

Abstract
The plant-derived sesquiterpene lactone parthenolide (PTL) was recently found to possess promising anticancer activity but elaboration of this natural product scaffold for optimization of its pharmacological properties has proven challenging via available chemical methods. In this work, P450-catalyzed C–H hydroxylation of positions C9 and C14 in PTL was coupled to carbamoylation chemistry to yield a panel of novel carbamate-based PTL analogs (‘parthenologs’). These compounds, along with a series of other C9- and C14-functionalized parthenologs obtained via O–H acylation, alkylation, and metal-catalyzed carbene insertion, were profiled for their cytotoxicity against a diverse panel of human cancer cell lines. These studies led to the discovery of several parthenologs with significantly improved anticancer activity (2–14 fold) compared to the parent molecule. Most interestingly, two PTL analogs with high cytotoxicity (LC50 ~ 1–3 μM) against T cell leukemia (Jurkat), mantle cell lymphoma (Jeko-1), and adenocarcinoma (HeLa) cells as well as a carbamate derivative with potent activity (LC50 = 0.6 μM) against neuroblastoma cells (SK-N-MC) were obtained. In addition, these analyses resulted in the identification of parthenologs featuring both a broad spectrum and tumor cell-specific anticancer activity, thus providing valuable probes for the future investigation of biomolecular targets that can affect cell viability across multiple as well as specific types of human cancers. Altogether, these results highlight the potential of P450-mediated chemoenzymatic C–H functionalization toward tuning and improving the anticancer activity of the natural product parthenolide.
Keywords: Parthenolide, Sesquiterpene lactones, Anti-cancer activity, Chemoenzymatic synthesis, Cytochrome P450BM3, Enzymatic hydroxylation, Late-stage C-H functionalization
Graphical Abstract
1. Introduction
Sesquiterpene lactones (SQLs) are a class of plant-derived natural products exhibiting a broad spectrum of biological activities including anti-inflammatory, anti-viral and anti-proliferative properties.1–2 These bioactive compounds commonly share an α-methylene-γ-lactone ring which is critical for their biological activity.2–3 Parthenolide (PTL), a SQL isolated from various plants in the Asteracea family, has attracted significant attention owing to its promising activity as an antileukemic agent. PTL was indeed shown to induce apoptosis in human acute myelogeneous leukemia (AML) cells as well as in AML stem cell subpopulations while sparing normal cells.3–5 Beside AML, PTL was found to possess cytotoxic activity against a variety of other human cancer cells.6–12 The anticancer activity of PTL involves a complex mechanism of action as a result of the ability of this compound to target and affect a broad range of cellular processes. These cellular processes primarily include inhibition of the NF-κB transcription factor complex,4,13–14 activation of the p53 pathway15 and generation of reactive oxygen species (ROS)4,16 and associated oxidative stress in cancer cells. Other studies have linked the anticancer activity of PTL to inhibition of the STAT3 transcription factor,17 activation of proapoptotic proteins,18 and epigenetic modulation (NSD1, SETD2, HDAC1).15,19–20
Owing to the promising activity of PTL as an antileukemic and anticancer agent, there has been a growing interest in modifying the PTL scaffold with the goal of improving its cytotoxicity and selectivity against cancer cells. By chemical means, functionalization of PTL structure has involved modification of the reactive C13 position (via Michael addition of amine nucleophiles21–24 or Heck coupling22) and of the C1–10 double bond9 (via cyclopropanation). These modifications have resulted in semi-synthetic PTL analogs with comparable or reduced anticancer activity compared to the parent natural product. Functionalization of the C13 position, in particular, has often led to a decrease or loss of anticancer activity, likely due to the critical importance of the α-methylene-γ-lactone ring for biological activity. As an exception, C13-dimethylamino-parthenolide (DMAPT) was found to maintain PTL-like antileukemic activity while exhibiting improved oral bioavailability, thus enabling an initial assessment of PTL therapeutic potential in animal models of AML.21,24 Subsequent studies suggested that DMAPT may act as a prodrug that releases PTL in the presence of glutathione.25
In the interest of expanding opportunities for manipulation of the PTL scaffold, our group has recently engineered variants of the bacterial fatty acid P450 monooxygenase CYP102A1 (P450BM3) that are capable of hydroxylating positions C9 and C14 in PTL with high regio- and stereoselectivity.26 The corresponding hydroxylated derivatives could be further elaborated via acylation or alkylation resulting in the identification of PTL analogs (referred to herein also as “parthenologs”) with enhanced cytotoxic activity and selectivity against AML cells and patient-derived primary AML specimens.26–27 In the present work, we first extended the scope of this chemoenzymatic approach to PTL functionalization by coupling P450-catalyzed hydroxylation with carbamoylation to yield a panel of carbamate-based parthenologs. Compared to the ester linkage in the acylated PTL analogs, the carbamate linkage provides superior hydrolytic stability while serving as both a H-bond donor and H-bond acceptor.28–29 Because of these properties, carbamate functionalities have found widespread use in medicinal chemistry and they are integral part of a number of marketed drugs (Figure 1).30–33
Figure 1.

Marketed drugs containing carbamate moieties.
The anticancer activity of C9- and C14-functionalized parthenologs has so far been investigated only in context of AML. Here, we report the profiling of the anticancer activity of of a large panel of C9- and C14-modified parthenologs, including the newly available carbamate derivatives, across a diverse spectrum of human cancer cell lines including those derived from T cell leukemia, lymphoma, and different types of solid tumors. Our results show that functionalization of the C9/C14 positions can dramatically alter the anticancer activity profile of this molecule, resulting in parthenologs that feature both a broad spectrum as well as cancer cell-specific cytotoxicity. In addition, these studies led to the discovery of PTL analogs with potent activity against T cell leukemia, mantle cell lymphoma, and neuroblastoma cells.
2. Results and discussion
2.1. Optimized protocol for synthesis of C9 and C14 oxyfunctionalized PTL analogs
We recently reported the development and application of engineered variants of CYP102A1 from Bacillus megaterium for the selective hydroxylation of position C9 and C14 in PTL.26,34 The products of these enzymatic reactions, namely 9(S)-hydroxy-PTL (2) and C14-hydroxy-PTL (3), can serve as valuable intermediates for the generation of C9- and C14-functionalized PTL analogs via chemical modification of the enzymatically installed hydroxyl group. Previously, 2 and 3 were prepared using CYP102A1 variants II-C5 and FL#46, respectively, in the presence of a NADPH cofactor regeneration system consisting of a thermostable phosphite dehydrogenase (PTDH)35–36, NADP+ and sodium phosphite as the sacrificial hydride donor. The enzymatic transformation could be carried using lysate of the P450-expressing E. coli cells directly, which bypasses the need for purification of the P450 enzyme.27 Attempts to further streamline the process by using a two-plasmid system for co-expression of both the P450 and PTDH in the host cells resulted however in reduced product conversion and isolated yield for both 2 and 3.27
To investigate the basis of this phenomenon, we conducted experiments in which either P450 or PTDH was supplemented at varying concentration (0.5–1 μM) to the lysate of E. coli cells co-expressing the P450 and PTDH enzymes. Insightfully, the hydroxylation reactions with P450-supplemented lysate did not show any improvement in PTL conversion to 2 (or 3), whereas those carried out with PTDH-supplemented lysate were found to reproduce the results obtained using our reference conditions (i.e., P450-expressing cell lysate with exogenously added PTDH at 2 μM). These results clearly indicated that the performance of the co-expression system is limited by the expression levels of PTDH. These conclusions were corroborated by additional experiments in which cells were induced with variable amounts of L-arabinose, which controls the transcription of the PTDH gene, and IPTG, which controls the transcription of the P450 gene. Specifically, a 2-fold increase in the L-arabinose concentration during induction (0.12% m/v), in combination with 0.5 mM IPTG (for P450 expression) and 0.3 mM (δ-aminolevulinic acid for heme biosynthesis), was found to result in adequate and reproducible levels of P450 and PTDH after cell lysis. Using the lysate of 1 L cell cultures prepared under these conditions, 0.1 g of PTL could be fully transformed resulting in the isolation of 2 and 3 in 44% and 54% yield, respectively (Scheme 1). By completely eliminating the need for purification of the enzymes required for both the hydroxylation reaction and NADPH cofactor regeneration, this procedure provides an improved and streamlined approach to the synthesis of these compounds for further functionalization.
