

Abstract
Background
Cathepsin K (CatK) is a widely expressed cysteine protease that has gained attention because of its enzymatic and non‐enzymatic functions in signalling. Here, we examined whether CatK‐deficiency (CatK−/−) would mitigate injury‐related skeletal muscle remodelling and fibrosis in mice, with a special focus on inflammation and muscle cell apoptosis.
Methods
Cardiotoxin (CTX, 20 μM/200 μL) was injected into the left gastrocnemius muscle of male wild‐type (CatK+/+) and CatK−/− mice, and the mice were processed for morphological and biochemical studies.
Results
On post‐injection Day 14, CatK deletion ameliorated muscle interstitial fibrosis and remodelling and performance. At an early time point (Day 3), CatK−/− reduced the lesion macrophage and leucocyte contents and cell apoptosis, the mRNA levels of monocyte chemoattractant protein‐1, toll‐like receptor‐2 and toll‐like receptor‐4, and the gelatinolytic activity related to matrix metalloproteinase‐2/‐9. CatK deletion also restored the protein levels of caspase‐3 and cleaved caspase‐8 and the ratio of the BAX to the Bcl‐2. Moreover, CatK deficiency protected muscle fibre laminin and desmin disorder in response to CTX injury. These beneficial muscle effects were mimicked by CatK‐specific inhibitor treatment. In vitro experiments demonstrated that pharmacological CatK inhibition reduced the apoptosis of C2C12 mouse myoblasts and the levels of BAX and caspase‐3 proteins induced by CTX.
Conclusions
These results demonstrate that CatK plays an essential role in skeletal muscle loss and fibrosis in response to CTX injury, possibly via a reduction of inflammation and cell apoptosis, suggesting a novel therapeutic strategy for the control of skeletal muscle diseases by regulating CatK activity.
Keywords: Cathepsin K, Apoptosis, Skeletal muscle, Injury, Remodelling, Fibrosis
Introduction
Age‐related loss of skeletal muscle mass and function can result in decreased quality of life.1 The causes of age‐associated muscle disease (i.e. sarcopenia) are multifactorial and include biological and environmental factors.1, 2, 3 Skeletal muscle cell apoptosis associated with various injuries has been shown to contribute to muscle remodelling and fibrosis in aged animals and humans.3 The pathogenesis of injury‐associated skeletal muscle disorder involves extensive extracellular matrix (ECM) remodelling, which requires the participation of extracellular proteases.1 The matrix metalloproteinase (MMP) family has been shown to participate in muscle disease inception and progression.4, 5, 6 However, genetic and pharmacological interventions aimed at MMP family members resulted in incomplete prevention of muscle remodelling and fibrosis,7, 8, 9 suggesting that other proteases may also contribute to such remodelling in injury‐related muscle disorders.
The cathepsins known as cysteine proteases were discovered in the second half of the 20th century.10 In humans, the cathepsin family consists of 11 members [i.e. cathepsin K (CatK), CatS, CatL, CatB, etc.].11 Initially, cathepsins were recognized to function as scavengers for protein recycling in lysosomes and endosomes of mammalian cells under physiological and pathological conditions.10, 11, 12, 13, 14, 15 However, the recognition of the cysteinyl cathepsins led to the exploration of their functions in metabolic and inflammatory cardiovascular disorders.16, 17, 18, 19, 20 Like the members of the MMP family, most of the cathepsins have been shown to be regulatory exoproteases that are widely expressed in various tissues or cells by inflammatory cytokines and growth factors.21, 22, 23, 24 CatK is one of the most potent of the mammalian elastases and collagenases.25 Previous studies have demonstrated that CatK expression is increased in the atherosclerotic lesions of animals and humans.26, 27 In our own previous report, we clearly showed that CatK deletion confers resistance to carotid artery injury via a reduction in the levels of toll‐like receptors (TLR)‐2/‐4.28 Moreover, CatK has been shown to control ischaemia‐induced neovascularization via the modulation of Notch‐1 activation and a downstream signalling pathway.29 Laboratory studies revealed that cathepsins are involved in apoptosis and proliferation in cancer and immune‐inflammatory cells. Comprehensive review articles have highlighted the functional significance of CatK in growth‐related tumours and cardiovascular disease.13, 30 Despite this body of research, however, there is limited information about the role of CatK in age‐related and injury‐related skeletal muscle disorders.
In the present study, we used CatK‐deficient (CatK−/−) mice and an experimental skeletal injury model to test our hypothesis that CatK activity controls muscle remodelling and dysfunction in response to injury.
Materials and methods
Antibodies and reagents
The following commercially available antibodies were used: Bcl‐2 (#2870), BAX (#2772), cleaved caspase‐8 (#9429), cleaved caspase‐3 (#9661), and caspase‐3 (#9662) (all from Cell Signaling Technology, Beverly, MA); CatK (#3368; BioVision, Milpitas, CA); CD45 (clone 30‐F11; Biolegend, San Diego, CA); CD68 (clone KP1, ab955; AbCam, Cambridge, UK); Laminin‐5 (BS‐7713R; Bioss, Woburn, MA); Desmin (Clone 33; Dako, Carpinteria, CA); Zenon rabbit and mouse IgG labelling kits (Molecular Probes, Eugene, OR); and GAPDH (G8795) as loading controls (Sigma‐Aldrich, St. Louis, MO). Cardiotoxin (CTX) was from Sigma‐Aldrich (Naja pallida, L8102, Latoxan). CatK inhibitor (ONO‐KK1‐300‐01) was provided by Ono Pharmaceutical (Osaka, Japan).
Pentobarbital sodium was from Dainippon Pharmaceutical Co. (Osaka, Japan), and the RNA PCR Core kit was from Applied Biosystems (Foster City, CA). The RNeasy Fibrous Tissue Mini‐Kit was from Qiagen (Hilden, Germany). The In Situ Cell Death Detection Kit (reference no. 11684795910) was from Roche Diagnostics (Mannheim, Germany). C2C12 mouse myoblasts were obtained from the American Type Culture Collection (Manassas, VA). Dulbecco's modified Eagle medium (DMEM) was from GIBCO Life Technologies (Grand Island, NY). Horse serum was also from GIBCO Life Technologies (Auckland, New Zealand).
Animal care and use
The male CatK−/− and wild‐type (C57BL/6, CatK+/+) littermates used in this study had approximately the same body weights (22–24 g).28 The animals were housed in a room with a controlled temperature (22°C ± 2°C) and a 12 h light–dark cycle, with ad libitum access to food and water. All mouse experiments were performed with the approval of the Institutional Animal Care and Use Committee at Nagoya University and were in accordance with the U.S. National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Model of the skeletal muscle injury and tissue collection
For evaluation of the role of CatK in muscle remodelling, 10 ± 2‐week‐old CatK+/+ and CatK−/− mice were used. The hair of both legs was shaved, and CTX (20 μM/200 μL) was injected into the left gastrocnemius muscle. For a separate specific CatK inhibitor experiment, CatK+/+ mice that received an injection of the CTX (20 μM/200 μL) were randomly assigned to one of the three groups and administered (by oral gavage) vehicle (200 μL of 0.5% carboxymethylcellulose; Cont) or a low dose or high dose of a CatK inhibitor (ONO‐KK1‐300‐01: 3 mg/kg, low‐dose CatK inhibitor; 30 mg/kg, high‐dose CatK inhibitor ; Ono Pharmaceutical) twice daily from 2 days before the injury to 14 days after the injury.
At the indicated time points, all animals were anaesthetized with an intraperitoneal injection of pentobarbital sodium (50 mg/kg), and the tissue (muscles) and arterial blood samples were collected. For the biological analysis, the skeletal muscle was isolated and kept in RNAlater solution (for the gene assay) or liquid nitrogen (for the protein assay). For the morphological analysis, after being immersed in fixative at 4°C, the skeletal muscles were embedded in optimal cutting temperature compound (Sakura Finetechnical, Tokyo) and stored at −20°C. After the blood was poured into an EDTA‐2Na blood collection tube (VenojectII; Terumo, Kakamigahara, Japan), it was centrifuged, and the plasma was collected and stored at −80°C.
Evaluation of muscle endurance capacity
A motorized rodent treadmill (S‐Con Mini‐Z: Tokyo Engineering, Tokyo) was used to determine the endurance capacity for running, as described previously.31 In the preliminary training sessions, mice at Day 1 before the CTX injection were made to run on the treadmill at an inclination of 0° as follows: warm‐up (5 min), 7 m/min; exercise (35 min), 17 m/min; cool down (5 min), 7 m/min. For the measurement of endurance capacity, the mice at Days 3 and 14 after CTX injury were placed on the treadmill, and the warm‐up was started at 6 m/min with the treadmill's tilt angle at 0°. After 5 min, the tilt angle of the treadmill was set to 10°, and the speed was gradually increased by 2 m/min every 2 min until reaching the maximum speed of 20 m/min and then maintained at the maximum speed.
During the running period, the mouse was prevented from resting by a shock bar placed behind the treadmill, which was set at 20 V. The calculations of the running distance and workload were finished when the mouse stopped moving for >10 s. The running distance was calculated as a product of the running time and speed. The workload in the vertical direction was calculated with consideration for the weight of the mouse (=weight × gravitational acceleration × mileage in the vertical direction). All items are expressed as the ratio of the observed values to the data obtained on Day 0 before the CTX injection.
Evaluation of grip strength
We also studied the animals' grip strength by using a small animal grip strength meter (Columbus, Largo, FL) as described previously.31 The forelimb of the mouse was placed on the limb grip of the meter, and its tail was gently pulled in the opposite direction. We measured the maximum value of the grip force that the mouse exerted before it released its grip. The grip strength was measured over five times for each mouse on Days 0, 3, and 14, and the values were averaged as the grip strength value for each of these days. To exclude the influence of exercise, the mice that underwent the grip strength and endurance examinations were not subjected to the histological or biological analyses.
