absent in FXIaiPD. Unlike the complete FXIa molecule, FXIaiPD exhibited no binding to FIX at the maximum concentration tested (30 μg/ml; 200 nM) (Figure S3). Additionally, addition of FXIaiPD at 100 μg/ml (660 nM) in the absence of DEF neither inhibited nor enhanced thrombin generation in the standard FXIIa-induced thrombin generation assay (Figure 7A). To increase sensitivity for detecting increased thrombin generation, we determined the effect of FXIaiPD addition on thrombin generation in the absence of FXIIa or tissue factor or at very low concentrations of these triggers (Figure S4). No significant differences were detected with FXIaiPD treatment. However, thrombin generation was quite variable at very low concentrations of FXIIa or TF and there was a trend toward increased thrombin generation under such conditions. This phenomenon would merit further study if a FXIaiPD-like molecule were considered for clinical development.
FXIaiPD reverses effects of DEF in an in vivo rabbit dosing experiment
To investigate the effects of FXIaiPD on reversal of DEF-mediated FXIa inhibition in an in vivo setting, we next carried out a sequential dosing experiment in rabbits. Rabbits received a bolus injection of 1 mg/kg DEF followed 30 minutes later by a bolus injection of 1 mg/kg FXIaiPD (Figure 8). Plasma samples were obtained before dosing and after each injection for measurement of activated partial thromboplastin time (APTT) and prothrombin time (PT), standard assays of intrinsic and extrinsic pathway-dependent coagulation, respectively. As expected (David et al., 2016), administration of DEF at 1 mg/kg prolonged APTT (Figure 8A) but not PT (Figure 8B). While APTT remains prolonged for >2 weeks after administration of 1 mg/kg DEF in the absence of reversal (David et al., 2016), DEF-driven APTT prolongation was completely reversed 30 minutes after dosing with 1 mg/kg FXIaiPD (Figure 8A). At 1 mg/kg for both molecules, the ratio of FXIaiPD to DEF binding sites would be ~3:1 considering that each DEF IgG1 has two FXIaiPD binding sites. Dosing of rabbits with FXIaiPD neither prolonged nor shortened APTT in the absence of DEF (Figure 8C), consistent with its failure to bind FIX or alter thrombin generation in plasma ex vivo (Figures 7, S3 and S4). These results suggest that FXIaiPD can sequester DEF in vivo and prevent its anticoagulant effect.
Figure 8. FXIaiPD reverses DEF anticoagulation in rabbits.

(A, B) Four rabbits received a 1 mg/kg bolus intravenous injection of DEF mAb followed 30 minutes later by a bolus intravenous injection of 1 mg/kg FXIaiPD. Just prior to each injection, and 30 minutes after the final (FXIaiPD) injection, blood was drawn to make plasma for APTT and PT clotting time assays. Data is plotted to show sequential effects on DEF IgG and decoy injection on APTT (A) and PT (B) coagulation times.
(C) Three rabbits received a 1 mg/kg bolus intravenous injection of FXIaiPD; blood samples for APTT determination were obtained before and 30 minutes after.
Discussion
FXI recently became a focus of drug development as multiple lines of evidence support the idea that inhibiting this intrinsic pathway coagulation protease may lead to antithrombotic benefit with less bleeding than current strategies (Fredenburgh et al., 2017; Gailani et al., 2015). We recently described the generation and functional characterization of a high-affinity anti-FXIa mAb designated DEF (David et al., 2016). DEF does not bind to the FXI zymogen or inhibit other coagulation proteases or plasma kallikrein and prevents thrombosis in two in vivo models at clinically relevant doses without a detectable increase in spontaneous or induced bleeding. A high-dose toxicology study in cynomolgus macaques with DEF did not reveal adverse effects, suggesting that clinical development of this molecule might be possible.
We report here a high-resolution co-crystal structure of DEF bound to the protease domain of FXIa that explains many properties of DEF. This structure reveals an extensive interaction between DEF and the FXIa substrate binding pocket and active site. These interactions would deny access of small and large substrates to the FXIa S1 subsite and active site serine. Further, interactions between DEF light chain residue Gln27 and FXIa catalytic triad component Asp462 (t.n. Asp102) might impair catalytic triad function. These observations explain the ability of DEF to prevent cleavage of FIX by FXIa and hence its anticoagulant effects. Additionally, because S1 subsite forms and the FXIa active conformation becomes stable after proteolytic conversion of the FXI zymogen to FXIa, the extensive interactions of DEF CDR L1 with the FXIa active site explain the inability of DEF to recognize the FXI zymogen. We have viewed this specificity as a theoretically desirable feature, in that DEF would act only during active coagulation, dosing would not need to match new FXI synthesis, DEF would not deplete the FXI zymogen, and its actions could be reversed without FXI replacement (see below).