Scheme 1.

Chemoenzymatic synthesis of C9- and C14-functionalized PTL analogs via P450-catalyzed hydroxylation and carbamoylation chemistry. (a–b) 1 mM parthenolide, cell lysate containing ~ 0.1 mol% P450 and ~ 2 μM PTDH, 150 μM NADP+, 50 mM sodium phosphite in 50 mM potassium phosphate buffer (pH 8.0), room temperature, 14 hours. P450 variant II-C5 was used for (a), and P450 variant FL#46 for (b); (c) R–NCO, DBTDL, DCM, room temperature (3–12 hours).
2.2. Synthesis of PTL analogs via carbamoylation and other chemistries
Previously, we found that the installation of aromatic substituents at either the C9 or C14 position of PTL were most effective toward improving the antileukemic activity of this natural product.26 Building upon these findings, we designed and targeted the synthesis of a panel of carbamate PTL analogs that bears varying substituted aryl groups at those positions. To afford these compounds, we were attracted by the mild reaction conditions provided by the carbamoylation of primary and secondary alcohols with isocyanates in the presence of tin-based catalyst.37–38 In particular, this reaction proceeds under neutral conditions, which was deemed important in view of the relative instability of the PTL scaffold under acidic or basic conditions. Gratifyingly, the desired carbamate-based parthenologs could be successfully prepared starting from 2 or 3 in the presence of the appropriate isocyanate reagent and catalytic amounts of dibutyltin dilaurate (DBTDL) (Scheme 1). The optimized protocol for these transformations involved dissolving the hydroxylated PTL precursor in dry dichloromethane followed by addition of the isocyanate reagent (1.4 equiv) and 20 mol% DBTDL. In general, faster reaction kinetics (3–4 hours) were observed for functionalization of 3 vs. 2, which bears a more sterically hindered, secondary alcohol, or in the presence of electronically activated isocyanates (e.g., p-nitrophenyl isocyanate to give 12–13). More extended reaction times (12–15 hrs) and excess isocyanate (4 equiv.) were required for functionalization of 2 in the presence of more sterically reagents such as 1-isocyanatonaphthalene (to give 11). In any case, both the C9- and C14-functionalized PTL derivatives could be prepared and isolated in good to excellent yields (35–90%; Scheme 1).
In addition to 4–13, a series of C9- and C14-functionalized parthenologs were prepared via acylation, alkylation, or metal-catalyzed carbene O–H insertion of 2 and 3, respectively, according to our previously reported protocols (Scheme 2).26–27 These compounds were selected from among those exhibiting most promising activity against AML cells (LC50 = 2–10 μM).26–27 In addition, these analogs were chosen to provide a range of different functionalization patterns and linkages (i.e., ester or ether linkage) at the C9 and C14 position, in order to examine the effect of these structural features on their anticancer activity.
Scheme 2.

Synthesis of C9- and C14-functionalized PTL analogs via acylation, alkylation and O-H carbene insertion chemistry.
2.3. Anticancer activity profiling of PTL analogs
In addition to antileukemic activity, PTL has been reported to display promising cytotoxicity against other human cancers, including breast,39–40 prostate,41 and pancreatic42 cancer. Whereas our previous studies demonstrated the beneficial effect of functionalization at the C9 or C14 sites for improving PTL cytotoxicity against AML cells, the potential of these structural modifications toward modulating and improving its activity against other forms of human cancer was not assessed.
To this end, the parthenologs described in Schemes 1 and 2 were screened for their activity against a broad panel of cancer cell lines, including HeLa (cervical cancer), MDA-MB-231 (breast), SK-N-MC (neuroblastoma), SK-MEL-2 (melanoma), SJSA-1 (osteosarcoma), A549 (lung cancer), HCT-116 (colon cancer), PC-3 (prostate cancer), Jurkat (acute T cell leukemia) and JeKo-1 (B cell lymphoma) cells. Cell viability was determined by a standard colorimetric assay using a tetrazolium dye, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) which correlates metabolic activity with cell viability. Using this assay, PTL was determined to exhibit a half-maximal lethality dose (LD50) between 10 and 45 μM, depending on the cell line (Table 1). Accordingly, an initial screening of the in vitro anticancer activity of the PTL analogs was carried out at a fixed concentration of 10 μM.
Table 1.
Cytotoxicity of PTL and selected parthenologs against various cell lines. LC50 values are mean ± SD (n = 6).
| Compound | Jurkat (blood) | Jeko-1 (blood) | HeLa (cervix) | A549 (lung) | HCT-116 (colon) | SJSA-1 (bone) | SK-N-MC (brain) | PC-3 (prostate) | MDA-MB-231 (breast) | SK-MEL-2 (skin) |
|---|---|---|---|---|---|---|---|---|---|---|
| PTL | 16.9 ± 3.0 | 10.0 ± 1.1 | 9.0 ± 0.8 | 29.7 ± 1.7 | 18.0 ± 1.7 | 42.4 ± 3.9 | 4.7 ± 0.8 | 20.5 ± 1.3 | 18.3 ± 1.1 | 24.9 ± 3.8 |
| 6 (9–32) | - | - | - | - | 11.8 ± 2.7 | - | - | - | - | - |
| 7 (14–32) | - | 6.7 ± 0.5 | - | - | - | - | - | - | - | - |
| 8 (9–33) | 2.8 ± 0.2 | 3.5 ± 0.3 | - | - | 7.9 ± 0.3 | - | - | - | 7.2 ± 0.7 | - |
| 9 (14–33) | 3.3 ± 0.2 | 3.4 ± 0.4 | 1.5 ± 0.1 | 5.5 ± 0.4 | 5.9 ± 0.2 | 9.2 ± 0.0 | 0.6 ± 0.2 | 5.5 ± 0.2 | 6.1 ± 0.6 | 4.3 ± 0.9 |
| 13 (14–35) | - | - | 1.7 ± 0.1 | - | 7.8 ± 0.4 | - | - | 5.7 ± 0.2 | - | - |
| 16 (9–12) | - | - | 2.1 ± 0.1 | - | - | - | 5.7 ± 0.2 | 5.3 ± 0.2 | 5.7 ± 0.9 | - |
| 17 (14–12) | - | - | 2.9 ± 0.1 | - | 8.2 ± 0.6 | 7.7 ± 0.6 | 5.6 ± 0.2 | 6.7 ± 0.5 | - | - |
| 18 (9–13) | 1.2 ± 0.1 | 1.2 ± 0.1 | - | - | - | - | 4.7 ± 0.5 | 3.1 ± 0.2 | 3.8 ± 0.4 | 2.6 ± 0.2 |
| 19 (14–13) | 1.9 ± 0.1 | 1.7 ± 0.2 | 1.5 ± 0.1 | 4.9 ± 0.2 | 5.7 ± 0.2 | 6.4 ± 0.8 | 0.6 ± 0.2 | 4.7 ± 0.4 | 6.2 ± 0.7 | - |
| 24 (14–18) | - | - | - | - | 11.8 ± 1.6 | - | - | - | - | - |
| 25 (9–19) | - | - | - | - | 8.3 ± 0.3 | - | - | - | - | - |
| 27 (14–20) | 2.3 ± 0.3 | - | 1.7 ± 0.1 | - | 7.1 ± 0.3 | 6.7 ± 0.5 | - | - | - | - |
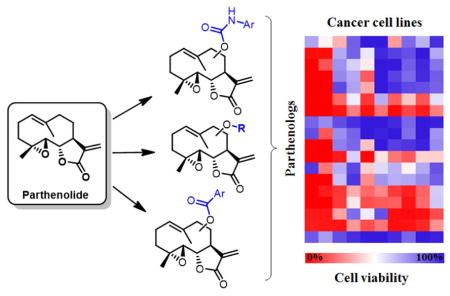
From these experiments, a heat map was generated to highlight compounds showing high (70–100% cell death, red), moderate (40–60% cell death, white to light red/blue) and low (0–30% cell death, blue) cytotoxicity against the various cancer cell lines (Figure 2). As shown by the plot, the hydroxylated parthenolide derivatives 2 and 3 show low or no cytotoxicity (0–10% cell death) against all of the cell lines, with the exception of the HeLa cells. In contrast, many of the parthenologs were determined to possess enhanced anticancer activity compared to PTL. Notably, most of the analogs (>85%) were found to exhibit high cytotoxicity against acute T cell leukemia (Jurkat) and B cell lymphoma cells (Jeko-1), causing >80–100% cell death at 10 μM. These results clearly indicate that the beneficial effect of C9 and C14 substitutions are not restricted to improving PTL activity against AML cells26–27 but extend to other forms of leukemia (Jurkat) and to lymphoma (Jeko-1). To further discriminate the most potent analogs against these two cancer cell lines, the cytotoxicity assay was repeated at a 5-fold lower compound dose (i.e., 2 μM). Under these conditions, the most promising analogs (>20–40% reduction in cell viability) were identified as the carbamate analogs 6–9 and ester derivatives 17–19 (Table S1).