Quantitative real‐time gene expression assay
Total RNA was extracted from the tissue, or the cell extracts with an RNeasy Fibrous Tissue Mini‐Kit in accord with the manufacturer's instructions. The mRNA was reverse‐transcribed to cDNA with an RNA polymerase chain reaction (PCR) Core kit. Quantitative gene expression was studied using an ABI 7300 PCR system with Universal PCR Master Mix (Applied Biosystems). All experiments were performed in triplicate. The sequences of the primers used for CatK, CatS, CatL, MMP‐2, MMP‐9, tissue inhibitor of MMP‐1 (TIMP‐1), TIMP‐2, TLR‐2, TLR‐4, monocyte chemoattractant protein‐1 (MCP‐1), tumour necrosis factor‐alpha (TNF‐α), and GAPDH genes are shown in Table S1. The transcription of target genes was normalized to GAPDH.
Morphometry and immunohistochemistry analyses
Serial cross‐cryosections (4 μm thick) were collected at the injured regions of the skeletal muscle at a rate of 3–4 sections every 40 μm. The sections at Day 3 after injury were used to evaluate the inflammation and cell apoptosis in response to injury. For the immunohistochemistry, corresponding sections on separate slides were treated with mouse monoclonal antibodies against CD45 for leucocytes or CD68 for macrophages (1:50 for each).32 The sections were then visualized with an ABC substrate kit (SK‐4400; Vector Laboratories, Burlingame, CA) in accord with the manufacturer's instructions.
Apoptotic staining was performed as follows: the sections were subjected to terminal deoxynucleotidyl transferase‐mediated dUTP nick end labelling (TUNEL) using a Fluorescein In Situ Cell Death Detection Kit (Sigma‐Aldrich). The sections at Day 14 were used to examine muscle remodelling and fibrosis. Masson's trichrome staining for fibrosis was performed as described previously.33 The slides of the muscles and the cells were mounted in glycerol‐based Vectashield medium (Vector Laboratories) containing the nucleus stain 4,6′‐diamidino‐2‐phenylindole.
For quantification of the positive cell staining, we took six to seven images for one section using a ×20 objective, and we counted the numbers of CD45+, CD68+, and TUNEL+ cells. For the quantifications of muscle fibres and fibrosis, we took seven to nine images at 90 000 μm2 for one section using a ×20 objective, and we measured cross‐sectional area of a muscle fibre with the central nucleus and the volumes of interstitial fibrosis in this field by using a microscope (BZ9000; Keyence, Osaka, Japan). For negative controls, the primary antibodies were replaced with non‐immune immunoglobulin G or Zenon‐labelled rabbit or mouse IgG.
Immunofluorescence
To examine the changes in the muscle fibre structural properties and healing capacity, double immunofluorescence labelling of the laminin and desmin was performed as described previously.31 In brief, the muscle sections were treated with a rabbit polyclonal antibody to laminin‐5 and a mouse monoclonal antibody to desmin (1:100 for each). Then, the sections were visualized using Zenon rabbit and mouse IgG labelling kits (1:200) according to the manufacturer's instructions. Staining sections were visualized with a BZ‐X700 microscope (Keyence, Osaka, Japan) using ×20 or ×40 objectives. We measured the average intensity of desmin protein expression for six fibres in one section by using the Image J software program.
Gelatin zymography
Gelatin zymography was performed as described previously.34 Each sample was mixed with an equal volume of sample buffer without a reducing agent. Forty micrograms of each muscle tissue protein extract was then loaded onto a 10% SDS–polyacrylamide gel containing 1 mg/mL gelatin as a substrate. After electrophoresis, the gels were washed twice with 2.5% Triton X‐100 and then incubated overnight at 37°C with substrate buffer (pH 8.0, 50 mM NaCl, 50 mM Tris, and 10 mM CaCl2). Finally, the gels were stained with Coomassie Brilliant blue and bleached to reveal the gelatinolytic activities as clear bands against a blue‐stained background. Quantitative data were obtained by densitometry.35
Biochemical analyses
To evaluate the muscle and cell injuries, the lactate dehydrogenase (LDH) levels in mouse plasma on Day 3 after injury and the C2C12 cell‐conditioned medium after treatment with CTX at the indicated concentrations for 24 h were measured at a commercial laboratory (SRL, Tokyo, Japan).
Simple western analysis
Simple western analysis is a well‐known protein detection method used in conjunction with the western blotting assay.36 WES™ instrumentation is one of the SimpleWestern™ systems provided by ProteinSimple. We performed the WES™ analysis according to the ProteinSimple user manual. In brief, samples were adjusted to an appropriate protein concentration in 4 μL with 0.1% sample buffer and further diluted 1:4 by adding 1.0 μL of ×5 fluorescent master mix (containing 20 μL of 10× sample buffer and 20 μL of 400 mM DTT). Five microlitres of each final sample and Biotin ladder (containing 16 μL of deionized water, 2 μL of 10× sample buffer, and 2 μL of 400 mM DTT) was heated at 95°C for 5 min, added to a 1:1 mixture of luminol‐S and peroxide (150 μL), and stored in ice until use. The primary antibodies (i.e. anti‐cleaved caspase‐8, anti‐caspase‐3, anti‐BAX, anti‐Bcl‐2, and anti‐GAPDH) were diluted 1:50 to 1:100 with antibody diluent II, and the secondary antibody was used without dilution. After applying the appropriate samples and reagents to each well, we centrifuged the plate for 5 min at 150 g at room temperature, and then set it and a capillary cartridge in WES™. The data obtained by SimpleWestern™ assay were automatically analysed to determine the molecular weight of the detected protein and to subject it to quantitative analysis using the attached Compass software. In addition, we also performed a general western blotting assay to evaluate the levels of cleaved caspase‐3, caspase‐3, and GADPH proteins in the muscles.
Cell culture
C2C12 cells were grown in DMEM containing 10% foetal bovine serum and antibiotics at 37°C with 5% CO2. At confluence, the myoblasts were induced to fuse by changing the medium to medium containing 2% horse serum (called ‘differentiation medium’ herein) as described previously.37 After 5–6 days of differentiation, when the myoblasts had lengthened, fused, and become multinucleated myotubes, the differentiated C2C12 cells were cultured with serum‐free DMEM for 6 h. Following pre‐treatment with a CatK‐specific inhibitor (ONO‐KK1‐300‐01) at the indicated concentrations, the cells were cultured in the presence or absence of CTX at the indicated concentrations and time points, and the conditioned medium and the cells were subjected to the biological analysis.
TUNEL staining
After the differentiation of C2C12 cells, the cells were cultured in serum‐free DMEM containing 0.5 μM CTX in the presence and absence of the CatK inhibitor ONO‐KK1‐300‐01 for 24 h and then subjected to TUNEL staining as described previously.31 The apoptotic cells in the muscles were also evaluated by TUNEL staining.
Collagenase assay
After the differentiation of C2C12 cells, the cells were cultured in serum‐free DMEM containing 0.5 μM CTX in the presence and absence of the CatK inhibitor ONO‐KK1‐300‐01 for 24 h and then subjected to collagenase assay used FITC‐Bovine Type I Collagen (Cat: #4001; Chondrex, Inc., Redmond, WA) as described previously.26
Statistical analysis
The data are expressed as the mean ± standard deviation. Student's t‐test (for comparisons between two groups) or one‐way analysis of variance (for comparisons of three or more groups) followed by Tukey post hoc tests was used for the statistical analyses. The muscle performance data were subjected to two‐way repeated‐measures analysis of variance and Bonferroni post hoc tests. SPSS software ver. 17.0 (SPSS, Chicago, IL) was used. P values <0.05 were considered significant.
Results
The changes in cathepsin expression in response to cardiotoxin injection
Figure S1A illustrates the severe muscle damage (e.g. oedema, haemorrhage, and muscle fibre loss). At first, we examined the influence of CTX on cathepsin family expression. We isolated total RNA from the muscles at the indicated time points after injury and used it in a quantitative real‐time gene assay to determine the CatK, CatS, and CatL mRNA levels. We observed that the CatK mRNA levels were significantly increased in CTX‐injured muscles throughout the follow‐up period and reached a 50‐fold peak at Day 3 after injury (Figure S1B). Similarly, the CatS and CatL mRNA levels had significantly increased between 3 and 7 days after injury, with the highest expression occurring at Day 3 (Figure S1B). We also observed that the injured muscles had increased levels of MMP‐2 and MMP‐9 mRNAs and increased levels of their endogenous inhibitor (TIMP‐1 and TIMP‐2) mRNAs compared with before the injury (Figure S1C,D). The gelatin zymography revealed that the gelatinolytic activities of MMP‐2 and MMP‐9 were increased in the injured muscles compared with those in the non‐injured muscles at Day 3 after the CTX injection (Figure S1E).
Cathepsin K deficiency protects against cardiotoxin‐induced muscle damage
To explore whether CatK modulates muscle remodelling and fibrosis in response to injury, we created a skeletal muscle CTX injury model using CatK+/+ and CatK−/− mice to monitor muscle morphological and functional changes in the gastrocnemius muscles. On post‐injection Day 14, the histological analysis revealed that the CatK−/− mice had better preserved muscle fibre size (450 ± 115 vs. 308 ± 52 μm2, P < 0.05) and lower levels of interstitial fibrosis (30661 ± 5077 vs. 45081 ± 4159 μm2, P < 0.01) compared with the wild‐type (CatK+/+) mice, respectively (Figure 1A and 1C). Desmin is known to be an intermediate filament protein that is highly expressed in immature muscle fibre during foetal life and regeneration.38 To further visualize the healing process, we performed double immunofluorescence labelling with desmin and laminin‐5. As shown in Figure 1D and 1E, CatK−/− mice exposed to CTX injury had markedly higher intracellular intensity of desmin expression (32.9 ± 3.4 vs. 12.8 ± 1.4, P < 0.01) than CatK+/+ mice, which suggests that CatK deletion preserved the structural properties of the muscle fibre and restored tissue healing in response to CTX injury.38 The quantitative analysis of muscle performance demonstrated that except grip strength, CatK deletion prevented CTX‐induced impairment of running distance and workload at Day 14 after injury (Figure 2A and 2B). We also observed that the running time and distance were better preserved in the CatK−/− mice compared with those in the CatK+/+ mice at Day 14 after injury (Figure 2C and 2D), indicating that CatK deficiency ameliorates muscle remodelling and dysfunction because of CTX injury.
