

Abstract
Background
Exercise rehabilitation is demonstrated to improve the prognosis of patients with coronary heart disease (CHD). Statins, as the key medicine to lower cholesterol in CHD, result in skeletal muscle injury and impair exercise training adaptation. Energy metabolism dysfunction is identified as the potential mechanism underlying statin‐induced skeletal muscle injury. In this study, we investigated the effects of the metabolic modulator trimetazidine on skeletal muscle energy metabolism and statin‐associated exercise intolerance.
Methods
High‐fat fed apolipoprotein E knockout (ApoE−/−) mice were given aerobic exercise and administrated simvastatin, trimetazidine, or simvastatin plus trimetazidine by gavage. Exercise capacity was evaluated at the end of the treatment by hanging grid test, forelimb grip strength, and running tolerance test. Plasma glucose, lipid, and creatine kinase concentrations were measured at the end of the treatment. After sacrifice, gastrocnemii were stored for assessment of muscle morphology and fibre type. Energy metabolism was estimated by plasma lactic acid concentration, ragged red fibres, and glycogen stores. Activities of mitochondrial complex III, citrate synthase activity, and membrane potential were measured to assess mitochondrial function. Oxidative stress was also evaluated by superoxide in mitochondria, superoxide dismutase activity, and glutathione redox state.
Results
In high‐fat fed ApoE−/− mice, exercise training had no effect on lipid concentrations. Lower lipid concentrations with increased creatine kinase were observed with additional simvastatin treatment. Exercise capacity increased significantly in response to exercise training alone but was blunted by the addition of simvastatin. Similarly, cross‐sectional area of muscle fibres and the proportion of slow‐twitch fibres increased in the exercise group but decreased in the simvastatin plus exercise group. Additionally, simvastatin increased centronucleated fibres and induced energy metabolism dysfunction by inhibiting complex III activity and thus promoted oxidative stress in gastrocnemius. We demonstrated that trimetazidine could reverse simvastatin‐induced exercise intolerance and muscle damages. We also found the ability of trimetazidine in restoration of muscle fibre hypertrophy and facilitating fast‐to‐slow type shift. The energy metabolism dysfunction and oxidative stress in gastrocnemii were rescued by trimetazidine.
Conclusions
Trimetazidine alleviated statin‐related skeletal muscle injury by restoration of oxidative phenotype and increasing fibre cross‐sectional areas in response to exercise training. Correspondingly, the exercise training adaptation were improved in high‐fat fed ApoE−/− mice. Moreover, trimetazidine is able to exert its positive effects without affecting the beneficial lipid‐lowering properties of the statins. Thus, trimetazidine could be prescribed to remedy the undesirable statins‐induced exercise intolerance during cardiac rehabilitation in patients with CHD.
Keywords: Simvastatin, Trimetazidine, Exercise, Skeletal muscle, Metabolism modulation
Introduction
Exercise rehabilitation, as the important component of heart rehabilitation,1 has been confirmed to improve parameters of cardiorespiratory fitness and exercise capacity.2 Meta‐analysis showed that exercise rehabilitation reduced hospital admission and improved the quality of life.3 Statins, as the foundation of the primary and secondary prevention of coronary heart disease (CHD), could nevertheless impair exercise training adaptation.4 Epidemiologic studies indicated that 7–29% of patients complained of statin‐associated muscle symptoms including myalgia, fatigued, and spasm.5 It has been observed that 38% of statin‐treated patients with myalgia avoided even moderate physical exertion during daily life.6
Energy metabolism abnormality was proved to be the key mechanism underlying statin‐induced myopathies. Statins appeared to be strong inhibitors of mitochondrial complex III7 and induced mitochondrial dysfunction that lead to cytoplasm Ca2+ overload8 and mitochondrial DNA depletion.9 In skeletal muscle cells, statins reduced respiratory capacity. The statin‐associated energy metabolism disorders induced muscle fibre atrophy and resulted in decreased skeletal muscle function.10 Hence, procedures that ameliorate energy metabolism‐related myotoxicity by statins could improve exercise capacity.
In 1969, the Servier Laboratories synthesized trimetazidine, 1‐(2,3,4‐trimethoxybenzyl)‐piperazine, as an energy modulator for angina pectoris. It inhibits activity of 3‐ketoacyl‐CoA thiolase (ATP), thereby increasing glucose utilization for adenosine triphosphate production.11 Moreover, trimetazidine was shown to preserve mitochondrial function12 and improved skeletal muscle performance.13 Trimetazidine protected muscle cells from starvation or inflammation‐induced atrophy.14 In patients with stable CHD, additional beneficial antianginal effects and improvement in exercise capacity were observed with a combination of trimetazidine and traditional pharmacotherapy.15 Trimetazidine increased the exercise tolerance of exercise training patients with ischaemic cardiomyopathy.16 However, studies examining the benefits of trimetazidine in statin‐induced exercise intolerance are limited.
In this study, the statin‐induced skeletal muscle injury apolipoprotein E knockout (ApoE−/−) mice model was constructed. And we elucidated whether trimetazidine could improve statin‐related exercise intolerance via skeletal muscle energy metabolism modulation.
Materials and methods
Animals
ApoE−/− mice show marked elevation in cholesterol and also fatty streaks, and the addition of a high‐fat diet presumably would exacerbate the phenotype substantially; thus, it would appear to be a model with a severe phenotype and was chosen in this experiment. Four‐week‐old male ApoE−/− (C57BL/6J background) mice were purchased from Beijing HFK Bio‐Technology. The mice were fed a high‐fat diet (40% calories from fat, 15% animal fat, 5% glucose, 0.25% cholate, 0.25% sodium, 1% cholesterol, and 78.5% basic forage). The mice were housed at 22°C and maintained on a 12/12 h light–dark cycle. After 4 weeks, the mice were randomized into five groups: sedentary, exercise, exercise + simvastatin (SMV), exercise + trimetazidine (TMZ), and exercise + SMV + TMZ. The mice in the exercise, exercise + SMV, exercise + TMZ, and exercise + SMV + TMZ groups were trained for 8 weeks at a speed of 13 m/min, 30 min/day, 5 days a week using a mouse treadmill (Zhishuduobao DB030, China). During the first week of exercise regimen, the training speed started at 8 m/min and increased 1 m/min each day until 13 m/min and the training time increased from 15 min to 30 min. In the remainder of the protocol (weeks 2–8), mice exercised for 30 min at 13 m/min. No warm‐up was provided before the training session. Mice in exercise + SMV group received gavage of 20 mg/kg/day of simvastatin for 8 weeks. Mice in exercise + TMZ group received gavage of 30 mg/kg/day of trimetazidine for 8 weeks. Mice in exercise + SMV + TMZ group received gavage of 20 mg/kg/day of simvastatin and 30 mg/kg/day of trimetazidine for 8 weeks. All experiments were approved by ethics boards of Qilu Hospital of Shandong University.