Figure 2.

Cytotoxicity of PTL and of the parthenologs against the panel of cancer cell lines at the standard dose of 10 μM (n = 5). Standard deviation is within 10%. Reported values corresponds to: (% cell viability)/100. See Table S1 for complete data and for cytotoxicity data against Jurkat and JeKo-1 cells at the lower compound dose (2 μM).
Compared to the blood cancer cell lines, a rather different scenario emerged from profiling of the compounds’ activity against cell lines derived from solid tumors. In this case, a relatively smaller albeit still sizeable fraction of the analogs (~30%) were found to display improved cytotoxicity activity compared to PTL (i.e., >2-fold reduction in % cell viability) toward one or more cancer cell lines. Interestingly, the majority of the improved analogs corresponds to either carbamate or ester derivatives (Figure 2). In comparison, only one compound out of the eight ether-based analogs (32) showed noticeably improved anticancer activity compared to PTL. Since some of these analogs share a structurally related or even identical aryl group at the same functionalization site (e.g., 29 and 33 vs. 15), these results point at the critical importance of the ester/carbamate linkage for anticancer activity in the context of solid tumors. This is in contrast to the high activity exhibited by many of the ether-based PTL analogs against leukemia and lymphoma cells as determined here (Figure 2) and previously27 for AML cells.
Other interesting trends emerged from a more in-depth analysis of structure-activity data relative to the carbamate- and ester-based parthenologs. Within the carbamate group, an increase in cytotoxicity against multiple cancer cell lines is observed upon installation of fluoro- and trifluoromethyl-substituted phenyl groups at either functionalization site (C9 or C14) of the PTL scaffold (i.e., 8–9 > 6–7 > 4–5). A similar trend is observed across the ester series of PTL analogs (i.e., 16–19 > 14–15). These results indicate that an increase in lipophilicity in the appended aryl group is generally beneficial toward enhancing the anticancer activity of these compounds. At the same time, a marked dependence of such activity on the size of the aryl group is also evident, as indicated by the moderate activity of 10, 11, 20, and 21, which carry a napthyl substituent at position C9/C14.
Particularly interesting is the effect of the C9/C14 functionalizations on the overall spectrum of anticancer activity exhibited by the parthenologs, especially in the context of the solid tumor cell lines. In this regard, two general activity profiles can be recognized from the data in Figure 2, corresponding to compounds showing either a broad spectrum or a cancer cell-selective activity profile. Relevant representatives of the first group are the fluorinated PTL analogs 9, 16, 17 and 19, which show high cytotoxicity (>70–80% reduction in cell viability) against most if not all of the cancer cell lines tested. Belonging to this group is also the carbamate analog 13 and the pyrazole-containing analog 27. Interestingly, the broad anticancer activity profile of the latter appears to be directly linked to the presence of the pyrazole ring, as structurally related analogs such as 23 and 26 fail to present such behavior. The pyrazole group is a well known privileged scaffold in medicinal chemistry due to its ability to mediate interactions with proteins via hydrogen bonding (donor/acceptor), π-π interactions and cation-π interactions.43–44
In contrast to the compounds mentioned above, a subset of the parthenologs was found to feature tumor type selectivity, exhibiting high activity against only a few or even just one of the solid cancer cell lines. Among these, compounds 8 and 18, which share a (3,5-bis-trifluoromethyl)phenyl substituent at position C9, show high cytotoxicity (>80% reduction in cell viability at 10 μM) against breast, prostate, cervical, and brain cancer cells, but moderate or low cytotoxicity (<30–50% reduction in cell viability) against lung, bone, and skin cancer cells. Of note, 8 is more active than 18 against colon cancer cells (HCT-116) but less active against skin cancer cells, which also highlights the functional importance of the carbamate vs. ester linkage for the observed biological activity. Interestingly, the colon cancer cell line HCT-116 was uniquely sensitive, compared to the other types of solid cancers, to the carbamate analog 6 or to the ester derivatives 23, 24, and 25. The latter compounds share a heterocycle-containing biaryl group, which thus represents a key structural motif for enhancing the anticancer activity of PTL in colon cancer cells. Noteworthy is also the activity profile of the carbamate analog 11, which shows high cytotoxicity only against B cell lymphoma cells within the entire panel of cancer cell lines tested (Figure 2).
Valuable structure-activity insights emerged also from comparison of the data relative to PTL analogs carrying an identical substitution at the C9 vs. C14 position. Whereas regioisomeric ‘pairs’ such as 6/7, 14/15, and 16/17 share a very similar anticancer activity profile, a distinctive impact of the site of functionalization on anticancer activity is apparent for others, including 8/9, 12/13, 18/19, 25/26 and 32/33. Particularly striking is the case of the carbamate analogs 12 and 13, for which functionalization of the C14 site results in a broad spectrum anticancer activity (13), whereas the same substitution at position C9 leads to enhanced cytotoxicity only against colon cancer (HCT-116) and blood cancer cells (Jurkat, Jeko-1). Also notable is the high selectivity of the C14-functionalized 11 against lymphoma cells when compared to the C9-substituted counterpart 10. In other cases, modification of the C9 vs. C14 position is critical for the observed anticancer activity, as shown by significantly higher cytotoxicity of 25 and 32 (compared to 26 and 33, respectively) against colon cancer cells.