The observation that DEF CDR L1 penetrates deeply into the FXIa active site with contacts near the catalytic serine explains our previous observation that antibody binding to FXIa is surprisingly sensitive to modification of the FXIa active site by PMSF. Indeed, the addition of a simple phenylmethyl group to the active site serine hydroxyl causes clear clashes with the central part of the L1 loop (Figure 3D). We showed the same sensitivity of DEF binding to PPACK-treated FXIa here and demonstrated that both observations may be explained by steric clashes with DEF CDR L1. The close apposition of DEF to the FXIa active site also explains the specificity of DEF for FXIa over other serine proteases (David et al., 2016). Overlaying the structure of the most homologous protease to FXIa, plasma kallikrein, reveals numerous clashes between PK and DEF CDR L2 and L3 (Figure 3E).
IgG inhibitors of serine proteases appear to function by allosteric modulation of protease activity or by preventing protease interaction with substrates by occupancy of the protease active site (Ganesan et al., 2010). Allosteric blockers include Ab40 to the hepatocyte growth factor activator (Ganesan et al., 2009) and the anti-FXIa IgG 076D-M007-H04 (Wilmen A, 2013). Active site blockers include the IgG inhibitor of plasma kallikrein, DX-2930 (Kenniston et al., 2014) and Fab E2 to matriptase (MT-SP1) (Farady et al., 2008). Like DEF CDR L1 in the active site of FXIa, CDR H3 of Fab E2 penetrates deeply into the matriptase active site and places an arginine side chain into its S1 subsite like the P1 amino acid of a substrate. The next amino acid (P1′) in E2 CDR H3 does interact on the prime side of the active site but the loop then turns toward the S2 and S3 subsites. This orientation may explain the observation that CDR H3 of Fab E2 is cleaved by matriptase at the P1 Arg but with very low efficiency (Farady et al., 2007). Similar considerations may explain our observation that DEF is not cleaved by FXIa (Figure S1). These include the ~5.5Å predicted distance from the FXIa active site serine to the closest scissile bond in DEF (the Gly28 carbonyl in DEF CDR L1) and the “backward” local geometry of the L1 loop in the active site (Figure 2D) as well as perhaps altered catalytic triad function due to the Gln27-Asp462 interaction.
The DEF-FXIaiPD structure also revealed features that explain the observed affinity maturation gains (Figure 4). Progressive accumulation of changes in CDR-H2 during affinity maturation (David et al., 2016) (Figure 4) places an increasing number of negatively charged residues near (~6Å) a positively charged surface on FXIa external to the active site. These substitutions appear to contribute long-range electrostatic interactions that assist docking of the IgG to FXIa (Figure 4) and coordinate water molecules between DEF and FXIa in the structure. This mode of Fab binding enhancement stands in contrast to affinity maturation changes reported for the anti-plasma kallikrein antibody DX-2930 (Kenniston et al., 2014). Although both DEF and DX-2930 insert a CDR loop into the active site to block substrate access to their respective protease active sites, affinity maturation changes to DX-2930 all occurred in the active-site penetrating CDR loop (Kenniston et al., 2014). The demonstration that the DEF binding properties can be improved without affecting the main recognition element suggests that other changes distant from the CDR L1-active site interface might be sites for further improvement. Among these, the network surrounding the two buried anions that coordinate R507 (Figure 5) is a leading candidate. The anion-binding sites might bind waters or phosphate under physiological conditions and the apparent partial occupancy of these sites by sulfate despite the high-sulfate environment used for crystallization suggests equilibrium with anion-unbound state(s). Mutations that optimize or replace anion binding might enhance affinity for FXIa. Additionally, the long-distance interactions between DEF and FXIa at this site might be improved.
To prevent autoproteolysis and proteolytic degradation during the crystallization phase, we mutated the FXIa catalytic serine to alanine. This site is required for procoagulant function. To generate a minimal protein to favor crystallization, we deleted the apple domains, which are required for FXI homodimerization and interactions with its natural activators and substrates (Emsley et al., 2010; Zucker et al., 2009). The ability of this molecule, designated FXIaiPD, to bind DEF with high affinity confirmed that DEF binding was attributable to binding to the FXIa protease domain (as borne out in the structure) and suggested that FXIaiPD might serve as a decoy to bind DEF and reverse its anticoagulant effect. Additionally, its lack of protease activity, procoagulant function and site for interaction with FXI substrates or activators suggested that FXIaiPD might not interfere with normal coagulation. Considered in this way, FXIaiPD became a candidate reversal agent of FXIa active-site inhibitors analogous to andexanet alpha, a reversal agent for FXa active-site inhibitors (Lu et al., 2013; Siegal et al., 2015). Although FXIa inhibition is predicted to be safer than current anticoagulants based on animal studies and the phenotype of human FXI-deficiency (Bane and Gailani, 2014; Fredenburgh et al., 2017; Muller et al., 2011), the latter is associated with increased bleeding in certain surgical and other settings and a reversal agent for FXIa inhibitors may be desirable for use in emergency surgery or bleeding events should FXIa inhibitors come into common use.
