

Abstract
Background
Muscle wasting is observed in the course of many diseases and also during physiological conditions (disuse, ageing). Skeletal muscle mass is largely controlled by the ubiquitin‐proteasome system and thus by the ubiquitinating enzymes (E2s and E3s) that target substrates for subsequent degradation. MuRF1 is the only E3 ubiquitin ligase known to target contractile proteins (α‐actin, myosins) during catabolic situations. However, MuRF1 depends on E2 ubiquitin‐conjugating enzymes for ubiquitin chain formation on the substrates. MuRF1‐E2 couples are therefore putative targets for preventing muscle wasting.
Methods
We focused on 14 E2 enzymes that are either expressed in skeletal muscle or up‐regulated during atrophying conditions. In this work, we demonstrated that only highly sensitive and complementary interactomic approaches (surface plasmon resonance, yeast three‐hybrid, and split green fluorescent protein) allowed the identification of MuRF1 E2 partners.
Results
Five E2 enzymes physically interacted with MuRF1, namely, E2E1, E2G1, E2J1, E2J2, and E2L3. Moreover, we demonstrated that MuRF1‐E2E1 and MuRF1‐E2J1 interactions are facilitated by telethonin, a newly identified MuRF1 substrate. We next showed that the five identified E2s functionally interacted with MuRF1 since, in contrast to the non‐interacting E2D2, their co‐expression in HEK293T cells with MuRF1 led to increased telethonin degradation. Finally, we showed that telethonin governed the affinity between MuRF1 and E2E1 or E2J1.
Conclusions
We report here the first MuRF1‐E2s network, which may prove valuable for deciphering the precise mechanisms involved in the atrophying muscle programme and for proposing new therapeutical approaches.
Keywords: E3 ubiquitin ligase, muscle wasting, ubiquitin‐conjugating enzyme, UBE2, Tcap, split‐GFP
Introduction
Muscle wasting prevails in many physiological (disuse, ageing) or pathological conditions [cancer cachexia, trauma, sepsis, chronic diseases (such as kidney failure, cardiovascular diseases, and chronic obstructive pulmonary disease), diabetes, and muscular dystrophies] and results in reduced mobility and impaired quality of life.1, 2 Indeed, skeletal muscle represents ~40% of the body protein mass and is the major reservoir of amino acids mobilized during disease and/or nutritional disorders for providing energy, synthesis of acute phase proteins, and the immune response. Short‐term muscle wasting is a key metabolic adaptation that presents numerous benefits. However, long‐term muscle wasting leads to impaired strength and mobility that contribute to decreased immune response, increased periods of hospitalization, long recovery periods, and increased morbidity and mortality. Consequently, reducing muscle protein loss during catabolic states is a major public health issue. A better understanding of the mechanisms involved is necessary to set up appropriate therapeutic strategies for reducing/preventing muscle wasting.
Telethonin, also known as titin‐cap protein or Tcap, was first identified as a structural protein in skeletal muscle, titin‐telethonin complex anchoring filaments to the Z‐disc of the sarcomere. Telethonin gene mutations or reduction of telethonin protein levels lead to muscular dystrophy LGMD2G (limb‐girdle muscular dystrophy type 2G),3 cardiomyopathies,4, 5, 6 or intestinal pseudo‐obstructions.7 Telethonin is present in cytosolic and nuclear compartments8 and now appears as a multifaceted protein. Telethonin may be involved in the development and/or maintenance of transverse tubule organization.9, 10 Cardiac telethonin is a member of a Z‐disc stretch sensor complex including titin and muscle LIM protein (MLP).5 Furthermore, the identification of telethonin partners such as myostatin,11 calsarcin,12 Ankrd2,13 and BMP1014 suggests potential signalling roles. This is supported by findings showing that telethonin promotes Mdm2‐mediated degradation of the pro‐apoptotic transcription factor p53 in the nuclear compartment upon biomechanical stress.8
The ubiquitin‐proteasome system (UPS), together with autophagy, controls skeletal muscle proteolysis and protein mass. Proteins to be degraded by the 26S proteasome are tagged by an ubiquitin (Ub) chain. This highly regulated process depends on the sequential action of the E1 ubiquitin‐activating enzyme, an E2 ubiquitin‐conjugating enzyme, and an E3 ubiquitin ligase. The process is initiated by E1 that activates and transfers Ub to the active site cysteine of an E2. The latter interacts with an E3 ligase that recruits specific substrates for catalyzing ubiquitination on a lysine residue via an isopeptide linkage. A specific E2 can interact with several E3s and vice versa. While numerous E2s have been identified in mammals (32 Ub‐specific, 6 Ub‐like specific), several hundreds of E3s have been predicted (>700 in the human genome), allowing infinite combinations. Each of them modulates the ubiquitination of a distinct set of substrates. Ubiquitination is a versatile post‐translational protein modification that includes several signals, from monoUb to structurally distinct polyUb chains that confer different functional properties to the tagged proteins: degradation, localization, or roles in DNA repair, cell‐cycle progression, innate immunity, and inflammation.15, 16, 17
E3 ligases have been extensively studied because they recognize the substrates to be degraded. However, E2s are considered as central players of the enzymatic cascade and are in most cases the only depository of the catalytic activity18 except for HECT E3s. E2s not only catalyze the attachment of Ub onto substrates but also define the type of modification (monoUb, polyUb) and thus the fate of the substrate.19, 20 Indeed, E2s direct the ubiquitination process to distinct subsets of Lys residues used.21 In other words, deciphering E2–E3 couples and their physiological substrates is a key goal for understanding physiologically relevant mechanisms and represents new avenues for the development of therapeutic approaches.
MuRF1 is a muscle‐specific RING‐type E3 Ub ligase systematically overexpressed in muscle‐wasting conditions. Interestingly, MuRF1 knockout partially protected skeletal muscles from atrophy in denervated or immobilized animals.22 MuRF1 targets major myofibrillar proteins (MHCI and MHCIIa, troponin‐I and α‐actin) for subsequent degradation.23, 24, 25, 26 Because other MuRF1 targets do not seem to be linked to muscle atrophy,27, 28, 29 blocking whole MuRF1 activity will likely affect other pathways and may be deleterious for muscle. Inhibiting MuRF1‐E2 couples dedicated to contractile protein targeting may prove to be more efficient for fighting against muscle atrophy without side effects.
While MuRF1 is a crucial player in muscle wasting, little is known about its E2 partners. Frameworks of E2‐RING E3 interactions of the human UPS have been reported using yeast two‐hybrid (Y2H) approaches.30, 31, 32 However, a single study included the E3 MuRF1, and experimental data did not fit with the conclusions drawn by the authors, so that more work is clearly needed.30 In skeletal muscle, UBE2B, formerly E2 14‐kDa, is the only E2 that was more deeply addressed. This E2 is regularly overexpressed at the mRNA level in skeletal muscle upon atrophy.33, 34, 35 We recently reported an important role of UBE2B on muscle protein homeostasis but independently of MuRF1. Indeed, we showed that UBE2B is recruited even upon mild atrophying conditions and targets the cytoplasmic pool of α‐actin and myosin heavy chain.36 UBE2D2 and other members of the family (D1 and D3) have been repeatedly used with MuRF1 for in vitro ubiquitination assays.23, 24, 25 However, we recently reported that UBE2D2 is neither an MuRF1 partner nor a key player in muscle wasting.37 Therefore, results with in vitro ubiquitination assays using UBE2D2 were probably artefactual because this E2 often interacts with RING‐type E3s32 and is known for its intrinsic high‐catalytic activity in vitro.18
Overall, the E2 enzyme(s) that catalyze(s) Ub chains with MuRF1 in muscle wasting and potentially lead(s) to muscle atrophy is (are) unknown. In this work, we screened for muscle E2s interacting with MuRF1. Amongst different strategies, a highly sensitive interactomic approach such as surface plasmon resonance (SPR) led to the identification of five E2 enzymes interacting with MuRF1, namely, E2E1, E2EG1, E2J1, E2J2, and E2L3. We also report differential E2‐MuRF1 interactions regarding strength, affinity, and kinetics parameters. Moreover, we show that a third partner, such as telethonin, can stabilize and reinforce such interactions and identified telethonin as a new MuRF1 target. The MuRF1‐E2 framework we describe here may be a promising way for developing new therapeutics specifically protecting the contractile apparatus.
Materials and methods
Reagents
Anti‐MuRF1 (C20) and anti‐telethonin (G11) antibodies were obtained from Santa Cruz Biotechnology. Untagged recombinant proteins E2C, E2D2, and E2K were from Enzo Life Sciences, and E2G2, E2L3, E2N, and E2Z from Life Sensors.
Yeast strains
The Saccharomyces cerevisiae yeast strains used for yeast two‐hybrid and three‐hybrid experiments were from Clontech: (i) Y2HGold: MATa, trp1‐901, leu2‐3, 112, ura3‐52, his3‐200, gal4Δ, gal80Δ, LYS2::GAL1UAS–Gal1TATA–His3, GAL2UAS–Gal2TATA–Ade2, URA3::MEL1UAS–Mel1TATA‐AUR1‐C, MEL1. (ii) Y187: MATα, ura3‐52, his3‐200, ade2‐101, trp1‐901, leu2‐3, 112, gal4Δ, gal80Δ, met–, URA3::GAL1UAS–Gal1TATA–LacZ, MEL1. In S. cerevisiae, AUR1‐C expression confers strong resistance to the highly toxic drug Aureobasidin A. This drug reporter gene alone exhibits very little background activity.
Constructs
Rat telethonin, UBE2B and MuRF1, and murine MuRF3, UBE2D2, UBE2E1, UBE2J2, UBE2J2c (cytosolic part of UBE2J2), UBE2L3, and UBE2N cDNAs were amplified by RT–PCR from either rat soleus muscles or murine skeletal muscle cells C2C12 mRNA, using Superscript II and Platinum Pfx DNA polymerase (Invitrogen). E2D2 was used as negative control because this E2 does not interact with MuRF1.35 Human MAFbx and UBE2G1 and UBE2G2 cDNAs were kindly supplied by Dr S. Leibovitch (University of Montpellier, France) and Dr A. Navon (The Weizmann Institute of Science, Rehovot, Israel), respectively. Human cDNAs of UBE2A and UBE2J1 were purchased (AddGene: pDEST17‐UBE2A came from the W. Harpers' laboratory, Boston. ATCC Research Center: pET22‐UBE2J1). Please refer to Table 1 for other E2 enzymes nomenclature. cDNAs were cloned in yeast two‐hybrid vectors pGADT7, producing a fusion protein with the activation domain (AD) of GAL4 transcription factor, or in pGBKT7 and pBridge vectors, producing a fusion protein with the binding domain (BD) of GAL4 (Clontech). E2J1 and E2J2 have a C‐terminal membrane domain and are predicted to be located at the endoplasmic reticulum membrane. Their cytosolic part, named J1c and J2c, were cloned to avoid putative false negative results with the full‐length E2J in Y2H. The pBridge vector allows the expression of 2 proteins, the second one being cloned into a distinct Multiple Cloning Site (MCS II). MuRF1 was cloned in pBridge, in the first MCS (MCS I), in fusion with the BD of GAL4 and telethonin in MCSII leading to the pBridge::MuRF1/telethonin plasmid. cDNAs were also cloned in the expression vectors pET28a (Novagen) for the production of recombinant proteins in Escherichia coli and in pcDNA3.1 (Invitrogen) for expression in mammalian cells. E2J1 and E2E1 cDNA were cloned into BspeI:XbaI sites of pcDNA_GFP10‐Nter fusion vector; MuRF1 cDNA was cloned into NotI‐ClaI sites of pcDNA_GFP11‐Cter fusion vector.38 GFP10 was replaced with mCherry into pcDNA_GFP10‐Nter fusion vector, and telethonine was cloned into BspeI:XbaI restriction sites.
