

Abstract
Glycosylation regulates functional responses mediated by the interaction of IgG with their receptors. Multiple analytical methods have been designed for the determination of the IgG N-glycan microheterogeneity, including MS methods for the analysis of site specific glycoforms of IgG. However, measurement of low abundant glycoforms remains challenging in complex samples like serum without enrichment of the IgG. We present a workflow for quantitative analysis of site specific glycoforms of IgG based on data independent acquisition (DIA) of Y-ions generated under “minimal” fragmentation conditions. The adjusted collision induced dissociation (CID) conditions generate specific Y-ions in the yield of up to 60% precursor ion intensity. These selective fragments, measured in high resolution, improve specificity of detection compared to the typically quantified B-ions which have higher overall intensity but lower signal-to-noise ratios. Under optimized conditions, we achieve label-free quantification of the majority of previously reported glycoforms of IgG (26 glycoforms of IgG1, 22 glycoforms of IgG 2/3, and 19 glycoforms of IgG4) directly in unfractionated samples of human plasma and we detect traces of previously unreported glycoforms of IgG1, including doubly fucosylated glycoforms. The SWATH data independent quantification of IgG glycoforms in pooled plasma samples of patients with liver cirrhosis detects reliably the expected changes in the quantity of major glycoforms compared to healthy controls. Our results show that optimized CID fragmentation enables DIA of IgG glycoforms and suggest that such workflow may enable quantitative analyses of the glycoproteome in complex matrixes.
Graphical abstract
N-glycosylation is a common and diverse modification of proteins.1,2 The common core of N-linked glycans is extended by specific glycosyltransferases in the ER and Golgi compartments, which leads to substantial diversity in the composition of monosaccharides and their linkages in the mature structures.3 Distribution of the heterogeneous N- glycoforms at specific sites of protein attachment changes in the context of diseases;4–6 however, the quantitative changes in microheterogeneity have been determined in detail for only a limited set of proteins and disease conditions.7,8 Perhaps the best characterized is the state of N-glycosylation of IgG because N-glycosylation is known to adjust IgG function in the context of both therapeutic interventions9 and human pathophysiology.10 It is therefore of considerable interest to characterize and quantify comprehensively the distribution of IgG glycoforms in various therapeutic and pathophysiological contexts.
Analysis of the glycoforms of therapeutic antibodies by chromatographic separation of detached N-glycans or glycopeptides followed by diverse methods of detection,11 including mass spectrometric methods,12 has been recently reviewed. Limitations of these methods in the resolution of isobaric glycan structures or glycoforms of the IgG1-4 subclasses were described and are generally considered acceptable.13–15 Analysis of IgG in the context of diseases is complicated by the variable background of the biological samples which limits, to some degree, the ability of the methods to quantify comprehensively the distribution of glycoforms.15 It is therefore most common to analyze IgG isolated from the biological samples, typically by affinity enrichment on protein A or G resins.13,16 This enrichment step enables the use of the methods developed for the analysis of therapeutic IgG provided that sufficient amounts of representative IgG glycoforms are accessible.14,17–19
Mass spectrometric methods for the analysis of IgG glycopeptides have the advantage of resolving, at least in part, the glycoforms of the IgG1-4 subclasses; it is important because the responses of the IgG subclasses differ in various disease contexts.14,19 The available MS methods are based most frequently on the quantification of precursor ion intensities13,20 because the signal, especially in the case of isolated IgG, is typically sufficient for adequate coverage of the glycoform precursors.14,21 Several groups reported also MRM quantification of oxonium ions generated by collision induced dissociation (CID) of glycopeptides as reviewed in ref 15. These common glycan fragments (m/z 138, 204, 366) have high sensitivity but low specificity because they are generated from virtually all N-glycopeptides and have interferences from some peptide fragments as well.22,23 Nonetheless, these B-ions, in the Domon/Costello nomenclature,24 were used for quantification of IgG glycoforms in both samples of isolated IgG19,25 and in complex biological samples.26 Abundance of IgG in biological samples is sufficient to allow label free quantification of at least the more abundant glycoforms using the MRM of B-ions; however, coverage of the low-abundance glycoforms is lower in the complex biological matrix.
In this study, we explore the possibility to use Y-ions, the large peptide-glycan fragments, in the analysis of glycopeptides and we describe CID conditions optimized for the analysis of the Y-ions of IgG. Our results document that high specificity of the Y-ions allows label free quantification of the glycoforms of IgG in complex samples with improved coverage of the low- abundance species. This is to our knowledge the first report describing the use of Y-ions in the analysis of glycopeptides of IgG using a SWATH data independent acquisition (DIA) liquid chromatography–tandem mass spectrometry (LC–MS/MS) workflow.