Hanging grid test
The hanging grid test was performed at the end of experiment. Mice were individually placed at the centre of a wire mesh screen (2 mm wire thickness). The screen was held 50 cm above a pad. The grid was inverted upside down with the head declining first. The duration of hanging was recorded in three independent trials conducted at least 20 min apart. The data of all three trials were averaged.
Forelimb grip strength test
At the end of experiment, a forelimb grip strength test was performed using a grip strength metre (Handpi HP‐5N, China). Mice were held by the tail and grasped a grid with fore paws. The mice were gently pulled by the tail until they released their grip. The forces from three trials were recorded and averaged.
Running tolerance test
Running tolerance test was performed at the end of experiment. The running speed was started at 13 m/min and increased 2 m/min every 3 min. The slop of track was started at 0°, increased 2° every 3 min, and maintained at 14° because of limit of the treadmill. No warm‐up was provided before the assessment of running tolerance. The time and distance were recorded when the mice were exhausted.
Tissue processing
Mice were weighed and then anaesthetized with amobarbital (80 mg/kg) via intraperitoneal injection. Once a surgical plane of anaesthesia was reached, a thoracotomy was performed and blood was obtained via cardiac puncture into heparinized tubes. Plasma was separated and stored at −80°C until required. Trans‐cardiac perfusion with normal saline was performed. Both gastrocnemii of each mouse were excited.
Haematoxylin–eosin stain
Gastrocnemius was fixed in 4% paraformaldehyde for 24 h. Subsequently, the fixed muscles were dehydrated through an ethanol series, treated with xylenes, embedded in paraffin sections, and cut into 5 μm sections. Dewaxing was performed through xylene and ethanol series to deionized water. Stain the nuclei with haematoxylin for 3 min and differentiate with acid ethanol for 30 s. Rinse in the running water until blue up. Then the slides were stained with eosin for 2 min. Dehydrate the slides through ethanol, clear in xylene, and mount. The imaging was performed using Olympus DP72 digital imaging system (Olympus Corporation, Tokyo, Japan). Cross‐sectional area (CSA) of muscle fibres was measured using Image Pro Plus. The analysis of CSA was conducted by single investigator blinded to sample.
Modified Gomori stain
After dewaxing, the slides were stained with haematoxylin for 5 min and washed in running water for 10 min. Slides were then incubated in Gomori solution for 30 min and washed in 0.2% acetic acid for 1 min. Dehydrate the slides through ethanol, clear in xylene, and mount. Fibres with accumulation of abnormal mitochondria were classified as ragged red fibres (RRF), considered a marker of mitochondrial dysfunction. Quantification was performed by measuring the ratio of RRF to total muscle fibres using Image Pro Plus. The analysis of images was conducted by single investigator blinded to sample.
Periodic acid–Schiff stain
After dewaxing, the slides were incubated in 0.5% periodic acid for 8 min and washed three times for 5 min in distilled water. Slides were then incubated in Schiff's reagent for 20 min and washed in running water for 10 min. Then stain the nuclei with haematoxylin for 1 min and differentiate with acid ethanol for 10 s. Finally, dehydrate the slides through ethanol, clear in xylene and mount. The decline of periodic acid–Schiff staining was associated with glycogen degradation. The mean values for periodic acid‐Schiff staining density was calculated using Image Pro Plus. The analysis of images was conducted by single investigator blinded to sample.
Immunofluorescence
Slides were dewaxed through xylene and ethanol series to deionized water. Antigen was repaired with citric acid buffer in microwave for 20 min (fast myosin heavy chain) or proteinase K in 37°C for 30 min (slow myosin heavy chain and laminin). Non‐specific binding was blocked with 5% bovine serum albumin (BSA). The slides were incubated with primary antibody of fast myosin heavy chain (fast MyHC) (Abcam ab51263, UK), slow myosin heavy chain (slow MyHC) (Abcam ab11083, UK), or laminin (Abcam ab11575, UK) at 4°C overnight. The slides were washed three times in phosphate‐buffered saline (PBS) for 5 min and incubated with secondary antibody of anti‐mouse fluor594 or anti‐rabbit fluor488 for 30 min. Slow and fast fibres both stained red. The nuclei were stained with 40,6‐diamidino‐2‐phenylindole (DAPI). DAPI‐labelled nuclei were defined as sky blue inclusions 2 μm in diameter, approximately. Quantification of slow or fast MyHC positive fibres was performed by measuring the ratio to total muscle fibres. Quantification of central nuclei fibres was performed by measuring the ratio to total muscle fibres. The quantification was performed using Image Pro Plus. The analysis of images was conducted by single investigator blinded to sample.
Plasma testing
Plasma was analysed for glucose (Glu), total cholesterol (TC), low‐density lipoprotein cholesterol (LDL‐C), high‐density lipoprotein cholesterol, triglyceride, non‐esterified fatty acid using the Bayer 1650 blood chemistry analyser (Bayer AG, Leverkusen, Germany). Plasma was analysed for creatine kinase (CK) and lactic acid using the colorimetry kit (Nanjing Jiancheng Biological Engineering Institution, China), according to the manufacturer's protocol.
Mitochondrial assay
After excised, gastrocnemii were immediately snap‐frozen in liquid nitrogen and stored at −80°C. Mitochondria of gastrocnemius were isolated with the Mitochondria Isolation Kit for Tissue (Abcam ab110168, UK) according to the manufacturer's instruction. Briefly, gastrocnemius was washed with washing buffer, homogenized in isolation buffer, and centrifuged at 1000 g for 10 min. The supernatant was centrifuged again at 12 000 g for 15 min. Pellets (mitochondrial fraction) were washed with isolation buffer with protease inhibitor cocktail (Abcam ab201111, UK) twice and resuspended with isolation buffer with protease inhibitor cocktail. Mitochondria were quantified by bicinchoninic acid (BCA) assay. Complex III activity was determined by measuring the reduction of cytochrome c at 550 nm with complex III Activity Assay Kit (Solarbio bc 3240, China) according to the manufacturer's instruction. Citrate synthase (CS) activity was determined by measuring coenzyme A formation at 412 nm with Citrate Synthase Activity Assay Kit (Solarbio bc 1060, China) according to the manufacturer's instruction. Mitochondrial membrane potential was determined by investigating fluorescence of 5,5′,6,6′‐tetrachlore‐1,1′,3,3′‐tetraethylbenziml‐dazolylcarbocyanine iodide (JC‐1) (Solarbio J8030, China) using a microplate fluorometer at an excitation/emission wavelength of 485/590 nm. Mitochondrial hydrogen peroxide level was measured using Amplex Red–horseradish peroxidase (Abcam, UK). Fluoroscence was measured using a microplate fluorometer at an excitation/emission wavelength of 535/587 nm.