Table 1.
Overview of skeletal muscle E2s: expression, in vitro activity, and interaction with MuRF1a
| UBE2 (other name) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C a | D2 | E1 | G1 | G2 | J1 | J2 | K a | L3 | N | Z | |
| (RAD6A) | (E2‐14 kDa) | (UbcH10) | (UbcH5B) | (UbcH6) | (UBC7) | (NCUBE1) | (NCUBE2) | (UbcH1) | (UbcH7) | (Ubc13) | (Use1) | ||
| Expressed in skeletal muscle | |||||||||||||
| + | + | − | + | + | + | + | + | + | − | + | + | + | |
| Over expression in muscle during some catabolic states | |||||||||||||
| → | ↑ | NA | → | → | ↑ | ↑ | ↑ | ND | NA | → | → | ND | |
| In vitro substrate ubiquitination with MuRF1 | |||||||||||||
| ND | ND | − | + | − | ND | ND | ND | ND | + | − | + | ND | |
| In vitro auto‐ubiquitination of MuRF1 | |||||||||||||
| − | − | − | + | − | ND | − | ND | ND | − | − | + | ND | |
| Interaction with MuRF1 (this work) | |||||||||||||
| Y2H | ND | − | ND | − | − | +/− | − | +/− | +/− | ND | + | − | ND |
| SPR | − | ND | − | − | + | + | − | ND | + | − | + | − | − |
| Y3H a | ND | − | ND | − | + (×100) | + (×4) | − | + (×7) | + (×10) | ND | + (×2) | − | ND |
NA, not adapted; ND, not determined; Y2H, yeast two‐hybrid; SPR, surface plasmon resonance; Y3H, yeast three‐hybrid; ↑, overexpression; →, mRNA stable level.
UBE2 enzymes involved in ubiquitination (excluding Ub‐like modification) and expressed in mouse's muscle according to NextBio (http://www.nextbio.com) and Genomatix (https://www.genomatix.de) websites. E2C and E2K, not expressed in muscle, were considered as negative controls. Details about references, the catabolic situations studied, and the substrates ubiquitinated are provided in the more complete Table S1.
Fold increase when compared with Y2H is indicated in parentheses.
Yeast two‐hybrid and yeast three‐hybrid experiments
We used the ‘Matchmaker™ Gold Yeast Two‐Hybrid System’ (Y2H) from Clontech, based on the reconstitution of the GAL4 transcription factor. Haploids Y2HGold clones containing pGBKT7 and pBridge constructs were mated against haploids Y187 clones containing pGADT7 constructs on YPDA medium for a period of 16 h. Diploids were then selected after replication on a selective medium lacking leucine, tryptophan, and methionine (Met) (‐LTM). Diploids were replicated on medium lacking leucine, tryptophan, histidine, and adenine (‐LTHAd, highly stringent medium) or lacking leucine, tryptophan, and histidine and supplemented with 20 mM Aureobasidin A and 2.5 mM 3‐Amino‐1,2,4‐triazole (‐LTH + Aureo + 3‐AT). 3‐AT is a competitive inhibitor of the product of the HIS3 gene. 3‐AT concentration was determined to avoid non‐specific interaction between MuRF1 and non‐interacting proteins (e.g. Large‐T) and to avoid non‐specific yeast growth. Interactions were assayed by the activation of HIS3, ADE2, and/or AUR1‐C reporter genes. Growth on selective plates was followed over a period of 21 days, Large‐T antigen, and p53 (from Clontech) being used as control. The pBridge vector was used to perform yeast three‐hybrid (Y3H) experiments, in combination with the AD fusion vector pGADT7.
Yeast proteins extraction
pBridge::MuRF1/telethonin transformant yeast strains were inoculated in liquid selective medium‐containing various Met concentrations and grown at 30°C. At OD600nm = 0.8, yeast were harvested and proteins extracted using a protocol adapted from Dualsystems Biotech firm, using alkaline lysis of cells followed by trichloroacetic acid precipitation. Protein extracts were submitted to immunoblot for detecting exogenous expressed proteins.
Protein expression and purification
GST and GST‐MuRF1 were expressed and purified using sepharose 4B affinity matrix (GE Healthcare) as described by Polge et al.26 UBE2A, UBE2B, UBE2E1, UBE2G1, and UBE2J2c were expressed in E. coli BL21(DE3) as his‐tag fusion proteins and purified on Ni‐NTA agarose matrix (Qiagen). The recombinant proteins were eluted, and the his‐tag removed by incubation with thrombin overnight, in [NaH2PO4 50 mM, pH 8.0, NaCl 300 mM], at 16°C. Thrombin was inhibited by 200 μM PMSF. Incubation with an MOPS buffer pH 7.0 (MOPS 25 mM, NaCl 150 mM) allowed the recovery of homogenous untagged proteins as confirmed by SDS‐PAGE stained with blue Coomassie.
GST‐MuRF1 and His‐telethonin were co‐expressed in E. coli. Briefly, E. coli BL21(DE3) were first transformed with GST‐MuRF1, and an isolated colony was then grown in 500 mL of liquid LB (Luria‐Bertani) growth medium with ampicillin (60 mg/mL) until 0.5 OD600. Bacteria were then centrifuged at 5000 g for 5 min and rendered competent using calcium chloride as previously described.39 The competent bacteria were then transformed with pET28a::telethonin plasmid, and double transformants were selected on LB plates with ampicillin (60 mg/mL) and kanamycin (25 mg/mL). Ten isolated colonies were then individually grown on 5 mL LB (two tubes per colony) containing both antibiotics. Using half of the culture, expression of proteins was induced using IPTG (100 μM) following the manufacturer's instructions, and the expression of MuRF1 and telethonin was verified by immunoblotting. The other half of the culture was prepared as glycerol stock and frozen at −80°C.
GST pulldown
GST pulldown experiments were performed as described by Polge et al.26
Crosslink
GST‐MuRF1 and His6‐telethonin were co‐expressed as described previously and purified using Sepharose 4B beads according to the manufacturer's instructions. MuRF1‐telethonin complexes were eluted using 10 mM reduced glutathione, 50 mM HEPES pH 8. Final concentration of proteins was 0.3 mg/mL. An aliquot of the eluate was treated with formaldehyde (0.0625% final concentration) for 2 min at room temperature. Crosslinking was stopped by adding 0.1 volume of 1.25 M glycine for 20 min at room temperature. The sample was then dialyzed against HEPES buffer (50 mM, pH 7.3) and used for Biacore experiments.
Samples loaded onto SDS‐PAGE for verifying the efficiency of crosslinking were incubated in Laemmli buffer at 65°C for 5 min. Conversely, reversal of crosslinking was performed by incubating the crosslinked proteins at 95°C for 10 min.
Size exclusion chromatography
Purified E2G1 protein (30–50 μg) was applied to an HiPrep 16/60 Superdex 200 gel filtration column (Mr 10 000–600 000; GE Healthcare) equilibrated with 25 mM MOPS (pH 7.0), 150 mM NaCl. Flow rate was 1 mL/min. The column was calibrated with the following markers: thyroglobulin (670 kDa), β‐amylase (200 kDa), alcohol dehydrogenase (150 kDa), bovine serum albumin (66 kDa), and carbonic anhydrase (29 kDa).
Surface plasmon resonance
Surface plasmon resonance experiments were performed with a BIAcore T200 instrument (GE Healthcare), at 25°C. GST‐MuRF1 and GST were covalently immobilized on a CM5 sensor chip by standard amine‐coupling generating multiple orientations of GST‐MuRF1 on the surface. Interaction measurements were carried out in running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 0.05% (v/v) surfactant P20) at a flow rate of 30 μL/min. For MuRF1‐E2 interaction screen, E2 proteins were diluted to 500 nM and 1 μM and injected in parallel onto the GST and GST‐MuRF1 surfaces for 70 s at 30 μL/min. For single cycle kinetics (SCK) experiments, serial dilutions of an E2 solution were successively injected at 30 μL/min, during 120 s and final dissociation was monitored during 600 s. Concentration range was chosen according to the level reached in pilot SPR screen: 250 nM, 500 nM, 666 nM, 1 μM, and 2 μM for UBE2L3; 750 nM, 1 μM, 1.5 μM, 2 μM, and 3 μM for UBE2G1 and 125 nM, 250 nM, 500 nM, 1 μM, and 2 μM for UBE2E1. For kinetic analysis, fitting of association, and dissociation curves was performed using BIAevaluation software (GE Healthcare).
Cell culture
MuRF1, telethonin, and E2 coding sequences were sub‐cloned in pcDNA3.1. HEK293T cells were cultured in Dulbecco's Modified Eagle Medium and 10% (v/v) foetal bovine serum. Cells were plated in 6‐well dishes and transfected by the calcium phosphate co‐precipitation method. Cells were transfected or co‐transfected with plasmid(s) encoding for green fluorescent protein (GFP) (Mock), MuRF1, telethonin, and E2 and were harvested after 48 h of transfection. Cells were lyzed, and soluble proteins were obtained as previously described.37 Overexpressed protein levels were analyzed by immunoblotting using anti‐telethonin, MuRF1 (SantaCruz) and E2 (Sigma) antibodies. Three independent experiments were performed.
Split‐GFP complementation assays
HEK293_GFP1‐9 cells38 were cultured using Dulbecco's Modified Eagle Medium and 10% (v/v) foetal bovine serum (Lonza, Basel, Switzerland). Transfection of plasmids was performed using jetPRIME (Polyplus‐transfection, Illkirch, France) according to manufacturer's instructions. GFP10‐E2J1, E2E1‐GFP10, E2G1‐GFP10, E2L3‐GFP10, E2D2‐GFP10, and MuRF1‐GFP11 constructs were co‐transfected at a 1:1 ratio with jetPRIME reagent (Polyplus‐transfection, Illkirch, France) in HEK293 cells stably expressing the GFP1‐9 fragment (HEK_GFP1‐9). We verified that these constructs gave clear background with non‐relevant partners or alone ( Figure S1). As an example, co‐expression of GFP1‐9, GFP10‐E2J1, and a leucine zipper domain C‐terminally fused to GFP11 did not produce fluorescence. Expression of the constructs was checked by immunostaining using an antibody raised against the C‐terminal part of GFP (Santa Cruz anti‐GFP (T‐19); d1/250), recognizing both GFP10 and GFP11 fragments, and Alexa 594 (Santa Cruz; d1/500) ( Figure S1).
For telethonin co‐expression experiments, 0.3 μg of a mCherry‐telethonin encoding plasmid was included in the co‐transfection mix. Eighteen hours after transfection, cells were fixed with 3.7% paraformaldhedyde in 1X PBS (phosphate buffered saline) and mounted with Mowiol (Calbiochem, EMD Millipore) supplemented with DAPI for nuclei staining.