EXPERIMENTAL SECTION
Study Population
Applicability of the method was documented on plasma samples of healthy controls (n = 10) and patients with cirrhosis of the liver (n = 10) recruited and processed as described in detail in the supplement and previously.27
Sample Processing
Blood samples were collected using EDTA Vacutainer tubes (BD Diagnostics, Franklin Lakes, NJ). Plasma was isolated according to the manufacturer’s protocol within 6 h of blood draw and was stored at −80 °C until use. Aliquots of thawed plasma were diluted 1:20 with sodium bicarbonate and processed as described previously28 with minor modifications. Briefly, diluted plasma was reduced with 5 mM DTT for 60 min at 60 °C and alkylated with 15 mM iodoacetamide for 30 min in the dark. Trypsin Gold (Promega, Madison, WI) digestion (2.5 ng/μL) was carried out at 37 °C in Barocycler NEP2320 (Pressure BioSciences, South Easton, MA) for 1 h, samples were evaporated using vacuum concentrator (Labconco), and dissolved in mobile phase A (2% ACN, 0.1% FA). Tryptic peptides were analyzed without further processing to ensure reliable quantification of the glycoforms.
Glycopeptide Analysis by Nano LC–MS/MS
Glycopeptide separation was achieved on a Nanoacquity LC (Waters, Milford, MA) using capillary trap, 180 μm × 0.5 mm, and analytical 75 μm × 150 μm Atlantis DB C18, 3 μm, 300 Å columns (Water, Milford, MA) interfaced with 5600 tripleTOF (Sciex, Framingham, MA). A 1 min trapping step using 2% ACN, 0.1% formic acid at 15 μL/min was followed by chromatographic separation at 0.4 μL/min as follows: starting conditions 5% ACN, 0.1% formic acid; 1–35 min, 5–50% ACN, 0.1% formic acid; 35–37 min, 50–95% ACN, 0.1% formic acid; 37–40 min 95% ACN, 0.1% formic acid followed by equilibration to starting conditions for additional 20 min. For all runs, we have injected 1 μL (1 μg of human plasma proteins derived from 14.3 nl of plasma) of tryptic digest directly on column. We have used an Information Independent Acquisition (DIA) workflow with one MS1 full scan (400– 1800 m/z) and n MS/MS fragmentations (700–1250), dependent on the isolation window (5 or 25 Da), with fixed or rolling collision energy. Our analysis targeted previously reported glycoforms of IgG (Table 1)29–31 and some additional glycoforms with compositions we expected to observe.13,30 Precursor masses were further verified with precursor isolation width 0.7 Da. MS/MS mass spectra were recorded in the range 100–2000 m/z with resolution 30 000 and mass accuracy less than 15 ppm (Table T-1) using the following experimental parameters: declustering potential 80 V, curtain gas 30, ion spray voltage 2 300 V, ion source gas 1 11, interface heater 150 °C, entrance potential 10 V, collision exit potential 11 V. Glycopeptide identities were assigned manually and the identified glycopeptides were quantified using peak area of the extracted ion chromatogram (XIC) of the product (Y) ions. Peak integration was performed manually using MultiQuant 2.0 software (Sciex) using two methods with variable window width as follows: full cluster method used window width of 1.2 Da, separated cluster methods used the sum of 3 major isotopes with accuracy 0.1 Da around m/z of the theoretical monoisotopic precursor. DIA quantification was carried out under similar conditions with minor differences in chromatography detailed in the supplement. Internal peptide (GPSVFPLAPSSK) 2+ (m/z = 593.82) derived from IgG1 was used to normalize intensity of the glycoforms.
Table 1.
Previously Reported Glycoforms of IgG Detected in This Studya
| Formula | Structure | IgGl P01857 EEQYN297STYR | IgG2/IgG3 P01859 EEQFN297STFR | IgG4 P01861 EEQFN297STYR | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Precursor | Product | Precursor | Product | Precursor | Product | |||||
| [M+3H]3+ | [M+2H]2+ | S/N | [M+3H]3+ | [M+2H]2+ | S/N | [M+3H]3+ | [M+2H]2+ | S/N | ||
| G0 |
|
830.001 | 1142.958 | 175 | 819.338 | 1126.963 | 19 | 824.669 | 1134.960 | 9 |
| G0N |
|
897.694 | 1244.497 | 108 | 887.031 | 1228.503 | 8 | 892.362 | 1236.500 | 8 |
| G0FN |
|
946.380 | 1317.526 | 1650 | 935.717 | 1301.532 | 525 | 941.048 | 1309.529 | 142 |
| G0F |
|
878.687 | 1215.987 | 5025 | 868.024 | 1199.992 | 1500 | 873.355 | 1207.989 | 225 |
| G1 |
|
884.018 | 1142.958 | 191 | 873.355 | 1126.963 | 113 | 878.687 | 1134.960 | 7 |
| G1N |
|
951.712 | 1244.497 | 52 | 941.048 | 1228.503 | 11 | ND | ND | ND |
| G1FN |
|
1000.398 | 1317.526 | 412 | 989.734 | 1301.532 | 225 | 995.066 | 1309.529 | 68 |
| G1FNS |
|
1097.429 | 1317.526 | 91 | 1086.766 | 1301.532 | 74 | 1092.098 | 1309.529 | 42 |
| G1NS |
|
1048.743 | 1244.497 | 37 | ND | ND | ND | ND | ND | ND |
| G1F |
|