Figure 1.
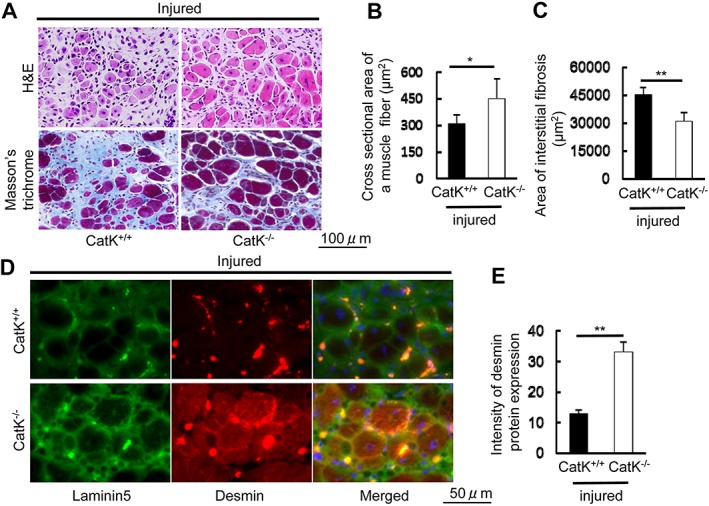
CatK deficiency (CatK−/−) alleviated the cardiotoxin‐induced skeletal muscle remodelling and fibrosis at Day 14 after injury. (A) Representative images of haematoxylin and eosin (H&E) and Masson's trichrome staining of the injured gastrocnemius muscle sections of wild‐type (CatK+/+) and CatK−/− mice. Scale bar, 100 μm. (B, C) Quantitative data showing the cross‐sectional area of a muscle fibre and interstitial fibrosis (90 000 μm2). (D, E) Representative immunofluorescence images and quantitative data for the intensity of desmin protein expression in CatK+/+ and CatK−/− mice at Day 14 after injury. Scale bar, 50 μm. Results are the mean ± SD (n = 5–6). Significance was estimated using Student's t‐test (*P < 0.05, **P < 0.01).
Figure 2.
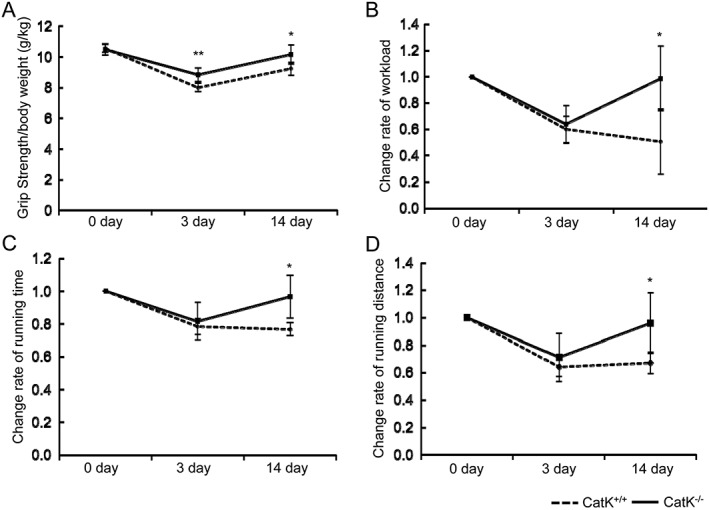
CatK deficiency prevented cardiotoxin‐induced muscle dysfunction. (A) Grip strength was measured. (B) Workload in the vertical direction was calculated as described in the Materials and Methods. (C, D) Running time and distance were calculated from the results of the running time and speed. All items are expressed as the ratio of observed values to the data obtained on Day 1 before cardiotoxin injection. Results are the mean ± SD (n = 6). *P < 0.05, **P < 0.01 vs. the corresponding controls by two‐way repeated‐measures analysis of variance and Bonferroni post hoc tests.
Cathepsin K deficiency mitigated the cardiotoxin‐induced cell apoptosis
The TUNEL staining showed marked TUNEL+ apoptotic cells in the injured muscles of CatK+/+ mice on Day 3 after the CTX injection, and this change was prevented by CatK deletion (Figure 3A and 3B). Plasma LDH levels have often been used to predict muscle injury in humans and animals.39 To further examine the difference in muscle injury, the plasma LDH levels were analysed by enzyme‐linked immunosorbent assay. The quantitative biological analysis revealed significantly lower levels of plasma LDH from the CatK−/− mice compared with that from the CatK+/+ mice (Figure 3C). To examine the possible participation of CatK in the protection against CTX‐induced cell apoptosis, we assessed the levels of pro‐apoptotic and anti‐apoptotic molecules by performing a simple western blotting analysis. As anticipated, the levels of caspase‐3, cleaved caspase‐8, the ratio of cleaved caspase‐3 to caspase‐3, and the ratio of BAX to Bcl‐2 proteins were all significantly lower in the injured muscles of CatK−/− mice compared with those in the CatK+/+ mice at Day 3 after injury (Figure 3D–3G, Figure S2A,B). CatK deficiency thus exhibited a muscle benefit via the reduction of CTX‐induced cell apoptosis in the mice.
Figure 3.
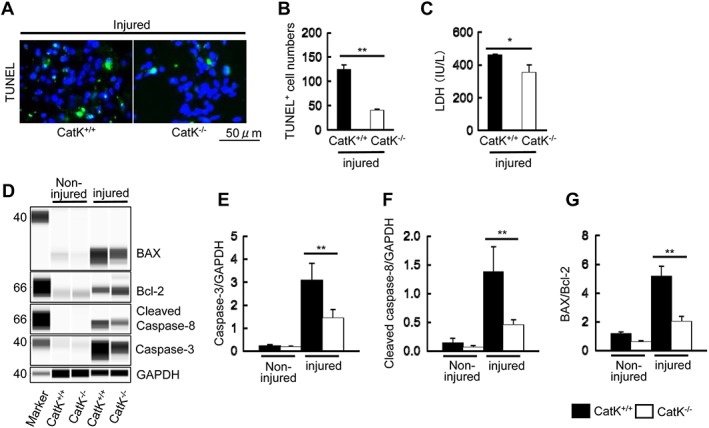
CatK deletion mitigated cardiotoxin‐induced apoptosis. (A, B) Representative images of terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) immunofluorescence and combined quantitative data show the numbers of TUNEL+ apoptotic cells in the injured gastrocnemius of wild‐type (CatK+/+) and CatK deficiency (CatK−/−) mice at Day 3 after cardiotoxin injection. (C) CatK deletion reduced the plasma lactate dehydrogenase (LDH) level. (D–G) Representative images of simple western blots and the combined quantitative data show the levels of caspase‐3 and cleaved caspase‐8 proteins and the ratio of BAX protein to Bcl‐2 protein in the injured muscles of CatK+/+ and CatK−/− mice. Results are the mean ± SD (n = 3–4). *P < 0.05, **P < 0.01 vs. the corresponding control groups by Student's t‐test or one‐way analysis of variance followed by Tukey post hoc tests.
Cathepsin K deficiency ameliorated inflammatory actions in response to cardiotoxin
It is known that inflammation plays a fundamental role in all stages of wound healing after injury. As inflammation activation seemed to be tightly associated with increased CatK expression, we extended our examination to inflammatory cell infiltration at Day 3 after the CTX injection. The results demonstrated that CatK deletion ameliorated the macrophage and leucocyte infiltrations (CD68: 46 ± 10 vs. 111 ± 13; CD45: 94 ± 17 vs. 207 ± 13, cell numbers: P < 0.01 for each) (Figure 4A–4C). The quantitative real‐time PCR data demonstrated that CatK deletion decreased the levels of inflammatory genes such as TNF‐α, TLR‐2, TLR‐4, and MCP‐1 in injured muscles compared with CatK+/+ mice (Figure 4D–4G), suggesting that CatK deficiency‐mediated anti‐inflammatory actions may contribute to the muscle benefit in this CTX injury model.
Figure 4.
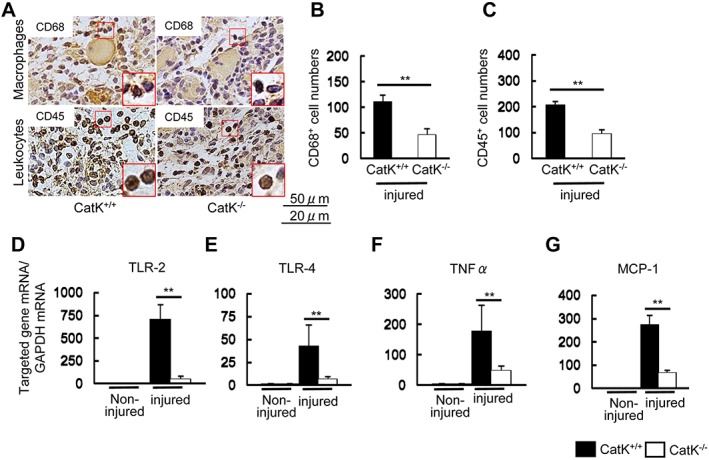
CatK deficiency (CatK−/−) ameliorated inflammatory actions in the injured muscles at Day 3 after cardiotoxin injection. (A) Representative images of the immunostaining using CD68 antibody (for infiltrated macrophages) and CD45 antibody (for infiltrated leucocytes) in the injured gastrocnemius sections of wild‐type (CatK+/+) and CatK−/− mice. Scale bars: A, 50 μm; B, 25 μm. (B, C) Quantitative data for CD68‐positive and CD45‐positive cell numbers. (D–G) Quantitative real‐time polymerase chain reaction data show the levels of TLR‐2, TLR‐4, TNF‐α, and MCP‐1 in the non‐injured and injured gastrocnemius muscles of CatK+/+ and CatK−/− mice. *P < 0.05, **P < 0.01 vs. the corresponding control groups by Student's t‐test or one‐way analysis of variance followed by Tukey post hoc tests.