Individual cells were imaged using LSM 780 microscope (Zeiss, Oberkochen, Germany). Split‐GFP complementation signal was achieved using a 488 Argon laser with a 490–553 nm emission filter (Zeiss). mCherry and DAPI labelling were acquired with Argon and 405 UV diode lasers respectively (561 nm: LSM 710). Image analysis and quantification of split‐GFP fluorescence intensities were performed for the various complexes by measuring pixel intensity of individual cells (n = 15–30) with ImageJ 1.47v software (National Institute of Health, Bethesda, MD, USA).
Statistical analysis
Results are expressed as means +/− SEM. Statistical analysis was performed using Student's t‐test.
Results
Yeast two‐hybrid screen fails to clearly identify E2 enzymes interacting with MuRF1
For simplification in this report, UBE2 proteins will be named E2, for example, UBE2A will be E2A. To identify E2 proteins interacting with the muscle‐specific E3 ubiquitin ligase MuRF1, we first selected nine E2s (i) involved in ubiquitination (excluding ubiquitin‐like modification) and (ii) expressed in muscle [compiled in Tables 1 and S1,40 NextBio (http://www.nextbio.com), and genomatix (https://www.genomatix.de) websites]. We performed yeast two‐hybrid (Y2H) experiments using these 9 E2s vs. MuRF1. Five transformations for each haploid strain were performed, and 20 to 30 diploid clones were replicated on selection plates. Co‐expression of MuRF1 and Large‐T (LT) was set as the background level and was used as negative control throughout the experiments. The correct expression and folding of MuRF1 was assessed by its co‐expression with MuRF3, the hetero‐dimerization of MuRF proteins in diploid yeast resulting in the activation of reporter genes (Figure 1A). Except for positive control (MuRF1‐MuRF3), no MuRF1‐E2 interaction was detected using the most stringent medium (‐LTHAd) (data not shown). Screens on the less stringent medium (‐LTH + Aureo + 3‐AT) gave few positive colonies for E2G1, E2J1c, and E2J2c. However, only few percentages of the colonies plated were positive, 15.6% for E2G1 and 9.1% for the cytosolic part E2J1c and E2J2c (Figure 1A). Only E2L3 exhibited a somehow consistent interaction (42.3% positive clones) with MuRF1. For E2G1, E2J1c, E2J2c, and E2L3, the colonies grew very slowly, requiring ~3–4 weeks for being detected. We concluded that, except for E2L3, these results were not clear enough to conclude that E2G1, E2J1, and E2J2 were real MuRF1 partners. Moreover, putative MuRF1‐interacting E2s could have been missed because of suboptimal interaction conditions.
Figure 1.
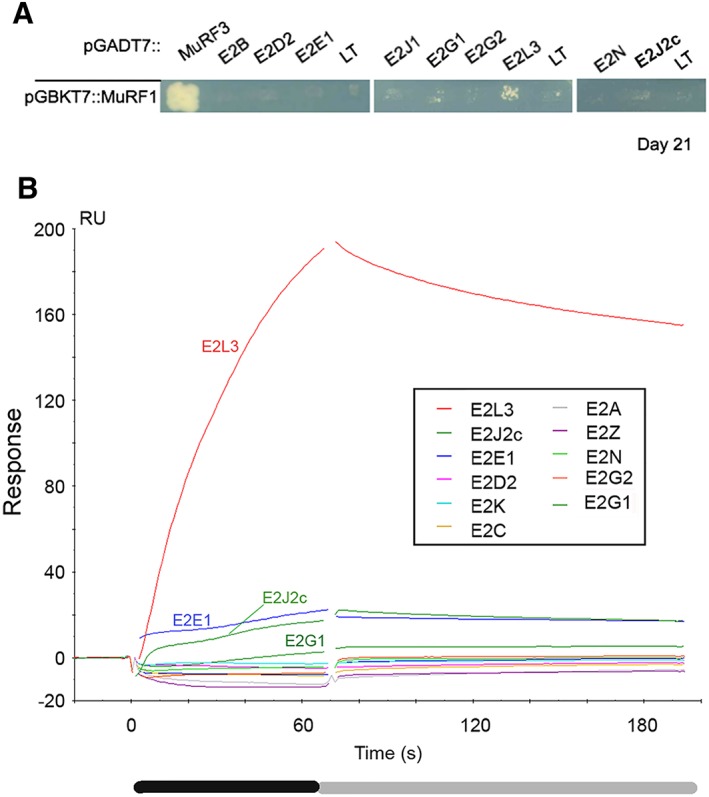
Only surface plasmon resonance screen reveals MuRF1 interacting E2s (A) yeast two‐hybrid (Y2H) screen. Y2HGold strain containing MuRF1 was mated with Y187 strain expressing MuRF3, E2, or LT (Large‐T antigen). LT construct was used as negative control against MuRF1 to estimate MuRF1 potential background level. Colonies are considered positive when bigger than this background. Colonies were plated on selective medium [‐LTH + Aureo + 3‐AT] (Experimental section) and monitored during 21 days. Three independent transformation experiments were performed, and 11 to 32 colonies were analyzed for each E2. (B) MuRF1‐E2s interactions were screened by surface plasmon resonance (SPR), using a BIAcore T200 (GE Healthcare). GST‐MuRF1 and GST were covalently immobilized on CM5 chips. E2s diluted to 1 μM (or 0.5 μM for E2J2c) were injected in parallel onto GST‐MuRF1 and GST surfaces at 30 μL/min. GST surface was used as a reference to subtract non‐specific binding of E2 on GST and/or on the CM5 surface. Only subtracted sensorgrams are shown. Black box, injection/association phase; grey box, dissociation phase; RU, arbitrary response units.
Surface plasmon resonance screen reveals E2 enzymes interacting with MuRF1
The Y2H results suggested that MuRF1‐E2 interactions were probably transient and labile. We next used a more sensitive technique (i.e. SPR) to detect weaker interactions. GST‐MuRF1 (≈600 RU) was immobilized on a CM5 sensor chip surface. Immobilized GST was used as reference surface to subtract non‐specific binding of E2 on GST and/or on the CM5 surface. Around 230 RU of GST were bound onto the reference surface to have similar number of ‘GST‐molecules’ on both surfaces. Twelve E2s were assayed in this SPR screen: E2A, E2C, E2D2, E2E1, E2G1, E2G2, E2J2c, E2K, E2L3, E2N, E2V2, and E2Z (Figure 1B). E2J1, identified as putative partner in Y2H, was not assayed because of technical problems to produce either the recombinant full‐length or the cytosolic portion of the protein. E2C and E2K, not detected in muscle, were used as negative controls. Untagged E2 proteins were used because an N‐terminal tag could hinder the E3‐BD localized at the N‐terminus of E2s (41). SPR replicates (n = 2–6) were reproducible, and as expected, no interaction was detected between MuRF1 and the negative controls E2C and E2K (Figures 1B and S2). Amongst the 12 E2s tested, a clear interaction was detected with E2L3, confirming Y2H screen data. Weaker interactions were also detected with E2J2c and E2G1 in agreement with Y2H screen, but also with E2E1, which was not detected first (Figures 1B and S2, Tables 1 and S1). In contrast, the other E2s tested, that is, E2A, E2D2, E2G2, E2N, E2V2, and E2Z did not interact with MuRF1. Therefore, the SPR screen proved to be a more sensitive and suitable approach than Y2H to identify E2–E3 interactions. These data also revealed that E2s exhibit different affinities for MuRF1 with E2L3 >> E2E1 = E2J2c > E2G1.
Characterization of MuRF1‐E2 interactions by surface plasmon resonance
To appreciate more quantitatively the MuRF1‐E2 interaction kinetics and affinities, we performed a series of in vitro experiments using SPR technology. We first focused on the more affine E2, that is, E2L3. Solutions of E2L3 diluted to 250 nM, 500 nM, 666 nM, 1 μM, and 2 μM were injected onto GST‐MuRF1 and GST control surfaces to perform SCK experiments (Figure 2A). The kinetics did not fit perfectly to a 1:1 Langmuir interaction model. We attributed this to non‐specific interaction of E2L3 with the sensor chip surface that we failed to eliminate. SCKs data were then analyzed using the ‘heterogeneous ligand’ model to artificially remove this ‘sticky’ component. Kinetics fitted well with this model, as witnessed by the low residuals of the fit, that is, the discrepancy between experimental and calculated data points (Figure 2B). The evaluation software distinguished between non‐specific interaction with the surface and specific E2L3‐MuRF1 interaction. Binding affinity constant (KD) of E2L3 for MuRF1 was thus estimated to be around 50 nM.
Figure 2.
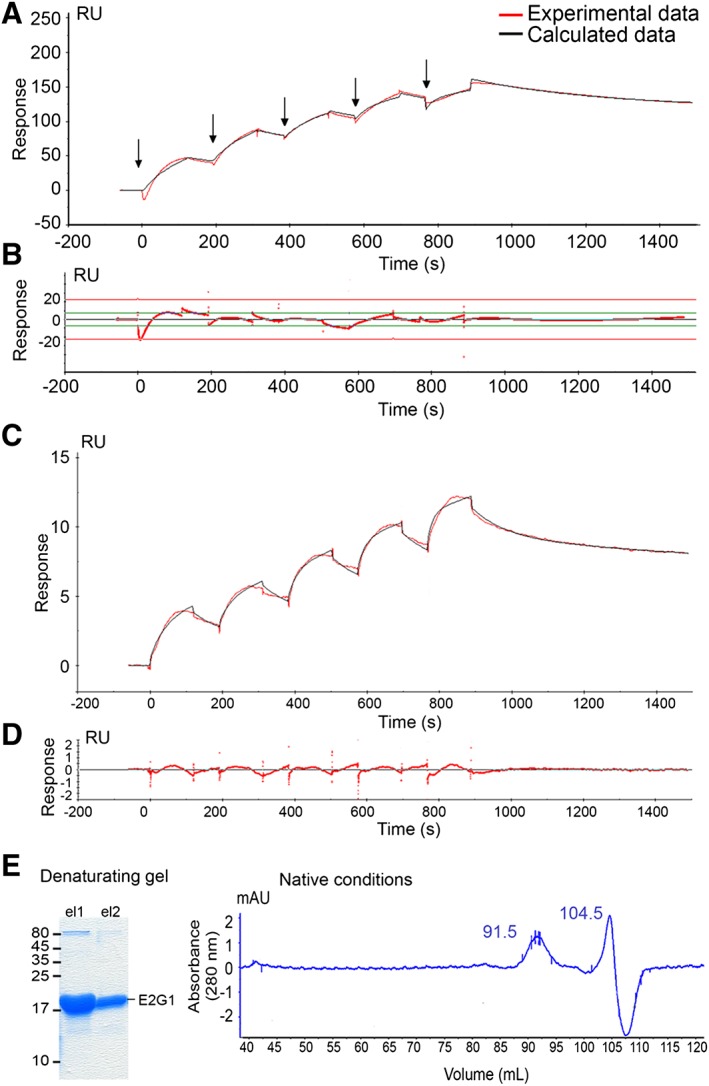
Determination of the binding affinity constant (KD) of E2L3‐MuRF1 and E2G1‐MuRF1 interactions MuRF1‐E2L3 (A, B) and MuRF1‐E2G1 (C, D, E) interactions was characterized using SPR analysis. (A) Sensorgram of a representative single cycle kinetics (SCK) experiment obtained by the sequential injection of serial dilutions of E2L3 (250 nM, 500 nM, 666 nM, 1 μM, and 2 μM) onto GST‐MuRF1 and GST control surfaces. Flow rate was 30 μL/min. Arrows denote sample injections. (B) Low residuals (<10% of the response, red line) indicated that the kinetics fitted well. Binding affinity constant (KD) of E2L3 for MuRF1 was estimated to be ≈50 nM. (C) Sensorgram of a representative single cycle kinetics obtained by the injection of serial dilutions of E2G1 (750 nM, 1 μM, 1.5 μM, 2 μM, and 3 μM) onto the GST‐MuRF1 and GST control surfaces at 30 μL/min. Red curve, experimental data; black curve, calculated data when using the ‘heterogeneous analyte’ model. (D) Residuals. The kinetics fitted to this model (heterogeneous solution of E2G1 monomers and dimers) as seen by the low residuals of the fit. (E) E2G1 protein preparation contained monomeric and dimeric forms. The E2G1 recombinant protein produced was pure as shown by the Coomassie staining of the denaturating gel (left). E2G1 recombinant protein was submitted to size exclusion chromatography (hiload 16/600 Superdex 200; GE Healthcare), performed in native conditions (right). E2G1 protein elution pattern confirmed the presence of monomers (at 104.5 mL corresponding to ≈18 kDa) and dimers (at 91.5 mL, corresponding to ≈46 kDa).