932.704 | 1215.987 | 3225 | 922.041 | 1199.992 | 829 | 927.373 | 1207.989 | 450 |
| G1FS |
|
1029.736 | 1215.987 | 91 | 1019.073 | 1199.992 | 225 | 1024.405 | 1207.989 | 11 |
| G2 |
|
938.036 | 1223.984 | 183 | 927.373 | 1207.989 | 12 | 932.704 | 1215.987 | 8 |
| G2N |
|
1005.729 | 1325.524 | 26 | 995.066 | 1309.529 | 8 | 1000.398 | 1317.526 | 26 |
| G2FN |
|
1054.415 | 1398.553 | 121 | 1043.752 | 1382.558 | 225 | 1049.084 | 1390.555 | 41 |
| G2NS |
|
1102.761 | 1325.524 | 15 | ND | ND | ND | ND | ND | ND |
| G2F |
|
986.722 | 1297.013 | 2475 | 976.059 | 1281.018 | 765 | 981.390 | 1289.016 | 304 |
| G2FS |
|
1083.754 | 1297.013 | 371 | 1073.091 | 1281.018 | 255 | 1078.422 | 1289.016 | 27 |
| G2S |
|
1035.068 | 1223.984 | 52 | 1024.405 | 1207.989 | 30 | 1029.736 | 1215.987 | 9 |
| G2S2 |
|
1132.100 | 1369.532 | 6 | ND | ND | ND | ND | ND | ND |
| Man6 |
|
856.667 | 1392.591* | 12 | 846.003 | 1360.602* | 5 | ND | ND | ND |
| Man8 |
|
964.703 | 1392.591* | 37 | 954.039 | 1360.602* | 8 | 959.371 | 1376.597* | 8 |
| Man9 |
|
1018.720 | 1392.591* | 121 | 1008.057 | 1360.602* | 11 | 1013.389 | 1376.597* | 9 |
| Man9Glc |
|
1072.778 | 1392.591* | 81 | ND | ND | ND | ND | ND | ND |
| G0F-N |
|
810.994 | 1114.447 | 11 | 800.331 | 1098.452 | 6 | 805.662 | 1106.449 | 11 |
| G1F-N |
|
865.0113 | 1114.447 | 19 | 854.348 | 1098.452 | 11 | 859.680 | 1106.450 | 8 |
| G1FS-N |
|
962.0431 | 1114.447 | 7 | 951.380 | 1098.452 | 16 | ND | ND | ND |
Legend: identified glycoforms of IgG with calculated mass of the “soft” fragments. The “soft” fragments are defined by their monoisotopic m/z and charge state 1– compared to the precursor ion; signal-to-noise (S/N) was defined as described in methods.
Indicates high mannose structures where the “soft” fragment represents peptide-GlcNAc (Y1-ion).
Optimization of the Fragmentation Conditions and SWATH Window
Fragmentation conditions were optimized using LC–MS SWATH analysis of the tryptic digest of unfractionated plasma (1 μg on column). Isolation window was set to a 5 Da step with 1 Da overlap. Collision energy (CE) optimization was carried out at a 2 eV step starting at 15 eV. CE was optimized and interval SWATH fragmentation conditions were determined using 100 mDa XIC of selected Y-ions of 10 glycoforms of IgG1. Optimized conditions for fragmentation using rolling CE (CE3+ = 0.03 × 10−3) were used to acquire data with SWATH isolation window set to 5 and 25 Da. Ions for quantification were extracted using a 10 ppm window and the method based on the sum of three isotopes described above.
Data Analysis
Multiquan 2.0 software was used for data processing. Processing methods were created for ion extraction from each SWATH window, average of noise was determined manually from zoomed chromatograms, and integrations of peaks was manually adjusted. Signal to noise (S/N) (Table 1) represents XIC of the “soft” fragment’s monoisotopic mass with 0.1 Da window extracted from the DIA analysis (60 min gradient) of tryptic digests of unfractionated human plasma. The Y-ion isotope cluster with isolation of a window of 1.2 Da, extracted from the SWATH MS/MS with a 5 Da step window, was used for analysis of the glycopeptide intensities (Table T-2). Further data processing and graphing was carried out in Microsoft Excel.
RESULTS AND DISCUSSION
Quantification of glycopeptides is frequently carried out by integration of precursor ion intensities.13,32–35 This is well- suited for quantification of relative intensities of glycoforms in the case of digests of isolated glycoproteins. The analysis of precursor ions eliminates potential influence of structure on the fragmentation of glycopeptides but has inherently lower sensitivity than the quantification of glycopeptide fragments by LC–MS/MS-MRM methods.15 Several groups documented the feasibility to quantify intensity of oxonium B-ions of glycopeptides in the digests of isolated proteins19,25 and, for the abundant glycoforms, even in the background of serum.26,36 The low mass B-ions have high intensity under CID conditions optimized for the fragmentation of glycopeptides, comparable to the intensity of peptide b- and y-ions. However, the B-ions have low specificity which limits their use for quantification of less abundant glycoforms in complex samples.15 Recent studies discuss optimization of CID conditions for improved yield of more selective fragments37,38 with focus on higher yield of peptide b- and y-ions for glycopeptide quantification under high CE fragmentation conditions.39 In this paper, we evaluate selectivity of Y-ions produced under low CE fragmentation conditions for improved quantification of site-specific glycoforms in complex samples.