Our in vitro studies confirmed that the FXIaiPD can indeed serve as a decoy to restore FXIa activity in the presence of DEF, related anti-FXIa mAbs, or a small molecule FXIa active site inhibitor. The observation that a 5-fold molar ratio of FXIaiPD to DEF (2.5-fold ratio of FXIaiPD to DEF binding sites) reversed the inhibitory effects of DEF on FXIIa-triggered thrombin generation in human plasma and reversed prolongation of APTT in DEF-treated rabbits suggests that FXIaiPD is an effective decoy for active-site FXIa inhibitors in vivo. The lack of FXIaiPD binding to FIX, the lack of prolongation or shortening of APTT in rabbits in the absence of DEF, and the apparent lack of a significant effect of FXIaiPD on thrombin generation in human plasma in the absence of DEF suggests that it may not interact substantially with coagulation factors or inhibitors at the concentrations likely to be used. This issue will require substantial additional examination if FXaiPD is ever considered for clinical development.
While the current generation inhibitors of factor Xa and thrombin have more predictable drug properties and confer benefits over early generation anticoagulants such as warfarin, agents that better separate the antithrombotic efficacy from bleeding risk remain desirable (Bane and Gailani, 2014; Fredenburgh et al., 2017; Muller et al., 2011). FXI inhibitors currently in development, including the active-site binding anti-FXIa antibody DEF — the structure of which is described in this report — may permit testing of the hypothesis that FXI inhibition will provide such an advance. However, situations will arise in which reversal of anti-FXI anticoagulants is desired. The FXIaiPD decoy molecule, described in this report, represents a possible candidate reversal agent for antibodies and small molecules that target the FXIa active site.
STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human anti-FXIa clone DEF IgG | David et al., 2016 | N/A |
| Human anti-FXIa clone C24 IgG | David et al., 2016 | N/A |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| TriniCAL Reference Plasma | Tcoag | T5101 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FPR-chloromethylketone (PPACK) | Haematologic Technologies | Cat# FPRCK01 |
| Human FXIa | Haematologic Technologies | Cat# HCXIA-0160 |
| Human FIX | Haematologic Technologies | Cat# HCIX-0040 |
| D-VPR-ANSNH-C4H9.2HCL | Haematologic Technologies | Cat# SN-59 |
| Thrombin calibrator | Diagnostica Stago | Cat# 86192 |
| Human Factor Alpha-XIIa | Enzyme Research Laboratories | Cat# HFXIIa 1212a |
| PPP-Reagent low | Diagnostica Stago | Cat# 86194 |
| Critical Commercial Assays | ||
| Classic Suite 1 (crystallization conditions) | Qiagen | Cat# 130901 |
| Classic Suite 2 (crystallization conditions) | Qiagen | Cat# 130923 |
| Protein Complex (crystallization conditions) | Qiagen | Cat# 130915 |
| AmSO4 Suite (crystallization conditions) | Qiagen | Cat# 130905 |
| ExpiFectamine 293 Transfection Kit | Thermofisher | Cat# A14525 |
| Biotin CAPture Kit (Biacore assay) | GE Life sciences | Cat# 28-9202-33 |
| Deposited Data | ||
| FXIa/DEF Fab complex coordinates | This paper | PDB: 6AOD |
| Experimental Models: Cell Lines | ||
| Expi293F cells (protein expression) | Thermofisher | Cat# A14527 |
| Experimental Models: Organisms/Strains | ||
| New Zeland White Rabbits (female, 4–6 Kg, age 4–6 months) | Charles River | Strain Code 571 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Expression vector: pRY19-DEF-Fab-LC | David et al., 2016 | N/A |
| Expression vector: pRY19-DEF-Fab-HC | David et al., 2016 | N/A |
| Expression vector: pRY19-FXIaPD | This paper | N/A |
| Expression vector: pRY19-FXIaiPD | This paper | N/A |
| Expression vector: pRY19-FXIaiPD-agly | This paper | N/A |
| Software and Algorithms | ||
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org/download/ |
| CCP4 suite | Murshudov et al., 2011 | http://www.ccp4.ac.uk/download/index.php#os=mac |
| COOT | Emsley and Cowtan, 2004 | http://www2.mrc-lmb.cam.ac.uk/Personal/pemsley/coot/devel/build-info.html |
| PISA | Krissinel and Henrick, 2007 | http://www.ccp4.ac.uk/download/index.php#os=mac |
| Biacore T200 Evaluation software 3.0 | GE Healthcare | Provided with instrument |
| Thrombinoscope software program | Diagnostica Stago | Cat# 86191 |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Author, Dr. Shaun Coughlin (Shaun.Coughlin@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
FXIaiPD reversal of DEF activity in rabbits
All procedures were performed in accord with protocols approved by an Institutional Animal Care and Use Committee or through an ethical review process. Four rabbits were anesthetized and treated as follows: at time 0, each animal received a 1 mg/kg intravenous bolus injection of the DEF IgG; 30 minutes later each animal received an intravenous bolus of 1 mg/kg FXIaiPD. Just prior to each injection, and 30 minutes after the final (FXIaiPD) injection, blood was drawn to make plasma for APTT and PT clotting assays. In a separate experiment, three rabbits were anesthetized and received an intravenous bolus injection of 1 mg/kg FXIaiPD, with plasma drawn for APTT and PT measurement before and 30 minutes after the dose.