The MuRF1‐E2E1 and MuRF1‐E2J2c couples never presented stable association on sensorgrams, despite several experiments. Indeed, association phase always presented a wave‐shaped profile (Figures 1B and S2), preventing correct determination of affinity and kinetic parameters for these interactions. These profiles indicate that E2E1 and E2J2c interacted with MuRF1, but they strongly suggest that something was missing to stabilize MuRF1‐E2 complexes.
MuRF1‐E2G1 interaction was further analyzed by SCK (Figure 2C). Dilutions of E2G1 (750 nM, 1 μM, 1.5 μM, 2 μM, and 3 μM) were injected on to GST‐MuRF1 and GST surfaces. Kinetics fitted to a ‘heterogeneous analyte’ model (Figure 2C and 2D), the evaluation software, suggesting that E2G1 interacted with MuRF1 as both a monomer and a dimer. To verify this hypothesis, E2G1 recombinant protein preparations were then analyzed using size exclusion chromatography (HiLoad 16/600 superdex 200 pg). As shown in Figure 2E, two elution peaks appeared at 91.5 and 104.5 mL corresponding respectively to ≈18.3 and ≈45 kDa, that is, the size of the monomeric (19 kDa) and dimeric (38 kDa) E2G1. Our data suggest that recombinant E2G1 spontaneously dimerizes in vitro and that both the monomer and the dimer were able to interact with MuRF1 with different kinetics parameters. The E2G1 monomer exhibited a slightly better affinity (KD ≈ 5.9 μM) when compared with the dimeric form (KD ≈ 22.0 μM). Monomeric E2G1 associated slowly with MuRF1 (ka ≈ 4 × 103 M−1 s−1) and dissociated quickly (kd ≈ 2.3 × 10−2 s−1). The dimeric form of E2G1 interacted more slowly with MuRF1 (ka ≈ 3 × 102 M−1 s−1), approaching the limits of the Biacore T200, while the MuRF1‐(E2G1)2 complex was more stable, once established, with a kd ~7.4 × 10−4 s−1. These in vitro observations clearly require further investigations for confirming the existence of dimers in vivo and their potential physiological significance.
Stabilization of MuRF1‐E2 interaction by a third partner
With the exception of E2L3, Y2H and SPR data indicated that MuRF1‐E2 interactions were weak and suggested that something was missing for stabilizing MuRF1‐E2 couples such as post‐translational modification(s) and/or a third partner. To test the latter hypothesis, we moved to a tripartite interaction experiment, the yeast three‐hybrid (Y3H), which allowed to detect the positive or negative impact of a third protein on MuRF1‐E2 interaction. Amongst proteins that could stabilize E2–E3 interactions, ubiquitin and already described binding partners represented first choices. However, ubiquitin was present in yeast assays and was obviously not missing. We thus chose an already described MuRF1 partner, amongst substrates or associated proteins. Because MuRF1 can be both in the soluble and myofibrillar fractions,41, 42, 43 we retained, as a first criterion, a partner present in both fractions. Another important point was the specificity of interaction. Indeed, MuRF1 shares some properties with the isoform MuRF3, as both isoforms target myosin heavy chains for degradation.24 To simplify the analysis, we chose telethonin that interacts with MuRF1 but not with MuRF344 and is present in both the myofibrillar and soluble fractions.8 We first confirmed telethonin‐MuRF1 specific interaction using Y2H analysis ( Figure S3a). Co‐expression of the positive controls, Large‐T and p53, led to fast growth on selective medium as these two proteins strongly interacted ( Figure S3a). MuRF1‐MuRF3 and MuRF3‐MuRF3 interactions were visualized at day 6 confirming that MuRF fusion proteins were correctly produced and folded in yeast ( Figure S3a, right panel). Interaction with telethonin was only observed with MuRF1 and not with MuRF3 or MAFbx, another muscle‐specific E3 ubiquitin ligase ( Figure S3a). Specific telethonin‐MuRF1 interaction was further confirmed by GST pulldown experiments using GST‐MuRF1 and His6‐telethonin co‐expressed in E. coli ( Figure S3b). As shown in this figure, the two proteins were efficiently produced (first lane Lys) and telethonin co‐eluted with GST‐MuRF1 (lane 5 Elu). In contrast, telethonin was not pulled down with GST alone (lane 10 Elu), confirming the specific interaction between MuRF1 and telethonin.
We next investigated the potential role of telethonin on MuRF1‐E2 interactions. We first verified that telethonin did not interact with the selected E2 enzymes, using Y2H assay with the less stringent medium (Figure 3A). This prompted us to select telethonin as the third partner for Y3H experiments, that is, MuRF1/telethonin /E2.
Figure 3.
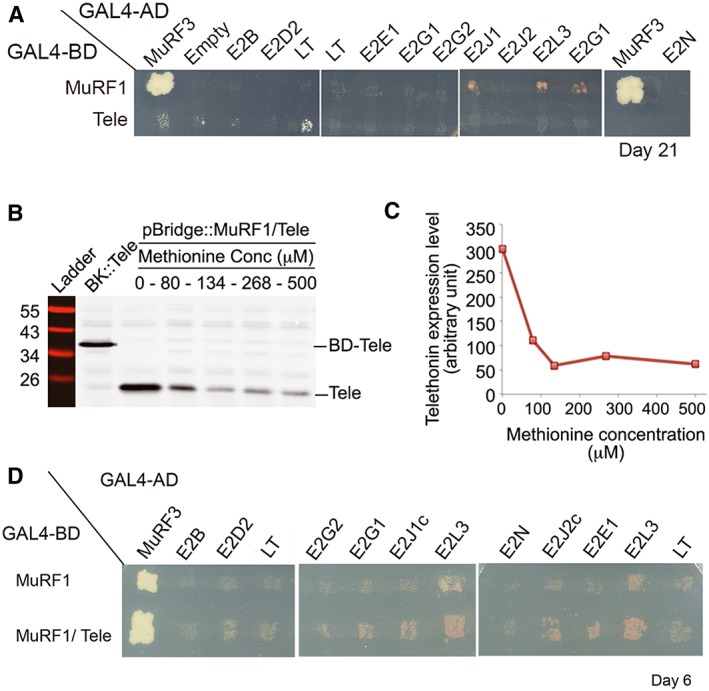
E2E1, E2G1, E2J1, E2J2, and E2L3 interact with MuRF1 in the presence of telethonin (A) Telethonin does not interact with E2s. Y2H experiments were performed using telethonin as a bait to confirm that this protein cannot directly interact with the E2 enzymes used in this work. The empty vector and the vector containing the LT construct were used as negative controls against telethonin to estimate potential background level. Signals above ‘empty’ and ‘LT’ lanes were considered as positive. Colonies were plated on selective medium [‐LTH + Aureo + 3‐AT] (Experimental section) and monitored during 21 days. LT, Large‐T antigen; Tele, telethonin. (B) Telethonin expression level in yeast varies according to methionine (Met) concentration in the medium. BD‐Tele, fusion protein between the binding domain of GAL4 and telethonin; Tele, telethonin. (C) Densitometry analysis from the immunoblot presented in (B). (D) Yeast three‐hybrid (Y3H) experiments revealed E2E1, E2G1, E2J1, E2J2, and E2L3 as MuRF1 partners in the presence of telethonin. E2‐expressing yeasts were mated against strains expressing either MuRF1 alone or MuRF1 and telethonin. Colonies were plated on selective medium [‐LTH + Aureo + 3‐AT] containing 134 mM Met. Results were observed at day 6. Three to four independent transformation experiments were performed and 11 to 32 colonies were analyzed for each E2.
Identification of E2 enzymes interacting with the MuRF1/telethonin complex
The pBridge::MuRF1/Tele vector was used for MuRF1/telethonin co‐expression, telethonin expression being specifically controlled by the MET25 promoter. As recommended by the manufacturer (Clontech), plating the yeast on media containing 1 mM Met should repress telethonin expression. In contrast, the absence of Met should allow telethonin expression. However, the growth of MuRF1‐expressing yeast was strongly slowed down at 0 mM and completely stopped at 1 mM Met (data not shown). Keeping in mind that yeast classical media contain 134 μM Met, we thus analyzed telethonin expression at different Met concentrations (0, 80, 134, 268, and 500 μM) to determine the optimal Met concentrations allowing normal growth of MuRF1‐expressing yeast and expression of telethonin. As expected, telethonin expression was maximal at 0 mM Met, decreased progressively up to 134 μM Met and then remained stable up to 500 μM (Figure 3B and 3C). This means that (i) the MET25 promoter did not give a black and white answer and that (ii) a significant amount of telethonin was produced in yeasts in the presence of 134 μM Met. Y3H screen was thus performed at this concentration, using pBridge::MuRF1/Tele or pBridge::MuRF1 alone against E2B, E2D2, E2E1, E2G1, E2G2, E2J1 E2J1c, E2J2c, E2L3, and E2N. Three to four independent transformation experiments were performed and 11 to 32 colonies were analyzed for each E2 (Figure 3D). For E2B, E2D2, E2G2, and E2N, Y3H yeast growth was similar to the negative control (LT), confirming that these E2 enzymes have no affinity for MuRF1. In contrast, E2E1, E2G1, E2J1, E2J1c, E2J2c, and E2L3 interacted with MuRF1 (Figure 3D), confirming SPR data and further indicating that the Y2H approach alone was poorly efficient for identifying MuRF1‐E2 interactions. When compared with Y2H (i.e. MuRF1‐E2 interactions), the presence of telethonin in Y3H assays (i.e. MuRF1/telethonin/E2 interactions) sharply increased the percentage of positive clones and strongly reduced the lag time for detecting the positive clones. Indeed, the percentage of positive clones increased in Y3H vs. Y2H assays from 0% to 93% for E2E1 (black and white answer), from 9% to 62% for E2J1c, from 9% to 88% for E2J2c, from 16% to 58% for E2G1 and from 42% to 81% for E2L3, respectively (Tables 1 and S1). In addition, yeast growth was improved for positive Y3H clones, as MuRF1‐telethonin‐E2 interactions were detected between days 4 and 14, while 3–4 weeks were required within Y2H assays with MuRF1 alone (compare data in Figure 3A obtained at week 3 and in Figure 3D obtained at day 6 in the presence of telethonin). These results indicated that the presence of an MuRF1 partner either stabilized MuRF1 and/or favoured MuRF1‐E2 interactions by an unknown mechanism.