Soft Fragmentation under Low CE Settings
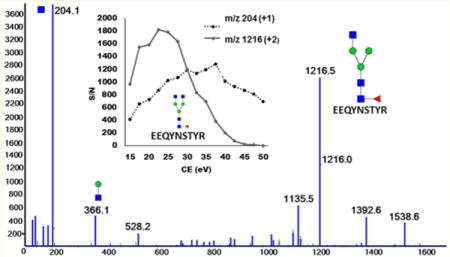
Low CE fragmentation maximizes the yield of Y-ions derived from minimal fragmentation of the glycopeptides (Figure 1); these large fragments have by definition higher specificity than the low mass B-ions previously used by us and others for MRM quantification of glycopeptides.40,41 Figure 1A shows the spectrum of the G2F glycoform of IgG1 obtained under rolling CE conditions optimized for the charge state and precursor mass of the peptide. Under these conditions, intense oxonium B-ions are accompanied by low intensity Y1 fragments with/out fucose. Figure 1B depicts the same G2F precursor (m/z 986.722, 3+ charge) fragmented under rolling CE optimal for recovery of the Y-ion corresponding to the loss of one arm (m/z 1297.013, 2+ charge). Under the soft CID conditions, the loss of one arm accompanied by the loss of one charge is, in fact, a general trend for the fragmentation of all glycoforms of IgG1, IgG2/3, and IgG4 examined in this study (Table 1 and Figure 4). The exception to this rule are the high mannose glycoforms which yield Y1 fragment as the major product; this fragmentation is also accompanied by loss of one charge compared to its precursor. The yield of these ions under the optimized CE conditions is high; the dominant Y-ions represent up to 60% of the precursor ion intensity (range 30–60%, 45% in case of the G2F glycoform of IgG1 in Figure 1).
Figure 1.
Fragmentation spectra of the G2F glycoform of IgG1 obtained under the following conditions: (A) optimal rolling CE based on the charge state and precursor mass of the peptide and (B) optimal rolling CE for maximum recovery of the Y-ion representing loss of one arm. The yield of the “soft” fragment m/z 1297.0 is, in this case, approximately 45% of the precursor ion intensity.
Figure 4.
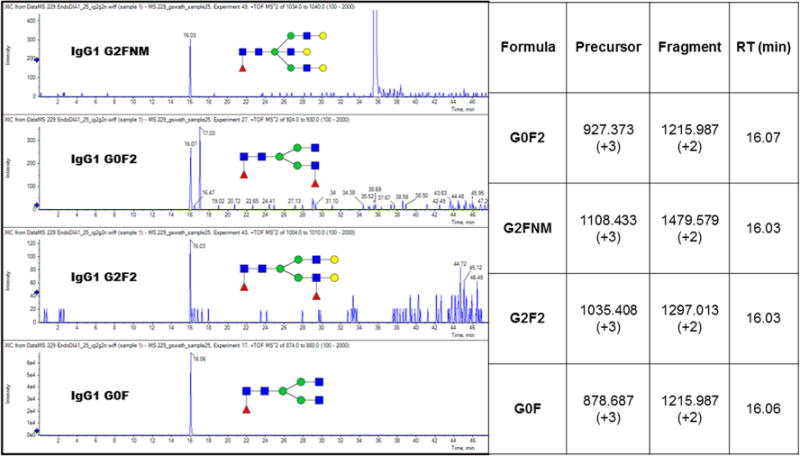
XIC chromatograms of the coeluting minor glycoforms of IgG1, aligned in retention time (RT) to the G0F glycoform, in a SWATH DIA of a tryptic digest of unfractionated plasma of a pool of cirrhotic patients using a 5 Da mass window.
Structure-Dependent Optimization of the Rolling CE
We carried out optimization of the CE for maximal yield of the Y-ion corresponding to the loss of an arm of the N-glycan. To this end, we examined intensity of the Y-ion at CE increasing from 15 to 50 eV in 2.5 eV steps (Figure 2). The fragmentation of four glycoforms of the tryptic glycopeptide of IgG1 (3+ charge state) shows dependence of the optimal rolling CE on precursor mass (G0 ≤ G0F < G2F < G2FS). The relationship is linear (CE3+ = 0.03 × 10−3) and the CE is approximately half of the CE required for the fragmentation of peptides of IgG1 of the same charge (CE3+ = 0.063 × 10−5). The CE optimal for the soft fragmentation of glycopeptides has a relatively narrow range and peptides virtually do not fragment in this CE range; further fragmentation of the N-glycans is also minimized as seen on the low intensity of the B-ions other than the fragment complementary to the dominant Y-ion (e.g., HexNAc-Hex in Figure 1). This minimizes the interferences typically encountered in the MS/MS analysis of glycopeptides and contributes to the high selectivity of the Y-ions generated under the “soft” CID conditions. All experiments were measured in parallel and RSD values were below 15% (Table T-3).
Figure 2.