2.4. LC50 determination
Based on the data summarized in Figure 2, the most promising PTL analogs were selected for more quantitative analysis of their anticancer activity via dose-response curves. Selection criteria were a ≥ 80% reduction in cell viability for any of solid cancer cell lines after incubation with the compound at the standard 10 μM concentration, and/or ≥ 25% reduction in viability of leukemia (Jurkat) or lymphoma cells (JeKo-1) at the reduced compound dose of 2 μM. Half-maximal lethal concentration (LC50) values were obtained from dose-dependent cell viability curves after normalization to control cells treated with vehicle only (media with 1% DMSO).
As summarized in Table 1, the different cancer cell lines show a variable degree of sensitivity to PTL, with SK-N-MC, Jeko-1, and HeLa cells showing LC50 values between 4.7 and 10.0 μM and A459 and SJSA-1 cells being about 5-fold less sensitive to this compound (LC50 = 29.7–42.4 μM). Consistent with the results from the initial activity profiling, the LC50 values corresponding to the tested PTL analogs demonstrate a significant increase in anticancer activity compared to PTL (Figure 2). The largest activity enhancement is exhibited by 18 against Jurkat cells, which corresponds to a 14-fold decrease in LC50 (1.2 μM vs. 16.9 μM for PTL). In general, the analogs containing trifluoromethyl-substituted aryl rings (9, 17, 18, 19) emerged as the most active compounds, displaying low micromolar to submicromolar LC50 values across all of the cancer cell lines tested. Within this subset of parthenologs, the impact of the functionalization chemistry (carbamoylation vs. esterification) on the compound anticancer properties are evident from comparison of 8 vs. 18 and 9 vs. 19. While these compounds share very similar LC50 values against cancer cells derived from solid tumors, 18 and 19 show a two-fold higher cytotoxicity compared 8 and 9, respectively, against Jurkat and Jeko-1 cells. Comparison of the activity data relative to 16 vs. 18 and to 17 vs. 19 revealed the functional implications of subtle structural variations in the appended aryl group (i.e., meta, meta- vs. ortho, para bis-trifluoromethylation). Particularly notable in this case is the higher activity (2- to 3-fold lower LC50) of 19 vs. 17 against HeLa cells, and of 18 vs. 16 against breast (MDA-MB-231) and prostate (PC-3) cancer cells.
As it became evident from the data of Figure 2, the functionalization site (C9 vs. C14) has a variable effect on the anticancer activity of the compounds, depending on the nature of the substituent and on the type of cancer cell. For example, 16 and 17 are equally active against HeLa, SK-N-MC, and PC-3 cells. In contrast, important differences in anticancer activity are observed for the C9-functionalized analogs 8 and 18 when compared to their C14-functionalized counterparts, 9 and 19, respectively. For example, 18 displays a >2–4 fold lower LC50 than 19 (1.5–6.4 vs. >10 μM) against epithelial (HeLa), lung (A459), colon (HCT-116), and bone (SJSA-1) cancer cells, whereas it exhibits 2-fold less cytotoxicity than 19 against prostate (PC-3) and breast (MDA-MB-231) cancer cells. To gain further insights into this aspect, the three-dimensional structure of a representative C9-functionalized analog (14) was determined by X-ray crystallography (Figure 3). These analyses show how functionalization of the pro-(S) C–H bond in C9 places the appended aryl group in the same region of the C14 methyl group. Accordingly, C9- and C14-functionalized analogs are expected to feature subtle differences with respect to the orientation of the appended functionality. Intriguingly, these structural differences are important for inducing high cytotoxicity in the context of certain cancer cells but not in others. Since PTL is known to target multiple proteins in cancer (AML) cells,45 this phenomenon is likely to reflect how these structural variations (i.e., C9 vs. C14 functionalization) and those mentioned earlier (e.g., carbamate vs. ester linkage) affect the interaction of the parthenologs with their biomolecular targets. In this regard, particularly interesting are the results with the neuroblastoma cells. In this case, modification of the C14 position with a (3,5-bis-trifluoromethyl)phenyl group (9 and 19) is uniquely beneficial for potentiating PTL cytotoxicity in the context of these cancer cells, resulting in the most potent analogs (LC50 = 0.6 μM) identified over the course of this study.
Figure 3.

X-ray crystal structure of compound 14.
Finally, it is worth noting how chemoenzymatic elaboration of the PTL scaffold have resulted in a parthenolog, 9, possessing potent antiproliferative activity (LC50 = 4.9–6.4 μM) also against lung (A549) and osteosarcoma cells (SJSA-1), which are considerably less sensitive to treatment with PTL (LC50 > 30–40 μM) compared to the other types of cancer cells investigated here (Table 1) and earlier (AML)26–27.
3. Conclusions
A series of C9- and C14-functionalized parthenologs were prepared by chemoenzymatic synthesis and evaluated for their activity against a diverse panel of cancer cells derived from blood and solid human tumors. Using this approach, PTL analogs exhibiting low micromolar activity (LC50 ~ 1.2–6.4 μM) against each of these different cancer cells were identified, along with parthenologs possessing high cytotoxicity again neuroblastoma cells (LC50 = 0.6 μM). The anticancer activity of these semisynthetic PTL derivatives correspond to up to 14-fold increase compared to the parent natural product, and extend to cancer cells that are relatively insensitive to this molecule (i.e., lung and osteosarcoma cells). Importantly, this study demonstrates how P450-mediated late-stage C–H functionalization of the PTL scaffold have proven effective not only toward potentiating the anticancer properties of this natural product, but also toward modulating the spectrum of activity across different types of cancer cells. As a result, parthenologs featuring both a broad spectrum of anticancer activity (e.g., 9, 19) as well as tumor-specific cytotoxicity (e.g., 6, 11, 25) could be identified. The results reported here are expected to aid the development of PTL-based anticancer agents as well as provide valuable probes for investigating biomolecular targets affected by these molecules across both multiple and specific types of human cancer. Further studies in these directions are currently ongoing in our laboratory.
4. Materials and Methods
4.1. Reagents and substrates
Chemical reagents and solvents were purchased from commercial suppliers (Sigma-Aldrich, TCI, Fluka, and Codexis) and used without additional purification. PTL was purchased from A Chemtek. All dry reactions were carried out under argon in oven-dried glassware with magnetic stirring using standard gas-tight syringes, cannulae and septa. Silica gel chromatography purifications were carried out using AMD Silica Gel 60 230–400 mesh. 1H and 13C NMR spectra were measured on a Bruker DPX-500 (operating at 500 MHz for 1H and 125 MHz for 13C). Mass spectrometry data were collected via direct infusion on an Advion Expression compact mass spectrometer in ESI mode.
4.2. Protein expression and enzymatic hydroxylation
Plasmids containing the P450BM3 (pCWori vector) and PTDH (pEVOL vector) gene were co-transformed in E. Coli DH5α cells. A single colony was transferred into 5–7 ml LB medium containing 100 μg/ml ampicillin and 25 μg/ml chloramphenicol. The culture was incubated overnight at 37°C, 200rpm and transferred into 1 L of TB media with 100 μg/ml AMP and 25 μg/ml CAM. The cells were grown at 37°C (200rpm) and induced with 0.5mM IPTG, 0.12% m/v L-arabinose and 0.3mM, δ-ALA at OD600= 0.8. The cells were harvested after induction for 24 hours at 27°C (150 rpm), resuspended in 50mM potassium phosphate buffer (pH 8.0) and stored at −80°C for up to 4 months. The frozen suspension was thawed at room temperature and lysed by sonication with the ampitude set at 50% and a pulse sequence of 1 seconds on and 4 seconds off for 12–15 minutes. The lysate was centrifuged at 14,000rpm at 4°C for 40 minutes and the clarified cellular lysate was used for the production of 2 and 3 as described previously.27
4.3.1. General procedure for carbamoylation reactions
To a 0.5 dram vial containing 2 or 3 (4–10 mg) in anhydrous DCM (0.5 ml) was added the corresponding phenyl isocyanate (1.4 eq) and DBTDL (dibuthyltin dilaurate) (20% mol). The reaction was stirred at room temperature for 4–12 hours till completion. A saturated solution of NaHCO3 was added and the aqueous phase was extracted with DCM (3x) in a conical vial. The collected organic layers were dried over Na2SO4, concentrated in vacuo and purified by flash chromatography on silica gel (eluting mixture 20–50% EtOAc in n-Hexane) to obtain the desired product as a solid.