METHOD DETAILS
Nomenclature
FXIa residues are designated in italics, with residue 1 the first amino acid in the FXI protein. Trypsin homology numbering (t.n.) is also provided. DEF light and heavy chain residues are in regular font, with residue 1 the first amino acid in the mature protein after cleavage of the signal peptide.
Protein expression and purification
The catalytic domain of human FXI from Ile370 to Ala606 (t.n. Ile16 to Ala 244) and its derivatives were expressed in Expi293F cells (Life Technologies, CA, USA). Expression plasmids encoded a mammalian signal peptide derived from mouse IgG, followed by the catalytic domain of human FXI and an octa-histidine tag (His8) at the C-terminus to facilitate detection and purification. Cys482 (t.n. Cys123) was replaced by Ser to remove a predicted unpaired Cys normally contributing a disulfide bond between the protease domain and apple domain 3 in the full-length protein. The catalytically inactive variant (FXIaiPD) added a Ser557Ala substitution. The aglycosylated FXIaiPD construct contained substitutions at predicted N-linked glycosylated sites (Asn432 and Asn473 (t.n. Asn73 and Asn113) were substituted with Gln) and a C-terminal hexa-histidine tag (His6). Suspension Expi293F cell cultures were transiently transfected using ExpiFectamine 293 (Life Technologies). Conditioned media was collected 4 days post transfection. Cells were pelleted by centrifugation and the protein was purified from the supernatant using Ni Sepharose resin per the manufacturer’s instructions (GE Healthcare). Captured protein was eluted using imidazole buffer and the buffer was exchanged via dialysis into phosphate buffer saline (PBS). The purity and size of the protein was analyzed by SDS-PAGE and the protein concentration measured using the absorbance at 280 nm.
The DEF Fab was expressed by co-transfecting Expi293F cells with two mammalian expression plasmids, one encoding the Fab heavy chain with a His6 tag and one the light chain, into Expi293F cells. Supernatants were harvested 3 days post transfection and the recombinant proteins captured on Ni Sepharose resin (GE Healthcare, PA, USA).
Surface plasmon resonance
Binding affinity was analyzed by surface plasmon resonance (SPR) using a Biacore T200 (GE Healthcare)(David et al., 2016). Recombinant FXIaiPD and blood-derived FXIa (Haematologic Technologies) were biotin labeled via primary amines and captured on a Biacore CAP chip (GE Healthcare). Experiments used a 30 μl/min flow rate in 10 mM HEPES (pH 7.4), 150 mM NaCl and 0.005% v/v Surfactant P20 (HBSp) buffer (GE Healthcare) at 37 °C. After each DEF Fab injection, the chip surface was regenerated with 6 M guanidine-HCl and 0.25 M NaOH, coated with the CAP reagent, and new FXIaiPD or FXIa was captured. For PPACK complexes, biotinylated FXIa was pre-incubated with 0.2 mM PPACK (Haematologic Technologies). Data were analyzed using Biacore T200 Evaluation software and background and buffer subtracted data fit to a 1:1 Langmuir binding model.
To assess the binding of FIX to FXIa and FXIaiPD, experiments were performed at 25 °C using a 50 μl/min flow rate in HBSp buffer (GE H ealthcare). After each FIX injection (5–200 nM), the chip surface was regenerated with a solution of 6 M guanidine-HCl and 0.25 M NaOH, coated with the CAP reagent and new FXIaiPD and FXIa was captured. All data was analyzed using the Biacore T200 Evaluation software and background and buffer subtracted. Results reported are representative of two experiments.
Structure of the DEF Fab bound to FXIaiPD
Complex formation and crystallization
Purified FXIaiPD protein was mixed with DEF Fab in a 1.5-fold molar excess and concentrated in a centrifugal filter unit at 4 °C. The complex was purified using a Superdex 200 gel filtration column (GE Healthcare Bio-Sciences) equilibrated in 10 mM Tris (pH8.0), 150 mM sodium chloride buffer (TBS) on an AKTA Avant FPLC instrument (GE Healthcare Bio-Sciences). Fractions corresponding to peaks in 280 nm absorbance were run on SDS-PAGE under reducing and non-reducing conditions. Fractions containing the DEF Fab – FXIa catalytic domain complex were pooled and concentrated for crystallization trials.