To further examine the consequence of CatK silencing on other cathepsin family members, we analysed the total RNA extraction of the muscles in a gene expression assay. The results indicated that CatK deletion had no effect on the CatS or CatL expression not only in non‐injured muscles but also in injured muscles (Figure S3A). However, compared with those of CatK+/+ mice, the injured muscles of CatK−/− mice had decreased levels of MMP‐2 and MMP‐9 as well as their endogenous inhibitor TIMP‐1 and TIMP‐2 genes (Figure S3B,C). Consistently, we observed that CatK deficiency reduced the gelatinolytic net activities of MMP‐2 and MMP‐9 (Figure S3D,E).
Pharmacological cathepsin K inhibition suppressed muscle remodelling and apoptosis
Figure 5A shows non‐injured and injured muscle fibre size (haematoxylin and eosin) and fibrosis (Masson's trichrome) of mice treated with vehicle (CONT), low‐dose CatK inhibitor, and high‐dose CatK inhibitor. Similarly to the CatK deletion, the pharmacological inhibition of CatK with ONO‐KK1‐300‐01 preserved the muscle fibre size and the intracellular intensity of desmin expression and reduced the interstitial fibrosis in a dose‐dependent manner (Figure 5B). Similarly, it also prevented cell apoptosis in injured muscles in a dose‐dependent manner (Figure 6A and 6B). Consistently, the CatK inhibition suppressed plasma LDH levels (Figure 6C). Both doses of CatK inhibitor reduced the expression of CatK protein in the injured muscles (Figure 6D and 6E). Moreover, CatK inhibition improved the changes in the levels of caspase‐3, cleaved caspase‐8, the ratio of cleaved caspase‐3 to caspase‐3, and the ratio of BAX to Bcl‐2 in a dose‐dependent manner (Figure 6D and 6F–H, Figure S2C,D). Thus, CatK inhibition also exhibited a protective effect against CTX.
Figure 5.
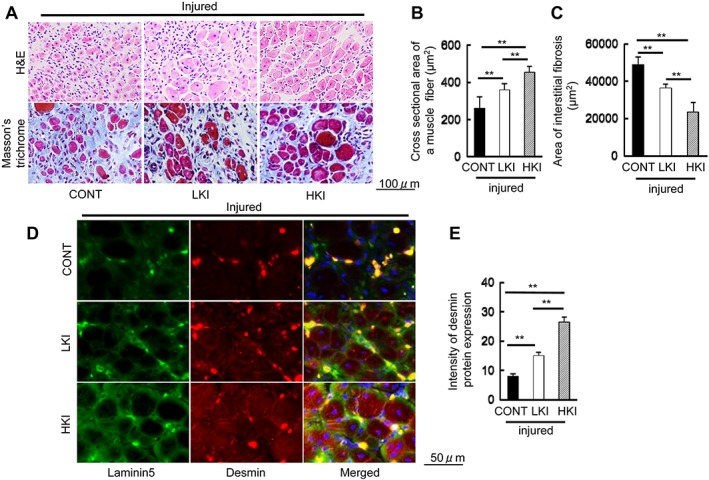
Cat K inhibition alleviated cardiotoxin‐induced skeletal muscle remodelling and fibrosis at Day 14 after injury. (A) Representative image of haematoxylin and eosin (H&E) and Masson's trichrome staining of the injured gastrocnemius sections of the mice treated with vehicle (200 μL of 0.5% carboxymethylcellulose; CONT mice) or with a low dose or high dose of CatK inhibitor [ONO‐KK1‐300‐01: 3 mg/kg, low‐dose CatK inhibitor (LKI) mice; 30 mg/kg, high‐dose CatK inhibitor (HKI) mice] at Day 3 after injury. Scale bar, 100 μm. (B, C) Quantitative data showing the cross‐sectional area of a muscle fibre and interstitial fibrosis (90 000 μm2). (D, E) Representative immunofluorescence images and quantitative data show the intensity of desmin protein expression in the three groups. Scale bar, 50 μm. Results are the mean ± SD (n = 6). *P < 0.05, **P < 0.01 vs. the controls by one‐way analysis of variance followed by Tukey post hoc tests.
Figure 6.
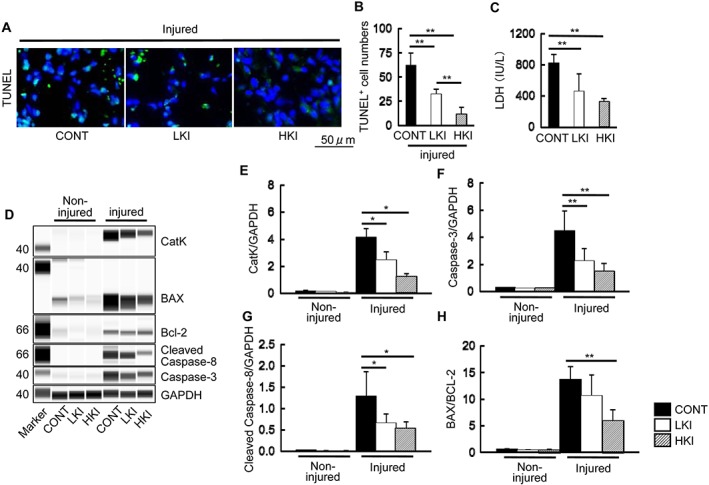
Pharmacological CatK inhibition mitigated apoptosis in the injured muscles of CONT (vehicle), LKI (low‐dose CatK inhibitor), and HKI (high‐dose CatK inhibitor) mice at Day 3 after injury. (A, B) Representative images of terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) immunofluorescence and combined quantitative data show the numbers of apoptotic cells in the injured gastrocnemius of the three experimental groups (n = 6). (C) CatK inhibition suppressed the plasma lactate dehydrogenase (LDH) levels in response to cardiotoxin injury in a dose‐dependent manner (n = 6). (D–H) Representative images of simple western blots and combined quantitative data show the levels of caspase‐3 and cleaved caspase‐8 proteins and the ratio of BAX protein levels to Bcl‐2 protein levels in the injured muscles of the three experimental groups (n = 3). Results are the mean ± SD. *P < 0.05, **P < 0.01 vs. the controls by one‐way analysis of variance followed by Tukey post hoc tests.
Cathepsin K inhibition mitigated the inflammatory response to cardiotoxin
Representative immunostaining and the related quantitative analysis demonstrated that the numbers of infiltrated macrophages and leucocytes were lower in the injured muscles of the CatK+/+ mice administered a CatK inhibitor compared with those of the non‐treated control mice (Figure 7A–C). Interestingly, compared with the control mice, the CatK inhibition also ameliorated the levels of TLR‐2, TLR‐4, TNF‐α, and MCP‐1 mRNAs in the injured muscles of the CatK+/+ mice (Figure 7D–G). Thus, the CatK inhibition‐mediated anti‐inflammatory effects might have contributed to the muscle protective actions in this mouse CTX injury model.
Figure 7.
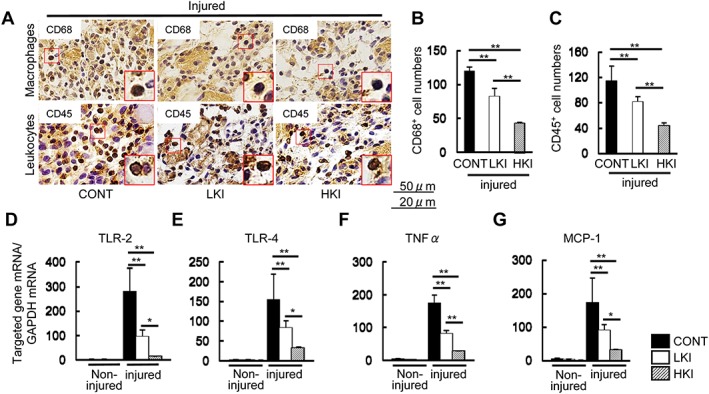
CatK inhibition ameliorated the inflammatory actions of the injured muscles of the CONT (vehicle), LKI (low‐dose CatK inhibitor), and HKI (high‐dose CatK inhibitor) mice at Day 3 after cardiotoxin injection. (A–C) Representative images of the immunostaining and combined quantitative data show the numbers of infiltrated macrophages (CD68+ cells) and leucocytes (CD45+ cells) in the injured gastrocnemius of the three experimental groups. Scale bar: A, 50 μm; B, 25 μm. (D–G) Quantitative real‐time polymerase chain reaction data show the levels of TLR‐2, TLR‐4, TNF‐α, and MCP‐1 in the non‐injured and injured muscles of the three experimental groups. Results are the mean ± SD (n = 6). *P < 0.05, **P < 0.01 vs. the corresponding control groups by Student's t‐test or one‐way analysis of covariance followed by Tukey post hoc tests. NS, not significant.
We next performed gene expression assays to examine the effects of CatK inhibition on the cathepsin and MMP gene expressions. Although there were no changes in the CatS or CatL gene levels in the non‐injured or injured muscles among the three experimental groups, we observed that CatK inhibition exerted a reduction of the CatK mRNA expression of the injured muscles in a dose‐dependent manner (Figure S4A,B). Real‐time PCR data showed that the levels of the MMP‐2/‐9 and TIMP‐1/‐2 genes were lower in the CatK+/+ mice that underwent CatK inhibitor treatment in a dose‐dependent manner (Figure S4C,D). Consistently, the quantitative gelatin zymography assays revealed that CatK inhibition suppressed the levels of MMP‐2 and MMP‐9 gelatinolytic activities in the injured muscles of the CatK+/+ mice (Figure S4E,F).
Cathepsin K inhibition mitigated impaired muscle performance induced by cardiotoxin
We also investigated whether CatK inhibition could improve CTX‐induced muscle dysfunction at the indicated time points. As expected, we observed that the muscle performance (i.e. grip strength and change rate of workload, running time, and distance) was preserved in the CatK+/+ mice subjected to CatK inhibitor treatment compared with that in the control CatK+/+ mice at Day 14 after injury (Figure S5).