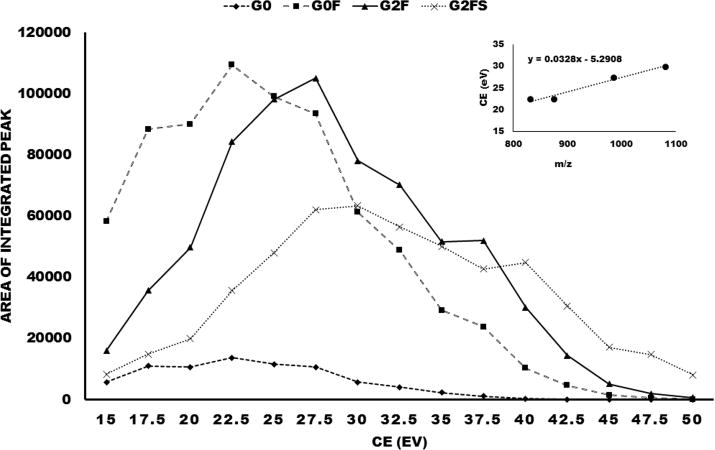
Optimization of CE for fragmentation of the glycoforms G0, G0F, G2F, and G2FS of the tryptic glycopeptide of IgG1 using an isolation window of 5 Da and step of 2.5 eV. Inset: Graph of dependence of the rolling CE (eV) on the m/z (Da) of precursor ion; the derived linear equation is inserted.
High selectivity of the Y-ions allowed us to examine DIA of SWATH data sets of previously described glycoforms of IgG (Table 1).13,30 We compared acquisition under SWATH mass windows of 5 and 25 Da (Figure S-1) and found that both settings have comparable signal-to-noise (S/N). The S/N at 5 Da is for some structures somewhat higher but the difference is at most 50%; this is documented in Figure S-1 on the G0, G0F, G2F, and G2FS glycoforms of IgG1. To further improve specificity of detection, we compared two data analysis methods; the soft CID fragment was quantified as either area of the entire isotopic cluster (C) or as the sum of 0.1 Da XIC (first isotope + second isotope + third isotope) (I) (Figure S-1). We found that addition of the narrow isotope segments increases S/N of the glycoforms in the range of 39% (G2F) to 73% (G0) compared to the integration of the entire cluster.
Figure 3 compares selectivity of detection of the G0F glycoform of IgG1 using the B-ion (204 Da) and Y-ion (1260 Da) under CE optimal for each ion (Figure 4a). In this experiment, we report Y-ions measured as full isotopic cluster CE in the range of 15–50 eV. The results document that the S/N of the Y-ion is higher at the lower CE and overall higher than the S/N of the B-ion at any CE. This is true in general as we show on 11 glycoforms of IgG2 whose B- and Y-ions are compared in Figure 4b. For each glycoform, the S/N of the Y- ion is higher than the S/N of the B-ion at its optimal CE. The optimal CE of the B-ions is in general higher (range 25–40) than the optimal CE for the Y-ions (range 22–27.5) of the same glycoform. In addition, the noise derived from the fragmentation of peptide and glycan bonds is minimized under the soft CID conditions optimal for Y-ions. This allows selective detection of the Y-ions under the soft fragmentation conditions even in the complex background of unfractionated plasma (Figure S-2) as documented above and on the coverage of low-abundance glycoforms described below.
Figure 3.
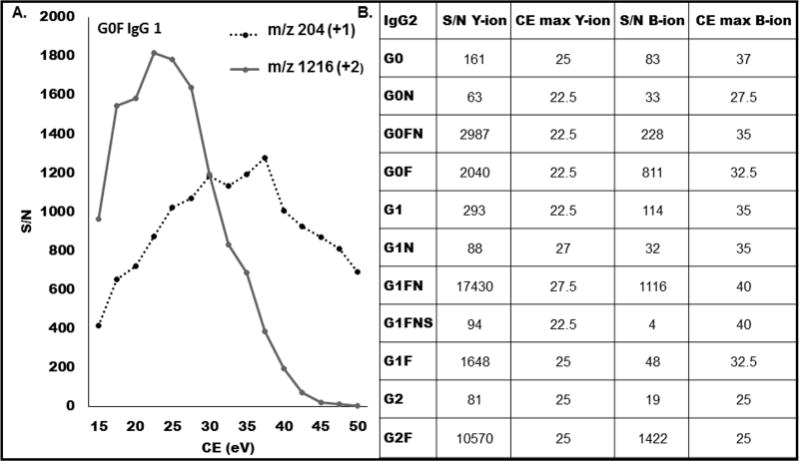
Dependence of the S/N on CE: (A) comparison of the yield of B-ion (204 Da, 1+) with the Y-ion (1216 Da, 2+) of the G0F glycoform of IgG1. The Y-ion was quantified as area of the entire isotopic with SWATH window 5 Da. (B) Table of the S/N of 11 IgG2 glycoforms based on Y- ions or B-ions obtained under CE settings optimal for each fragment.
