

Abstract
Cyclooxygenase-derived thromboxane (TxA2) and prostacyclin (PGI2) regulate atherogenesis in preclinical models. However, the relationship between TxA2 and PGI2 biosynthesis, vascular inflammation, and atherosclerotic cardiovascular disease (ASCVD) progression in humans remains unclear. The association between stable urine metabolites of thromboxane (TxA2-M) and prostacyclin (PGI2-M), circulating levels of cellular adhesion molecules (CAMs: E-selectin, P-selectin), chemokines and C-reactive protein, and the incidence of major adverse cardiovascular events (MACE) were evaluated in 120 patients with stable ASCVD on aspirin therapy. Urinary TxA2-M levels were significantly correlated with circulating P-selectin (r=0.319, p<0.001) and E-selectin (r=0.245, p=0.007) levels, and associated with higher risk of MACE (p=0.043). In contrast, PGI2-M levels were not significantly associated with CAM levels or MACE. These results provide insight into the contribution of TxA2 biosynthesis to ASCVD progression in humans, and suggest that patients with elevated TxA2-M levels may be predisposed to advanced platelet and endothelial activation and higher risk of adverse cardiovascular outcomes.
Keywords: thromboxane, prostacyclin, cyclooxygenase, eicosanoids, inflammation, atherosclerotic cardiovascular disease, prognosis, humans
INTRODUCTION
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of morbidity and mortality worldwide despite recent advances in its management and treatment [1]. Inflammation is a key driver of the pathogenesis and progression of ASCVD in preclinical models and humans [2]. It is well established that elevated circulating biomarkers of systemic (C-reactive protein) and vascular (chemokines, cellular adhesion molecules [CAMs]) inflammation are predictive of poor prognosis in patients with established ASCVD [3–6]. Therefore, ASCVD patients with persistent vascular inflammation, despite treatment with current evidence-based medications, may be candidates for adjunct therapeutic strategies. Identification of the key biomarkers and pathways that regulate these phenotypes in humans, however, is essential to facilitate the development of novel targeted therapies for these high-risk subsets of ASCVD patients [7].
It is well-established that cyclooxygenase (COX)-mediated metabolism of arachidonic acid to bioactive eicosanoids, most notably COX-1 derived thromboxane (TxA2) in platelets and COX-2 derived prostacyclin (PGI2) in endothelial cells, play a critical role in the regulation of platelet activation, vascular tone, and cardiovascular inflammation in preclinical models and in humans [8–11]. Suppression of the potent anti-platelet, vasodilatory and anti-inflammatory cardiovascular protective effects of PGI2 accelerate the pathogenesis and progression of ASCVD in preclinical models, and is a key mechanism underlying the cardiovascular hazard of non-steroidal anti-inflammatory drugs (NSAIDs) [11,12]. On the other hand, low-dose aspirin selectively abolishes COX-1 mediated TxA2 biosynthesis in platelets without significantly suppressing COX-2 mediated PGI2 biosynthesis in endothelial cells, inhibits platelet activation and significantly improves outcomes in ASCVD patients [11,13].
Although platelets are the predominant source of TxA2 biosynthesis, approximately 30% of systemic TxA2 production derives from extra-platelet sources, including monocytes/macrophages and vascular endothelial cells, and extra-platelet TxA2 biosynthesis increases in inflammatory conditions such as ASCVD [10,14–18]. Preclinical studies have demonstrated that TxA2 exerts potent vascular pro-inflammatory effects, in addition to promoting platelet activation, that promote the development, growth and destabilization of atherosclerotic plaques [14,19]. Furthermore, elevated urinary 11-dehydro-thromboxane B2 levels, a stable metabolite and biomarker of systemic TxA2 biosynthesis, have been associated with higher risk of adverse cardiovascular outcomes in humans with ASCVD [20–22]. However, the functional relationship between TxA2 and PGI2 biosynthesis, vascular inflammation, and prognosis in stable ASCVD patients receiving low-dose aspirin remains unknown.
Therefore, the primary objective of this study was to characterize the relationship between inter-individual variation in biomarkers of TxA2 and PGI2 biosynthesis and vascular inflammation phenotypes in patients with stable ASCVD, and determine whether the subset of individuals with the highest TxA2 or lowest PGI2 metabolite levels exhibit advanced inflammation and higher risk of future adverse cardiovascular events.
MATERIALS AND METHODS
Study Population
A cohort of 123 adult patients with angiographically confirmed ASCVD (defined as ≥50% stenosis in one or more major coronary arteries), were identified in the University of North Carolina (UNC) Cardiac Catheterization Laboratory, as described [23,24], from October 2007 to December 2011. Exclusion criteria included pregnancy, atrial fibrillation, left-ventricular systolic dysfunction (ejection fraction ≤35%), use of long-acting nitrates or insulin, active autoimmune disease, history of severe aortic stenosis, history of solid organ transplant or dialysis, or history of cancer within the previous 5 years. A parallel cohort of 39 healthy volunteers from the local community, defined as no history of cardiovascular disease, no risk factors for ASCVD and taking no medications for a chronic medical condition, were also enrolled as described [24].
Eligible participants completed a single research study visit that was conducted in the morning after fasting overnight. Participants were instructed to withhold taking their morning medications until after the study visit. During the study visit, participants underwent a medication history interview, and medication adherence was documented by patient self-report. A blood and spot urine sample was collected in each participant, aliquoted and stored at −80°C pending analysis. Participants were also instructed to refrain from tobacco products, caffeine, and vigorous exercise the morning of the study visit, and from use of vitamin C, vitamin E, fish oil, niacin or arginine supplements, oral decongestants, NSAIDs, or erectile dysfunction medications for at least 7 days prior to the study visit. Individuals experiencing a respiratory tract infection within 4 weeks of the study visit were not eligible to participate. The ASCVD patients completed their study visit 63±34 days after their cardiac catheterization, and were confirmed to be clinically stable and chest pain free at the study visit. The study protocol was approved by the UNC Biomedical Institutional Review Board, and all study participants provided written informed consent.
Quantification of Inflammatory Biomarkers
High sensitivity C-reactive protein (hs-CRP) was quantified in fresh serum using the VITROS® 5600 Chemistry System (Ortho-Clinical Diagnostics, Inc., Rochester, NY), and plasma cellular adhesion molecules (CAMs: E-selectin and P-selectin), neutrophil chemokine (epithelial neutrophil-activating protein [ENA]-78; also known as CXCL5) and monocyte chemokine (monocyte chemoattractant protein [MCP]-1; also known as CCL2) concentrations were quantified using Multi-Analyte Profiling Kits (R&D Systems, Minneapolis, MN), as described [23]. These circulating biomarkers of inflammation were selected since elevated hs-CRP (a systemic inflammatory mediator produced in the liver that correlates with cytokines including interleukin-6 [6,25]), CAMs (inflammatory mediators expressed on endothelial cells that mediate leukocyte and platelet adhesion [4]) and MCP-1 (a chemokine synthesized in monocytes and endothelial cells that drives monocyte recruitment to the vascular wall [3]) levels, and genetic predisposition to higher ENA-78 levels (a chemokine synthesized in neutrophils and endothelial cells that drives neutrophil recruitment to the vascular wall [5]) have each been associated with poorer prognosis in patients with established ASCVD.