Purified DEF Fab-aglycosylated FXIaiPD complex was concentrated to 25.7 mg/ml. A 0.25 μl drop of protein sample was mixed with 0.25 μl of reservoir solution (1.86 M ammonium sulfate, 8 mM CoCl2, 30 mM K/Na phosphate, pH 6.5) and crystallized in hanging-drop configuration over 100 μl of reservoir solution at 20 °C. Crystals appeared overnight and grew to full size in 1 week. Crystals were harvested and cryoprotected with a saturated ammonium sulfate solution containing 8 mM CoCl2, 30 mM K/Na phosphate, pH 6.5 and 3% glycerol and then flash frozen in liquid nitrogen.
Data collection and processing
Two datasets were collected from a single crystal at 100 K using synchrotron radiation (APS GM/CAT beamline 23-IDB, Chicago, IL). A native dataset was collected at λ=1.033 Å with 180° rotation. An additional dataset was collected near the cobalt Kedge (λ=1.606 Å) in order to maximize the cobalt anomalous signal present in the crystallization solution. The two datasets were processed with XDS, scaled and merged with Aimless and a resolution cutoff of 1.8 Å and 2.5 Å for the native and derivative datasets respectively, was applied accordingly to the CC1/2 criterion (Diederichs and Karplus, 2013; Karplus and Diederichs, 2012). Crystals belong to the I422 space group.
Structure determination and refinement
The structure of the DEF Fab-aglycosylated FXIaiPD complex was solved by molecular replacement using the Fab fragment of the human antibody 5-51/O12 (PDB code: 4KMT) as search model. Once the Fab fragment was placed, the electron density for the missing FXIa catalytic domain was clearly visible. It was finally placed with a second MR round using PDB code 3SOS as search model. Iterative rounds of model building using COOT (Emsley and Cowtan, 2004) and refinement using REFMAC5 (Collaborative Computational Project, 1994, Murshudov et al., 2011) and PHENIX (Adams et al., 2010) were carried out to improve the electron density map.
To help assign the residual positive peaks, an anomalous map was generated using the dataset collected at cobalt K-edge. The wavelength used was sufficient to get a strong signal from the sulfate ions, helping us to model 4 sulfate anions and 2 cobalt cations.
X-ray structure-based epitope mapping
The complex of DEF Fab and aglycosylated FXIaiPD crystallized as 1 copy of the complex per asymmetric unit. Residues of the DEF Fab (paratope) in contact with FXIa (epitope) were determined with PISA (Protein Interactions, Surfaces and Assemblies) (Krissinel and Henrick, 2007).
Inhibition of anti-FXIa antibody by FIXaiPD
Enzyme activity was determined as cleavage of a fluorogenic peptide substrate. Reactions were initiated by mixing blood-derived FXIa (0.11 μg/ml) (Haematologic Technologies Inc.) with 100 μM SN-59 peptide substrate (Haematologic Technologies Inc) in assay buffer containing 50 mM Tris-HCl (pH7.4), 250 mM NaCl and 1 mM EDTA. To confirm the activity of the catalytic domains, 50 nM purified FXI catalytic domain was mixed with the SN-59 peptide in assay buffer. The reaction was monitored in a SpectraMax fluorescent plate reader (Molecular Devices) at 37 °C with an excitation of 352 nm and emission of 370 nm. Data was collected every 60 seconds and results reported as reaction rate (units/sec) for 50–1000 sec. Results are reported as the mean of duplicate data points.
The ability of FXIaiPD to inhibit FXIa activity was assessed by premixing blood-derived FXIa (0.11 μg/ml) and FXIaiPD (10, 20, 100 and 500 nM) before adding the DEF antibody (10 nM) for 5 minutes and initiating the reaction with the peptide substrate SN-59 as described above.
The ability of FXIaiPD to inhibit the activity of different anti-FXIa antibodies with different CDR sequences and different affinities for FXIa was assessed by premixing the individual antibodies with FXIaiPD prior to adding human FXIa (300 pM) and SN-59 substrate (100 μM; Haematologic Technologies Inc) to start the reaction. FXIaiPD catalytic domain was included at a 10:1 ratio to IgG over the IgG dose range of 25.6, 8.6, 2.85, 0.95, 0.31, 0.1, 0.35 μg/ml. Assays with the small molecule FXIa active site inhibitor SM1 were carried out in similar fashion, with indicated concentrations of decoy premixed with either 500 nM or 2000 nM SM1, prior to addition of human FXIa (1 nM and SN-59 substrate (100 uM) to start the reaction. IC50 for SM1 in above FXIa activity assay was carried out prior to monitoring of decoy effects on assay.