4.4. Compound characterization data
4.4.1. (3aS,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl phenylcarbamate (4)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (8.4 mg, 0.032 mmol), DBTDL (0.006 mmol), and substituted phenyl isocyanate (0.045 mmol). Isolated PTL-09–31: 10.2 mg, 84% yield. 1H NMR (500 MHz, CDCl3): δ = 7.37–7.31 (m, 2H), 7.28 (t, J = 7.6 Hz, 2H), 7.05 (t, J= 7.6 Hz, 1H), 6.34 (d, J = 3.4 Hz, 1H), 5.70 (d, J = 3.4 Hz, 1H), 5.51 (dd, J = 3.4, 11.9 Hz, 1H), 5.19 (d, J = 10.5 Hz, 1H), 3.84 (t, J = 8.9 Hz, 1H), 2.95–2.88 (m, 1H), 2.68 (d, J = 8.5 Hz, 1H), 2.51–2.39 (m, 1H), 2.33–2.21 (m, 2H), 2.17–2.11 (m, 1H), 2.06–1.98 (m, 1H), 1.73 (s, 3H), 1.63–1.51 (m, 1H), 1.30 (s, 3H) ppm, 13C NMR (125 MHz, CDCl3): δ = 168.7, 158.7, 138.3, 133.1, 133.0, 129.2, 127.8, 123.8, 122.2, 121.0, 92.3, 81.7, 66.1, 61.4, 44.1, 36.4, 36.0, 23.8, 17.4, 11.8 ppm, MS (ESI) calculated for C22H25NO5 [M+Na]+ m/z: 406.43; found: 406.5.
4.4.2. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10] cyclodeca [1,2-b]furan-6-yl)methyl phenylcarbamate (5)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (6 mg, 0.023 mmol), DBTDL (0.005 mmol), and substituted phenyl isocyanate (0.032 mmol). Isolated PTL-14–31: 7.3 mg, 84 % yield. 1H NMR (500 MHz, CDCl3): δ = 7.37–7.30 (m, 2H), 7.26 (t, J = 7.4 Hz, 2H), 7.05 (t, J= 7.4Hz, 1H), 6.30 (d, J = 3.4 Hz, 1H), 5.58 (d, J = 3.4 Hz, 1H), 5.47 (dd, J = 3.3, 12.5 Hz, 1H), 4.87 (d, J = 12.5 Hz, 1H), 4.65 (d, J = 11.7 Hz, 1H), 3.96 (t, J= 8.6 Hz, 1H), 2.81–2.71 (m, 2H), 2.67–2.61 (m, 1H), 2.57–2.46 (m, 1H), 2.34–2.23(m, 1H), 2.21–2.09 (m, 3H), 1.83–1.71 (m, 1H), 1.25–1.22 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 169.0, 158.6, 139.1, 132.1, 132.0, 129.3, 128.1, 123.9, 121.6, 121.5, 82.3, 66.1, 61.2, 61.1, 47.4, 36.6, 36.3, 31.4, 23.9, 17.0 ppm, MS (ESI) calculated for C22H25NO5 [M+Na]+ m/z: 406.43; found: 406.4.
4.4.3. (3aS,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl (2,4-difluorophenyl)carbamate (6)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (7.6 mg, 0.029 mmol), DBTDL (0.006 mmol), and substituted phenyl isocyanate (0.12 mmol). Isolated PTL-09–32: 10.8 mg, 90% yield. 1H NMR (500 MHz, CDCl3): δ = 6.87–6.79 (m, 2H), 6.71 (s, 1H), 6.35 ( d, J = 4.3 Hz, 1H), 5.69 (d, J = 3.5 Hz, 1H), 5.52 (dd, J = 3.9, 12.6 Hz, 1H), 5.19 (d, J = 11.3 Hz, 1H), 3.84 (t, J = 9.1 Hz, 1H), 2.95–2.88 (m, 1H), 2.69 (d, J = 20 Hz, 1H), 2.51–2.41 (m, 1H), 2.33–2.22 (m, 2H), 2.18–2.12 (m, 1H), 2.08–1.99 (m, 1H), 1.73 (s, 3H), 1.64–1.54 (m, 1H), 1.30 (s, 3H) ppm, 13C NMR (125 MHz, CDCl3): δ = 178.6, 168.7, 154.6, 138.0, 132.9, 128.0, 122.3, 122.2, 111.4, 111.2, 103.7, 82.1, 81.6, 66.0, 61.3, 44.1, 36.2, 36.0, 24.7, 17.4, 11.7 ppm, MS (ESI) calculated for C22H23F2NO5 [M+Na]+ m/z: 442.41; found: 442.4.
4.4.4. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-6-yl)methyl (2,4-difluorophenyl)carbamate (7)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (5.9 mg, 0.022 mmol), DBTDL (0.004 mmol), and substituted phenyl isocyanate (0.031 mmol). Isolated PTL-14–32: 8 mg, 85% yield. 1H NMR (500 MHz, CDCl3): δ = 6.87–6.79 (m, 2H), 6.68 (s, 1H), 6.31 (d, J = 3.4 Hz, 1H), 5.59 (d, J = 2.3 Hz, 1H), 5.50 (dd, J = 3.1, 12.8 Hz, 1H), 4.91 (d, J = 11.3Hz, 1H), 4.62 (d, J = 13.2 Hz, 1H), 3.85–3.76 (m, 1H), 2.81–2.72 (m, 2H), 2.69–2.61 (m, 1H), 2.56–2.45 (m, 1H), 2.34–2.24 (m, 1H), 2.21–2.07 (m, 3H), 1.77–1.68 (m, 1H), 1.25–1.19 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 174.9, 169.0, 150.5, 138.9, 133.3, 128.1, 122.2, 121.6, 111.5, 111.3, 103.9, 82.5, 66.2, 62.0, 61.0, 47.2, 36.3, 36.1, 31.1, 24.0, 17.1 ppm, MS (ESI) calculated for C22H23F2NO5 [M+Na]+ m/z: 442.41; found: 442.3.
4.4.5. (3aS,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl (3,5-bis(trifluoromethyl)phenyl)carbamate (8)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (7.5 mg, 0.028 mmol), DBTDL (0.006 mmol), and substituted phenyl isocyanate (0.11 mmol). Isolated PTL-09–33: 11.1 mg, 75% yield. 1H NMR (500 MHz, CDCl3): δ = 7.87 (s, 2H), 7.54 (s, 1H), 6.36 ( d, J = 4.1 Hz, 1H), 5.69 (d, J = 4.1 Hz, 1H), 5.55 (dd, J = 3.4, 12.3 Hz, 1H), 5.22 (d, J = 11.1 Hz, 1H), 3.85 (t, J = 8.4 Hz, 1H), 2.96–2.89 (m, 1H), 2.69 (d, J = 9 Hz, 1H), 2.52–2.41 (m, 1H), 2.34–2.24 (m, 2H), 2.20–2.13 (m, 1H), 2.11–2.02 (m, 1H), 1.74 (s, 3H), 1.63–1.55 (m, 1H), 1.30 (s, 3H) ppm, 13C NMR (125 MHz, CDCl3): δ = 168.7, 159.4, 139.4, 139.3, 137.6, 132.4, 128.2, 122.5, 118.4, 117.1, 82.3, 81.4, 65.9, 61.3, 44.3, 36.3, 36.0, 23.8, 17.4, 11.9 ppm, MS (ESI) calculated for C24H23F6NO5 [M+Na]+ m/z: 542.43; found: 542.5.