Quantification of Stable Urine Prostanoid Metabolites
Systemic TxA2 and PGI2 levels were assessed by measuring concentrations of the stable metabolites 11-dehydro-thromboxane B2 (TxA2-M) and 2,3-dinor-6-keto-prostaglandin F1α (PGI2-M), respectively, in urine using a commercially available ELISA assay (TxA2-M: assay #519510, PGI2-M: assay #515121; Cayman Chemical, Ann Arbor, MI, USA) according to manufacturer’s instructions. TxA2 and PGI2 are rapidly converted to more chemically stable and relatively biologically inactive metabolites thromboxane B2 and 6-keto-prostagladin F1α, respectively, which are then excreted in urine as the stable metabolites 11-dehydro-thromboxane B2 (TxA2-M) and 2,3-dinor-6-keto-prostaglandin F1α (PGI2-M) [26,27]. Thus, TxA2-M and PGI2-M serve as reliable surrogate measures of total TxA2 and PGI2 formation in vivo [21,26,28]. The TxA2-M and PGI2-M concentrations were normalized to urinary creatinine concentrations, to account for inter-individual differences in urine dilution, and expressed as pg/mg Cr. The urinary ratio of TxA2-M to PGI2-M was also calculated in each participant.
Longitudinal Assessment of Adverse Cardiovascular Outcomes
The incidence of major adverse cardiovascular events (MACE) over time were retrospectively abstracted from the electronic medical record from the time of the research study visit and sample collection (baseline) through December 2015. Eleven of the 123 study participants (8.9%) did not present to the UNC health care system for either follow-up clinic visits or emergent care after the study visit, were considered lost to follow-up, and were excluded from the clinical outcome analysis. The pre-specified cardiovascular outcome endpoint (MACE) was defined as a composite of death, hospitalization for a non-fatal acute coronary syndrome (ACS) event (unstable angina, non-ST segment elevation myocardial infarction [NSTEMI], ST segment elevation myocardial infarction [STEMI]), and hospitalization for a nonfatal cerebrovascular event (ischemic stroke or transient ischemic attack [TIA]). Clinician reported outcomes were verified from the electronic medical record by two individuals. The time from the research study visit (baseline) to the first occurrence of MACE or the last follow-up encounter in the medical record was recorded for each participant.
Statistical Analysis
Data are presented as mean ± standard deviation, median (interquartile range), or count (%) unless otherwise indicated. Urinary prostanoid metabolites and circulating biomarkers of inflammation were not normally distributed, and therefore were log-transformed prior to analysis. In order to account for batch variation in the quantification of inflammatory biomarkers, data within each batch were standardized using a z-score [i.e. (subject value – batch mean)/batch standard deviation], as described [23], which are normally distributed. All analyses were performed using JMP Pro 12.0.1 or SAS Version 9.4 (SAS Institute, Cary, NC). A P-value <0.05 was considered to be statistically significant.
Three ASCVD patients reported not taking daily aspirin therapy at their research study visit. Due to the known effects of aspirin on TxA2 biosynthesis, the primary study analyses were conducted in the 120 ASCVD patients taking daily aspirin therapy at the time of sample collection. The ASCVD study population characteristics were compared across TxA2-M and PGI2-M tertiles using one-way ANOVA for continuous data and chi-squared test or Fisher’s exact test for categorical data as appropriate. For the primary cross-sectional analysis, associations between urine prostanoid metabolite levels (TxA2-M, PGI2-M and TxA2-M:PGI2-M ratio) and each inflammatory biomarker (MCP-1, ENA-78, hs-CRP, P-selectin, E-selectin) at baseline were evaluated by Pearson correlation. A secondary analysis was conducted using a model that adjusted for potential demographic (age, race, gender) and clinical confounders (smoking status, diabetes, obesity, multivessel disease, hypertension, renin-angiotensin system inhibitor use, aspirin dose) that associated with urinary prostanoid levels and/or the phenotypic indices of vascular inflammation in our population. In order to further assess the prostanoid-vascular inflammation relationships, vascular phenotypes were compared across TxA2-M and PGI2-M tertiles by ANOVA and a post-hoc Tukey-Kramer test.
The relationship between baseline urinary TxA2-M and PGI2-M levels (tertiles) and time to occurrence of a MACE was evaluated using Cox proportional hazard regression in the 109 ASCVD patients on aspirin therapy with available follow-up data. Kaplan-Meier curves were generated using GraphPad Prism 6.0. Time-to-event analyses were completed after adjusting for demographic and clinical covariates.
RESULTS
Study Population
The baseline characteristics of the ASCVD study population are shown in Table 1. The study participants were, on average, 60 years old, 70% male and 17% African-American, and exhibited risk factors for ASCVD such as hypertension (84%), obesity (46%), and diabetes (24%). The majority of patients had advanced ASCVD, with 33% experiencing an acute coronary syndrome at the time of coronary angiography, 63% exhibiting multivessel coronary artery disease, and 69% undergoing a revascularization procedure. Medication utilization and revascularization rates were consistent with current clinical practice guidelines. At the study visit, 120 (98%) of the ASCVD cohort participants were receiving aspirin therapy (81 or 325 mg/day).
Table 1.