Superoxide dismutase activity assay
Frozen muscle samples were homogenized in cold saline and centrifuged at 12 000 g for 5 min. The supernatant was analysed for total superoxide dismutase (SOD) activity by xanthine oxidase method with SOD Assay Kit (KeyGEN BioTECH KGT001100, China) according to manufacturer's instruction.
Glutathione redox state
Glutathione redox state was measured by total and oxidized glutathione (GSH and GSSG) respectively using a commercially available kit (Solarbio BC1170 and BC1185, China).
Data analysis
Data are presented as mean ± SEM. Statistical significance was determined using one‐way analysis of variance followed by Tukey's multiple comparison test. Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, California, America). Figures 1, 2, 3 compare sedentary, exercise, and exercise + simvastatin to verify that simvastatin treatment impairs adaptation to exercise training in high‐fat fed ApoE−/− mice. Figures 4, 5, 6 compare exercise, exercise + simvastatin, exercise + trimetazidine, and exercise + simvastatin + trimetazidine to verify that trimetazidine mitigates the effect of simvastatin and restores the positive adaptation to exercise training.
Figure 1.
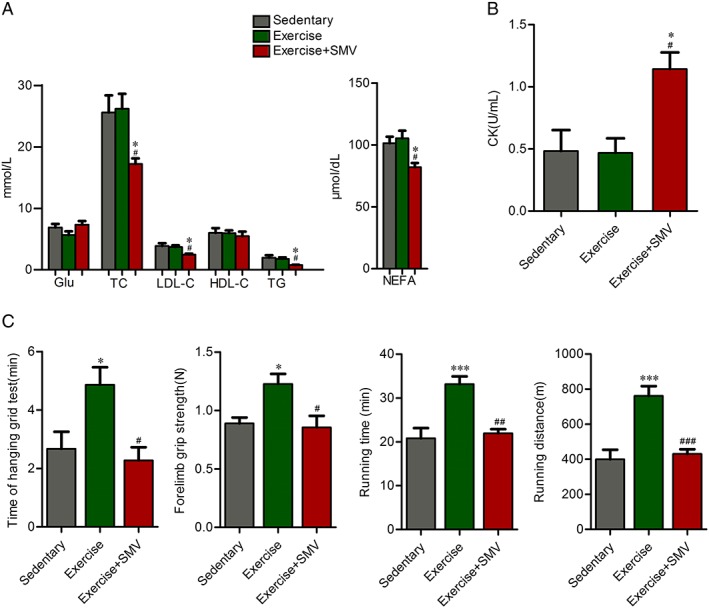
Simvastatin lowered plasma lipid and attenuated increases in exercise capacity in response to exercise in ApoE−/− mice. ApoE−/− mice were assigned to three groups: sedentary, exercise, and exercise + simvastatin (SMV). (A) Plasma glucose and lipid concentrations were measured before sacrifice. (B) Plasma creatine kinase (CK) concentrations were measured before sacrifice. (C) Exercise capacity tests were performed at the end of experiment. An inverted grip‐hanging test was performed and time to fall off was recorded. Forelimb grip strength was measured by using a grip strength metre. Running distance and running time were measured by a treadmill. N = 6. * P < 0.05 and *** P < 0.001 vs. sedentary; # P < 0.05, ## P < 0.01 and ### P < 0.001 vs. exercise. Glu, glucose; TC, total cholesterol; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; NEFA, non‐esterified fatty acid; TG, triglyceride.
Figure 2.
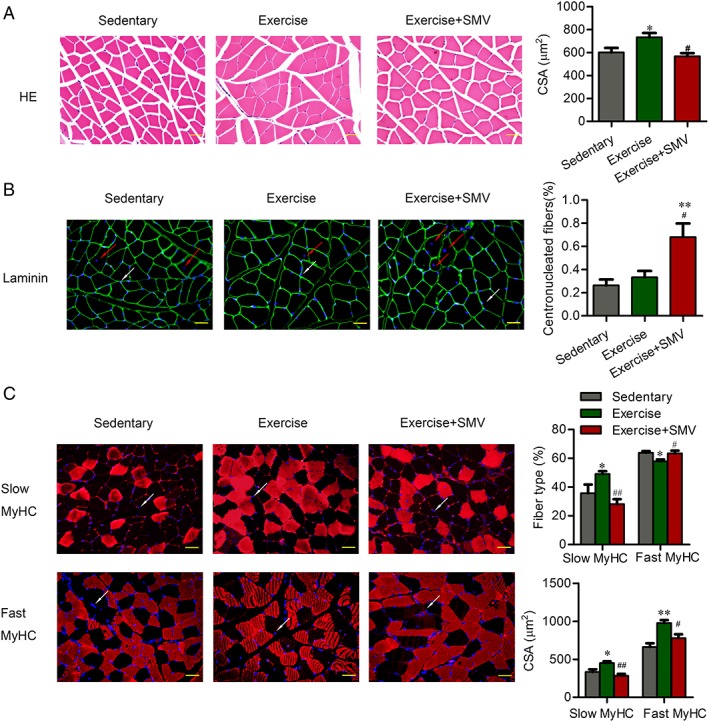
Simvastatin induced muscle damage and fibre type shift in ApoE−/− mice. ApoE−/− mice were assigned to three groups: sedentary, exercise, and exercise + simvastatin (SMV). (A) The morphologic changes of gastrocnemius were presented by haematoxylin and eosin (HE) staining. Histogram shows the cross‐sectional areas (CSA) of muscle fibres. (B) Gastrocnemius sections were stained with antibody of laminin and 40, 6‐diamidino‐2‐phenylindole (DAPI). The red arrows point to the centronucleated fibres. The white arrows point to the examples of DAPI‐labelled nuclei. Histogram shows the percentage of centronucleated fibres with respect to the total number of fibres. (C) Gastrocnemius sections were stained with antibody of slow or fast myosin heavy chain [slow myosin heavy chain (MyHC) or fast MyHC]. DAPI was used to detect nuclei. The white arrows point to the examples of DAPI‐labelled nuclei. Histograms show the percentage of fibre types with respect to the total number of fibres and the CSA of each type fibres. N = 6. * P < 0.05 and ** P < 0.01 vs. sedentary; # P < 0.05 and ## P < 0.01 vs. exercise. Scale bar: 20 μm.
Figure 3.