Telethonin favours MuRF1 interactions with E2E1 and E2J1
Telethonin may act either as a stabilizer of MuRF1 or as a cooperative protein that will more specifically favour interactions with specific E2s. In the latter case, we expected a dose‐dependent effect of telethonin on yeast growth in Y3H screen and modification of the kinetic parameters using SPR. We performed Y3H assays at different Met concentrations, that is, with different telethonin levels in yeast. However, telethonin level rapidly reached a constant level in the range of 0–134/268 μM methionine concentrations (Figure 3B and 3C). However, only the 75–134/268 μM allowed similar yeast growth and thus enabled us to make valid comparisons. Yeasts containing pBridge::MuRF1/Tele plus one E2 were replicated on plates containing 75 μM Met vs. 134 or 268 μM Met. These concentrations allowed (i) comparable yeast growth within the different conditions and (ii) differential expression levels of telethonin. Moreover, to avoid any potential bias due to the replica plating order, we performed serial replica by switching from low to high and high to low Met concentrations. Dose‐dependent telethonin experiments were carried out with both MuRF1‐interacting (E2E1, E2G1, E2J1, E2L3) and non‐MuRF1‐interacting E2s (E2B, E2G2, E2D2, and E2N). Telethonin concentration did not affect growth of positive and negative controls (MuRF1‐MuRF3 and MuRF1‐LT) (Figure 4A). For MuRF1‐E2L3 (Figure 4A and 4(B) right panels) and MuRF1‐E2G1 (data not shown), the growth rate of colonies was not affected by the telethonin expression level. Thus, telethonin probably did not affect the interaction strength for MuRF1‐E2L3 and MuRF1‐E2G1 couples. In contrast, MuRF1‐E2J1 and MuRF1‐E2E1 interactions seemed depend on the telethonin level (Figure 4A and 4B, left and middle panels), suggesting that telethonin may influence MuRF1 preference for these E2s.
Figure 4.
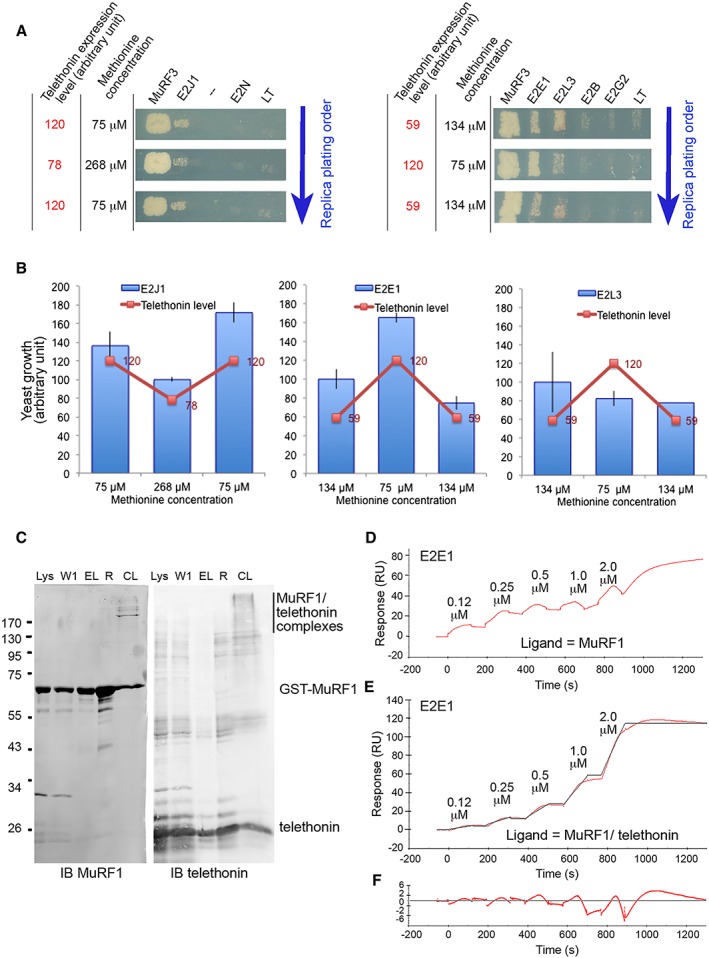
Telethonin favoured MuRF1‐E2E1 or MuRF1‐E2J1 interactions (A) dose‐dependent effect of telethonin on the growth of yeasts expressing MuRF1 and E2J1 or MuRF1 and E2E1. Yeasts expressing pBridge::MuRF1/telethonin were mated with yeasts expressing different E2s or MuRF3 (positive control) or LT (negative control). Y3H assays were carried out at different Met concentrations, that is, with different telethonin levels in yeast. Serial replica were performed by switching from low to high and high to low Met concentrations to avoid any bias due to the replica plating order. LT, Large‐T antigen; Tele, telethonin. (B) Yeast growth quantification from (A) is parallel to telethonin expression level (red curve). (C) Production of MuRF1/telethonin stable complexes. Immunoblots show the different steps of the production of crosslinked MuRF1/telethonin complexes that were thereafter bound on CM5 sensorchip for subsequent SPR experiments. IB, Immunoblot; L, lysat; W1, wash 1; El, eluted proteins; R, proteins remained on matrix; CL, cross linked proteins. (D, E, F) Telethonin stabilized MuRF1/E2E1 interaction. SPR experiments were performed using a single cycle kinetics method (SCK). Serial E2E1 solutions were injected in parallel over GST‐MuRF1 (D), GST‐MuRF1/telethonin complexes (E), and GST (reference) surfaces. Red curves, experimental data; black curve, calculated data for a fit using the ‘heterogeneous ligand model’. (F) Residual of the fit from (E).
To further confirm this hypothesis, we compared the affinity of E2E1 for MuRF1 either alone or as an MuRF1/telethonin complex using SPR approach. E2J1 was not assayed because protein production of this E2 failed in bacteria. MuRF1 and telethonin recombinant proteins were co‐produced in BL21(DE3) E. coli (Lanes Lys Figure 4C), co‐purified (Lanes R Figure 4C), and stabilized as a complex using chemical crosslinking (Lanes CL Figure 4C). Immunoblots revealed the presence of MuRF1/telethonin complexes of different sizes using mild crosslinking conditions, suggesting that homo‐oligomeric MuRF1 probably interacted with several molecules of telethonin (Figure 4C). The presence of MuRF1 oligomers was in agreement with the literature,45, 46 and we used the MuRF1/telethonin complexes for SPR analyses. Using the SCK method, we injected different concentrations of E2E1 (125 nM, 250 nM, 500 nM, 1 μM, and 2 μM) in parallel onto GST (reference), GST‐MuRF1, and GST‐MuRF1/telethonin surfaces (Figure 4D and 4E). The MuRF1‐E2E1 interaction was greatly improved in the presence of telethonin as no kinetic parameters could be calculated in the absence of telethonin (compare Figure 4D and 4E). Because heterologous MuRF1/telethonin complexes were present on the surface, we fitted the SCK obtained with MuRF1/telethonin complexes using a ‘heterogeneous ligand model’. The fit was good and presented low residuals, thus validating the method (Figure 4F). Association was slow, ka being around 5 × 103 M−1 s−1. Dissociation was slightly disturbed for the two higher concentrations leading to an under‐estimation of kd and thus mildly overestimating KD at around 5 nM. Altogether, these data indicate that telethonin greatly strengthened MuRF1‐E2E1 interaction.
Telethonin co‐localizes with MuRF1/E2 complexes in mammalian cells
We next tested whether MuRF1‐E2E1, MuRF1‐E2G1, MuRF1‐E2J1, and MuRF1‐E2L3 complexes could be visualized in mammalian cells using the split‐GFP system.38 The assay is based on tripartite association between 22 amino‐acids long GFP tags, GFP10 and GFP11, fused to interacting protein partners, and the complementary GFP1‐9 detector. When proteins interact, GFP10 and GFP11 self‐associate with GFP1‐9 to reconstitute a functional GFP. The system is likely to detect in cells weak and transient protein complexes thanks to the irreversibility of the split‐GFP association and long‐term accumulation of signal (18 h). As shown in Figures 5A and S4, co‐transfection of HEK293‐GFP1‐9 cells with E2‐GFP10 [E2E1‐GFP10, E2G1‐GFP10, E2L3‐GFP10, or GFP10‐E2J1 (full‐length)], and MuRF1‐GFP11 constructs produced a green fluorescent signal, in a perinuclear region, confirming that MuRF1 interacted with these E2s. In contrast, neither the transfection with the different constructions alone ( Figure S1) nor the co‐transfection of cells with the non‐interacting E2D2 and MuRF1 (Figure 5B) produced fluorescence. This clearly highlighted the specificity of the split‐GFP assay and indicated that MuRF1 did not interact with non‐cognate E2s in this cell assay.
Figure 5.