Study population characteristics.
| Baseline Characteristic | |
|---|---|
| N | 123 |
| Age (years) | 59.9 ± 9.9 |
| Female (%) | 37 (30.1%) |
| African-American (%) | 21 (17.1%) |
| Body mass index (kg/m2) | 29.8 ± 5.5 |
| Obese (%) | 57 (46.3%) |
| Current smoker (%) | 29 (23.6%) |
| Diabetes (%) | 29 (23.6%) |
| Hypertension (%) | 103 (83.7%) |
| Previous myocardial infarction (%) | 49 (39.8%) |
| Recent acute coronary syndrome (%)a | 40 (32.5%) |
| Multivessel disease (%) | 77 (62.6%) |
| Recent revascularization procedure (%)b | 85 (69.1%) |
| Systolic blood pressure (mmHg) | 133 (22) |
| Diastolic blood pressure (mmHg) | 78 (12) |
| Total cholesterol (mg/dL) | 158 (55) |
| LDL cholesterol (mg/dL) | 86 (42) |
| HDL cholesterol (mg/dL) | 48 (17) |
| Triglycerides (mmol/L) | 97 (85) |
| ACE inhibitor or ARB use (%) | 76 (64.2%) |
| Beta-blocker use (%) | 100 (81.3%) |
| Statin use (%) | 115 (93.5%) |
| Aspirin use (%) | 120 (97.6%) |
| 81 mg/day (%) | 42 (34.1%) |
| 325 mg/day (%) | 78 (63.4%) |
| Clopidogrel use (%) | 99 (80.5%) |
ACE = angiotensin-converting enzyme, ARB=angiotensin receptor blocker, HDL=high density lipoprotein, LDL=low density lipoprotein
Data presented as mean ± standard deviation, median (interquartile range) or count (proportion).
21/123 (17.1%) and 19/123 (15.4%) were experiencing an unstable angina and myocardial infarction event, respectively, in the cardiac catheterization laboratory at the time of screening. At the time of the study visit, participants were confirmed to be clinically stable and chest pain free.
75/123 underwent a percutaneous coronary intervention and 10/123 underwent a coronary artery bypass grafting procedure between screening and the study visit.
Urinary Thromboxane and Prostacyclin Metabolite Levels
Within the ASCVD cohort, aspirin use was associated with significantly lower urinary TxA2-M levels (P=0.011), such that the three patients who were not taking aspirin had significantly higher TxA2-M levels compared to those taking either 81 mg/day or 325 mg/day of aspirin (Table 2). TxA2-M levels were not significantly different between the two dose levels. Compared to the ASCVD cohort, the cohort of healthy volunteers (who were not taking aspirin) had significantly higher urinary TxA2-M levels (P<0.001). In contrast, PGI2-M levels were not associated with aspirin use or dose (Table 2).
Table 2.
Urine thromboxane and prostacyclin metabolite levels by aspirin dose and atherosclerotic cardiovascular disease (ASCVD) status.
| ASCVD No aspirin |
ASCVD Aspirin 81mg |
ASCVD Aspirin 325mg |
P-valuea | ASCVD All |
Healthyb Volunteer |
P-valuec | |
|---|---|---|---|---|---|---|---|
| N | 3 | 42 | 78 | 123 | 39 | ||
|
TxA2-M (pg/mg Cr) |
2,383 (1,166–2,781) |
688* (455–1,090) |
651* (396–929) |
0.011 | 679 (436–1,038) |
1,990 (1,372–2,422) |
<0.001 |
|
PGI2-M (pg/mg Cr) |
10,339 (5,379–22,869) |
9,871 (5,368–17,819) |
10,076 (6,163–16,096) |
0.856 | 9,991 (6,001–16,765) |
12,794 (7,022–27,114) |
0.074 |
Data presented as median (interquartile range). Data were log-transformed prior to analysis.
Comparison of prostanoid concentration across aspirin dose (no aspirin versus 81 mg/day versus 325 mg/day) within the ASCVD cohort by ANOVA.
Post-hoc P<0.05 versus no aspirin. The post-hoc comparison of aspirin 81 versus 325 mg/day for TxA2-M was not statistically significant (P=0.377)
Parallel cohort of healthy volunteers (defined as no cardiovascular disease history or risk factors and taking no medications, including aspirin).
Comparison of prostanoid concentration across the ASCVD cohort and healthy volunteer cohort.
Multiple demographic and clinical factors significantly differed across TxA2-M and PGI2-M tertiles (Supplemental Table S1 and S2). Female gender, African-American race, current cigarette smoking and cholesterol levels were associated with higher TxA2-M levels. African-American race and a recent revascularization procedure was associated with lower and higher PGI2-M levels, respectively.
Urine Prostanoid Metabolites and Inflammatory Biomarkers
The relationships between inter-individual variation in urinary prostanoid stable metabolite levels and circulating biomarkers of vascular inflammation in the ASCVD cohort are provided in Table 3. Higher urinary TxA2-M levels were significantly associated with higher circulating levels of the CAMs P-selectin (r=0.319, p<0.001, Figure 1A) and E-selectin (r=0.245, p=0.007, Figure 1C). These significant positive associations were also observed after adjusting for clinical and demographic covariates and aspirin dose (Table 3). Furthermore, the subset of ASCVD patients with urinary TxA2-M levels in the highest tertile exhibited significantly higher circulating P-selectin levels compared to those in the lowest TxA2-M tertile (Figure 1B). Similarly, a stepwise trend of higher E-selectin levels was observed across increasing TxA2-M tertiles (Figure 1D, P for trend=0.056); however, statistically significant differences were not observed between each tertile.
Table 3.
Correlation between urinary thromboxane and prostacyclin metabolite levels and circulating biomarkers of inflammation in stable atherosclerotic cardiovascular disease patients on aspirin therapy.
| Prostanoid | hsCRP | ENA-78 | P-Selectin | E-Selectin | MCP-1 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| r | P | r | P | r | P | r | P | r | P | |
| TxA2-M | ||||||||||
| Unadjusted | 0.222 | 0.015 | 0.225 | 0.014 | 0.319 | <0.001 | 0.245 | 0.007 | 0.065 | 0.486 |
| Adjusted* | 0.077 | 0.425 | 0.129 | 0.181 | 0.376 | <0.001 | 0.277 | 0.004 | −0.004 | 0.967 |
| PGI2-M | ||||||||||
| Unadjusted | −0.190 | 0.039 | −0.303 | 0.001 | −0.049 | 0.601 | −0.088 | 0.344 | −0.086 | 0.357 |
| Adjusted* | −0.180 | 0.062 | −0.275 | 0.004 | −0.060 | 0.540 | −0.119 | 0.219 | −0.046 | 0.634 |
| TxA2-M: PGI2-M | ||||||||||
| Unadjusted | 0.272 | 0.003 | 0.357 | <0.001 | 0.231 | 0.012 | 0.212 | 0.021 | 0.100 | 0.282 |
| Adjusted* | 0.183 | 0.057 | 0.283 | 0.003 | 0.276 | 0.004 | 0.260 | 0.007 | 0.033 | 0.732 |
Data presented as Pearson correlation coefficient (r) and P-value. Analyses were conducted in the 120 study participants on aspirin therapy at the study visit.
Partial Pearson correlations were evaluated after adjusting for age, race, gender, obesity, ACE inhibitor or ARB use, diabetes, multivessel disease, smoking status, hypertension, and aspirin dose (81 or 325 mg)
Figure 1. Association between urinary thromboxane metabolite levels and circulating biomarkers of vascular inflammation.