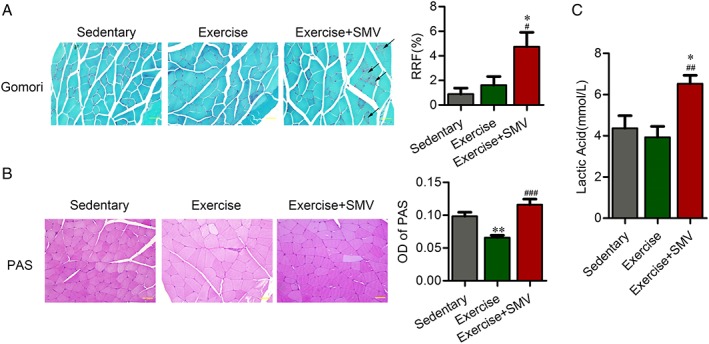
Simvastatin induced increased ragged red fibres and decreased glycogen consumption in ApoE−/− mice. ApoE−/− mice were assigned to three groups: sedentary, exercise, and exercise + simvastatin (SMV). (A) The change of ragged red fibres (RRF) was presented by Gomori staining. Histogram shows the percentage of RRF with respect to the total number of fibres. The black arrows indicate examples of RRF. (B) The content of glycogen was presented by periodic acid–Schiff (PAS) staining. Histogram shows the optical density (OD) of PAS staining. (C) Plasma concentrations of lactic acid were measured before sacrifice. N = 6. * P < 0.05 and ** P < 0.01 vs. sedentary; # P < 0.05, ## P < 0.01, and ### P < 0.001 vs. exercise. Scale bar: 20 μm.
Figure 4.
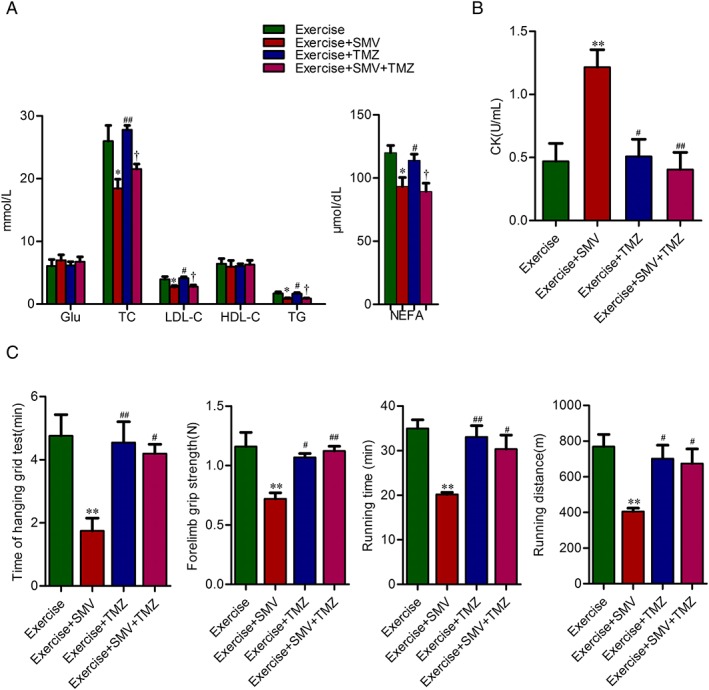
Trimetazidine improved the exercise intolerance induced by simvastatin in ApoE−/− mice. ApoE−/− mice were assigned to four groups: exercise, exercise + simvastatin (SMV), exercise + trimetazidine (TMZ), and exercise + SMV + TMZ. (A) Plasma concentrations of glucose and lipid. (B) Plasma concentrations of creatine kinase (CK). (C) Time of hanging grid test, forelimb grip strength, running time, and running distance are presented. N = 6. * P < 0.05 and ** P < 0.01 vs. exercise; # P < 0.05 and ## P < 0.01 vs. exercise + SMV; † P < 0.05 vs. exercise + TMZ. NEFA, non‐esterified fatty acid.
Figure 5.
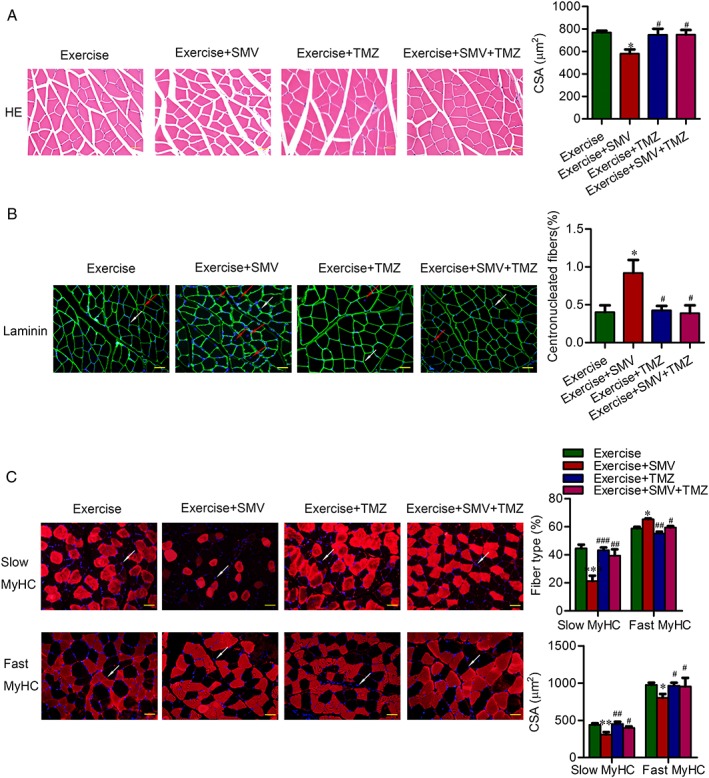
Trimetazidine protected muscle from atrophy and fibre type shift induced by simvastatin in ApoE−/− mice. ApoE−/− mice were assigned to four groups: exercise, exercise + simvastatin (SMV), exercise + trimetazidine (TMZ), and exercise + SMV + TMZ. Representative images of gastrocnemius sections are shown. (A) Representative images of haematoxylin and eosin (HE) staining and histogram of muscle fibres' cross‐sectional areas (CSA). (B) Representative images of immunofluorescent images for laminin and histograms of percentage for centronucleated fibres. The red arrows point to the centronucleated fibres. The white arrows point to the examples of 40, 6‐diamidino‐2‐phenylindole (DAPI)‐labelled nuclei. (C) Representative images of immunofluorescent images for slow or fast myosin heavy chain [slow myosin heavy chain (MyHC) or fast MyHC]. The white arrows point to the examples of DAPI‐labelled nuclei. Histograms show the percentage of fibre types and the CSA of each type fibres. N = 6. * P < 0.05, ** P < 0.01 and *** P < 0.001 vs. exercise; # P < 0.05, ## P < 0.01, and ### P < 0.001 vs. exercise + SMV. Scale bar: 20 μm.
Figure 6.