In vitro thrombin generation
Thrombin generation was measured using a fluorogenic thrombin substrate in a multi-well automated fluorescent plate reader (ThrombinoSCOPE, Maastricht, the Netherlands) according to the manufacturer’s protocol. Briefly, 5 μL of anti-FXIa DEF antibody (16 μg/ml) containing the FXIaiPD decoy molecule at 0, 3.1, 6.3, 12.5, 25, 50, 100 μg/ml) was mixed with 20 μL PBS, containing 60 nM human Factor XIIa (Enzyme Research Laboratories, South Bend, IN, USA) and phosphatidylcholine/phosphatidylserine (Phospholipid-TGT, DiaPharma, West Chester, OH, USA) in a 96-well plate. 75 μL citrated human plasma (Triclinical Reference Plasma, TCoag, Wicklow, Ireland) was added, and thrombin generation was triggered by the addition of 20 μL calcium chloride buffer containing fluorogenic thrombin substrate. The amount of thrombin generated in the reaction was measured over time. Due to lot-to-lot variability, the concentration of each lot of phospholipid reagent was adjusted to achieve an ~10-minute lag to onset and ~125 nM peak thrombin activity.
To assess the impact of FXIaiPD on thrombin generation in human plasma, 10 μL of FXIaiPD in PBS (60 μg/ml) or PBS only was mixed with 20 μL PBS containing Factor XIIa (Enzyme Research Laboratories, South Bend, IN, USA) at 1, 10, 100, 1000, 10000 pM and phosphatidylcholine/phosphatidylserine (Phospholipid-TGT, DiaPharma, West Chester, OH, USA) in a 96-well plate. Alternatively, 10 μL of FXIaiPD in PBS (60 μg/ml) or PBS only was mixed with 20 μL of PPP reagent (ThrombinoSCOPE, Maastricht, the Netherlands) diluted with PBS to achieve TF concentration of 0.2, 0.5, 1.7 and 5 pM. 70 μL citrated human plasma (Triclinical Reference Plasma, TCoag, Wicklow, Ireland) was added, and thrombin generation was triggered by the addition of 20 μL calcium chloride buffer containing fluorogenic thrombin substrate. Endogenous thrombin potential (area under curve of thrombin activity vs. time), lag time (time to onset of increase in thrombin activity) and time to peak thrombin activity were measured using a ThrombinoSCOPE.
Assay for cleavage of DEF by FXIa
Proteins were incubated for 24 hours at 37 °C in as say buffer comprised of 50 mM Tris-HCl pH7.4, 250 mM NaCl, 1 mM EDTA. The final volume for each reaction was 25 μl and containing either 10 μg FXIa (~2.5 μM), 10 ug FXIa and 10 ug DEF IgG (~2.7 μM), or 10 ug DEF IgG. Samples were prepared for SDS-PAGE by adding sample loading buffer with or without reducing reagent to 10 μl of reaction and heated to 95 °C for 3 min. Samples were run on a 4–12% Bis-Tris gel (Invitrogen, Thermo Fisher Scientific) and visualized with a Coomassie G-250 based stain.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical analysis for each experiment is documented in the figure legends and methods where appropriate.
DATA AND SOFTWARE AVAILABILITY
Coordinates and structure factors have been deposited in the PDB under the ID code 6AOD.
Supplementary Material
Highlights.
Anticoagulants that better separate prevention of thrombosis and bleeding are needed.
DEF, an MAb to FXIa, prevents thrombosis without detectable bleeding in animals.
DEF is highly specific for FXIa over its zymogen and other coagulation proteases.
A DEF-FXIa structure explains DEF activity and specificity and provides an antidote.
Acknowledgments
We thank E. Chu, C. Layla for technical assistance and I. Rondon, J. Dal Porto, R. Lindberg, and A. Coyle for helpful discussions. This work was funded by Pfizer Centers for Therapeutic Innovation and NIH/NHLBI R01HL080050 to D.L.M.
Footnotes
Author Contributions: L.K.E., M.L., T.D., K.L., Y.C.K., D.S., P.B., J.L.G., and T.M. orchestrated the experiments. T.M., D.L.M. and S.R.C. conceived the studies, supervised the work, and wrote the paper with L.K.E. and M.L.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica. Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. The New England journal of medicine. 1991;325:153–158. doi: 10.1056/NEJM199107183250303. [DOI] [PubMed] [Google Scholar]
- Bane CE, Jr, Gailani D. Factor XI as a target for antithrombotic therapy. Drug discovery today. 2014;19:1454–1458. doi: 10.1016/j.drudis.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A [letter] Nature genetics. 1995;10:119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- Bugge TH, Xiao Q, Kombrinck KW, Flick MJ, Holmbäck K, Danton MJ, Colbert MC, Witte DP, Fujikawa K, Davie EW, et al. Fatal embryonic bleeding events in mice lacking tissue factor, the cell-associated initiator of blood coagulation. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6258–6263. doi: 10.1073/pnas.93.13.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, Segers A, Verhamme P, Weitz JI. Factor XI antisense oligonucleotide for prevention of venous thrombosis. The New England journal of medicine. 2015;372:232–240. doi: 10.1056/NEJMoa1405760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David T, Kim YC, Ely LK, Rondon I, Gao H, O’Brien P, Bolt MW, Coyle AJ, Garcia JL, Flounders EA, et al. Factor XIa-specific IgG and a reversal agent to probe factor XI function in thrombosis and hemostasis. Science translational medicine. 2016;8:353ra112. doi: 10.1126/scitranslmed.aaf4331. [DOI] [PubMed] [Google Scholar]