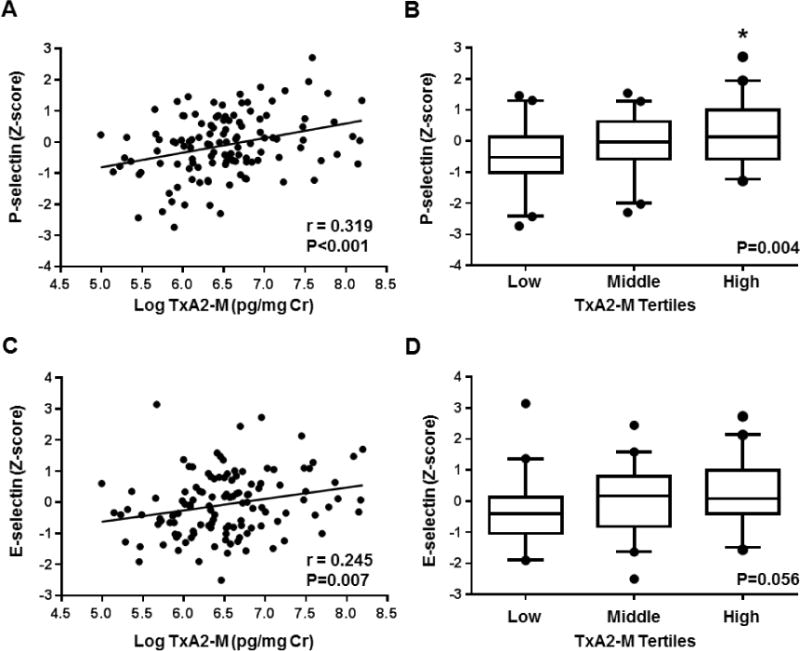
The correlation between urinary TxA2-M levels (log-transformed) and plasma (A) P-selectin and (B) E-selectin levels (z-score) is displayed. The unadjusted Pearson correlation coefficient (r) and corresponding P-value are provided. The distribution of plasma (B) P-selectin and (D) E-selectin levels (z-score) across TxA2-M tertiles is displayed using box plots (low tertile: 147–496 pg/mg Cr, middle tertile: 507–815 pg/mg Cr, high tertile: 818–3,629 pg/mg Cr). Data are presented as median (midline), interquartile range (box), and 95th percentile (whiskers). The ANOVA p-value for the association is provided. *P<0.05 versus the low tertile.
A positive association was also observed between urinary TxA2-M levels and circulating levels of hs-CRP (r=0.222, p=0.015) and ENA-78 (r=0.225, p=0.014); however, these associations were not statistically significant in the adjusted model (Table 3). No relationship was observed between TxA2-M and MCP-1 levels. Consistent with the observed associations between urinary TxA2-M levels and circulating biomarkers of inflammation, the urinary ratio of TxA2-M to PGI2-M levels also exhibited a positive association with P-selectin, E-selectin, ENA-78 and hs-CRP (Table 3).
In contrast, higher urinary PGI2-M levels were associated with lower circulating levels of the neutrophil chemokine ENA-78 (r=-0.303, p=0.001; Figure 2A), such that the subset of ASCVD patients with urinary PGI2-M levels in the highest tertile exhibited significantly lower ENA-78 levels compared to those in the lowest tertile (Figure 2B). A significant inverse association was also observed after adjusting for demographic and clinical covariates (Table 3). An inverse association was also observed with circulating hs-CRP levels (r=-0.190, p=0.039); however, this relationship was not statistically significant in the adjusted model. No relationship was observed between urinary PGI2-M levels and circulating CAMs or MCP-1 (Table 3).
Figure 2. Association between urinary prostacyclin metabolite levels and circulating biomarkers of vascular inflammation.
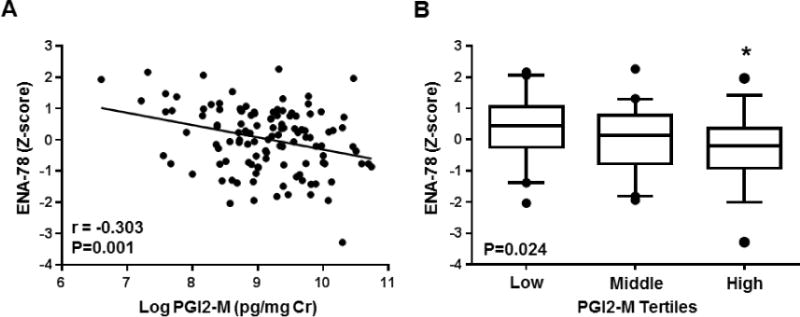
(A) The correlation between urinary PGI2-M levels (log-transformed) and plasma ENA-78 levels (z-score) is displayed. The unadjusted Pearson correlation coefficient (r) and corresponding P-value are provided. (B) The distribution of plasma ENA-78 levels (z-score) across PGI2-M tertiles is displayed using box plots (low tertile: 738–6,932 pg/mg Cr, middle tertile: 7,034–13,260 pg/mg Cr, high tertile: 13,639–46,139 pg/mg Cr). Data are presented as median (midline), interquartile range (box), and 95th percentile (whiskers). The ANOVA p-value for the association is provided. *P<0.05 versus the low tertile.
Urine Prostanoid Metabolites and MACE Outcomes
Follow-up data was available in 109 (91%) of the enrolled participants with stable ASCVD on aspirin therapy. The median (IQR) follow-up was 5.6 (4.2–6.7) years. Thirty-one participants (28.4%) experienced a MACE outcome during the follow-up period (4 deaths, 23 non-fatal ACS events, and 4 non-fatal stroke/TIA events). Urinary TxA2 metabolite levels at baseline exhibited a positive association with future occurrence of a MACE, such that a stepwise increase in the proportion of patients experiencing a future MACE was observed across the lowest (16.2%), middle (30.6%) and highest (38.9%) TxA2-M tertiles (adjusted log rank P=0.043) (Figure 3A). Compared to the lowest TxA2-M tertile, the highest TxA2-M tertile was associated with a significantly higher risk of MACE (adjusted hazard ratio [HR] 2.92, 95% CI 1.08–8.70, p=0.035). Although the middle tertile also exhibited a higher frequency of MACE, the risk of MACE was not significantly higher than the lowest tertile (adjusted HR 2.54, 95% CI 0.93–7.63, p=0.069).
Figure 3. Urinary thromboxane and prostacyclin metabolite levels and risk of subsequent major adverse cardiovascular events (MACE).