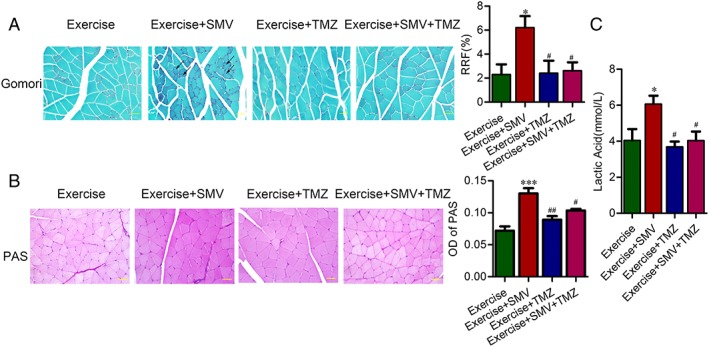
Trimetazidine counteracted ragged red fibres and glycogen storage induced by simvastatin in ApoE−/− mice. ApoE−/− mice were assigned to four groups: exercise, exercise + simvastatin (SMV), exercise + trimetazidine (TMZ), and exercise + SMV + TMZ. Representative images of gastrocnemius sections are shown. (A) Representative images of Gomori staining and histogram of percentage for ragged red fibres (RRF). The black arrows indicate examples of RRF. (B) Representative images of PAS staining and histogram of optical density (OD). (C) Plasma concentration of lactic acid. N = 6. * P < 0.05 and *** P < 0.001 vs. exercise; # P < 0.05 and ## P < 0.01 vs. exercise + SMV. Scale bar: 20 μm.
Results
Simvastatin lowered plasma lipid and attenuated increase in exercise capacity in response to exercise in ApoE−/− mice
Fasting plasma glucose and lipid concentrations were compared between groups (Figure 1A). The glucose concentration was moderately lower in exercise group compared with sedentary group and slightly increased in simvastatin group. But these differences did not achieve statistical significance. There were not any differences in plasma concentrations of lipid between sedentary and exercise groups, which might be related to the particular high‐fat fed ApoE−/− mouse model. Simvastatin reduced concentrations of TC (26.23 ± 2.41 mmol/L vs. 17.24 ± 0.91 mmol/L), LDL‐C (3.70 ± 0.26 mmol/L vs. 2.47 ± 0.14 mmol/L), triglyceride (1.78 ± 0.23 mmol/L vs. 0.76 ± 0.06 mmol/L), and non‐esterified fatty acid (105.30 ± 6.17 μmol/dL vs. 82.00 ± 3.38 μmol/dL) (Figure 1A). Besides hypolipidaemic effect, simvastatin is also linked to elevated CK. The elevated CK levels are suggestive of muscle injury because CK is released into blood from damaged muscle and therefore is a marker of muscle damage. Compared with sedentary or exercise group, the plasma CK level was increased in simvastatin‐treated mice (sedentary vs. exercise + SMV, 0.48 ± 0.17 U/mL vs. 1.14 ± 0.13 U/mL; exercise vs. exercise + SMV, 0.47 ± 0.12 U/mL vs. 1.14 ± 0.13 U/mL) (Figure 1B).
Then we assessed the exercise capacity of ApoE−/− mice (Figure 1C). As expected, we observed that exercise induced an increase in time of hanging grid test (2.67 ± 0.58 min vs. 4.87 ± 0.60 min) and forearm grip strength (0.89 ± 0.05 N vs. 1.23 ± 0.09 N). Compared with exercise group, time of hanging grid test (4.87 ± 0.60 min vs. 2.28 ± 0.45 min) and forearm grip strength (1.23 ± 0.09 N vs. 0.85 ± 0.10 N) were decreased in exercise plus simvastatin group. In addition, exercise triggered a significant improvement in running tolerance (running time, 22.80 ± 2.36 min vs. 33.15 ± 1.79 min; running distance, 399.50 ± 54.43 m vs. 761.10 ± 56.13 m), whereas simvastatin counteracted the effect of exercise on running tolerance (running time, 33.15 ± 1.79 min vs. 21.94 ± 0.97 min; running distance, 761.10 ± 56.13 m vs. 430.30 ± 26.54 m).
Simvastatin induced muscle damage and counteracted the change in myosin heavy chain stimulated by exercise in ApoE−/− mice
Haematoxylin and eosin staining of skeletal muscles revealed that exercise increased fibres cross‐sectional area (601.90 ± 38.15 μm2 vs. 733.70 ± 37.06 μm2). Simvastatin abrogated exercise‐induced fibre hypertrophy (733.70 ± 37.06 μm2 vs. 567.90 ± 27.10 μm2) (Figure 2A). An increased number of fibres containing centrally located nuclei suggested that simvastatin induced the degeneration–regeneration processes in muscle (sedentary vs. exercise + SMV, 0.26% ± 0.05% vs. 0.68% ± 0.12%; exercise vs. exercise + SMV, 0.33% ± 0.05% vs. 0.68% ± 0.12%) (Figure 2B).
Muscle function is closely related to fibre type shifts. Using myosin heavy chain (MHC) immunofluorescence staining, we found that exercise had up‐regulated slow‐twitch fibres (35.78% ± 5.96% vs. 49.07% ± 1.97%). Slow‐twitch fibres significantly decreased upon simvastatin treatment (49.07% ± 1.97% vs. 28.16% ± 3.44%), whereas fast‐twitch fibres increased (57.95% ± 1.24% vs. 63.37% ± 2.00%). Consistent with improvement of force generating capacity and exercise endurance, exercise ApoE−/− mice showed an increase in size of both slow (334.30 ± 34.08 μm2 vs. 449.90 ± 22.87 μm2) and fast‐twitch (661.70 ± 48.95 μm2 vs. 977.40 ± 38.44 μm2) muscle fibres. Simvastatin administration blunted the exercise‐induced enlargement of slow (449.90 ± 22.87 μm2 vs. 284.20 ± 23.53 μm2) and fast (977.40 ± 38.44 μm2 vs. 780.20 ± 49.96 μm2) fibre CSA (Figure 2C).
Simvastatin induced energy metabolism dysfunction of skeletal muscle
The RRF was significantly increased upon simvastatin treatment (sedentary vs. exercise + SMV, 0.89% ± 0.49% vs. 4.74% ± 1.16%; exercise vs. exercise + SMV, 1.62% ± 0.70% vs. 4.74% ± 1.16%), which suggested that simvastatin disturbed mitochondrial function (Figure 3A). Glycogen content, as an important substrate of energy metabolism in skeletal muscle, decreased in exercised ApoE−/− mice. Simvastatin increased muscular glycogen in exercise ApoE−/− mice (Figure 3B). Simvastatin also caused the elevation of the plasma lactic acid in exercise ApoE−/− mice (3.93 ± 0.53 mmol/L vs. 6.53 ± 0.41 mmol/L), which suggested the impaired aerobic metabolism of skeletal muscle (Figure 3C).