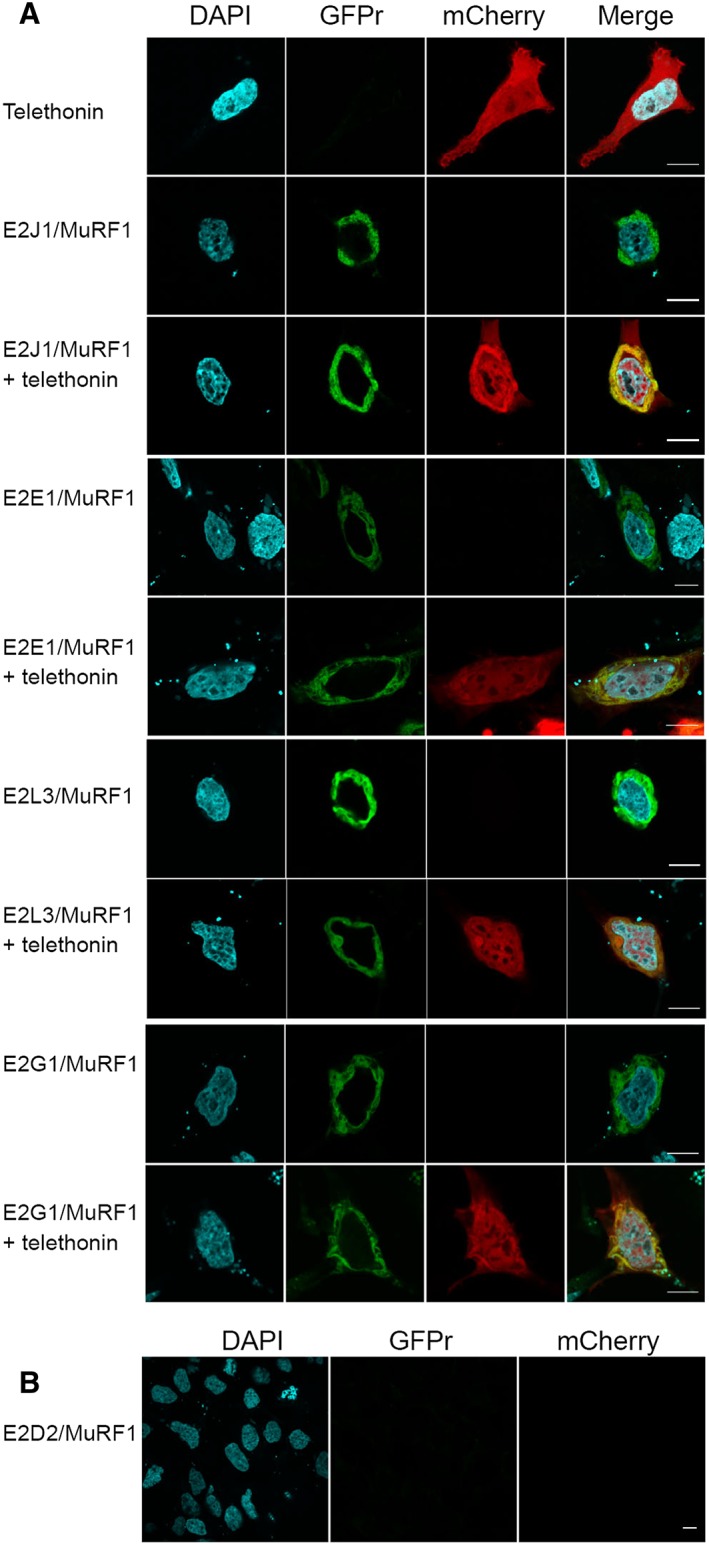
Telethonin co‐localized with MuRF1/E2 complexes in mammalian cells (A) interaction and localization of MuRF1‐E2s complexes in HEK_GFP1‐9 cells. Green fluorescent signal only results from E2‐MuRF1 interactions. (B) E2D2, an E2 that did not interact with MuRF1 (Y2H, Y3H, SPR) did not generate green fluorescence signal. Representative confocal microscopy images of split‐GFP fluorescence (rGFP). + telethonin, additional co‐expression of a mCherry‐telethonin (red signal) fusion construct. GFPr fluorescence was visualized in the FITC channel (488 nm); DAPI nuclear labelling (cyan) and mCherry fluorescence (561 nm). Merge images indicate co‐localization (yellow) of telethonin with MuRF1‐E2 complexes. Signal was acquired over 18 h and represented the sum of all interaction events over this period. (B) Negative control with the non‐interacting E2D2. Scale bars: 10 μm.
Cells transfected with mCherry‐telethonin fusion construct presented a homogenous staining in the cytosol and the nucleus (Figures 5A and S4). Additional co‐transfection with MuRF1‐E2J1 (but also E2E1, E2G1, or E2L3) led to telethonin co‐localization with perinuclear MuRF1‐E2 complexes (Figures 5A and S2). In addition, in presence of E2J1, telethonin was clearly re‐localized and concentrated in the perinuclear area. It should be pointed out that split‐GFP is not a degradation assay because interactions are stabilized by the irreversible split‐GFP association. This interferes with the correct processing of substrate ubiquitination and subsequent degradation.39 Thus, split‐GFP assay demonstrated that MuRF1‐E2‐telethonin associated in cells and we moved to another assay to test whether this association led to telethonin degradation.
Telethonin is an MuRF1 substrate and is degraded when MuRF1 is combined with its cognate E2s
We previously identified telethonin as a 26S proteasome substrate in atrophying rat muscles.47 We thus investigated whether MuRF1 could drive telethonin degradation within a cellular context. Telethonin was co‐transfected with MuRF1 or MuRF1 plus one E2 in HEK293T cells (Figure 6). The main advantage when using these cells, over muscle cell lines, is that they do not express telethonin or MuRF1 (data not shown). This means that results will not be biased by endogenous protein production. E2D2 was used as a negative control because this E2 did not interact with MuRF1. As expected, E2D2 co‐transfection with telethonin and MuRF1 did not depress telethonin levels. In contrast, co‐transfection with E2s identified as MuRF1 partners (E2E1, E2G1, E2J1, or E2L3) greatly induced telethonin degradation, suggesting that telethonin was an MuRF1 substrate (Figure 6). These results also showed that the physical MuRF1‐E2 interactions identified in this report are functional in cells.
Figure 6.

Telethonin is degraded in presence of MuRF1 and its E2 partners (A) telethonin level is depressed in HEK293 cells co‐transfected with MuRF1, telethonin and an E2 interacting with MuRF1 but not with the negative control E2D2. Immunoblotting was performed on cell lysates against telethonin. IB, immunoblot; Load: membranes were stained using Blot‐FastStain dye (a portion of the membrane is shown). (B) Densitometric analysis was used to correct for uneven loading. *P < 0.05, n = 6; **P < 0.01, n = 6.
Discussion
To set up efficient therapeutic strategies for reducing/preventing muscle wasting, a better understanding of the mechanisms involved in muscle wasting is necessary. Skeletal muscle protein mass is largely under the control of the UPS and thus of ubiquitinating enzymes. MuRF1 is the only muscle‐specific E3 ligase known to target contractile proteins (troponin‐I, α‐actin, myosin heavy chains, etc.) for degradation by the UPS during catabolic situations (disuse, chronic diseases, etc.). MuRF1 is thus a first choice for pharmacological targeting to ameliorate atrophying conditions. However, MuRF1 alone is not sufficient to cause muscle wasting and degradation of myosin when overexpressed in skeletal muscle,29 which suggests that another co‐factor (e.g. E2 enzymes) is necessary. Indeed, RING E3 ligases like MuRF1 are tightly dependent on cognate E2s for catalysis of Ub chains as their role is limited to the recruitment of the substrate and the E2. However, MuRF1 cognate E2(s) are not yet known. E2–E3 interaction networks represent an emergent field with the growing although limited number of in vitro structural and mechanistic studies, but none including muscle‐specific E3. Using complementary approaches (SPR, Y3H screens, and in cellulo assays), we report that five E2 enzymes physically and functionally interact with MuRF1 (E2E1, E2G1, E2J1, E2J2, E2L3). Moreover, we report that MuRF1‐E2E1 and MuRF1‐E2J1 interactions are facilitated by telethonin, a new MuRF1 substrate, by a potential allosteric mechanism.
E2 enzymes have been largely neglected (with the exception of E2B), and only relatively recent studies have emphasized their importance for defining the fate of the substrate. This means that only few addressed the expression levels of (a limited number of) E2 enzymes during catabolic states.48 It thus hard to depict a clear picture of E2 expression levels and of their individual implication in the development of muscle atrophy. We compile in this work available data (Tables 1 and S1), but the limited number of studies clearly impedes drawing any definitive conclusion. Amongst the E2s preferentially expressed in skeletal muscle and screened in this work, E2B, E2G1, and E2J1 and to a lesser extent E2G2 and E2L3 are up‐regulated in various atrophying conditions (Tables 1 and S1). E2B is so far the only E2 enzyme that has been extensively studied owing to its systematic overexpression in wasting conditions. Unfortunately, E2B possesses an MuRF1‐independent role downstream of the sarcomere disassembly by targeting the cytoplasmic pool of α‐actin and myosin heavy chain.36 As E2B‐MuRF1 interaction was never addressed using highly sensitive approaches, we included E2B in the present study and confirmed that it does not interact with MuRF1.
Y2H is a common approach for screening interactions, but it failed in this work to efficiently identify MuRF1‐E2 couples. This is in line with previous works reporting frameworks of E2‐RING E3 interactions using such approach.30, 31, 32 These reports revealed that (i) a limited number of E2 enzymes trusted all the interactions observed and (ii) for most E3s no interaction could be detected with any E2. This strongly suggests that for most RING E3 ligases, Y2H is not well suited for the identification of E2 partners.
Using the surface plasmon resonance approach, which is highly sensitive, we identified the first known MuRF1 E2 partners: E2E1, E2G1, E2J1, E2J2, and E2L3. Our data confirmed that MuRF1‐E2 interactions were weak and labile (except for E2L3), hence the lack of efficiency of Y2H approach for their identification. Our data are consistent with previous studies reporting that E2–E3 interactions range from moderate to weak.18 An exception is UBE2L3 that tightly bound (~200 nM KD) with the RING‐between‐RING (RBR) region of the HHARI, an RBR‐type E3 ligase.49 Interestingly, adding a third protein that interacted with MuRF1 (telethonin) greatly enhanced the strength of interaction between MuRF1 and E2s. Altogether, MuRF1‐E2 interactions with or without telethonin gave complementary results. We found that MuRF1 interacted differentially with its E2 partners in term of kinetic parameters, affinity (high or low), and requirement of telethonin for stabilization. A rough classification was made based on affinity with MuRF1 in the absence of telethonin: E2L3 >> E2E1 = E2J1 = E2J2 > E2G1. E2L3 binds to many RING domains but the strong interaction observed between E2L3 and MuRF1 was surprising. Indeed, E2L3 is supposed to only function with HECT‐type and RBR‐type E3 ligases that possess intrinsic catalytic activities.18 We report here not only that E2L3 interacted with the RING E3 ligase MuRF1 but also that co‐expression of E2L3 and MuRF1 in HEK293T cells promoted telethonin degradation. These results imply either (i) that E2L3 can function with an RING E3 ligase or (ii) that another protein (e.g. an E2 or an E3) acts as a co‐factor in HEK293T cells. In line with the first hypothesis, another RING‐type E3, c‐Cbl, has been reported to interact and function with E2L3 to ubiquitinate one of its substrates, the epidermal growth factor receptor.50, 51
A recent study revealed that E2s of the E2E family mainly catalyze monoUb but could also form K63 and K48 polyUb chains under particular conditions.52 Accordingly MuRF1‐E2E1 co‐transfection led to telethonin degradation in cellulo. This suggests that the MuRF1‐E2E1 couple synthetized polyUb chains on telethonin, even though the cooperation with another E2 present in HEK293 cells cannot be ruled out. Division of labour between two E2s has already been described, for example, UBE2C catalyzed the attachment of the first Ub on the substrate and UBE2S elongated the chain for the degradation of APC‐dependent targets during the cell‐cycle progression.53
E2J1 and E2J2 are membrane proteins anchored to the endoplasmic reticulum. In this work, in cellulo split‐GFP assay performed with E2J1 and MuRF1 confirmed that these proteins collaborate in a perinuclear region (Figure 5). This is in agreement with reports showing that MuRF1 and telethonin are present in different muscle compartments and that MuRF1 possesses other targets than contractile proteins, for example, enzymes linked to energy metabolism.27, 29 Future work is clearly needed to explain the functional role and significance of these interactions and their localization.