4.4.6. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-6-yl)methyl (3,5-bis(trifluoromethyl)phenyl)carbamate (9)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (5.6 mg, 0.021 mmol), DBTDL (0.004 mmol), and substituted phenyl isocyanate (0.030 mmol). Isolated PTL-14–33: 6.7 mg, 61% yield. 1H NMR (500 MHz, CDCl3): δ = 7.90 (s, 2H), 7.76 (s, 1H), 7.51 (s, 1H), 6.27 (d, J = 3.5 Hz, 1H), 5.62 (d, J = 2.9 Hz, 1H), 5.49 (dd, J = 3.5, 12.2 Hz, 1H), 4.81 (d, J = 12.2 Hz, 1H), 4.75 (d, J = 12.2 Hz, 1H), 3.99 (t, J = 8.6, 1H), 2.84–2.78 (m, 1H), 2.75 (d, J = 8.8 Hz, 1H), 2.63–2.55 (m, 1H), 2.55–2.43 (m, 1H), 2.34–2.24 (m, 1H), 2.24–2.11 (m, 3H), 2.03–1.92 (m, 1H), 1.31 (s, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 162.8, 153.1, 140.2, 139.7, 139.0, 132.9, 132.2, 122.3, 118.4, 116.6, 82.8, 66.1, 63.5, 63.2, 47.2, 36.3, 36.2, 24.2, 16.8 ppm, MS (ESI) calculated for C24H23F6NO5 [M+Na]+ m/z: 542.43; found: 542.5.
4.4.7. (3aS,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl naphthalen-1-ylcarbamate (10)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (3) (8.0 mg, 0.030 mmol), DBTDL (0.006 mmol), and substituted phenyl isocyanate (0.012 mmol). Isolated PTL-09–34: 7.2 mg, 55% yield. 1H NMR (500 MHz, CDCl3): δ = 7.88–7.81 (m, 2H), 7.69–7.63 (m, 1H), 7.54–7.42 (m, 3H), 6.96 ( m, 1H), 6.34 (d, J = 4.0 Hz, 1H), 5.72 (d, J = 3.9 Hz, 1H), 5.51 (dd, J = 3.4, 11.8 Hz, 1H), 5.23 (d, J = 10.6 Hz, 1H), 3.83 (t, J = 8.7 Hz, 1H), 2.95–2.87 (m, 1H), 2.68 (d, J = 11.3 Hz, 1H), 2.52–2.40 (m, 1H), 2.36–2.28 (m, 2H), 2.28–2.21 (m, 1H), 2.18–2.12 (m, 1H), 2.01 (s, 3H), 1.65–1.51 (m, 1H), 1.29 (s, 3H) ppm, 13C NMR (125 MHz, CDCl3): δ =168.9, 158.7, 146.6, 138.9, 135.0, 133.4, 133.1, 129.7, 128.7, 128.5, 127.8, 126.4, 126.1, 124.8, 121.2, 120.0, 82.7, 66.2, 61.4, 61.0, 47.1, 36.6, 36.0, 23.9, 17.2, 11.6 ppm, MS (ESI) calculated for C26H27NO5 [M+Na]+ m/z: 456.49; found: 456.5.
4.4.8. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-6-yl)methyl naphthalen-1-ylcarbamate (11)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (6 mg, 0.023 mmol), DBTDL (0.005 mmol), and substituted phenyl isocyanate (0.032 mmol). Isolated PTL-14–34: 8.4 mg, 85% yield. 1H NMR (500 MHz, CDCl3): δ = 7.91–7.85 (m, 2H), 7.76–7.69 (m, 1H), 7.55–7.41 (m, 3H), 7.01 (m, 1H), 6.37 (d, J = 3.4 Hz, 1H), 5.70 (d, J = 3.3 Hz, 1H), 5.61 (dd, J = 3.5, 12.1 Hz, 1H), 4.87 (d, J = 12.5 Hz, 1H), 4.65 (d, J = 11.7 Hz, 1H), 3.95 (t, J= 8.5 Hz, 1H), 2.81–2.72 (m, 2H), 2.72–2.67 (m, 1H), 2.57–2.47 (m, 1H), 2.34–2.25 (m, 1H), 2.22–2.10 (m, 3H), 1.86–1.78 (m, 1H), 1.29–1.26 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ 169.1, 157.9, 147.6, 139.2, 135.1, 133.7, 133.3, 129.7, 128.9, 128.6, 127.1, 126.3, 126.1, 124.9, 121.1, 120.0, 82.4, 66.3, 61.4, 61.2, 47.4, 36.8, 36.1, 23.9, 17.2 ppm, MS (ESI) calculated for C26H27NO5 [M+Na]+ m/z: 456.49; found: 456.4.
4.4.9. (3aS,5S,9aR,10aS,10bS,E)-6,9a-dimethyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-5-yl (4-nitrophenyl)carbamate (12)
Standard procedure was applied using 9(S)-hydroxy-parthenolide (2) (7.6 mg, 0.029 mmol), DBTDL (0.006 mmol), and substituted phenyl isocyanate (0.041 mmol). Isolated PTL-09–35: 4.3 mg, 35% yield. 1H NMR (500 MHz, CDCl3): δ = 8.17 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 9.3 Hz, 2H), 6.33 (d, J = 3.2 Hz, 1H), 5.71 (d, J = 3.1 Hz, 1H), 5.51 (dd, J = 4.1, 12.2 Hz, 1H), 5.20 (d, J = 10.4 Hz, 1H), 3.81 (t, J = 9.1 Hz, 1H), 2.94–2.89 (m, 1H), 2.71 (d, J = 10.1 Hz, 1H), 2.58–2.49 (m, 1H), 2.35–2.22 (m, 2H), 2.19–2.13 (m, 1H), 2.04–1.98 (m, 1H), 1.74 (s, 3H), 1.65–1.52 (m, 1H), 1.20 (m, 3H) ppm, 13C NMR (125 MHz, CDCl3): δ = 164.4, 152.4, 144.9, 144.1, 138.1, 133.3, 127.1, 124.3, 121.1, 118.1, 83.1, 82.1, 66.1, 61.2, 44.3, 36.2, 36.1, 23.9, 17.0, 11.4 ppm, MS (ESI) calculated for C22H24N2O7 [M+Na]+ m/z: 451.43; found: 451.4.