- De Caterina R, Husted S, Wallentin L, Andreotti F, Arnesen H, Bachmann F, Baigent C, Huber K, Jespersen J, Kristensen SD, et al. General mechanisms of coagulation and targets of anticoagulants (Section I). Position Paper of the ESC Working Group on Thrombosis--Task Force on Anticoagulants in Heart Disease. Thrombosis and haemostasis. 2013;109:569–579. doi: 10.1160/TH12-10-0772. [DOI] [PubMed] [Google Scholar]
- Dewerchin M, Liang Z, Moons L, Carmeliet P, Castellino FJ, Collen D, Rosen ED. Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thrombosis and haemostasis. 2000;83:185–190. [PubMed] [Google Scholar]
- Diederichs K, Karplus PA. Better models by discarding data? Acta crystallographica. Section D, Biological crystallography. 2013;69:1215–1222. doi: 10.1107/S0907444913001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggen CJ, Rosendaal FR, Meijers JC. Levels of intrinsic coagulation factors and the risk of myocardial infarction among men: Opposite and synergistic effects of factors XI and XII. Blood. 2006;108:4045–4051. doi: 10.1182/blood-2005-12-023697. [DOI] [PubMed] [Google Scholar]
- Emsley J, McEwan PA, Gailani D. Structure and function of factor XI. Blood. 2010;115:2569–2577. doi: 10.1182/blood-2009-09-199182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica. Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Erlich J, Parry GC, Fearns C, Muller M, Carmeliet P, Luther T, Mackman N. Tissue factor is required for uterine hemostasis and maintenance of the placental labyrinth during gestation. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8138–8143. doi: 10.1073/pnas.96.14.8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farady CJ, Egea PF, Schneider EL, Darragh MR, Craik CS. Structure of an Fab-protease complex reveals a highly specific non-canonical mechanism of inhibition. Journal of molecular biology. 2008;380:351–360. doi: 10.1016/j.jmb.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farady CJ, Sun J, Darragh MR, Miller SM, Craik CS. The mechanism of inhibition of antibody-based inhibitors of membrane-type serine protease 1 (MT-SP1) Journal of molecular biology. 2007;369:1041–1051. doi: 10.1016/j.jmb.2007.03.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredenburgh JC, Gross PL, Weitz JI. Emerging anticoagulant strategies. Blood. 2017;129:147–154. doi: 10.1182/blood-2016-09-692996. [DOI] [PubMed] [Google Scholar]
- Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. Journal of thrombosis and haemostasis: JTH. 2015 doi: 10.1111/jth.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailani D, Geng Y, Verhamme I, Sun MF, Bajaj SP, Messer A, Emsley J. The mechanism underlying activation of factor IX by factor XIa. Thrombosis research. 2014;133(Suppl 1):S48–51. doi: 10.1016/j.thromres.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan R, Eigenbrot C, Kirchhofer D. Structural and mechanistic insight into how antibodies inhibit serine proteases. The Biochemical journal. 2010;430:179–189. doi: 10.1042/BJ20100634. [DOI] [PubMed] [Google Scholar]
- Ganesan R, Eigenbrot C, Wu Y, Liang WC, Shia S, Lipari MT, Kirchhofer D. Unraveling the allosteric mechanism of serine protease inhibition by an antibody. Structure (London, England: 1993) 2009;17:1614–1624. doi: 10.1016/j.str.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Gruber A, Hanson SR. Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood. 2003;102:953–955. doi: 10.1182/blood-2003-01-0324. [DOI] [PubMed] [Google Scholar]
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston JA, Faucette RR, Martik D, Comeau SR, Lindberg AP, Kopacz KJ, Conley GP, Chen J, Viswanathan M, Kastrapeli N, et al. Inhibition of plasma kallikrein by a highly specific active site blocking antibody. The Journal of biological chemistry. 2014;289:23596–23608. doi: 10.1074/jbc.M114.569061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. Journal of molecular biology. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Jin L, Pandey P, Babine RE, Gorga JC, Seidl KJ, Gelfand E, Weaver DT, Abdel-Meguid SS, Strickler JE. Crystal Structures of FXIa Catalytic Domain in Complex with Ecotin Mutants Reveal Substrate-like Interations. The Journal of biological chemistry. 2005;280:4704–4712. doi: 10.1074/jbc.M411309200. [DOI] [PubMed] [Google Scholar]