No physical interaction was detected in our study between MuRF1 and E2K, although this E2 was reported to synthetize K48‐ubiquitin chains in vitro with MuRF1.54 This discrepancy can be explained by the capacity of E2K to build chains in the absence of an E3 enzyme under standard in vitro conditions.55 Moreover, E2K is not expressed in muscle according to the NextBio and Genomatix databases confirming the lack of physiological relevance of a functional partnership between MuRF1 and E2K. Similarly, we recently demonstrated that UBE2D2 is not an MuRF1 cognate E2 despite several MuRF1‐E2D2 in vitro ubiquitination assays on contractile proteins.37 A recent review18 pointed out the difficulty when working in vitro with highly active and promiscuous E2 enzymes like E2K and E2Ds.18, 55 Altogether, this questions the physiological relevance of in vitro ubiquitination assays.
We report in the present work that the binding of an MuRF1 substrate greatly improved MuRF1‐E2 interactions. However, we report two different behaviours of MuRF1 depending on the E2. Using complementary approaches (SPR, Y3H, Split‐GFP), we found that the presence of telethonin not only stabilized MuRF1‐E2E1 (or J1) interaction but also increased the affinity in a concentration‐dependent manner. This is in agreement with a potential allosteric regulation by the substrate itself. To our knowledge, this is the first time that such a regulation is described. In contrast, the presence of telethonin only stabilized MuRF1‐E2G1 or MuRF1‐E2L3 interactions but did not increase the complex affinities. Other MuRF1 partners and/or post‐translational modifications may modify MuRF1 sensitivity towards these two E2s. The identification of biological E2/E3/substrate trio is extremely difficult and few have been described. One example is the complex formed by the Ring1B/Bmi1 RING heterodimer, E2D2, and the nucleosome core particle (NCP, substrate), which is the only E2/E3/substrate complex crystallized so far.56 We hypothesize that MuRF1/E2J1/telethonin and MuRF1/E2E1/telethonin represent physiologically relevant newly identified complexes. The plurality of roles and localization of telethonin in the stability of the myofibrillar architecture, myostatin antagonism in the cytosol, and regulation of p53 in the nucleus is in favour of multiple E2s involved in its ubiquitination. Future work should focus on (i) deciphering the role of each E2 on telethonin during standard and catabolic conditions in skeletal muscle, (ii) identifying Ub‐chain linkages depending on the E2 involved, and (iii) potential E2s cooperation.
Potential role of the substrate on E2–E3 interaction may represent a new mechanism that could open new avenues for potential therapies at least at two levels. First, the E2–E3 interface is a highly specific area that may be targeted by new drugs for modulating sarcomeric protein degradation without interfering with the degradation of other proteins. Second, the role of the substrate may also be considered for fighting against muscle atrophy and rationally designed therapies should focus on the E2–E3‐substrate interconnections for achieving even more specific drugs. Indeed, the crucial challenge in drug development is to ensure safety while being efficient, and the more specific the drug, the better it will. Thus, our pioneering work should prove highly important for achieving these goals. We are aware that our work needs further investigation for confirming in skeletal muscle the role of these E2 enzymes and of the substrate for its own degradation. Future work will have to address this point first by using animal models of atrophy and ultimately to confirm MuRF1‐E2 partnership in human skeletal muscle, for example, from cachectic patients.
The present work reports the first MuRF1‐E2s network. This necessary groundwork was possible only by using in vitro and heterologous in cellulo approaches. Although interesting, these approaches may have hidden the involvment of other MuRF1 partners and/or post‐translational modifications in muscle. Future work will thus have to confirm in muscle the role of these E2 enzymes on contractile proteins (α‐actin, MHC, etc.) targeting and thus on muscle atrophy. The MuRF1‐E2‐substrate interface is the ultimate step that finely tunes protein degradation and represents a potential target for drug action. Preventing discrete E2‐MuRF1‐contractile protein interactions may thus be useful to avoid drug side effects and open the way for elaborating new therapeutic strategies to limit muscle atrophy. In that way, our work paves the way for researchers dealing with clinical and preclinical studies.
Supporting information
Supplemental figure 1 Representative wide field images of negative controls of split‐GFP assays GFP10‐E2J1, E2E1‐GFP10, E2G1‐GFP10, E2L3‐GFP10, E2D2‐GFP10 and MuRF1‐GFP11 constructs were transfected with jetPRIME reagent (Polyplus‐transfection) in HEK293 cells stably expressing the GFP1‐9 fragment (HEK_GFP1‐9). These constructs alone gave clear background. Scale bars = 10 μm.
{kind=link}
Supplemental figure 2 Zoom of E2s mildly and non‐interacting with MuRF1 during the Surface Plasmon Resonance screen
{kind=link}
Supplemental figure 3 Specific interaction of telethonin with the E3 ligase MuRF1 (a) Yeast two‐hybrid assay. Y2HGold strain expressing GAL4‐BD fusions and Y187 strain expressing GAL4‐AD fusions were mated. Diploids were plated on selective medium missing Leu and Trp (‐LT) to control homogeneous growth of the yeast and on medium missing Leu, Trp, His and adenine (‐LTHAd) to detect interactions. MuRF1‐MuRF3, MuRF3‐MuRF3 and p53‐LargeT interactions were used as positive controls. GAL4‐AD, fusions with the Activation Domain of GAL4; GAL4‐BD, fusions with the DNA Binding Domain of GAL4; D1, day 1; D6, day 6. (b) Confirmation of GST‐MuRF1‐telethonin interaction using GST pulldown assay. His‐telethonine was co‐expressed with GST‐MuRF1 or GST in BL21 (DE3) E. coli. Bacteria were lyzed and extracts were submitted to GST pulldown. Immunoblotting against MuRF1 (upper panel) and telethonin (lower panel) showed that telethonin was pulled down with GST‐MuRF1 but not with GST alone (arrows). Lys, lysat; FT, Flowthrough; W1 and W3, first and third washes; Elu, eluate; IB, immunoblot
{kind=link}
Supplemental figure 4 Representative wide field images of split‐GFP assays performed in Fig. 5
{kind=link}
Supplemental table S1 Compilation of skeletal muscle E2s data
Acknowledgements
This work was supported by grants from the French Institut National de la Recherche Agronomique and the AFM‐Téléthon (Trampoline grant #16438, AFM‐Téléthon grant #19521). SC and CB were supported by the french Institut National de la Santé et de la Recherche Médicale. The authors declare that they have no conflict of interest. The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015.57
Polge, C. , Cabantous, S. , Deval, C. , Claustre, A. , Hauvette, A. , Bouchenot, C. , Aniort, J. , Béchet, D. , Combaret, L. , Attaix, D. , and Taillandier, D. (2018) A muscle‐specific MuRF1‐E2 network requires stabilization of MuRF1‐E2 complexes by telethonin, a newly identified substrate. Journal of Cachexia, Sarcopenia and Muscle, 9: 129–145. doi: 10.1002/jcsm.12249.
References
- 1. von Haehling S, Anker MS, Anker SD. Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: facts and numbers update 2016. J Cachexia Sarcopenia Muscle 2016. Dec;7:507–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tyrovolas S, Koyanagi A, Olaya B, Ayuso‐Mateos JL, Miret M, Chatterji S, et al. Factors associated with skeletal muscle mass, sarcopenia, and sarcopenic obesity in older adults: a multi‐continent study. J Cachexia Sarcopenia Muscle 2016. Jun;7:312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vainzof M, Moreira ES, Suzuki OT, Faulkner G, Valle G, Beggs AH, et al. Telethonin protein expression in neuromuscular disorders. Biochim Biophys Acta BBA ‐ Mol Basis Dis 2002. Oct 9;1588:33–40. [DOI] [PubMed] [Google Scholar]
- 4. Bos JM, Poley RN, Ny M, Tester DJ, Xu X, Vatta M, et al. Genotype—phenotype relationships involving hypertrophic cardiomyopathy‐associated mutations in titin, muscle LIM protein, and telethonin. Mol Genet Metab 2006. May;88:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Knöll R, Hoshijima M, Hoffman HM, Person V, Lorenzen‐Schmidt I, Bang M‐L, et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 2002. Dec 27;111:943–955. [DOI] [PubMed] [Google Scholar]
- 6. de Fuenmayor‐Fernández de la CP, Hernández‐Laín A, Olivé M, Fernández‐Marmiesse A, Domínguez‐González C. Novel mutation in TCAP manifesting with asymmetric calves and early‐onset joint retractions. Neuromuscul Disord NMD 2016. Nov;26:749–753. [DOI] [PubMed] [Google Scholar]
- 7. Mazzone A, Strege PR, Tester DJ, Bernard CE, Faulkner G, De Giorgio R, et al. A mutation in telethonin alters Nav1.5 function. J Biol Chem 2008. Jun 13;283:16537–16544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Knöll R, Linke WA, Zou P, Miocic S, Kostin S, Buyandelger B, et al. Telethonin deficiency is associated with maladaptation to biomechanical stress in the mammalian heart. Circ Res 2011. Sep 16;109:758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang R, Yang J, Zhu J, Xu X. Depletion of zebrafish Tcap leads to muscular dystrophy via disrupting sarcomere‐membrane interaction, not sarcomere assembly. Hum Mol Genet 2009. Nov 1;18:4130–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Candasamy AJ, Haworth RS, Cuello F, Ibrahim M, Aravamudhan S, Krüger M, et al. Phosphoregulation of the Titin‐cap protein telethonin in cardiac myocytes. J Biol Chem 2014. Jan 17;289:1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nicholas G, Thomas M, Langley B, Somers W, Patel K, Kemp CF, et al. Titin‐cap associates with, and regulates secretion of. Myostatin J Cell Physiol 2002. Oct;193:120–131. [DOI] [PubMed] [Google Scholar]
- 12. Frey N, Olson EN. Calsarcin‐3, a novel skeletal muscle‐specific member of the calsarcin family, interacts with multiple Z‐disc proteins. J Biol Chem 2002. Apr 19;277:13998–14004. [DOI] [PubMed] [Google Scholar]
- 13. Kojic S, Medeot E, Guccione E, Krmac H, Zara I, Martinelli V, et al. The Ankrd2 protein, a link between the sarcomere and the nucleus in skeletal muscle. J Mol Biol 2004. May 28;339:313–325. [DOI] [PubMed] [Google Scholar]
- 14. Nakano N, Hori H, Abe M, Shibata H, Arimura T, Sasaoka T, et al. Interaction of BMP10 with Tcap may modulate the course of hypertensive cardiac hypertrophy. Am J Physiol Heart Circ Physiol 2007. Dec;293:H3396–H3403. [DOI] [PubMed] [Google Scholar]
- 15. Ikeda F, Crosetto N, Dikic I. What determines the specificity and outcomes of ubiquitin signaling? Cell 2010. Nov 24;143:677–681. [DOI] [PubMed] [Google Scholar]
- 16. Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. EMBO Rep 2008. Jun 1;9:536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Polge C, Uttenweiler‐Joseph S, Leulmi R, Heng A‐E, Burlet‐Schiltz O, Attaix D, et al. Deciphering the ubiquitin proteome: limits and advantages of high throughput global affinity purification‐mass spectrometry approaches. Int J Biochem Cell Biol 2013. Oct;45:2136–2146. [DOI] [PubMed] [Google Scholar]