4.4.10. ((3aR,9aR,10aS,10bS,Z)-9a-methyl-3-methylene-2-oxo-2,3,3a,4,5,8,9,9a,10a,10b-decahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-6-yl)methyl (4-nitrophenyl)carbamate (13)
Standard procedure was applied using 14-hydroxy-parthenolide (3) (5.8 mg, 0.022 mmol), DBTDL (0.004 mmol), and substituted phenyl isocyanate (0.031 mmol). Isolated PTL-14–35: 5.2 mg, 62% yield. 1H NMR (500 MHz, CDCl3): δ = 8.16 (d, J = 8.0 Hz, 2H), 7.53 (d, J = 9.5 Hz, 2H), 6.29 (d, J = 3.2 Hz, 1H), 5.61 (d, J = 3.2 Hz, 1H), 5.51 (dd, J = 4.4, 12.6 Hz, 1H), 4.87 (d, J = 12.0 Hz, 1H), 4.71 (d, J = 12.9 Hz, 1H), 3.90 (t, J = 9.3 Hz, 1H), 2.84–2.78 (m, 1H), 2.75 (d, J = 10.8 Hz, 1H), 2.66–2.58 (m, 1H), 2.56–2.46 (m, 1H), 2.35–2.25 (m, 1H), 2.22–2.13 (m, 3H), 1.95–1.82 (m, 1H), 1.25–1.20 (m, 4H) ppm, 13C NMR (125 MHz, CDCl3): δ = 163.4, 152.3, 146.9, 144.8, 139.1, 133.3, 132.3, 125.3, 122.1, 118.0, 82.7, 66.1, 63.0, 61.3, 47.3, 36.9, 36.1, 31.6, 23.9, 16.9 ppm, MS (ESI) calculated for C22H24N2O7 [M+Na]+ m/z: 451.43; found: 451.4.
4.5. Synthesis of ester- and ether-based parthenologs
Compounds 14–35 were synthesized following our previously reported procedures26–27 at the 5–10 mg scale. The isolated yields were between 30 and 60% for the ester-based analogs and between 30 and 45% for the ether-based analogs.
4.6. X-ray crystallography
The structure of 14 was determined via single crystal X-ray diffraction analysis using a Bruker SMART APEX II CCD platform diffractometer. A detailed description of the procedures for X-ray diffraction data collection, analysis, and structure refinement are provided as Supporting Information and in Table S2. The crystal structure was deposited with the Cambridge Crystallographic Data Centre under the code CCDC 1546909.
4.7. Cell cultures
All cell lines were maintained in a 37 °C humidified incubator with 5% CO2. A549, MDA-MB-231, PC-3, SJSA-1, SK-N-MC, HeLa, HCT-116 and SK-MEL-2 cells were cultured in DMEM medium (Gibco) supplemented with 10% FBS (Gibco) and 100 I.U./ml penicillin-streptomycin (Sigma). JeKo-1 and Jurkat cells were cultured in α-MEM medium (Gibco) supplemented with 10% FBS and 100 I.U./ml penicillin-streptomycin. Adherent cells were dissociated using Accutase (Millipore).
4.8. Cytotoxicity assay
Adherent cells were seeded in a 96-well plate at a density of 500–10,000 cells per well in 90 μl complete DMEM media. Suspension cells were seeded in conical 96-well plates (Nunc) at a density of 30,000 cells per well in 90 ul of complete α-MEM media devoid of phenol red indicator. The plates were incubated in a humidified incubator for 15 hours prior to treatment. PTL analogs were prepared as concentrated stocks (10 mM) in DMSO and stored at −30 °C. Prior to testing, the analogs were diluted into complete cell culture medium and the media was supplemented with sterile DMSO (10% of final volume). For the initial cytotoxicity screening, the compounds were added to the cells in 10 μaliquots to give a final concentration of 10 μM parthenologs and 1% DMSO (n = 5). Following a 24-hour incubation, the culture media from the adherent cell plates (e.g., HeLa, A549) was aspirated and replaced with 100 μl of 1mg/ml Thiazolyl Blue Tetrazolium Bromide (MTT, Sigma) in complete culture medium devoid of phenol red. For the suspension cells (Jurkat, JeKo-1), 20 ul of a 5mg/ml Thiazolyl Blue Tetrazolium Bromide solution was added directly to the culture media. After incubation at 37 °C for 3 hours, the plates were centrifuged (4000 rpm, 5 minutes), the media was removed and 100 ul of DMSO was added to solubilize the formazan product. The resulting OD was measured at 550 nm using a multi-well plate reader (Tecan).
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Joshua N. Kolev and Dr. William Brennessel (U. Rochester) for assistance with crystallographic analyses. We thank Dr. Wei Hso (University of Rochester Medical Center, URMC) for the HCT-116 cell line, Dr. David Dean (URMC) for the A549 cell line, Dr. Ian Dickerson (URMC) for the SK-N-MC cell line, Dr. Vera Gorbunova for the PC-3 cell line and Dr. Andrew Terentis (Florida Atlantic University) for the HeLa, SK-MEL-2 and MDA-MB-231 cell lines. This work was supported by the Leukemia and Lymphoma Society Translational Research Grant LLS-6116-14 and in part by the U.S. National Institute of Health grant GM098628 awarded to R.F.. B.J.F. acknowledges support from the National Institute of Health grant CA180615. MS instrumentation was supported by the U.S. National Science Foundation grant CHE-0946653.
Abbreviations
- PTL
parthenolide
- AML
acute myelogenous leukemia
- CSCs
cancer stem cells
- DMAPT
dimethylamino-parthenolide
- PTDH
phosphite dehydrogenase
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced)
- IPTG
β-D-1-thiogalactopyranoside
- δ-ALA
δ-aminolevulinic acid
- TB
terrific broth
- DMSO
dimethylsulfoxide
- DBTDL
dibutyltin laurate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Breitmaier E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones. Wiley-VCH Verlag GmbH; Weinheim: 2006. [Google Scholar]
- 2.Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N. Drug Discov Today. 2010;15:668. doi: 10.1016/j.drudis.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Ghantous A, Sinjab A, Herceg Z, Darwiche N. Drug Discov Today. 2013;18:894. doi: 10.1016/j.drudis.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Guzman ML, Rossi RM, Karnischky L, Li XJ, Peterson DR, Howard DS, Jordan CT. Blood. 2005;105:4163. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guzman ML, Jordan CT. Expert Opin Biol Th. 2005;5:1147. doi: 10.1517/14712598.5.9.1147. [DOI] [PubMed] [Google Scholar]
- 6.An Y, Guo W, Li L, Xu C, Yang D, Wang S, Lu Y, Zhang Q, Zhai J, Fan H, Qiu C, Qi J, Chen Y, Yuan S. PLoS One. 2015;10:e0116202. doi: 10.1371/journal.pone.0116202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hexum JK, Becker CM, Kempema AM, Ohlfest JR, Largaespada DA, Harki DA. Bioorg Med Chem Lett. 2015;25:2493. doi: 10.1016/j.bmcl.2015.04.058. [DOI] [PubMed] [Google Scholar]