Cathepsin K inhibition prevented cardiotoxin‐induced C2C12 cell injury
Figure 8A shows the morphological changes of the differentiated C2C12 mouse myoblasts cultured in the presence and absence of CTX (0.5 μM) and CTX + CatK inhibitor ONO‐KK1‐300‐01 (25 μM, CTX + CatK‐I). As anticipated, CatK inhibition suppressed the levels of conditioned medium LDH in response to CTX (Figure 8B). We also observed that CatK inhibition prevented CTX‐induced C2C12 cell apoptosis (Figure 8C). The western blotting assays showed that CatK inhibition improved the changes in the levels of BAX and caspase‐3 proteins as well as the CatK protein levels (Figure 8D and 8E), indicating that CatK inhibition can protect against differentiated C2C12 apoptosis via a reduction of apoptosis‐related targeted protein expressions. In addition, we observed that CTX stimulated CatK gene expression in a time‐dependent and dose‐dependent manner, whereas the serum‐free medium had no effect on CatK gene expression (Figure S6). Moreover, quantitative collagenasse assay revealed that CatK inhibitor significantly suppressed CTX‐induced collagenolytic activity (14 896 ± 228 vs. 15 527 ± 246 intensity, P < 0.01).
Figure 8.
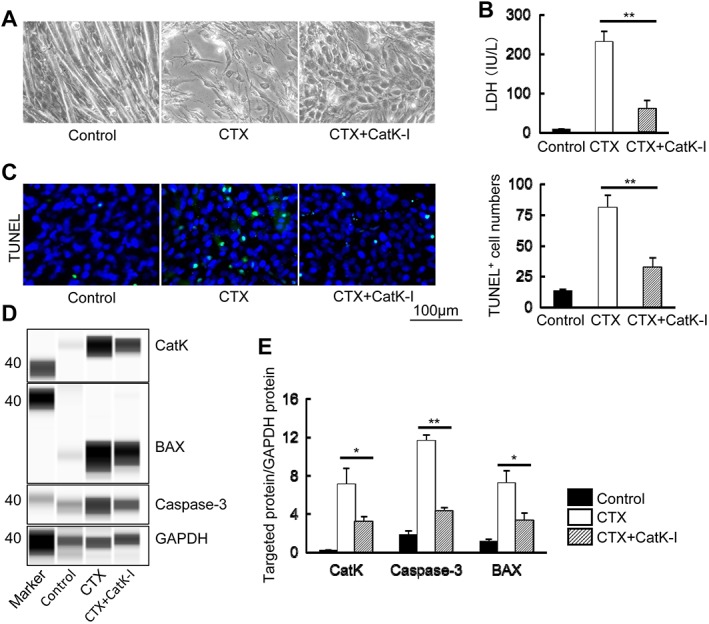
CatK inhibition prevented cardiotoxin (CTX)‐induced C2C12 cell apoptosis. (A) Representative images of morphological change of the differentiated C2C12 cells cultured in the presence and absence of cardiotoxin (0.5 μM) alone and in combination with the CatK inhibitor ONO‐KK1‐300‐01 (25 μM) for 24 h. (B) CatK inhibition suppressed the C2C12 cell‐conditioned medium lactate dehydrogenase (LDH) levels in response to CTX (n = 6). (C) Representative images of terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) immunofluorescence and combined quantitative data for the TUNEL+ cell numbers (n = 5). Scale bar, 100 μm. (D, E) Representative images of the simple western blots and combined quantitative data show the levels of CatK, caspase‐3, and BAX proteins in the lysates of the experimental groups (control, CTX, and CTX + CatK‐I; n = 3 per group). Results are the mean ± SD. *P < 0.05, **P < 0.01 vs. the corresponding controls by one‐way analysis of covariance followed by Tukey post hoc tests.
Discussion
Skeletal muscle apoptosis has been shown to participate in muscle weakness and wasting in response to injury. The identification of novel targets to prevent maladaptive skeletal muscle remodelling and fibrosis will contribute to therapeutic strategies to pre‐empt muscle mass wasting and dysfunction in response to various injuries. The significant finding of the present study is that mice lacking the CatK gene were resistant to acute CTX injury‐induced skeletal muscle loss and remodelling and the decline in muscle strength. At the molecular level, CatK deletion reduced CTX‐induced TLR‐2/‐4 and their downstream inflammatory gene expressions as well as inflammatory cell infiltrations (macrophages and leucocytes) and changes in apoptosis‐related molecules (i.e. caspase‐3, cleaved caspase‐3, cleaved caspase‐8, and the ratio of BAX to Bcl‐2). Our pharmacological intervention targeting CatK also resulted in a skeletal muscle protective action via the reduction of inflammation and cell apoptosis. In vitro, the specific CatK inhibitor attenuated the apoptosis of differentiated C2C12 muscle cells associated with the reductions of caspase‐3 and BAX proteins. To the best of our knowledge, this is the first study to provide evidence that genetic and pharmacological interventions targeting CatK confer skeletal muscle protection against acute CTX injury.
Cysteinyl cathepsins can degrade the basement membrane and surrounding ECM of cardiovascular walls.13, 14, 15 Cathepsin family members such as CatK and CatS have been shown to play a critical role in metabolic and inflammatory cardiovascular disorders.15, 16, 17 Several clinical studies reported that there is relationship between plasma CatK levels and cardiovascular disease stages and progression.18, 19, 20 Our research and studies by other groups have demonstrated the increased expression and activity of CatK in cardiovascular tissues in response to various injuries.15, 16, 17 The ability of CTX injury to increase CatK expression and activity probably contributed to the muscle repair under our experimental conditions. In agreement with reports that CatK deletion reduced cardiovascular remodelling in response to various injuries,24, 28 we observed that skeletal muscle loss and fibrosis and dysfunction after injuries are mitigated by CatK−/−. Our present findings showed that a pharmacological intervention targeting CatK also exhibited beneficial effects on skeletal muscle under the same experimental conditions. Collectively, these observations suggest that CatK may act as a key player to modulate skeletal muscle remodelling, fibrosis and dysfunction in response to CTX injury.
The engagement of TLRs on the cell surface by their specific ligands leads to an increase in the expressions of inflammatory chemokines and cytokines (e.g. MCP‐1, interleukin‐1β, and TNF‐α).40, 41, 42 In the present study, the injured muscles of CatK−/− mice contained much lower levels of TLR‐2 and TLR‐4 genes as well as TNF‐α and MCP‐1 genes compared with the control CatK+/+ mice. CatK deletion retarded the macrophage accumulation of the injured skeletal muscle tissues. The pharmacological inhibition of CatK also improved the inflammation‐related expressions of those genes and the inflammatory cell infiltration to the CTX injury in the CatK+/+ mice. Our previous study demonstrated a genetic or pharmacological inhibition of CatK ameliorated injury‐related vascular remodelling via the reduction of TLR‐2/‐4‐mediated inflammatory in the carotid artery.28 Thus, CatK−/− appears to mitigate macrophage infiltration and activation and inflammatory chemokine expression in injury‐stress states through its ability to reduce TLR‐2 and TLR‐4 expressions.
It is well established that among the members of the MMP family, MMP‐2 and MMP‐9, which are expressed and secreted mainly by inflammatory cells (i.e. macrophages and leucocytes), participate in muscle dystrophy and ECM remodelling.4, 5, 6 The ability of CTX injury to also increase both of these genes and their activities is likely to have contributed to the muscle interstitial fibrosis and dysfunction observed in the CTX‐treated mice in the present study. Data from the present and previous studies show that CatK inhibition by a genetic or pharmacological approach ameliorates not only MMP‐2 and MMP‐9 expression and activities but also targeted tissue/organ damage and dysfunction under conditions of ischaemic, surgical, and CTX injuries.28, 29 TLR‐2 and TLR‐4 have been shown to modulate monocyte/macrophage activation and inflammatory responses in animals.28 Other studies have demonstrated that both membrane‐type receptor activations are involved in the regulation of the CatK and MMP‐2/‐9 expressions in macrophages and endothelial cells by several intracellular signalling pathways.43, 44 Our present experiments revealed that the increased levels of TLR‐2 and TLR‐4 gene expression induced by CTX were alleviated by CatK inhibition in vivo. Thus, the ability of CatK inhibition to abolish TLR‐2/‐4‐mediated MMP‐2/‐9 expression and activities is attributable to the contribution of CatK inhibition to the muscle benefits in CatK−/− mice or ONO‐KK1‐300‐01‐treated CatK+/+ mice under our experimental conditions.
Given that the comprehensive reviews have focused on the roles of a cathepsin/cystatin system in cardiovascular and inflammatory cell events,11, 13 it is notable that CatK seems to be of particular importance for cell apoptosis in the cardiovascular repair process. Among the members of the lysosomal cathepsin family, CatC has been shown to facilitate lysosomal rupture and necrotic cell death.45 In addition, in vitro and in vivo experiments revealed that silencing of CatK inhibited cardiac cell apoptosis via reductions of mitochondrial cytochrome c production and proapoptotic molecule expression.24 Sun et al.43 showed that aneurysm lesions from CatK−/− mice contained fewer TUNEL+ apoptotic cells than did CatK+/+ mice.46 They also reported that a deficiency in the endogenous cathepsin inhibitor cystatin C resulted in epidermal hyperplasia via the modulation of epithelial apoptosis.47 Our present results showed that the muscle lesions in CatK−/− mice had lower numbers of TUNEL+ apoptotic cells compared with the muscle lesions of the control mice. ONO‐KK1‐300‐01 treatment also suppressed the cell apoptosis of the injured skeletal muscle tissues in the CatK+/+ mice. In vitro, CatK inhibition suppressed the CTX‐induced apoptosis of matured C2C12 cells. Because CTX injury induces CatK expression and activation in vivo and in vitro, we propose that CatK modulates skeletal muscle remodelling/loss and dysfunction in a CTX‐injury state through its ability to activate muscle apoptosis. BAX/Bcl‐2 and caspase‐3/‐8 molecules have been widely accepted as the critical modulators of various types of cell apoptosis. Our present findings indicate that genetic and pharmacological inhibitions of CatK reduced the protein levels of cleaved caspase‐3 and cleaved caspase‐8 and the ratio of BAX to Bcl‐2 in the muscle lesions. We also observed that the expressions of the TLR‐4 and TLR‐2 genes were decreased in injured muscles of CatK−/− mice and CatK+/+ mice treated with CatK‐I. In C2C12 cells, we have shown that CatK inhibition suppressed CTX‐induced BAX and caspase‐3 protein expressions. Several previous lines of investigation have demonstrated that TLR‐2 and TLR‐4 are required for cell apoptosis via the activation of the caspase‐8 and nuclear factor‐κB signalling pathway.48, 49 Further, it has been shown that TLR‐2/‐4 deficiency prevents oxygen‐induced endothelial apoptosis and vascular degeneration50 Caspase‐8 has been shown to be a key mediator in the interaction between the TLR‐2/‐4 axis.51 Taken together, these findings suggest that the attenuation of cell apoptosis by CatK inhibition via the improvement of TLR‐2/‐4‐mediated pro‐apoptotic and anti‐apoptotic molecule expressions may represent a common mechanism underlying the reduction of injury‐induced skeletal muscle remodelling and fibrosis under our experimental conditions.