Coverage and Quantification of IgG Glycoforms by DIA Analysis of Unfractionated Plasma
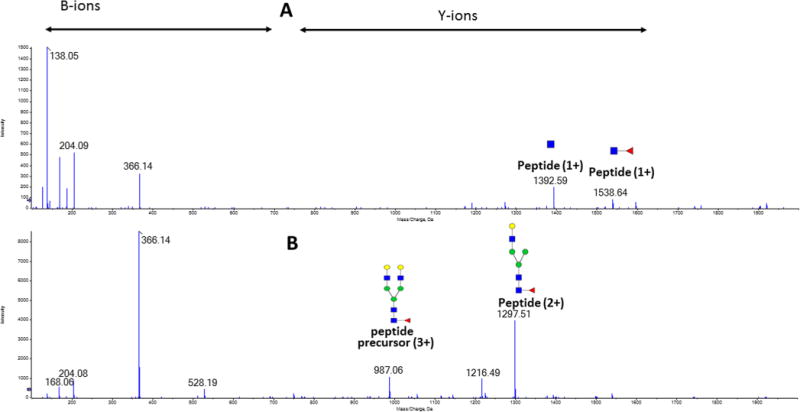
Table 1 summarizes previously described glycoforms of IgG10,19,20 detected by the optimized SWATH (5 Da window) DIA of the IgG1, IgG2/3, and IgG4 subclasses in a pooled sample of unfractionated plasma of healthy volunteers (n = 5). The S/N in this table is based on the integration of the monoisotopic peak of the dominant Y-ion of each glycoform. The coverage includes 26 glycoforms of IgG1, 22 glycoforms of IgG2/3, and 19 glycoforms of IgG4. This is comparable to reported analyses of purified IgG and more complete than previously reported analysis of serum.26 Identification of the glycoforms is based on mass of the precursor and fragment ions (at <15 ppm) (Table T-1) and alignment of retention times of the glycoforms; some of the glycoforms of IgG1 and IgG4 as well as IgG4 and IgG2/3 are isobaric and the use of retention time (RT) for their analysis is essential, as described previously.13 In our analysis it is even more important, because few additional fragments can be used to verify identity of the glycoforms under the soft CID conditions. The RTs of the subclasses differ by at least a minute (RT = 16.1 min for IgG1, RT = 19.4 min for IgG2/3, and RT = 17.1 min for IgG4) (Figure S-3). The alignment of the individual glycoforms of each subclass is excellent and falls within 0.2 min under our experimental conditions (Figures S-3 and S-4). We minimized the in source fragmentation and confirmed on other glycopeptides (e.g., glycopeptides of haptoglobin) that such in source fragments are not observed. We cannot exclude at this point that in source fragmentation contributes to the intensity of some structures but this is not likely; we have observed chromatographic separation of G0F and G0F-N glycoforms which confirms the presence of the G0F-N glycoform in the sample before it can be generated in source (Figure S-4). Spectra of the minor G2FN glycoform (Figure S-3B) show that the S/N of its dominant Y-ion is sufficient for confident identification, together with the RT alignment of the G2FN glycoform with the major G0F glycoform.
The selectivity of the Y-ions measured with high accuracy is, in fact, good enough to suggest presence of previously undescribed IgG glycoforms which align perfectly with the G0F glycoform of IgG1 (Figure 4). These glycoforms have low intensity and cannot be detected by precursor ion mass; only the Y-ion product corresponding to loss of an arm is clearly extracted from the background noise. The identification relies on imputed precursor masses and mass of the observed Y-ion which is clearly not sufficient for resolution of the structural details of these glycoforms. We cannot exclude, for example, that the postulated G2FNHex glycoform of IgG1 is a triantennary glycan (which is isobaric); however, the triantennary glycans are not considered feasible in the case of IgG due to structural constraints10 and the G2FNG glycoform was previously observed.29 This study does not focus on the resolution of isobaric structures and we did not pursue this topic in further detail. However, the presence of the doubly fucosylated glycoform (G2F2) of IgG1 is further supported by the detection of the 512 Da B-ion at the same RT which is consistent with the expected outer arm localization of the second fucose. To confirm our observation, we analyzed the same material with an isolation window 0.7 Da.
Quantitative Comparison of IgG1 Glycoforms
The DIA method is well suited for quantification of the glycopeptides as shown on the comparison of glycoforms of IgG1 in the pooled samples of healthy controls and cirrhotic patients (2 pools, n = 5 patients were analyzed in each group) (Supplemental Table 2). In this analysis, we compared 24 glycoforms of the IgG1 subclass using SWATH DIA setting with a 5 Da window. Our analysis confirms previously reported approximately 2–3-fold increase in the G0F and G0FN glycoforms19 in the cirrhotic patients using LC–MS/MS-MRM analysis of purified IgGs.40 In addition, we achieve better quantitative coverage of less abundant glycoforms even though we use unfractionated plasma without any IgG enrichment. These differences are not due to changes in IgG abundance as we normalize all intensities to the abundance of an IgG1 peptide. The majority of the observed changes are smaller than the well-known change in the agalactosylated structures but further analyses would be needed for any definitive conclusions. At this point we only wanted to document that the method can be used for label free quantitative comparisons of between samples even in the case of less abundant glycoforms.