- 8.Janganati V, Ponder J, Jordan CT, Borrelli MJ, Penthala NR, Crooks PA. J Med Chem. 2015;58:8896. doi: 10.1021/acs.jmedchem.5b01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kempema AM, Widen JC, Hexum JK, Andrews TE, Wang D, Rathe SK, Meece FA, Noble KE, Sachs Z, Largaespada DA, Harki DA. Bioorg Med Chem. 2015;23:4737. doi: 10.1016/j.bmc.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nasim S, Pei S, Hagen FK, Jordan CT, Crooks PA. Bioorg Med Chem. 2011;19:1515. doi: 10.1016/j.bmc.2010.12.045. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava SK, Abraham A, Bhat B, Jaggi M, Singh AT, Sanna VK, Singh G, Agarwal SK, Mukherjee R, Burman AC. Bioorg Med Chem Lett. 2006;16:4195. doi: 10.1016/j.bmcl.2006.05.083. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Lu Y, Ding Y, Zhai J, Ji Q, Ma W, Yang M, Fan H, Long J, Tong Z, Shi Y, Jia Y, Han B, Zhang W, Qiu C, Ma X, Li Q, Shi Q, Zhang H, Li D, Zhang J, Lin J, Li LY, Gao Y, Chen Y. J Med Chem. 2012;55:8757. doi: 10.1021/jm301064b. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Pineres AJ, Castro V, Mora G, Schmidt TJ, Strunck E, Pahl HL, Merfort I. J Biol Chem. 2001;276:39713. doi: 10.1074/jbc.M101985200. [DOI] [PubMed] [Google Scholar]
- 14.Hehner SP, Heinrich M, Bork PM, Vogt M, Ratter F, Lehmann V, Schulze-Osthoff K, Droge W, Schmitz ML. J Biol Chem. 1998;273:1288. doi: 10.1074/jbc.273.3.1288. [DOI] [PubMed] [Google Scholar]
- 15.Gopal YN, Chanchorn E, Van Dyke MW. Mol Cancer Ther. 2009;8:552. doi: 10.1158/1535-7163.MCT-08-0661. [DOI] [PubMed] [Google Scholar]
- 16.Pei S, Minhajuddin M, Callahan KP, Balys M, Ashton JM, Neering SJ, Lagadinou ED, Corbett C, Ye H, Liesveld JL, O’Dwyer KM, Li Z, Shi L, Greninger P, Settleman J, Benes C, Hagen FK, Munger J, Crooks PA, Becker MW, Jordan CT. J Biol Chem. 2013;288:33542. doi: 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung MS, Xuemin Q, Pramod U, Kwon HH, Fekadu K. Curr Cancer Drug Targets. 2014;14:59. [Google Scholar]
- 18.Zhang S, Ong CN, Shen HM. Cancer Letters. 2004;211:175. doi: 10.1016/j.canlet.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 19.Liu Z, Liu S, Xie Z, Pavlovicz RE, Wu J, Chen P, Aimiuwu J, Pang J, Bhasin D, Neviani P, Fuchs JR, Plass C, Li PK, Li C, Huang TH, Wu LC, Rush L, Wang H, Perrotti D, Marcucci G, Chan KK. J Pharmacol Exp Ther. 2009;329:505. doi: 10.1124/jpet.108.147934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakshatri H, Appaiah HN, Anjanappa M, Gilley D, Tanaka H, Badve S, Crooks PA, Mathews W, Sweeney C, Bhat-Nakshatri P. Cell Death Dis. 2015;6:e1608. doi: 10.1038/cddis.2014.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guzman ML, Rossi RM, Li XJ, Corbett C, Hassane DC, Bushnell T, Carroll M, Sullivan E, Neelakantan S, Crooks PA, Jordan CT. Blood. 2006;108:74a. [Google Scholar]
- 22.Han C, Barrios FJ, Riofski MV, Colby DA. J Org Chem. 2009;74:7176. doi: 10.1021/jo901533e. [DOI] [PubMed] [Google Scholar]
- 23.Nasim S, Crooks PA. Bioorg Med Chem Lett. 2008;18:3870. doi: 10.1016/j.bmcl.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 24.Neelakantan S, Nasim S, Guzman ML, Jordan CT, Crooks PA. Bioorg Med Chem Lett. 2009;19:4346. doi: 10.1016/j.bmcl.2009.05.092. [DOI] [PubMed] [Google Scholar]
- 25.Woods JR, Mo H, Bieberich AA, Alavanja T, Colby DA. J Med Chem. 2011;54:7934. doi: 10.1021/jm201114t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolev JN, O’Dwyer KM, Jordan CT, Fasan R. ACS Chem Biol. 2014;9:164. doi: 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyagi V, Alwaseem H, O’Dwyer KM, Ponder J, Li QY, Jordan CT, Fasan R. Bioorg Med Chem. 2016;24:3876. doi: 10.1016/j.bmc.2016.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosh AK, Brindisi M. J Med Chem. 2015;58:2895. doi: 10.1021/jm501371s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vacondio F, Silva C, Mor M, Testa B. Drug Metab Rev. 2010;42:551. doi: 10.3109/03602531003745960. [DOI] [PubMed] [Google Scholar]
- 30.Lam SW, Guchelaar HJ, Boven E. Cancer Treatment Reviews. 2016;50:9. doi: 10.1016/j.ctrv.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Brickner SJ, Barbachyn MR, Hutchinson DK, Manninen PR. J Med Chem. 2008;51:1981. doi: 10.1021/jm800038g. [DOI] [PubMed] [Google Scholar]
- 32.Ruane PJ, Brinson C, Ramgopal M, Ryan R, Coate B, Cho M, Kakuda TN, Anderson D. HIV Medicine. 2015;16:288. doi: 10.1111/hiv.12211. [DOI] [PubMed] [Google Scholar]
- 33.Sangal RB, Blumer JL, Lankford DA, Grinnell TA, Huang H. Pediatrics. 2014;134:e1095. doi: 10.1542/peds.2013-4221. [DOI] [PubMed] [Google Scholar]
- 34.Narhi LO, Fulco AJ. J Biol Chem. 1987;262:6683. [PubMed] [Google Scholar]
- 35.Johannes TW, Woodyer RD, Zhao H. Appl Environ Microbiol. 2005;71:5728. doi: 10.1128/AEM.71.10.5728-5734.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLachlan MJ, Johannes TW, Zhao H. Biotechnol Bioeng. 2008;99:268. doi: 10.1002/bit.21546. [DOI] [PubMed] [Google Scholar]
- 37.Cui J, Hao J, Ulanovskaya OA, Dundas J, Liang J, Kozmin SA. PNAS. 2011;108:6763. doi: 10.1073/pnas.1015253108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Labafzadeh SR, Kavakka JS, Vyavaharkar K, Sievanen K, Kilpelainen I. RSC Adv. 2014;4:22434. [Google Scholar]
- 39.Sweeney CJ, Mehrotra S, Sadaria MR, Kumar S, Shortle NH, Roman Y, Sheridan C, Campbell RA, Murry DJ, Badve S, Nakshatri H. Mol Cancer Ther. 2005;4:1004. doi: 10.1158/1535-7163.MCT-05-0030. [DOI] [PubMed] [Google Scholar]
- 40.Zhou J, Zhang H, Gu P, Bai J, Margolick JB, Zhang Y. Breast Cancer Res Treat. 2008;111:419. doi: 10.1007/s10549-007-9798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawasaki BT, Hurt EM, Kalathur M, Duhagon MA, Milner JA, Kim YS, Farrar WL. The Prostate. 2009;69:827. doi: 10.1002/pros.20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yip-Schneider MT, Nakshatri H, Sweeney CJ, Marshall MS, Wiebke EA, Schmidt CM. Mol Cancer Ther. 2005;4:587. doi: 10.1158/1535-7163.MCT-04-0215. [DOI] [PubMed] [Google Scholar]
- 43.Kumar V, Kaur K, Gupta GK, Sharma AK. Eur J Med Chem. 2013;69:735. doi: 10.1016/j.ejmech.2013.08.053. [DOI] [PubMed] [Google Scholar]
- 44.Naim MJ, Alam O, Nawaz F, Alam MJ, Alam P. J Pharm Bioall Sci. 2016;8:2. doi: 10.4103/0975-7406.171694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pei S, Minhajuddin M, D’Alessandro A, Nemkov T, Stevens BM, Adane B, Khan N, Hagen FK, Yadav VK, De S, Ashton JM, Hansen KC, Gutman JA, Pollyea DA, Crooks PA, Smith C, Jordan CT. J Biol Chem. 2016;291:25280. doi: 10.1074/jbc.A116.750653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.