Several limitations should be considered. First, the in vitro model used in the present study is not a proper model to represent CTX‐induced muscle fibre injury in vitro, especially considering that muscle fibres in vivo are generally replaced by fibrotic/connective tissue and show a high level of inflammatory cells that interact with muscle cells to reduce muscle cell function. Thus, our C2C12 cell experiments may not fully mimic all the muscle changes to address the related molecular mechanism in vivo. Second, CTX‐induced muscle injury model is not closely relevant to aging‐related muscle disease but rather to acute injury‐related muscle disease. Third, here, 10‐week‐old young male CatK+/+ and CatK−/− mice were applied to the CTX‐induced muscle injury model to study the role of CatK in age‐related muscle disease (i.e. sarcopenia/dynapenia). Aged both genotype mice will be more suitable to investigate our hypothesis. Fourth, we could not to deign to confirm our findings in generalizability to aging. Further studies should be look at aged animals and humans. Additionally, in 2015, Fry and colleagues demonstrated that inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia.52 Thus, injury‐induced muscle apoptosis appears to be one obvious contributory mechanism to age‐related muscle loss but not the be‐all and end‐all. Fifth, little or nothing is known about muscle in pycnodysostosis—the human disease caused by CatK deficiency. Sixth, it has been reported that ONO‐KK1‐300‐01, N‐{3‐[(2Z)‐2‐(3‐methyl‐1,3‐thiazolidin‐2‐ylidene)hydrazino]‐2,3‐dioxo‐1‐tetrahydro‐2H‐pyran‐4‐ylpropyl}cycloheptanecarboxamide hydrochloride, and ONO‐5334, N‐((1S)‐3‐{(2Z)‐2‐[(4R)‐3,4‐dimethyl‐1,3‐thiazolidin‐2‐ylidene]hydrazino}‐2,3‐dioxo‐1‐(tetrahydro‐2H‐pyran‐4‐yl)propyl)cycloheptanecarboxamide, were synthesized in ONO Pharmaceutical Co., Ltd. (Osaka, Japan).53, 54 The former is hydrochloride, and the latter has one more methyl group.53, 54 Both CatK inhibitors have shown to ameliorate bone density and/or bone resporption in several animal metabolic bone disease models.53, 54 Similarly, the data from a small scale clinical studies reported that ONO‐5334 mitigated bone mineral density without the change in bone size.55 However, we have pointed out one large clinical LOFT trial (the Long‐Term Odanacatib Fracture Trail) that was started to treat with ONO‐5334 post‐menopausal osteoporosis.56 Unfortunately, this trial has led to discontinuation of the development of odanacatib for the treatment of osteoporosis due to cardiovascular side effects.56 These negative data of CatK inhibitor prompt clinicians to look at some muscle/sarcopenia endpoints in existing clinical trial data.
Conclusions
The ability of injury to increase skeletal muscle apoptosis have been shown to contribute to the age‐associated muscle weakness and wasting in a wide range of mammals. However, the molecular mechanisms by which injury causes muscle cell apoptosis and remodelling are poorly understood. The expressions of the CatK gene and protein are known to be increased in injured muscle tissues. Here, CatK deletion was shown to prevent acute CTX‐induced muscle apoptosis, remodelling and fibrosis and muscle dysfunction. The pharmacological inhibition of CatK mimics the protective effects of genetic CatK ablation on skeletal muscle. It seems that selective CatK inhibitors may have potential utility in the treatment or control of injury‐related skeletal muscle apoptosis, remodelling and dysfunction in age‐associated muscle disorders. However, it remains to be determined whether pharmacological CatK inhibition has muscle benefits in humans, and whether or not, this will not be hampered by other unwanted side effects. Further studies will be also needed to investigate these issues.
Conflict of interest
M.K. received research support from the ONO Pharmaceutical Co., Ltd. (Osaka, Japan). However, ONO had no involvement in the process of manuscript development and did not influence the content of this manuscript in any manner. The other authors declare no conflicts of interest.
Author contribution statement
S.O.: Major contributor to the collection and assembly of data; manuscript writing; morphological analysis; western blotting and real‐time PCR analysis; collection and assembly of samples and data, data analysis and interpretation. X.W.C.: Major contributor to the collection and assembly of data; financial support; conception, design and editing of the manuscript. A.I. and H.G.: Collection and assembly of samples and data, data analysis and interpretation. L.H., L.P., C.Y., H.G., W.X., G.Z., L.Y., G.Y., and K.K.: CatK genotyping and bleeding. H.U. and G.P.S.: Reviewed/edited the manuscript. M.K.: Financial support; conception, design and editing of the manuscript.
Supporting information
Table S1. Primer sequences for mice used for quantitative real‐time PCR
Figure S1. Expressions of matrix metalloproteinase (MMP) and cathepsin family genes in the muscles of CatK+/+ mice at the indicated time points after cardiotoxin (CTX) injection. A: Photos of both gastrocnemius mass and representative microscopy images of H&E staining of the non‐injured and injured muscles of CatK+/+ mice on days 3 and 14 after injury. B–D: Quantitative real‐time PCR data show the levels of CatK, CatS, CatL, MMP‐2, MMP‐9, tissue inhibitor of metalloproteinase (TIMP)‐1, and TIMP‐2 at days 0, 3, 7, and 14 after injury. Results are the mean ± SD (n = 6). E: A representative result of gelatin zymography shows the levels of MMP‐2 and MMP‐9 gelatinolytic activities of the non‐injured and injured muscles of CatK+/+ mice at day 3 after CTX injection. *p < 0.05, **p < 0.01 vs. the corresponding day 0 by one‐way ANOVA followed by Tukey post hoc tests.
Figure S2. Effects of CatK inhibition on the levels of caspase‐3 and cleaved caspase‐3 in the muscles. A‐B: Representative images of Western blots and the combined quantitative data show the ratio of cleaved caspase‐3 to caspase‐3 band intensities in the injured muscles of CatK+/+ and CatK−/− mice. C‐D: Representative images of Western blots and the combined quantitative data show the ratio of cleaved caspase‐3 to caspase‐3 band intensities in the injured muscles of CONT, LKI (low‐dose CatK inhibitor) and HKI (high‐dose CatK inhibitor) mice.
Figure S3. The gene expressions of cathepsin and MMP families in the skeletal muscles of mice of both genotypes at day 3 after injury. A–C: Quantitative real‐time PCR data show the levels of CatS, CatL, MMP‐2, MMP‐9, TIMP‐1, and TIMP‐2 genes in the non‐injured and injured gastrocnemius muscles of mice of both genotypes. Results are the mean ± SD (n = 5). D,E: Representative images of gelatin zymography and combined quantitative data show the gelatinolytic activities of MMP‐2 and MMP‐9 in the non‐injured and injured gastrocnemius muscles of mice in both experimental groups. Results are the mean ± SD (n = 5). *p < 0.05, **p < 0.01 vs. the corresponding control groups by Student's t‐test or one‐way ANOVA followed by Tukey post hoc tests. NS: not significant.
Figure S4. Evaluation of cathepsin and MMP gene expressions in the non‐injured and injured gastrocnemius muscles of CatK+/+ mice treated with vehicle (0.5% carboxymethylcellulose; CONT mice) or a low or high dose of the specific CatK inhibitor ONO‐KK1–300‐01 (3 mg/kg, LKI mice; 30 mg/kg, HKI mice) at day 3 after CTX injury. A–D: Quantitative real‐time PCR data show the levels of CatS, CatK, CatL, MMP‐2, MMP‐9, TIMP‐1, and TIMP‐2 in the non‐injured and injured muscles of the three experimental groups. Results are the mean ± SD (n = 6). E,F: Representative images of gelatin zymography and combined quantitative data show the gelatinolytic activities of MMP‐2 and MMP‐9 of the injured gastrocnemius in the three experimental groups (n = 4). *p < 0.05, **p < 0.01 vs. the corresponding control groups by Student's t‐test or one‐way ANOVA followed by Tukey post hoc tests. NS: not significant.
Figure S5. CatK inhibition prevented CTX‐induced muscle function. A: Grip strength was measured over 5 times in each mouse on 0, 3 and 14 days after CTX injury and averaged to obtain the grip strength for each time point. B: Workload in the vertical direction was calculated in consideration of the weight of the mouse (= weight × gravitational acceleration × mileage in the vertical direction). C,D: Running time and distance were calculated from the product of running time and speed. All items were expressed as the ratio of observed values to data on day 0 before CTX injection. Results are the mean ± SD (n = 3). *P < 0.05, vs. the corresponding controls by 2‐way repeated‐measures ANOVA and Bonferroni post hoc tests.