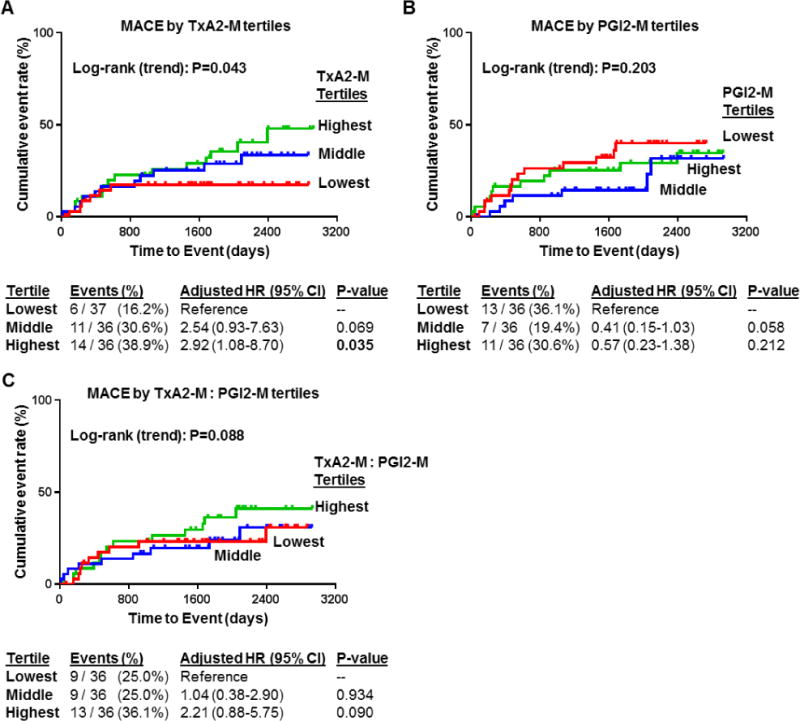
Kaplan-Meier curves were generated for incidence of MACE according to tertiles of (A) stable thromboxane metabolite (TxA2-M) levels, (B) stable prostacyclin metabolite (PGI2-M) levels, and (C) the ratio of TxA2-M: PGI2-M in urine at baseline. The log rank P-value for trend across tertiles (adjusted) for each Kaplan-Meier curve is provided. Below the curves, the number and frequency of events within each tertile group, along with the adjusted hazard ratio (HR), 95% confidence interval (CI) and P-value, are provided. Analyses adjusted for demographic (age >65, race, gender) and clinical covariates (diabetes, multivessel disease, aspirin dose).
In contrast, no significant association was observed between either baseline PGI2-M levels (Figure 3B) or the ratio of TxA2-M to PGI2-M levels (Figure 3C) and future occurrence of MACE. The highest TxA2-M to PGI2-M ratio appeared to exhibit a higher frequency of MACE compared to the lowest tertile (36.1% versus 25.0%, respectively); however, this difference was not statistically significant (adjusted HR 2.21, 95% CI 0.88–5.75, P=0.090).
DISCUSSION
Cyclooxygenase-derived TxA2 and PGI2 are key regulators of cardiovascular inflammation and the pathogenesis of ASCVD [8–11,13]. However, the relationship between inter-individual variation in systemic TxA2 and PGI2 levels, vascular inflammation phenotypes, and prognosis in patients with stable ASCVD on aspirin therapy remains unclear. This study demonstrated that elevated urinary 11-dehydro-TxB2 levels, a stable metabolite and biomarker of systemic TxA2 biosynthesis, were associated with higher circulating levels of the CAMs P-selectin and E-selectin and higher risk of a future MACE. In contrast, urinary levels of the stable PGI2 metabolite 2,3-dinor-6-keto-prostaglandin F1α exhibited no significant association with either CAM levels or prognosis.
The observed positive association between urinary TxA2-M levels with circulating levels of P-selectin and E-selectin is consistent with preclinical studies in which TxA2 receptor (TP) activation induced the expression of platelet and endothelial adhesion molecules and promoted atherogenesis [29–32]. Furthermore, and consistent with the known association between elevated circulating CAM levels and poorer prognosis in ASCVD patients [4], we also observed a significant relationship between elevated TxA2-M levels and higher risk of future adverse cardiovascular outcomes. These observations are consistent with previous studies that have reported a significant association between higher urinary TxA2-M levels and a higher risk of MACE in patients with stable ASCVD [20,21], a higher risk of MACE in acute MI patients undergoing PCI [22], and a higher risk of thrombotic events in patients following coronary artery bypass graft (CABG) surgery [33] on treatment with low-dose aspirin. Szczeklik et al. reported a positive correlation between urinary TxA2-M levels and systemic biomarkers of inflammation such as hs-CRP and white blood cell count in acute MI patients [22]. The positive correlation between TxA2-M and hs-CRP and the neutrophil chemokine ENA-78 in the current study of stable ASCVD patients were consistent with these results; however, these relationships were not statistically significant after adjusting for demographic and clinical factors, and were weak in comparison to the significant positive associations between TxA2-M and more specific biomarkers of platelet (P-selectin) and endothelial (E-selectin) inflammation. Moreover, these previous studies did not investigate urinary PGI2-M levels, which were quantified in parallel to TxA2-M in our study. Although an inverse correlation between urinary PGI2-M levels and circulating hs-CRP and ENA-78 were observed, PGI2-M levels were not significantly associated with either P-selectin and E-selectin levels or MACE outcomes, suggesting these relationships may be unique to TxA2-M in stable ASCVD. Taken together, these data provide insight into the pathologic contribution of TxA2 to ASCVD progression in humans, and suggest that the subset of stable ASCVD patients with elevated TxA2-M levels may be predisposed to advanced platelet and endothelial activation and higher risk of adverse cardiovascular outcomes.
It is well-established that the majority of systemic TxA2 is produced in platelets in a COX-1 dependent manner, which is irreversibly inhibited by low doses of aspirin. However, about 30% of TxA2 is synthesized from extraplatelet sources, such as inflammatory monocytes and macrophages and activated endothelial cells, via COX-2 [10,14–18]. Inhibition of these COX-2 derived sources of TxA2 requires much higher doses of aspirin than the low doses used clinically to inhibit platelet derived TxA2 and prevent MACE, and has been proposed as a mechanism of “aspirin resistance” [34]. The mechanisms driving excess extraplatelet TxA2 production, however, remain unclear. It has been reported that TxA2 synthase expression and TxA2 biosynthesis is upregulated in atherosclerosis plaques and positively correlated with the degree of inflammatory macrophage infiltration [14]. In our study, risk factors associated with vascular inflammation and ASCVD progression such as African-American race, cigarette smoking, and cholesterol levels, were associated with higher TxA2-M levels, whereas statin use was associated with a trend toward lower TxA2-M levels. Similarly, Eikelboom et al. reported that increasing age, cigarette smoking, treatment for diabetes, and advanced peripheral vascular disease were associated with higher TxA2-M levels, and statin use was associated with lower TxA2-M levels [21]. Additionally, the metabolic syndrome has been linked to higher TxA2-M levels [35], which is consistent with our finding that dyslipidemia were associated with higher levels of TxA2-M. Although these clinical factors were associated with higher TxA2-M levels, the observed association between TxA2-M and MACE was significant after adjusting for these covariates in the current and prior studies [21]. It is important to note that, a higher percentage of our cohort were also being treated with contemporary guideline-directed therapy for ASCVD, such as revascularization and statin medications, than previous studies evaluating the link between TxA2-M levels and prognosis in stable ASCVD patients [20,21], suggesting that additional interventions may be needed to mitigate the risk associated with elevated TxA2 biosynthesis.