Trimetazidine counteracted simvastatin‐induced exercise intolerance
The serum concentration of glucose did not change upon trimetazidine treatment. Trimetazidine neither affected plasma lipid level nor impacted the lipid‐lowering effect of simvastatin (Figure 4A). Whereas trimetazidine treatment restored the plasma CK elevation induced by simvastatin (1.21 ± 0.14 U/mL vs. 0.40 ± 0.14 U/mL) (Figure 4B). Although trimetazidine did not increase the exercise capacity of exercise ApoE−/− mice, it protected exercise ApoE−/− mice from the decline of grid‐hanging time (1.75 ± 0.41 min vs. 4.20 ± 0.29 min), forelimb grip (0.72 ± 0.05 N vs. 1.12 ± 0.04 N) and running tolerance induced by simvastatin (running time, 20.16 ± 0.47 min vs. 30.34 ± 3.15 min; running distance, 404.60 ± 19.79 m vs. 673.60 ± 82.63 m) (Figure 4C).
Trimetazidine removed the inhibitory effect of simvastatin on exercise‐induced muscle hypertrophy and fibre type shift in ApoE−/− mice
To confirm a direct and protective effect of trimetazidine on muscle, we observed the change of histomorphology. Simvastatin blunted the hypertrophic response to exercise training. And trimetazidine was effective at removing the inhibitory effect of simvastatin on exercise‐induced muscle growth (580.50 ± 37.15 μm2 vs. 748.80 ± 41.56 μm2) (Figure 5A). Trimetazidine alleviated simvastatin‐induced elevation of central nuclei fibres (0.92% ± 0.17% vs. 0.39% ± 0.10%) (Figure 5B). Compared with simvastatin treatment alone, simvastatin plus trimetazidine treatment induced an increase of slow‐twitch muscle fibres (21.18% ± 3.90% vs. 39.42% ± 4.53%). Consistent with the increased exercise performance, trimetazidine restored the responsiveness of slow (309.50 ± 31.75 μm2 vs. 398.6 ± 19.72 μm2) and fast (817.40 ± 39.47 μm2 vs. 995.40 ± 87.65 μm2) fibres to the growth promoting effect of exercise (Figure 5C).
Trimetazidine improved simvastatin‐induced energy metabolism dysfunction in skeletal muscle
We investigated whether trimetazidine protected muscle by modulation of energy metabolism. Trimetazidine treatment decreased simvastatin‐induced RRF in exercise ApoE−/− mice (6.21% ± 0.96% vs. 2.62% ± 0.69%) (Figure 6A). We also observed decreased muscle glycogen content upon trimetazidine treatment in exercise ApoE−/− mice with simvastatin gavage (Figure 6B). Compared with simvastatin treatment alone, simvastatin and trimetazidine treatment reduced the plasma lactic acid level of exercise ApoE−/− mice (6.06 ± 0.46 mmol/L vs. 4.03 ± 0.50 mmol/L) (Figure 6C).
Simvastatin acted on mitochondrial complex III to exert the inhibitory effects on mitochondrial function and promoted oxidative stress, which were reversed by trimetazidine
Direct testing of the effects on the individual enzymatic activities of all five mitochondrial complexes in vitro revealed significant inhibition by simvastatin only for complex III.7 Thus, we determined the complex III activity of mitochondria from gastrocnemius. Exercise increased complex III activity. Complex III activity of simvastatin‐treated mice decreased by 75% compared with mice in exercise group (P < 0.05) (Figure 7A). We tested whether the complex III inhibition affected membrane potential. The membrane potential increased in exercised ApoE−/− mice. Compared with mice in exercise group, the membrane potential decreased after simvastatin treatment (P < 0.05) (Figure 7B). Trimetazidine restored the decrease of complex III activity induced by simvastatin (Figure 7A) and then improved mitochondrial membrane potential (Figure 7B). To obtain an index of mitochondrial mass, we measured the biochemical activity of mitochondrial matrix enzyme CS. Exercise induced a 35% increase of CS activity (P < 0.05). CS activity of simvastatin‐treated mice decreased by 50% compared with mice in exercise group (P < 0.01). And trimetazidine ameliorated simvastatin‐induced decrease of mitochondrial mass (Figure 7C).
Figure 7.
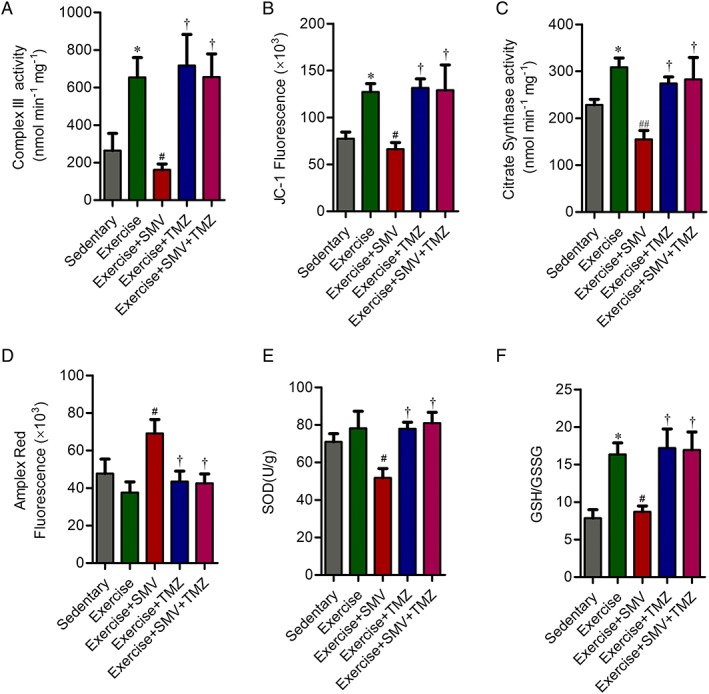
Trimetazidine reversed statin‐induced mitochondrial dysfunction via modulation of mitochondrial complex III activity and oxidative stress. (A) Enzymatic activity of the complex III in mitochondria isolated from gastrocnemius. The activity of complex III is expressed in nmol reduced cytochrome c per mg mitochondrial protein. (B) Fluorescence of JC‐1 for mitochondria isolated from gastrocnemius. (C) Enzymatic activity of the citrate synthase (CS) in mitochondria isolated from gastrocnemius. The activity of CS is expressed in nmol 2‐nitro‐5‐thiobenzoate (TNB) per mg mitochondrial protein. (D) Fluorescence of Amplex Red oxidation for mitochondria isolated from gastrocnemius. (E) The activity of superoxide dismutase (SOD) in gastrocnemius. (F) The glutathione redox state (GSH/GSSG) in gastrocnemius. N = 6. * P < 0.05 vs. sedentary; # P < 0.05 and ## P < 0.01 vs. exercise; † P < 0.05 vs. exercise + SMV.