Figure S6. Changes in the expression of CatK mRNA induced by CTX in a time‐ and dose‐dependent manner. A: Microscopy images and quantitative real‐time PCR data show the levels of CatK mRNA in the serum‐free medium at the indicated time points. B: Representative images and quantitative real‐time PCR data show changes in the CTX‐induced CatK mRNA expression over time. C: Representative images and quantitative real‐time PCR data show changes in the CTX‐induced CatK mRNA expression with dose. Scale bars: A, 50 μm. *p < 0.05, **p < 0.01 vs. the corresponding control groups by Student's t‐test or one‐way ANOVA followed by Tukey post hoc tests. NS: not significant.
Acknowledgements
We thank K. Suzuki and M. Hasegawa for their technical assistance. The authors certify that the study complies with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015.57 This work was supported in part by the National Natural Science Foundation of China (nos. 81560240 and 81770485); by the Ministry of Education, Culture, Sports, Science, and Technology of Japan (15K15270, 16K15410, 15H04801, and 15H04802), by the Novartis Aging and Geriatrics Research Foundation (25‐7778), and by an Ono Pharmaceutical Corporate Joint Research Grant (2615Dk‐21b).
Ogasawara, S. , Cheng, X. W. , Inoue, A. , Hu, L. , Piao, L. , Yu, C. , Goto, H. , Xu, W. , Zhao, G. , Lei, Y. , Yang, G. , Kimura, K. , Umegaki, H. , Shi, G.‐P. , and Kuzuya, M. (2018) Cathepsin K activity controls cardiotoxin‐induced skeletal muscle repair in mice. Journal of Cachexia, Sarcopenia and Muscle, 9: 160–175. doi: 10.1002/jcsm.12248.
Institution where the study was conducted: Nagoya University Graduate School of Medicine
References
- 1. Arthur ST, Cooley ID. The effect of physiological stimuli on sarcopenia; impact of Notch and Wnt signaling on impaired aged skeletal muscle repair. Int J Biol Sci 2012;8:731–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Girgis CM, Baldock PA, Downes M. Vitamin D, muscle and bone: integrating effects in development, aging and injury. Mol Cell Endocrinol 2015;410:3–10. [DOI] [PubMed] [Google Scholar]
- 3. Peake J, Della Gatta P, Cameron‐Smith D. Aging and its effects on inflammation in skeletal muscle at rest and following exercise‐induced muscle injury. Am J Physiol Regul Integr Comp Physiol 2010;298:R1485–R1495. [DOI] [PubMed] [Google Scholar]
- 4. Dahiya S, Bhatnagar S, Hindi SM, Jiang C, Paul PK, Kuang S, et al. Elevated levels of active matrix metalloproteinase‐9 cause hypertrophy in skeletal muscle of normal and dystrophin‐deficient mdx mice. Hum Mol Genet 2011;20:4345–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Urso ML, Wang R, Zambraski EJ, Liang BT. Adenosine A 3 receptor stimulation reduces muscle injury following physical trauma and is associated with alterations in the MMP/TIMP response. J Appl Physiol 2010;112:658–670. [DOI] [PubMed] [Google Scholar]
- 6. Zimowska M, Olszynski KH, Swierczynska M, Streminska W, Ciemerych MA. Decrease of MMP‐9 activity improves soleus muscle regeneration. Tissue Eng Part A 2012;18:1183–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bellayr I, Holden K, Mu X, Pan H, Li Y. Matrix metalloproteinase inhibition negatively affects muscle stem cell behavior. Int J Clin Exp Pathol 6:124–141. [PMC free article] [PubMed] [Google Scholar]
- 8. Ogura Y, Tajrishi MM, Sato S, Hindi SM, Kumar A. Therapeutic potential of matrix metalloproteinases in Duchenne muscular dystrophy. Front Cell Dev Biol 2014;2:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yeghiazaryan M, Zybura‐Broda K, Cabaj A, Wlodarczyk J, Slawinska U, Rylski M, et al. Fine‐structural distribution of MMP‐2 and MMP‐9 activities in the rat skeletal muscle upon training: a study by high‐resolution in situ zymography. Histochem Cell Biol 2010;138:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turk B, Turk V, Turk D. Structural and functional aspects of papain‐like cysteine proteinases and their protein inhibitors. Biol Chem 1997;378:141–150. [PubMed] [Google Scholar]
- 11. Cheng XW, Huang Z, Kuzuya M, Okumura K, Murohara T. Cysteine protease cathepsins in atherosclerosis‐based vascular disease and its complications. Hypertension 2011;58:978–986. [DOI] [PubMed] [Google Scholar]
- 12. Turk B, Turk D, Turk V. Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta 2000;1477:98–111. [DOI] [PubMed] [Google Scholar]
- 13. Cheng XW, Shi GP, Kuzuya M, Sasaki T, Okumura K, Murohara T. Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation 2012;125:1551–1562. [DOI] [PubMed] [Google Scholar]
- 14. Song H, Jiang H, Nan Y, Cheng X. Degesting the remodeled cardiovascular wall: role of cysteinyl cathepsins in cardiovascular disease. Minerva Cardioangiol 2015;63:525–531. [PubMed] [Google Scholar]
- 15. Cheng XW, Sasaki T, Kuzuya M. The role of cysteinyl cathepsins in venous disorders. Thromb Haemost 2014;112:216–218. [DOI] [PubMed] [Google Scholar]
- 16. Wu H, Cheng XW, Hu L, Takeshita K, Hu C, Du Q, et al. Cathepsin S activity controls injury‐related vascular repair in mice via the TLR2‐mediated p38MAPK and PI3K‐Akt/p‐HDAC6 signaling pathway. Arterioscler Thromb Vasc Biol 2016;36:1549–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu H, Cheng XW, Hu L, Hao CN, Hayashi M, Takeshita K, et al. Renin inhibition reduces atherosclerotic plaque neovessel formation and regresses advanced atherosclerotic plaques. Atherosclerosis 2014;237:739–747. [DOI] [PubMed] [Google Scholar]
- 18. Li X, Li Y, Jin J, Jin D, Cui L, Rei Y, et al. Increased serum cathepsin K in patients with coronary artery disease. Yonsei Med J 2014;55:912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao G, Li Y, Cui L, Li X, Jin Z, Han X, et al. Increased circulating cathepsin K in patients with chronic heart failure. PLoS One 2015;10: e0136093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Izumi Y, Hayashi M, Morimoto R, Cheng XW, Wu H, Ishii H, et al. Impact of circulating cathepsin K on the coronary calcification and the clinical outcome in chronic kidney disease patients. Heart Vessels 2017;31:6–14. [DOI] [PubMed] [Google Scholar]
- 21. Li X, Cheng XW, Hu L, Wu H, Guo P, Hao CN, et al. Cathepsin S activity controls ischemia‐induced neovascularization in mice. Int J Cardiol 2015;183:198–208. [DOI] [PubMed] [Google Scholar]
- 22. Cheng XW, Murohara T, Kuzuya M, Izawa H, Sasaki T, Obata K, et al. Superoxide‐dependent cathepsin activation is associated with hypertensive myocardial remodeling and represents a target for angiotensin II type 1 receptor blocker treatment. Am J Pathol 2008;173:358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheng XW, Obata K, Kuzuya M, Izawa H, Nakamura K, Asai E, et al. Elastolytic cathepsin induction/activation system exists in myocardium and is upregulated in hypertensive heart failure. Hypertension 2006;48:979–987. [DOI] [PubMed] [Google Scholar]
- 24. Hua Y, Zhang Y, Dolence J, Shi GP, Ren J, Nair S. Cathepsin K knockout mitigates high‐fat diet‐induced cardiac hypertrophy and contractile dysfunction. Diabetes 2013;62:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Z, Hou WS, Bromme D. Collagenolytic activity of cathepsin K is specifically modulated by cartilage‐resident chondroitin sulfates. Biochemistry 2000;39:529–536. [DOI] [PubMed] [Google Scholar]
- 26. Cheng XW, Kuzuya M, Sasaki T, Arakawa K, Kanda S, Sumi D, et al. Increased expression of elastolytic cysteine proteases, cathepsins S and K, in the neointima of balloon‐injured rat carotid arteries. Am J Pathol 2004;164:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest 1998;102:576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu L, Cheng XW, Song H, Inoue A, Jiang H, Li X, et al. Cathepsin K activity controls injury‐related vascular repair in mice. Hypertension 2014;63:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiang H, Cheng XW, Shi GP, Hu L, Inoue A, Yamamura Y, et al. Cathepsin K‐mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat Commun 2014;5:3838. [DOI] [PubMed] [Google Scholar]
- 30. Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J Clin Invest 2010;120:3421–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Inoue A, Cheng XW, Huang Z, Hu L, Kikuchi R, Jiang H, et al. Exercise restores muscle stem cell mobilization, regenerative capacity and muscle metabolic alterations via adiponectin/AdipoR1 activation in SAMP10 mice. J Cachexia Sarcopenia Muscle 2016. Nov 29;https://doi.org/10.1002/jcsm.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng XW, Kuzuya M, Nakamura K, Di Q, Liu Z, Sasaki T, et al. Localization of cysteine protease, cathepsin S, to the surface of vascular smooth muscle cells by association with integrin alphanubeta3. Am J Pathol 2006;168:685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuzuya M, Nakamura K, Sasaki T, Cheng XW, Itohara S, Iguchi A. Effect of MMP‐2 deficiency on atherosclerotic lesion formation in apoE‐deficient mice. Arterioscler Thromb Vasc Biol 2006;26:1120–1125. [DOI] [PubMed] [Google Scholar]
- 34. Cheng XW, Kuzuya M, Kim W, Song H, Hu L, Inoue A, et al. Exercise training stimulates ischemia‐induced neovascularization via phosphatidylinositol 3‐kinase/Akt‐dependent hypoxia‐induced factor‐1 alpha reactivation in mice of advanced age. Circulation 2010;122:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cheng XW, Kuzuya M, Nakamura K, Maeda K, Tsuzuki M, Kim W, et al. Mechanisms underlying the impairment of ischemia‐induced neovascularization in matrix metalloproteinase 2‐deficient mice. Circ Res 2007;100:904–913. [DOI] [PubMed] [Google Scholar]