Our analysis has limitations that must be acknowledged. First, although the observed associations were consistent with prior preclinical and clinical studies, the observational design does not allow us to establish a cause-and-effect relationship between TxA2-M levels, vascular inflammation and cardiovascular outcomes. Second, the vascular inflammatory phenotypes assessed were not comprehensive. For instance, we did not assess biomarkers of enhanced oxidative stress, such as 8-iso-PGF2α, which have been shown to positively correlate with aspirin-insensitive TxA2 production in prior studies [36–38]. Delineating the mechanisms underlying platelet and extraplatelet sources of TxA2 synthesis in the setting of aspirin use, and the effects on distinct inflammatory phenotypes and ASCVD progression, is beyond the scope of the current investigation and will require further study. Third, we cannot rule out the contribution of nonadherence to aspirin therapy on TxA2-M levels and the observed associations. Fourth, we took a single measurement of urinary metabolite levels at baseline rather than repeated measures over time, which was beyond the scope of this study. Furthermore, we used ELISA to quantify TxA2-M and PGI2-M concentrations instead of gold standard liquid chromatography/mass spectrometry (LC/MS) methods. It is important to note that the polyclonal ELISA assay used was also utilized in the HOPE and CHARISMA studies [20,21], and has previously been shown to be highly correlated with LC-MS methods and more specific for the 11-dehydro-TxB2 metabolite than available monoclonal antibody assays [39], which enhances confidence in our results. Lastly, our analysis included multiple statistical comparisons and was limited by its relatively small sample size, increasing the risk of false-positive findings. Future studies in larger populations will ultimately be necessary to validate the predictive utility of urinary TxA2-M levels as a biomarker of ASCVD progression and prognosis.
Novel therapeutic strategies that inhibit TxA2 biosynthesis and/or action in a more targeted manner than low-dose aspirin, namely TxA2 synthase inhibitors (TXSI), TP receptor antagonists (TXRA), and dual acting synthase-receptor antagonists, have been an active area of drug development [40–42]. The TXSIs suppress TxA2 biosynthesis downstream of COX by inhibiting conversion of PGH2 to the bioactive effector TxA2 with minimal effects on parallel prostanoids. The TXRAs, most notably terutroban, block the activation and downstream signaling of TP receptors, which are expressed in platelets, inflammatory cells, endothelial cells and atherosclerotic plaques, and activated by TxA2 as well as other ligands such as prostanoid endoperoxides and isoprostanes [40,41]. Dual TXSI/TXRAs picotamide and ridogrel have also been developed. However, despite promising results in preclinical studies, these novel agents have failed to improve cardiovascular outcomes compared to low-dose aspirin in clinical trials of patients at high risk for ischemic events [41–45]. Due to the established benefit and low cost of aspirin, the desire to continue their development has been stagnated. It is important to note, however, that these clinical trials have been conducted in broadly defined populations. The observed relationship between elevated TxA2-M and prognosis suggest that the subset of ASCVD patients adherent to aspirin therapy with elevated TxA2-M levels may have enriched potential for clinical benefit from targeted inhibition of TxA2 biosynthesis and action compared to the general population. Through application of precision medicine principles, future biomarker-guided interventional studies that evaluate the clinical effects of TXSI/TXRA in targeted subsets of ASCVD patients with elevated urinary TxA2-M levels should be considered.
Conclusions
In summary, we have demonstrated that elevated TxA2-M levels are associated with activated vascular inflammation phenotypes and higher risk of adverse cardiovascular outcomes in a population of patients with stable ASCVD treated with aspirin. These data indicate that the subset of stable ASCVD patients predisposed to elevated TxA2 levels biosynthesis may exhibit advanced platelet and endothelial inflammation and worse prognosis, and suggest that TxA2 biosynthesis from extraplatelet sources may be an important driver of ASCVD progression. Future studies are needed to validate these results in larger populations, investigate the causes of increased extraplatelet TxA2 biosynthesis in certain individuals, and develop therapeutic interventions that mitigate the risks associated with elevated TxA2 levels in ASCVD patients on aspirin therapy.
Supplementary Material
HIGHLIGHTS.