Amplex Red oxidation revealed that mitochondrial hydrogen peroxide production was elevated with simvastatin treatment. Mitochondrial hydrogen peroxide was lower in simvastatin plus trimetazidine treated mice when compared with mice treated with simvastatin alone (Figure 7D). Correspondingly, SOD activity and GSH/GSSG in skeletal muscle of simvastatin‐treated mice were significantly decreased compared with the trained mice. The activity of SOD and GSH/GSSG were increased in the gastrocnemius from simvastatin plus trimetazidine treated mice when compared with mice treated with simvastatin alone (Figure 7E and 7F). These data were allowed to conclude that trimetazidine could reverse simvastatin‐induced alterations in mitochondrial function, likely because of its actions on the improvements of complex III activity, mitochondrial mass, and corresponding inhibition of oxidative stress.
Discussion
The present study showed that simvastatin abated improvements in exercise capacity in ApoE−/− mice with exercise training. The underlying mechanism was the decline of energy metabolic efficiency caused by simvastatin. Energy metabolic modulation with trimetazidine alleviated skeletal muscle injury and increased exercise performance.
Simvastatin impairs benefits of exercise training. We confirmed that exercise training increased the exercise tolerance of high‐fat fed ApoE−/− mice. Exercise tolerance was demonstrated to be the significant predictor of prognosis of CHD. Every 1 MET (3.5 mL of oxygen per kg of body weight per min) increase in exercise tolerance is associated with an 18% reduction in cardiovascular mortality.17 In this study, we investigated the effect of simvastatin on exercise performance. The results revealed that simvastatin treatment decreased the exercise capacity in ApoE−/− mice with exercise training as well as the plasma LDL‐C level. It was observed that simvastatin attenuated the benefits of exercise training in overweight or obese patients.4 Thus, the mechanism underlying the statin‐associated exercise intolerance is urgent to be elucidated to improve the prognosis of patients with CHD.
In this study, increased fibre cross‐sectional area and slow‐twitching fibres were observed in skeletal muscles from ApoE−/− mice with exercise training, which were reversed by simvastatin intervention. Slow‐twitching fibres, which rely mostly on aerobic metabolism, specialize in long duration contractile activities and more efficient substrate utilization. Endurance exercise training improves slow‐twitch fibre development.18 Accordingly, the significantly lower proportion of slow‐twitch fibres in the mice treated with simvastatin in the present study contributes to the observed decrease in exercise endurance. To provide adenosine triphosphate and relieve fatigue, exercise stimulates changes in gene transcription that promote mitochondrial biogenesis or increase mitochondrial oxidative capacity.19 The adaptation, which leads to greater capacity of mitochondria, is the key component of exercise‐mediated improvements in exercise tolerance.4 Simvastatin‐induced mitochondrial dysfunction was revealed by increased RRF. Moreover, following simvastatin treatment, we detected upregulation of plasma lactic acid and decreased consumption of muscle glycogen, so confirming the decrease in energy metabolism. Decreased mitochondrial complexes activities have been demonstrated to contribute to inhibition of mitochondrial electron transport chain and mitochondrial dysfunction.20, 21 Simvastatin appeared to be a strong inhibitor of mitochondrial complex III in vitro,7 and a decreased maximal mitochondrial oxidative phosphorylation capacity was observed in simvastatin‐treated patients.22 In this study, simvastatin inhibited mitochondrial complex III activity in skeletal muscle of exercise trained mice. Inhibition of mitochondrial electron transport chain was proved to induce the generation of oxidative stress that plays a causal role in muscle injury.23, 24 And our study also verified that simvastatin induced the generation of oxidative stress in skeletal muscle. Simvastatin‐related mitochondrial dysfunction resulted in skeletal muscle injury that manifested as increased plasma CK and centronucleated muscle fibres. Thus, simvastatin was able to enhance reactive oxygen species (ROS) generation and muscle fibre necrosis, which indicates that the muscular adverse reaction of simvastatin is related to mitochondrial dysfunction.25, 26
Trimetazidine restores improvement in exercise capacity in response to exercise training by helping muscles recover from statin‐associated disorder of energy metabolism. In this study, trimetazidine reversed the simvastatin‐related mitochondrial dysfunction, which was presented as less RRF, lower plasma lactic acid level, increased glycogen consumption, and more slow‐twitch fibres. The positive effects of trimetazidine on energy metabolism are directly exerted acting on mitochondria. Trimetazidine protected cardiomyocyte from mitochondrial fission and dysfunction induced by palmitate.21 The beneficial effects of trimetazidine lay in improving activity of mitochondrial complex as well as preventing the elevated complex‐mediated electron leak.27 In the model of ischaemia, trimetazidine exerted cardioprotective effects, which were attributed to the preservation of mitochondrial structure and function via inhibition of oxidative stress.12 In this study, trimetazidine improved complex III activity and inhibited oxidative stress. Correspondingly, trimetazidine protected muscle in ApoE−/− mice from simvastatin‐induced injury via improving energy metabolism. In C2C12 myotubes, the improvement in metabolic efficiency caused by trimetazidine directly affected signalling pathways leading to protein synthesis and counteracted atrophy induced by tumor necrosis factor alpha (TNF‐α) and starvation.14 Muscular protective effect of trimetazidine was secondary to effects on improved mitochondrial function that resulted in increased exercise performance.13 In this study of ApoE−/− mice, treatment with trimetazidine significantly prevented the decrease in exercise capacity observed in simvastatin‐treated groups during exercise training.
In conclusion, we provided evidences that trimetazidine attenuated decrease in exercise capacity and simvastatin associated skeletal muscle injury in high‐fat fed ApoE−/− mice with exercise training. Further clinic studies are needed to confirm that trimetazidine minimizes the adverse effects of simvastatin on adaptation to exercise training and lower risk of cardiovascular disease morbidity and mortality. Our results could have important implications in reforming drug prescription in heart rehabilitation.
Conflict of interest
None declared.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia, and Muscle.28
This work was supported by the National Natural Science Foundation of China (81400285, 81670411, 81600633, 81570400, 81470560, 81471036, and 81400285); the Natural Science Foundation of Shandong Province (ZR2014HQ037 and ZR2016HP36); and Key Research and Development Program of Shandong Province (2015GSF118062).
Song, M. , Chen, F. , Li, Y. , Zhang, L. , Wang, F. , Qin, R. , Wang, Z. , Zhong, M. , Tang, M. , Zhang, W. , and Han, L. (2018) Trimetazidine restores the positive adaptation to exercise training by mitigating statin‐induced skeletal muscle injury. Journal of Cachexia, Sarcopenia and Muscle, 9: 106–118. doi: 10.1002/jcsm.12250.