- Lu G, DeGuzman FR, Hollenbach SJ, Karbarz MJ, Abe K, Lee G, Luan P, Hutchaleelaha A, Inagaki M, Conley PB, et al. A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nature medicine. 2013;19:446–451. doi: 10.1038/nm.3102. [DOI] [PubMed] [Google Scholar]
- Marder V, Aird WC, Bennett JS, Schulman S, White GC., II . Hemostasis and Thrombosis. 6. Philadelphia: Wolters Kluwer/Lipincott Williams & Wilkins; 2013. [Google Scholar]
- Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. The New England Journal of Medicine. 2000;342:696–701. doi: 10.1056/NEJM200003093421004. [DOI] [PubMed] [Google Scholar]
- Muller F, Gailani D, Renne T. Factor XI and XII as antithrombotic targets. Curr Opin Hematol. 2011;18:349–355. doi: 10.1097/MOH.0b013e3283497e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Setiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyvandi F, Kaufman RJ, Seligsohn U, Salomon O, Bolton-Maggs PH, Spreafico M, Menegatti M, Palla R, Siboni S, Mannucci PM. Rare bleeding disorders. Haemophilia. 2006;12(Suppl 3):137–142. doi: 10.1111/j.1365-2516.2006.01271.x. [DOI] [PubMed] [Google Scholar]
- Peyvandi F, Palla R, Menegatti M, Siboni SM, Halimeh S, Faeser B, Pergantou H, Platokouki H, Giangrande P, Peerlinck K, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. Journal of Thrombosis and Haemostasis: JTH. 2012;10:615–621. doi: 10.1111/j.1538-7836.2012.04653.x. [DOI] [PubMed] [Google Scholar]
- Polgar L. The catalytic triad of serine peptidases. Cellular and Molecular Life Sciences: CMLS. 2005;62:2161–2172. doi: 10.1007/s00018-005-5160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskob GE, Angchaisuksiri P, Blanco AN, Buller H, Gallus A, Hunt BJ, Hylek EM, Kakkar TL, Konstantinides SV, McCumber M, et al. Thrombosis: a major contributor to global disease burden. Semin Thromb Hemost. 2014;40:724–735. doi: 10.1055/s-0034-1390325. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Chan JC, Idusogie E, Clotman F, Vlasuk G, Luther T, Jalbert LR, Albrecht S, Zhong L, Lissens A, et al. Mice lacking factor VII develop normally but suffer fatal perinatal bleeding. Nature. 1997;390:290–294. doi: 10.1038/36862. [DOI] [PubMed] [Google Scholar]
- Siegal DM, Curnutte JT, Connolly SJ, Lu G, Conley PB, Wiens BL, Mathur VS, Castillo J, Bronson MD, Leeds JM, et al. Andexanet Alfa for the Reversal of Factor Xa Inhibitor Activity. The New England Journal of Medicine. 2015;373:2413–2424. doi: 10.1056/NEJMoa1510991. [DOI] [PubMed] [Google Scholar]
- Sun WY, Witte DP, Degen JL, Colbert MC, Burkart MC, Holmbäck K, Xiao Q, Bugge TH, Degen SJ. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7597–7602. doi: 10.1073/pnas.95.13.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zoppè M, Hackeng TM, Griffin JH, Lee KF, Verma IM. A factor IX-deficient mouse model for hemophilia B gene therapy. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11563–11566. doi: 10.1073/pnas.94.21.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Smith PL, Hsu MY, Gailani D, Schumacher WA, Ogletree ML, Seiffert DA. Effects of factor XI deficiency on ferric chloride-induced vena cava thrombosis in mice. Journal of Thrombosis and Haemostasis: JTH. 2006;4:1982–1988. doi: 10.1111/j.1538-7836.2006.02093.x. [DOI] [PubMed] [Google Scholar]
- Wendelboe AM, Raskob GE. Global Burden of Thrombosis: Epidemiologic Aspects. Circulation research. 2016;118:1340–1347. doi: 10.1161/CIRCRESAHA.115.306841. [DOI] [PubMed] [Google Scholar]
- Wilmen ASJ, Dittmer F, Strerath M, Buchnuller A, Grudzinska-Goebel J, Finnern R, Schafer M, Gerdes C, Joriben H, Itakura A, Leung PY, Tucker E. WIPO, editor . Antibodies capable of binding to the coagulation factor XI and its activated form factor XIa and uses thereof. 2013. [Google Scholar]
- Yang DT, Flanders MM, Kim H, Rodgers GM. Elevated factor XI activity levels are associated with an increased odds ratio for cerebrovascular events. Am J Clin Pathol. 2006;126:411–415. doi: 10.1309/QC259F09UNMKVP0R. [DOI] [PubMed] [Google Scholar]
- Yeh CH, Hogg K, Weitz JI. Overview of the new oral anticoagulants: opportunities and challenges. Arteriosclerosis, Thrombosis, and Vascular Biology. 2015;35:1056–1065. doi: 10.1161/ATVBAHA.115.303397. [DOI] [PubMed] [Google Scholar]
- Zucker M, Zivelin A, Landau M, Rosenberg N, Seligsohn U. Three residues at the interface of factor XI (FXI) monomers augment covalent dimerization of FXI. Journal of Thrombosis and Haemostasis: JTH. 2009;7:970–975. doi: 10.1111/j.1538-7836.2009.03353.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.