COX metabolites, inflammation biomarkers and outcomes were evaluated in human ASCVD
TxA2 metabolite levels positively correlated with P-selectin and E-selectin
Elevated TxA2 metabolite levels associated with higher risk of adverse CVD outcomes
The observed associations occurred in stable ASCVD patients treated with aspirin
Extraplatelet TxA2 biosynthesis may contribute to ASCVD progression in humans
Acknowledgments
The authors gratefully acknowledge the UNC Clinical and Translational Research Center staff for their assistance, and the UNC Cytokine Analysis facility for performing the inflammatory biomarker assays. The project described was supported by American Heart Association grant 16GRNT29300003 to Dr. Lee, American Heart Association predoctoral fellowship 11PRE7240059 to Dr. Schuck, a predoctoral training program in Integrative Vascular Biology supported by the NIH/NHLBI (T32 HL069768) to Dr. Schuck, and pilot grants to Dr. Lee supported by the UNC University Research Council and the NIH National Center for Advancing Translational Sciences through Grant Award Number 1UL1TR001111. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, O. behalf of the A.H.A.S.C. and S.S. Subcommittee Heart Disease and Stroke Statistics—2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.de Lemos JA, Morrow DA, Blazing MA, Jarolim P, Wiviott SD, Sabatine MS, Califf RM, Braunwald E. Serial measurement of monocyte chemoattractant protein-1 after acute coronary syndromes: results drom the A to Z trial. J Am Coll Cardiol. 2007;50:2117–2124. doi: 10.1016/j.jacc.2007.06.057. [DOI] [PubMed] [Google Scholar]
- 4.Blankenberg S, Rupprecht HJ, Bickel C, Peetz D, Hafner G, Tiret L, Meyer J. Circulating cell adhesion molecules and death in patients with coronary artery disease. Circulation. 2001;104:1336–1342. doi: 10.1161/hc3701.095949. [DOI] [PubMed] [Google Scholar]
- 5.Zineh I, Beitelshees AL, Welder GJ, Hou W, Chegini N, Wu J, Cresci S, Province MA, Spertus JA. Epithelial neutrophil-activating peptide (ENA-78), acute coronary syndrome prognosis, and modulatory effect of statins. PLOS ONE. 2008;3:e3117. doi: 10.1371/journal.pone.0003117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, Pfeffer MA, Braunwald E, Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) Investigators C-reactive protein levels and outcomes after statin therapy. N Engl J Med. 2005;352:20–28. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 7.Han Y, Li L, Zhang Y, Yuan H, Ye L, Zhao J, Duan DD. Phenomics of vascular disease: the systematic approach to the combination therapy. Curr Vasc Pharmacol. 2015;13:433–440. doi: 10.2174/1570161112666141014144829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oates JA, FitzGerald GA, Branch RA, Jackson EK, Knapp HR, Roberts LJ. Clinical implications of prostaglandin and thromboxane A2 formation (1) N Engl J Med. 1988;319:689–698. doi: 10.1056/NEJM198809153191106. [DOI] [PubMed] [Google Scholar]
- 9.Davidge ST. Prostaglandin H synthase and vascular function. Circ Res. 2001;89:650–660. doi: 10.1161/hh2001.098351. [DOI] [PubMed] [Google Scholar]
- 10.Vanhoutte PM. COX-1 and vascular disease. Clin Pharmacol Ther. 2009;86:212–215. doi: 10.1038/clpt.2009.108. [DOI] [PubMed] [Google Scholar]
- 11.Patrono C, Baigent C. Nonsteroidal anti-inflammatory drugs and the heart. Circulation. 2014;129:907–916. doi: 10.1161/CIRCULATIONAHA.113.004480. [DOI] [PubMed] [Google Scholar]
- 12.Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Puré E, Funk CD, FitzGerald GA. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4:132ra54. doi: 10.1126/scitranslmed.3003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patrono C, García Rodríguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353:2373–2383. doi: 10.1056/NEJMra052717. [DOI] [PubMed] [Google Scholar]
- 14.Gabrielsen A, Qiu H, Bäck M, Hamberg M, Hemdahl AL, Agardh H, Folkersen L, Swedenborg J, Hedin U, Paulsson-Berne G, Haeggström JZ, Hansson GK. Thromboxane synthase expression and thromboxane A2 production in the atherosclerotic lesion. J Mol Med. 2010;88:795–806. doi: 10.1007/s00109-010-0621-6. [DOI] [PubMed] [Google Scholar]
- 15.Frelinger AL, Li Y, Linden MD, Tarnow I, Barnard MR, Fox ML, Michelson AD. Aspirin “resistance”: role of pre-existent platelet reactivity and correlation between tests. J Thromb Haemost. 2008;6:2035–2044. doi: 10.1111/j.1538-7836.2008.03184.x. [DOI] [PubMed] [Google Scholar]
- 16.Cattaneo M. Letter by cattaneo regarding article, “incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: determinants and effect on cardiovascular risk”. Circulation. 2009;119:e594. doi: 10.1161/CIRCULATIONAHA.108.838888. author reply e595–e596. [DOI] [PubMed] [Google Scholar]
- 17.Santilli F, Davì G, Basili S, Lattanzio S, Cavoni A, Guizzardi G, De Feudis L, Traisci G, Pettinella C, Paloscia L, Minuz P, Meneguzzi A, Ciabattoni G, Patrono C. Thromboxane and prostacyclin biosynthesis in heart failure of ischemic origin: effects of disease severity and aspirin treatment. J Thromb Haemost. 2010;8:914–922. doi: 10.1111/j.1538-7836.2010.03820.x. [DOI] [PubMed] [Google Scholar]
- 18.Hui Y, Ricciotti E, Crichton I, Yu Z, Wang D, Stubbe J, Wang M, Puré E, FitzGerald GA. Targeted deletions of cyclooxygenase-2 and atherogenesis in mice. Circulation. 2010;121:2654–2660. doi: 10.1161/CIRCULATIONAHA.109.910687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cyrus T, Sung S, Zhao L, Funk CD, Tang S, Praticò D. Effect of low-dose aspirin on vascular inflammation, plaque stability, and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation. 2002;106:1282–1287. doi: 10.1161/01.cir.0000027816.54430.96. [DOI] [PubMed] [Google Scholar]
- 20.Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–1655. doi: 10.1161/01.cir.0000013777.21160.07. [DOI] [PubMed] [Google Scholar]
- 21.Eikelboom JW, Hankey GJ, Thom J, Bhatt DL, Steg PG, Montalescot G, Johnston SC, Steinhubl SR, Mak KH, Easton JD, Hamm C, Hu T, Fox KAA, Topol EJ, Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management and Avoidance (CHARISMA) Investigators Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: determinants and effect on cardiovascular risk. Circulation. 2008;118:1705–1712. doi: 10.1161/CIRCULATIONAHA.108.768283. [DOI] [PubMed] [Google Scholar]