CONCLUSIONS
We document that soft CID fragmentation conditions result in high yield of Y-ions corresponding to loss of an N-glycan arm (or Y1 ion in the case of high mannose N-glycans of IgG). This minimal fragmentation, carried out at CE lower than typical CE for the fragmentation of peptides, allows selective detection of glycoforms of IgG in complex background. This is to our knowledge the first report of successful analysis of glycoforms of any protein by SWATH DIA in the unfractionated digest of human plasma. DIA with SWATH window of 5 or 25 Da allowed detection of approximately 20 glycoforms of each of the IgG1, IgG2/3, and IgG4 subclasses and quantitative comparison of the IgG1 glycoforms in the context of liver cirrhosis. Selectivity of the SWATH DIA workflow allowed us to detect masses of Y-ions corresponding to the previously undetected doubly fucosylated glycoforms of IgG1 coeluting with other glycoforms of the IgG1 subclass. The results suggest that SWATH DIA under soft CID fragmentation conditions is a viable strategy for label free quantification of glycopeptides in complex samples.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants UO1 CA168926, UO1 CA171146, and RO1 CA135069 (to R.G.) and CCSG Grant P30 CA51008 (to Lombardi Comprehensive Cancer Center supporting the Proteomics and Metabolomics Shared Resource).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b02554.
Supplemental methods (study population; DIA analysis of the glycoforms of IgG1 glycopeptides), Table T-1 (table of mass accuracy of the measurement of IgG glycoforms by DIA), Table T-2 (quantitative DIA analysis of the glycoforms of IgG1), Table T-3 (reproducibility of the measurements of peak area of selected IgG glycoforms by DIA under optimized CE), Figure S-1 (optimization of the signal-to-noise (S/N) based on the isolation window (A) and based on the XIC method (B)), Figure S-2 (MS/MS spectra acquired under peptide based CE and “soft” CE optimal for glycopeptides), Figure S-3 (detection and fragmentation of IgG glycoforms using the DIA method), Figure S-4 (separation of G0F and G0F-N glycopeptides) (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Breitling J, Aebi M. Cold Spring Harbor Perspect Biol. 2013;5:a013359. doi: 10.1101/cshperspect.a013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mazumder R, Morampudi KS, Motwani M, Vasudevan S, Goldman R. PLoS One. 2012;7:e36212. doi: 10.1371/journal.pone.0036212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cummings RD. Mol BioSyst. 2009;5:1087–1104. doi: 10.1039/b907931a. [DOI] [PubMed] [Google Scholar]
- 4.Arnold JN, Saldova R, Hamid UM, Rudd PM. Proteomics. 2008;8:3284–3293. doi: 10.1002/pmic.200800163. [DOI] [PubMed] [Google Scholar]
- 5.Chandler K, Goldman R. Mol Cell Proteomics. 2013;12:836–845. doi: 10.1074/mcp.R112.026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clerc F, Reiding KR, Jansen BC, Kammeijer GS, Bondt A, Wuhrer M. Glycoconjugate J. 2016;33:309. doi: 10.1007/s10719-015-9626-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thaysen-Andersen M, Packer NH. Biochim Biophys Acta, Proteins Proteomics. 2014;1844:1437–1452. doi: 10.1016/j.bbapap.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Canis K, McKinnon TA, Nowak A, Haslam SM, Panico M, Morris HR, Laffan MA, Dell A. Biochem J. 2012;447:217–228. doi: 10.1042/BJ20120810. [DOI] [PubMed] [Google Scholar]
- 9.Reusch D, Tejada ML. Glycobiology. 2015;25:1325–1334. doi: 10.1093/glycob/cwv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. Annu Rev Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- 11.Reusch D, Haberger M, Maier B, Maier M, Kloseck R, Zimmermann B, Hook M, Szabo Z, Tep S, Wegstein J, Alt N, Bulau P, Wuhrer M. MAbs. 2015;7:167–179. doi: 10.4161/19420862.2014.986000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reusch D, Haberger M, Falck D, Peter B, Maier B, Gassner J, Hook M, Wagner K, Bonnington L, Bulau P, Wuhrer M. MAbs. 2015;7:732–742. doi: 10.1080/19420862.2015.1045173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selman MH, Derks RJ, Bondt A, Palmblad M, Schoenmaker B, Koeleman CA, van de Geijn FE, Dolhain RJ, Deelder AM, Wuhrer M. J Proteomics. 2012;75:1318–1329. doi: 10.1016/j.jprot.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Huffman JE, Pucic-Bakovic M, Klaric L, Hennig R, Selman MH, Vuckovic F, Novokmet M, Kristic J, Borowiak M, Muth T, Polasek O, Razdorov G, Gornik O, Plomp R, Theodoratou E, Wright AF, Rudan I, Hayward C, Campbell H, Deelder AM, Reichl U, Aulchenko YS, Rapp E, Wuhrer M, Lauc G. Mol Cell Proteomics. 