- 36. Harris VM. Protein detection by Simple Western™ analysis. Methods Mol Biol 2015;1312:465–468. [DOI] [PubMed] [Google Scholar]
- 37. Kimura K, Cheng XW, Inoue A, Hu L, Koike T, Kuzuya M. β‐Hydroxy‐β‐methylbutyrate facilitates PI3K/Akt‐dependent mammalian target of rapamycin and FoxO1/3a phosphorylations and alleviates tumor necrosis factor alpha/interferon gamma‐induced MuRF‐1 expression in C2C12 cells. Nutr Res 2014;34:368–374. [DOI] [PubMed] [Google Scholar]
- 38. Liu N, Nelson BR, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel‐Duby R, et al. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc Natl Acad Sci U S A 2014;111:4109–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mnafgui K, Hajji R, Derbali F, Gammoudi A, Khabbabi G, Ellefi H, et al. Anti‐inflammatory, antithrombotic and cardiac remodeling preventive effects of eugenol in isoproterenol‐induced myocardial infarction in Wistar rat. Cardiovasc Toxicol 2016;16:336–344. [DOI] [PubMed] [Google Scholar]
- 40. Saxena A, Rauch U, Berg KE, Andersson L, Hollender L, Carlsson AM, et al. The vascular repair process after injury of the carotid artery is regulated by IL‐1RI and MyD88 signalling. Cardiovasc Res 2016;91:350–357. [DOI] [PubMed] [Google Scholar]
- 41. Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, et al. Toll‐like receptor‐4 mediates vascular inflammation and insulin resistance in diet‐induced obesity. Circ Res 2007;100:1589–1596. [DOI] [PubMed] [Google Scholar]
- 42. Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll‐like receptor 2. J Clin Invest 2005;115:3149–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun Y, Ishibashi M, Seimon T, Lee M, Sharma SM, Fitzgerald KA, et al. Free cholesterol accumulation in macrophage membranes activates Toll‐like receptors and p38 mitogen‐activated protein kinase and induces cathepsin K. Circ Res 2009;104:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng XW, Song H, Sasaki T, Hu L, Inoue A, Bando YK, et al. Angiotensin type 1 receptor blocker reduces intimal neovascularization and plaque growth in apolipoprotein E‐deficient mice. Hypertension 2011;57:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brojatsch J, Lima H, Kar AK, Jacobson LS, Muehlbauer SM, Chandran K, et al. A proteolytic cascade controls lysosome rupture and necrotic cell death mediated by lysosome‐destabilizing adjuvants. PLoS One 2014;9: e95032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun J, Sukhova GK, Zhang J, Chen H, Sjoberg S, Libby P, et al. Cathepsin K deficiency reduces elastase perfusion‐induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol 2012;32:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu W, Liu J, Shi MA, Wang J, Xiang M, Kitamoto S, et al. Cystatin C deficiency promotes epidermal dysplasia in K14‐HPV16 transgenic mice. PLoS One 2010;5: e13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H, Zhu C, Wang B, Zhu W, Feng Y, Du F, et al. 17β‐Estradiol protects the retinal nerve cells suppressing TLR2 mediated immune‐inflammation and apoptosis from oxidative stress insult independent of PI3K. J Mol Neurosci 2016;60:195–204. [DOI] [PubMed] [Google Scholar]
- 49. Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A. The apoptotic signaling pathway activated by Toll‐like receptor‐2. EMBO J 2000;19:3325–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. He C, Lai P, Wang J, Zhou T, Huang Z, Zhou L, et al. TLR2/4 deficiency prevents oxygen‐induced vascular degeneration and promotes revascularization by downregulating IL‐17 in the retina. Sci Rep 2016;6: 27739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lemmers B, Salmena L, Bidere N, Su H, Matysiak‐Zablocki E, Murakami K, et al. Essential role for caspase‐8 in Toll‐like receptors and NFkappaB signaling. J Biol Chem 2007;282:7416–7423. [DOI] [PubMed] [Google Scholar]
- 52. Fry CS, Lee JD, Mula J, Kirby TJ, Jackson JR, Liu F, et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med 2015;21:76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ochi Y, Yamada H, Mori H, Kawada N, Tanaka M, Imagawa A, et al. Combination therapy with ONO‐KK1‐300‐01, a cathepsin K inhibitor, and parathyroid hormone results in additive beneficial effect on bone mineral density in ovariectomized rats. J Bone Miner Metab 2016;34:33–40. [DOI] [PubMed] [Google Scholar]
- 54. Ochi Y, Yamada H, Mori H, Nakanishi Y, Nishikawa S, Kayasuga R, et al. Effects of ONO‐5334, a novel orally‐active inhibitor of cathepsin K, on bone metabolism. Bone 2011;49:1351–1356. [DOI] [PubMed] [Google Scholar]
- 55. Engelke K, Nagase S, Fuerst T, Small M, Kuwayama T, et al. The effect of the cathepsin K inhibitor ONO‐5334 on trabecular and cortical bone in postmenopausal osteoporosis: the OCEAN study. J Bone Miner Res 2014;29:629–638. [DOI] [PubMed] [Google Scholar]
- 56. Bone HG, Dempster DW, Eisman JA, Greenspan SL, McClung MR, Nakamura T, et al. Odanacatib for the treatment of postmenopausal osteoporosis: development history and design and participant characteristics of LOFT, the Long‐Term Odanacatib Fracture Trial. Osteoporos Int 2014;26:699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences for mice used for quantitative real‐time PCR
Figure S1. Expressions of matrix metalloproteinase (MMP) and cathepsin family genes in the muscles of CatK+/+ mice at the indicated time points after cardiotoxin (CTX) injection. A: Photos of both gastrocnemius mass and representative microscopy images of H&E staining of the non‐injured and injured muscles of CatK+/+ mice on days 3 and 14 after injury. B–D: Quantitative real‐time PCR data show the levels of CatK, CatS, CatL, MMP‐2, MMP‐9, tissue inhibitor of metalloproteinase (TIMP)‐1, and TIMP‐2 at days 0, 3, 7, and 14 after injury. Results are the mean ± SD (n = 6). E: A representative result of gelatin zymography shows the levels of MMP‐2 and MMP‐9 gelatinolytic activities of the non‐injured and injured muscles of CatK+/+ mice at day 3 after CTX injection. *p < 0.05, **p < 0.01 vs. the corresponding day 0 by one‐way ANOVA followed by Tukey post hoc tests.
Figure S2. Effects of CatK inhibition on the levels of caspase‐3 and cleaved caspase‐3 in the muscles. A‐B: Representative images of Western blots and the combined quantitative data show the ratio of cleaved caspase‐3 to caspase‐3 band intensities in the injured muscles of CatK+/+ and CatK−/− mice. C‐D: Representative images of Western blots and the combined quantitative data show the ratio of cleaved caspase‐3 to caspase‐3 band intensities in the injured muscles of CONT, LKI (low‐dose CatK inhibitor) and HKI (high‐dose CatK inhibitor) mice.
Figure S3. The gene expressions of cathepsin and MMP families in the skeletal muscles of mice of both genotypes at day 3 after injury. A–C: Quantitative real‐time PCR data show the levels of CatS, CatL, MMP‐2, MMP‐9, TIMP‐1, and TIMP‐2 genes in the non‐injured and injured gastrocnemius muscles of mice of both genotypes. Results are the mean ± SD (n = 5). D,E: Representative images of gelatin zymography and combined quantitative data show the gelatinolytic activities of MMP‐2 and MMP‐9 in the non‐injured and injured gastrocnemius muscles of mice in both experimental groups. Results are the mean ± SD (n = 5). *p < 0.05, **p < 0.01 vs. the corresponding control groups by Student's t‐test or one‐way ANOVA followed by Tukey post hoc tests. NS: not significant.
Figure S4. Evaluation of cathepsin and MMP gene expressions in the non‐injured and injured gastrocnemius muscles of CatK+/+ mice treated with vehicle (0.5% carboxymethylcellulose; CONT mice) or a low or high dose of the specific CatK inhibitor ONO‐KK1–300‐01 (3 mg/kg, LKI mice; 30 mg/kg, HKI mice) at day 3 after CTX injury. A–D: Quantitative real‐time PCR data show the levels of CatS, CatK, CatL, MMP‐2, MMP‐9, TIMP‐1, and TIMP‐2 in the non‐injured and injured muscles of the three experimental groups. Results are the mean ± SD (n = 6). E,F: Representative images of gelatin zymography and combined quantitative data show the gelatinolytic activities of MMP‐2 and MMP‐9 of the injured gastrocnemius in the three experimental groups (n = 4). *p < 0.05, **p < 0.01 vs. the corresponding control groups by Student's t‐test or one‐way ANOVA followed by Tukey post hoc tests. NS: not significant.
Figure S5. CatK inhibition prevented CTX‐induced muscle function. A: Grip strength was measured over 5 times in each mouse on 0, 3 and 14 days after CTX injury and averaged to obtain the grip strength for each time point. B: Workload in the vertical direction was calculated in consideration of the weight of the mouse (= weight × gravitational acceleration × mileage in the vertical direction). C,D: Running time and distance were calculated from the product of running time and speed. All items were expressed as the ratio of observed values to data on day 0 before CTX injection. Results are the mean ± SD (n = 3). *P < 0.05, vs. the corresponding controls by 2‐way repeated‐measures ANOVA and Bonferroni post hoc tests.
Figure S6. Changes in the expression of CatK mRNA induced by CTX in a time‐ and dose‐dependent manner. A: Microscopy images and quantitative real‐time PCR data show the levels of CatK mRNA in the serum‐free medium at the indicated time points. B: Representative images and quantitative real‐time PCR data show changes in the CTX‐induced CatK mRNA expression over time. C: Representative images and quantitative real‐time PCR data show changes in the CTX‐induced CatK mRNA expression with dose. Scale bars: A, 50 μm. *p < 0.05, **p < 0.01 vs. the corresponding control groups by Student's t‐test or one‐way ANOVA followed by Tukey post hoc tests. NS: not significant.