- 22.Szczeklik W, Stodółkiewicz E, Rzeszutko M, Tomala M, Chrustowicz A, Żmudka K, Sanak M. Urinary 11‐dehydro‐thromboxane B2 as a predictor of acute myocardial infarction outcomes: results of Leukotrienes and Thromboxane In Myocardial Infarction (LTIMI) Study. J Am Heart Assoc. 2016;5:e003702. doi: 10.1161/JAHA.116.003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuck RN, Theken KN, Edin ML, Caughey M, Bass A, Ellis K, Tran B, Steele S, Simmons BP, Lih FB, Tomer KB, Wu MC, Hinderliter AL, Stouffer GA, Zeldin DC, Lee CR. Cytochrome P450-derived eicosanoids and vascular dysfunction in coronary artery disease patients. Atherosclerosis. 2013;227:442–448. doi: 10.1016/j.atherosclerosis.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CR, Bass A, Ellis K, Tran B, Steele S, Caughey M, Stouffer GA, Hinderliter AL. Relation between digital peripheral arterial tonometry and brachial artery ultrasound measures of vascular function in patients with coronary artery disease and in healthy volunteers. Am J Cardiol. 2012;109:651–657. doi: 10.1016/j.amjcard.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 26.Lahoz C, Alonso R, Ordovás JM, López-Farré A, de Oya M, Mata P. Effects of dietary fat saturation on eicosanoid production, platelet aggregation and blood pressure. Eur J Clin Invest. 1997;27:780–787. doi: 10.1046/j.1365-2362.1997.1860735.x. [DOI] [PubMed] [Google Scholar]
- 27.S M, Ea H. Arachidonate metabolism in blood cells and the vessel wall. Clin Haematol. 1986;15:273–292. [PubMed] [Google Scholar]
- 28.Oates JA, FitzGerald GA, Branch RA, Jackson EK, Knapp HR, Roberts LJ. Clinical implications of prostaglandin and thromboxane A2 formation (2) N Engl J Med. 1988;319:761–767. doi: 10.1056/NEJM198809223191206. [DOI] [PubMed] [Google Scholar]
- 29.Ishizuka T, Suzuki K, Kawakami M, Hidaka T, Matsuki Y, Nakamura H. Thromboxane A2 receptor blockade suppresses intercellular adhesion molecule-1 expression by stimulated vascular endothelial cells. Eur J Pharmacol. 1996;312:367–377. doi: 10.1016/0014-2999(96)00478-5. [DOI] [PubMed] [Google Scholar]
- 30.Bayat H, Xu S, Pimentel D, Cohen RA, Jiang B. Activation of thromboxane receptor upregulates interleukin (IL)-1beta-induced VCAM-1 expression through JNK signaling. Arterioscler Thromb Vasc Biol. 2008;28:127–134. doi: 10.1161/ATVBAHA.107.150250. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T, Arai H, Oida H, Yurugi-Kobayashi T, Yamashita JK, Katagiri H, Majima M, Yokode M, Kita T, Narumiya S. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J Clin Invest. 2004;114:784–794. doi: 10.1172/JCI21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Worth NF, Berry CL, Thomas AC, Campbell JH. S18886, a selective TP receptor antagonist, inhibits development of atherosclerosis in rabbits. Atherosclerosis. 2005;183:65–73. doi: 10.1016/j.atherosclerosis.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 33.Gluckman TJ, McLean RC, Schulman SP, Kickler TS, Shapiro EP, Conte JV, McNicholas KW, Segal JB, Rade JJ. Effects of aspirin responsiveness and platelet reactivity on early vein graft thrombosis after coronary artery bypass graft surgery. J Am Coll Cardiol. 2011;57:1069–1077. doi: 10.1016/j.jacc.2010.08.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halushka MK, Halushka PV. Why are some individuals resistant to the cardioprotective effects of aspirin? Circulation. 2002;105:1620–1622. doi: 10.1161/01.cir.0000015422.86569.52. [DOI] [PubMed] [Google Scholar]
- 35.Smith JP, Haddad EV, Taylor MB, Oram D, Blakemore D, Chen Q, Boutaud O, Oates JA. Suboptimal inhibition of platelet cyclooxygenase-1 by aspirin in metabolic syndrome. Hypertension. 2012;59:719–725. doi: 10.1161/HYPERTENSIONAHA.111.181404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cipollone F, Ciabattoni G, Patrignani P, Pasquale M, Gregorio DD, Bucciarelli T, Davı G, Cuccurullo FC, Patrono C. Oxidant stress and aspirin-insensitive thromboxane biosynthesis in severe unstable angina. Circulation. 2000;102:1007–1013. doi: 10.1161/01.cir.102.9.1007. [DOI] [PubMed] [Google Scholar]
- 37.Ames PRJ, Batuca JR, Muncy IJ, De La Torre IG, Pascoe-Gonzales S, Guyer K, Matsuura E, Lopez LR. Aspirin insensitive thromboxane generation is associated with oxidative stress in type 2 diabetes mellitus. Thromb Res. 2012;130:350–354. doi: 10.1016/j.thromres.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 38.Schwedhelm E, Bierend A, Maas R, Trinks R, Kom GD, Tsikas D, Böger RH. Redox-generated isoprostanes are associated with residual platelet activity in aspirin-treated patients with stable coronary heart disease. J Thromb Haemost. 2010;8:2662–2670. doi: 10.1111/j.1538-7836.2010.04117.x. [DOI] [PubMed] [Google Scholar]
- 39.Olson MT, Kickler TS, Lawson JA, McLean RC, Jani J, FitzGerald GA, Rade JJ. Effect of assay specificity on the association of urine 11-dehydro thromboxane B2 determination with cardiovascular risk. J Thromb Haemost. 2012;10:2462–2469. doi: 10.1111/jth.12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davì G, Santilli F, Vazzana N. Thromboxane receptors antagonists and/or synthase inhibitors. Handb Exp Pharmacol. 2012:261–286. doi: 10.1007/978-3-642-29423-5_11. [DOI] [PubMed] [Google Scholar]
- 41.Ferreiro JL, Angiolillo DJ. New directions in antiplatelet therapy. Circ Cardiovasc Interv. 2012;5:433–445. doi: 10.1161/CIRCINTERVENTIONS.111.966176. [DOI] [PubMed] [Google Scholar]
- 42.Ritter JM. TP receptor antagonists (TXRAs): expensive irrelevance or wonder drugs strangled at birth? Br J Clin Pharmacol. 2011;71:801–803. doi: 10.1111/j.1365-2125.2011.03985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bousser MG, Amarenco P, Chamorro A, Fisher M, Ford I, Fox KM, Hennerici MG, Mattle HP, Rothwell PM, de Cordoüe A, Fratacci MD, PERFORM Study Investigators Terutroban versus aspirin in patients with cerebral ischaemic events (PERFORM): a randomised, double-blind, parallel-group trial. Lancet. 2011;377:2013–2022. doi: 10.1016/S0140-6736(11)60600-4. [DOI] [PubMed] [Google Scholar]
- 44.Bots ML, Ford I, Lloyd SM, Laurent S, Touboul PJ, Hennerici MG, Prevention of Cerebrovascular and Cardiovascular Events of Ischemic Origin with Terutroban in Patients with a History of Ischemic Stroke or Transient Ischemic Attack Vascular Ultrasound Study Investigators Thromboxane prostaglandin receptor antagonist and carotid atherosclerosis progression in patients with cerebrovascular disease of ischemic origin: a randomized controlled trial. Stroke. 2014;45:2348–2353. doi: 10.1161/STROKEAHA.114.004775. [DOI] [PubMed] [Google Scholar]
- 45.Neri Serneri GG, Coccheri S, Marubini E, Violi F. Drug Evaluation in Atherosclerotic Vascular Disease in Diabetics (DAVID) Study Group. Picotamide, a combined inhibitor of thromboxane A2 synthase and receptor, reduces 2-year mortality in diabetics with peripheral arterial disease: the DAVID study. Eur Heart J. 2004;25:1845–1852. doi: 10.1016/j.ehj.2004.07.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.