2014;13:1598–1610. doi: 10.1074/mcp.M113.037465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldman R, Sanda M. Proteomics: Clin Appl. 2015;9:17–32. doi: 10.1002/prca.201400152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adamczyk B, Tharmalingam-Jaikaran T, Schomberg M, Szekrenyes A, Kelly RM, Karlsson NG, Guttman A, Rudd PM. Carbohydr Res. 2014;389:174–185. doi: 10.1016/j.carres.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 17.Mahan AE, Tedesco J, Dionne K, Baruah K, Cheng HD, De Jager PL, Barouch DH, Suscovich T, Ackerman M, Crispin M, Alter G. J Immunol Methods. 2015;417:34–44. doi: 10.1016/j.jim.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein A, Carre Y, Louvet A, Michalski JC, Morelle W. Proteomics: Clin Appl. 2010;4:379–393. doi: 10.1002/prca.200900133. [DOI] [PubMed] [Google Scholar]
- 19.Yuan W, Sanda M, Wu J, Koomen J, Goldman R. J Proteomics. 2015;116:24–33. doi: 10.1016/j.jprot.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selman MH, Hoffmann M, Zauner G, McDonnell LA, Balog CI, Rapp E, Deelder AM, Wuhrer M. Proteomics. 2012;12:1337–1348. doi: 10.1002/pmic.201100498. [DOI] [PubMed] [Google Scholar]
- 21.Zauner G, Selman MH, Bondt A, Rombouts Y, Blank D, Deelder AM, Wuhrer M. Mol Cell Proteomics. 2013;12:856–865. doi: 10.1074/mcp.R112.026005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carr SA, Huddleston MJ, Bean MF. Protein Sci. 1993;2:183–196. doi: 10.1002/pro.5560020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jebanathirajah J, Steen H, Roepstorff P. J Am Soc Mass Spectrom. 2003;14:777–784. doi: 10.1016/S1044-0305(03)00263-0. [DOI] [PubMed] [Google Scholar]
- 24.Domon B, Costello CE. Glycoconjugate J. 1988;5:397–409. [Google Scholar]
- 25.Toyama A, Nakagawa H, Matsuda K, Sato TA, Nakamura Y, Ueda K. Anal Chem. 2012;84:9655–9662. doi: 10.1021/ac3023372. [DOI] [PubMed] [Google Scholar]
- 26.Hong Q, Lebrilla CB, Miyamoto S, Ruhaak LR. Anal Chem. 2013;85:85. doi: 10.1021/ac4009995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benicky J, Sanda M, Pompach P, Wu J, Goldman R. Anal Chem. 2014;86:10716–10723. doi: 10.1021/ac502727s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanda M, Pompach P, Benicky J, Goldman R. Electrophoresis. 2013;34:2342–2349. doi: 10.1002/elps.201200658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harvey DJ, Crispin M, Scanlan C, Singer BB, Lucka L, Chang VT, Radcliffe CM, Thobhani S, Yuen CT, Rudd PM. Rapid Commun Mass Spectrom. 2008;22:1047–1052. doi: 10.1002/rcm.3470. [DOI] [PubMed] [Google Scholar]
- 30.Wang JR, Guan WD, Yau LF, Gao WN, Zhan YQ, Liu L, Yang ZF, Jiang ZH. Sci Rep. 2015;5:7648. doi: 10.1038/srep07648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wuhrer M, Stam JC, van de Geijn FE, Koeleman CA, Verrips CT, Dolhain RJ, Hokke CH, Deelder AM. Proteomics. 2007;7:4070–4081. doi: 10.1002/pmic.200700289. [DOI] [PubMed] [Google Scholar]
- 32.Stadlmann J, Pabst M, Kolarich D, Kunert R, Altmann F. Proteomics. 2008;8:2858–2871. doi: 10.1002/pmic.200700968. [DOI] [PubMed] [Google Scholar]
- 33.Jiang J, Tian F, Cai Y, Qian X, Costello CE, Ying W. Anal Bioanal Chem. 2014;406:6265–6274. doi: 10.1007/s00216-014-8037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurogochi M, Amano J. Molecules. 2014;19:9944–9961. doi: 10.3390/molecules19079944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loke I, Packer NH, Thaysen-Andersen M. Biomolecules. 2015;5:1832–1854. doi: 10.3390/biom5031832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song E, Pyreddy S, Mechref Y. Rapid Commun Mass Spectrom. 2012;26:1941–1954. doi: 10.1002/rcm.6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolli V, Dodds ED. Analyst. 2014;139:2144–2153. doi: 10.1039/c3an02342g. [DOI] [PubMed] [Google Scholar]
- 38.Kolli V, Roth HA, De La Cruz G, Fernando GS, Dodds ED. Anal Chim Acta. 2015;896:85–92. doi: 10.1016/j.aca.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 39.Hinneburg H, Stavenhagen K, Schweiger-Hufnagel U, Pengelley S, Jabs W, Seeberger PH, Silva DV, Wuhrer M, Kolarich D. J Am Soc Mass Spectrom. 2016;27:507–519. doi: 10.1007/s13361-015-1308-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan W, Sanda M, Wu J, Koomen J, Goldman R. J Proteomics. 2015;116:24–33. doi: 10.1016/j.jprot.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldman R, Sanda M. Proteomics: Clin Appl. 2015;9:17–32. doi: 10.1002/prca.201400152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.