References
- 1. Ades PA. Cardiac rehabilitation and secondary prevention of coronary heart disease. N Engl J Med 2001;345:892–902. [DOI] [PubMed] [Google Scholar]
- 2. Lavie CJ, Milani RV. Cardiac rehabilitation and exercise training in secondary coronary heart disease prevention progress in cardiovascular diseases. Prog Cardiovasc Dis 2011;53:397–403. [DOI] [PubMed] [Google Scholar]
- 3. Anderson L, Oldridge N, Thompson DR, Zwisler AD, Rees K, Martin N, et al. Exercise‐based cardiac rehabilitation for coronary heart disease Cochrane systematic review and meta‐analysis. J Am Coll Cardiol 2016;67:1–12. [DOI] [PubMed] [Google Scholar]
- 4. Mikus CR, Boyle LJ, Borengasser SJ, Oberlin DJ, Naples SP, Fletcher J, et al. Simvatatin impairs exercise training adaptations. J Am Coll Cardiol 2013;62:709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stroes ES, Thompson PD, Corsini A, Vladutiu GD, Raal FJ, Ray KK, et al. Statin‐associated muscle symptoms: impact on statin therapy—European Atherosclerosis Society Consensus Panel Statement on Assessment. Aetiology and Management Eur Heart J 2015;36:1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franc S, Dejager S, Bruckert E, Chauvenet M, Giral P, Turpin G. A comprehensive description of muscle symptoms associated with lipid‐lowering drugs. Cardiovasc Drugs Ther 2003;17:459–465. [DOI] [PubMed] [Google Scholar]
- 7. Schirris TJ, Renkema GH, Ritschel T, Voermans NC, Bilos A, van Engelen BG, et al. Statin‐induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab 2015;22:399–407. [DOI] [PubMed] [Google Scholar]
- 8. Sirvent P, Mercier J, Vassort G, Lacampagne A. Simvastatin triggers mitochondria‐induced Ca2+ signaling alteration in skeletal muscle. Biochem Biophys Res Commun 2005;329:1067–1075. [DOI] [PubMed] [Google Scholar]
- 9. Stringer HA, Sohi GK, Maguire JA, Côté HC. Decreased skeletal muscle mitochondrial DNA in patients with statin‐induced myopathy. J Neurol Sci 2013;325:142–147. [DOI] [PubMed] [Google Scholar]
- 10. Bonifacio A, Sanvee GM, Bouitbir J, Krähenbühl S. The AKT/mTOR signaling pathway plays a key role in statin‐induced myotoxicity. Biochim Biophys Acta 1853;2015:1841–1849. [DOI] [PubMed] [Google Scholar]
- 11. Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long‐chain 3‐ketoacyl coenzyme A thiolase. Circ Res 2000;86:580–588. [DOI] [PubMed] [Google Scholar]
- 12. Dehina L, Vaillant F, Tabib A, Bui‐Xuan B, Chevalier P, Dizerens N, et al. Trimetazidine demonstrated cardioprotective effects through mitochondrial pathway in a model of acute coronary ischemia. Naunyn Schmiedebergs Arch Pharmacol 2013;386:205–215. [DOI] [PubMed] [Google Scholar]
- 13. Ferraro E, Pin F, Gorini S, Pontecorvo L, Ferri A, Mollace V, et al. Improvement of skeletal muscle performance in ageing by the metabolic modulator trimetazidine. J Cachexia Sarcopenia Muscle. 2016;7:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferraro E, Giammarioli AM, Caldarola S, Lista P, Feraco A, Tinari A, et al. The metabolic modulator trimetazidine triggers autophagy and counteracts stress‐induced atrophy in skeletal muscle myotubes. FEBS J 2013;280:5094–5108. [DOI] [PubMed] [Google Scholar]
- 15. Szwed H. Clinical benefits of trimetazidine in patients with recurrent angina. Coron Artery Dis 2004;15:S17–S21. [PubMed] [Google Scholar]
- 16. Belardinelli R, Lacalaprice F, Faccenda E, Volpe L. Trimetazidine potentiates the effects of exercise training in patients with ischemic cardiomyopathy referred for cardiac rehabilitation. Eur J Cardiovasc Prev Rehabil 2008;15:533–540. [DOI] [PubMed] [Google Scholar]
- 17. Barlow CE, Defina LF, Radford NB, Berry JD, Cooper KH, Haskell WL, et al. Cardiorespiratory fitness and long‐term survival in “low‐risk” adults. J Am Heart Assoc 2012;1: e001354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilson JM, Loenneke JP, Jo E, Wilson GJ, Zourdos MC, Kim JS. The effects of endurance, strength, and power training on muscle fiber type shifting. J Strength Cond Res 2012;26:1724–1729. [DOI] [PubMed] [Google Scholar]
- 19. Ferraro E, Giammarioli AM, Chiandotto S, Spoletini I, Rosano G. Exercise‐induced skeletal muscle remodeling and metabolic adaptation: redox signaling and role of autophagy. Antioxid Redox Signal 2014;21:154–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lambertucci RH, Hirabara SM, Silveira Ldos R, Levada‐Pires AC, Curi R, Pithon‐Curi TC. Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J Cell Physiol 2008;216:796–804. [DOI] [PubMed] [Google Scholar]
- 21. Kuzmicic J, Parra V, Verdejo HE, Lopez‐Crisosto C, Chiong M, Garcia L, et al. Trimetazidine prevents palmitate‐induced mitochondrial fission and dysfunction in cultured cardiomyocytes. Biochem Pharmoacol 2014;91:323–336. [DOI] [PubMed] [Google Scholar]
- 22. Larsen S, Stride N, Hey‐Mogensen M, Hansen CN, Bang LE, Bundgaard H, et al. Simvastatin effects on skeletal muscle relation to decreased mitochondrial function and glucose intolerance. J Am Coll Cardiol 2013;61:44–53. [DOI] [PubMed] [Google Scholar]
- 23. Boengler K, Kosiol M, Mayr M, Schulz R, Rohrbach S. Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J Cachexia Sarcopenia Muscle. 2017;8:349–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Javadov S, Jang S, Rodriguez‐Reyes N, Rodriguez‐Zayas AE, Soto Hernandez J, Krainz T, et al. Mitochondria‐targeted antioxidant preserves contractile properties and mitochondrial function of skeletal muscle in aged rats. Oncotarget 2015;6:39469–39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carvalho AA, Lima UW, Valiente RA. Statin and fibrate associated myopathy: study of eight patients. Arq Neuropsiquiatr 2004;62:257–261. [DOI] [PubMed] [Google Scholar]
- 26. Gambelli S, Dotti MT, Malandrini A, Mondelli M, Stromillo ML, Gaudiano C, et al. Mitochondrial alterations in muscle biopsies of patients on statin therapy. J Submicrosc Cytol Pathol 2004;36:85–89. [PubMed] [Google Scholar]
- 27. Dedkova EN, Seidlmayer LK, Blatter LA. Mitochondria‐mediated cardioprotection by trimetazidine in rabbit heart failure. J Mol Cell Cardiol 2013;59:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle 2015;4:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]