- 18. Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS. E2 enzymes: more than just middle men. Cell Res 2016. Apr;26:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Wijk SJL, Timmers HTM. The family of ubiquitin‐conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J Off Publ Fed Am Soc Exp Biol 2010. Apr;24:981–993. [DOI] [PubMed] [Google Scholar]
- 20. Christensen DE, Klevit RE. Dynamic interactions of proteins in complex networks: identifying the complete set of interacting E2s for functional investigation of E3‐dependent protein ubiquitination. FEBS J 2009. Oct;276:5381–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. David Y, Ziv T, Admon A, Navon A. The E2 ubiquitin‐conjugating enzymes direct polyubiquitination to preferred lysines. J Biol Chem 2010. Mar 19;285:8595–8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001. Nov 23;294:1704–1708. [DOI] [PubMed] [Google Scholar]
- 23. Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone‐treated skeletal muscle. Cell Metab 2007. Nov;6:376–385. [DOI] [PubMed] [Google Scholar]
- 24. Fielitz J, Kim M‐S, Shelton JM, Latif S, Spencer JA, Glass DJ, et al. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Invest 2007. Sep;117:2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kedar V, McDonough H, Arya R, Li H‐H, Rockman HA, Patterson C. Muscle‐specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci U S A 2004. Dec 28;101:18135–18140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Polge C, Heng A‐E, Jarzaguet M, Ventadour S, Claustre A, Combaret L, et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J 2011. Nov 1;25:3790–3802. [DOI] [PubMed] [Google Scholar]
- 27. Zhao T‐J, Yan Y‐B, Liu Y, Zhou H‐M. The generation of the oxidized form of creatine kinase is a negative regulation on muscle creatine kinase. J Biol Chem 2007. Apr 20;282:12022–12029. [DOI] [PubMed] [Google Scholar]
- 28. Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, et al. Muscle RING‐finger protein‐1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J Mol Biol 2008. Mar 7;376:1224–1236. [DOI] [PubMed] [Google Scholar]
- 29. Hirner S, Krohne C, Schuster A, Hoffmann S, Witt S, Erber R, et al. MuRF1‐dependent regulation of systemic carbohydrate metabolism as revealed from transgenic mouse studies. J Mol Biol 2008. Jun 13;379:666–677. [DOI] [PubMed] [Google Scholar]
- 30. van Wijk SJL, de Vries SJ, Kemmeren P, Huang A, Boelens R, Bonvin AMJJ, et al. A comprehensive framework of E2‐RING E3 interactions of the human ubiquitin‐proteasome system. Mol Syst Biol 2009;5:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Markson G, Kiel C, Hyde R, Brown S, Charalabous P, Bremm A, et al. Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res 2009. Oct;19:1905–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Napolitano LM, Jaffray EG, Hay RT, Meroni G. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem J 2011. Mar 1;434:309–319. [DOI] [PubMed] [Google Scholar]
- 33. Temparis S, Asensi M, Taillandier D, Aurousseau E, Larbaud D, Obled A, et al. Increased ATP‐ubiquitin‐dependent proteolysis in skeletal muscles of tumor‐bearing rats. Cancer Res 1994. Nov 1;54:5568–5573. [PubMed] [Google Scholar]
- 34. Taillandier D, Aurousseau E, Meynial‐Denis D, Bechet D, Ferrara M, Cottin P, et al. Coordinate activation of lysosomal, Ca 2+‐activated and ATP‐ubiquitin‐dependent proteinases in the unweighted rat soleus muscle. Biochem J 1996. May 15;316:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Voisin L, Breuillé D, Combaret L, Pouyet C, Taillandier D, Aurousseau E, et al. Muscle wasting in a rat model of long‐lasting sepsis results from the activation of lysosomal, Ca2+‐activated, and ubiquitin‐proteasome proteolytic pathways. J Clin Invest 1996. Apr 1;97:1610–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Polge C, Leulmi R, Jarzaguet M, Claustre A, Combaret L, Béchet D, et al. UBE2B is implicated in myofibrillar protein loss in catabolic C2C12 myotubes: UBE2B and myofibrillar protein degradation. J Cachexia Sarcopenia Muscle 2016. Jun;7:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Polge C, Koulmann N, Claustre A, Jarzaguet M, Serrurier B, Combaret L, et al. UBE2D2 is not involved in MuRF1‐dependent muscle wasting during hindlimb suspension. Int J Biochem Cell Biol 2016. Oct;79:488–493. [DOI] [PubMed] [Google Scholar]
- 38. Cabantous S, Nguyen HB, Pedelacq J‐D, Koraïchi F, Chaudhary A, Ganguly K, et al. A new protein‐protein interaction sensor based on tripartite split‐GFP association. Sci Rep 2013. Oct 4;3:2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sambrook J, Russell DW. Preparation and transformation of competent E. coli using Calcium chloride In Sambrook J, Russell D, eds. Cold Spring Harb Protoc. New York: Cold Spring Harbor Laboratory Press; 2006. Jun. 2006(1):pdb.prot3932. [Google Scholar]
- 40. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J Off Publ Fed Am Soc Exp Biol 2004. Jan;18:39–51. [DOI] [PubMed] [Google Scholar]
- 41. Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol 2001. Mar 2;306:717–726. [DOI] [PubMed] [Google Scholar]
- 42. McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle‐specific RING finger‐1 interacts with titin to regulate sarcomeric M‐line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein‐1. J Cell Biol 2002. Apr 1;157:125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Arya R, Kedar V, Hwang JR, McDonough H, Li H‐H, Taylor J, et al. Muscle ring finger protein‐1 inhibits PKC{epsilon} activation and prevents cardiomyocyte hypertrophy. J Cell Biol 2004. Dec 20;167:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Witt SH, Granzier H, Witt CC, Labeit S. MURF‐1 and MURF‐2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF‐dependent muscle ubiquitination. J Mol Biol 2005. Jul 22;350:713–722. [DOI] [PubMed] [Google Scholar]
- 45. Mrosek M, Meier S, Ucurum‐Fotiadis Z, von Castelmur E, Hedbom E, Lustig A, et al. Structural analysis of B‐Box 2 from MuRF1: identification of a novel self‐association pattern in a RING‐like fold. Biochemistry (Mosc) 2008. Oct 7;47:10722–10730. [DOI] [PubMed] [Google Scholar]
- 46. Franke B, Gasch A, Rodriguez D, Chami M, Khan MM, Rudolf R, et al. Molecular basis for the fold organization and sarcomeric targeting of the muscle atrogin MuRF1. Open Biol 2014. Mar 26;4: 130172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heng AE, Ventadour S, Jarzaguet M, Pouch‐Pélissier MN, Guezennec CY, Bigard X, et al. Coordinate expression of the 19S regulatory complex and evidence for ubiquitin‐dependent telethonin degradation in the unloaded soleus muscle. Int J Biochem Cell Biol 2008;40:2544–2552. [DOI] [PubMed] [Google Scholar]
- 48. Polge C, Attaix D, Taillandier D. Role of E2‐Ub‐conjugating enzymes during skeletal muscle atrophy. Front Physiol [Internet] 2015. Mar; 10 [cited 2016 Nov 4];6 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4354305/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Duda DM, Olszewski JL, Schuermann JP, Kurinov I, Miller DJ, Nourse A, et al. Structure of HHARI, a RING‐IBR‐RING ubiquitin ligase: autoinhibition of an Ariadne‐family E3 and insights into ligation mechanism. Struct Lond Engl 1993 2013. Jun 4;21:1030–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yokouchi M, Kondo T, Houghton A, Bartkiewicz M, Horne WC, Zhang H, et al. Ligand‐induced ubiquitination of the epidermal growth factor receptor involves the interaction of the c‐Cbl RING finger and UbcH7. J Biol Chem 1999. Oct 29;274:31707–31712. [DOI] [PubMed] [Google Scholar]
- 51. Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c‐Cbl‐UbcH7 complex: RING domain function in ubiquitin‐protein ligases. Cell 2000. Aug 18;102:533–539. [DOI] [PubMed] [Google Scholar]
- 52. Nguyen L, Plafker KS, Starnes A, Cook M, Klevit RE, Plafker SM. The ubiquitin‐conjugating enzyme, UbcM2, is restricted to monoubiquitylation by a two‐fold mechanism that involves backside residues of E2 and Lys48 of ubiquitin. Biochemistry (Mosc) 2014. Jun 24;53:4004–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brown NG, VanderLinden R, Watson ER, Weissmann F, Ordureau A, Wu K‐P, et al. Dual RING E3 Architectures regulate multiubiquitination and ubiquitin chain elongation by APC/C. Cell 2016. Jun 2;165:1440–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, et al. Certain pairs of ubiquitin‐conjugating enzymes (E2s) and ubiquitin‐protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem 2007. Jun 15;282:17375–17386. [DOI] [PubMed] [Google Scholar]
- 55. Sheng Y, Hong JH, Doherty R, Srikumar T, Shloush J, Avvakumov GV, et al. A human ubiquitin conjugating enzyme (E2)‐HECT E3 ligase structure‐function screen. Mol Cell Proteomics MCP 2012. Aug;11:329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McGinty RK, Henrici RC, Tan S. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 2014. Oct 30;514:591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wenzel DM, Stoll KE, Klevit RE. E2s: structurally economical and functionally replete. Biochem J 2011. Jan 1;433:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wittwer M, Flück M, Hoppeler H, Müller S, Desplanches D, Billeter R. Prolonged unloading of rat soleus muscle causes distinct adaptations of the gene profile. FASEB J Off Publ Fed Am Soc Exp Biol 2002. Jun;16:884–886. [DOI] [PubMed] [Google Scholar]
- 60. Banduseela VC, Ochala J, Chen Y‐W, Göransson H, Norman H, Radell P, et al. Gene expression and muscle fiber function in a porcine ICU model. Physiol Genomics 2009. Nov 6;39:141–159. [DOI] [PubMed] [Google Scholar]
- 61. Lecker SH, Solomon V, Price SR, Kwon YT, Mitch WE, Goldberg AL. Ubiquitin conjugation by the N‐end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J Clin Invest 1999. Nov;104:1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Combaret L, Tilignac T, Claustre A, Voisin L, Taillandier D, Obled C, et al. Torbafylline (HWA 448) inhibits enhanced skeletal muscle ubiquitin‐proteasome‐dependent proteolysis in cancer and septic rats. Biochem J 2002. Jan 15;361:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1‐dependent ubiquitylation. J Cell Biol 2009. Jun 15;185:1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marblestone JG, Butt S, McKelvey DM, Sterner DE, Mattern MR, Nicholson B, et al. Comprehensive ubiquitin E2 profiling of ten ubiquitin E3 ligases. Cell Biochem Biophys 2013. Sep;67:161–167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1 Representative wide field images of negative controls of split‐GFP assays GFP10‐E2J1, E2E1‐GFP10, E2G1‐GFP10, E2L3‐GFP10, E2D2‐GFP10 and MuRF1‐GFP11 constructs were transfected with jetPRIME reagent (Polyplus‐transfection) in HEK293 cells stably expressing the GFP1‐9 fragment (HEK_GFP1‐9). These constructs alone gave clear background. Scale bars = 10 μm.
Supplemental figure 2 Zoom of E2s mildly and non‐interacting with MuRF1 during the Surface Plasmon Resonance screen
Supplemental figure 3 Specific interaction of telethonin with the E3 ligase MuRF1 (a) Yeast two‐hybrid assay. Y2HGold strain expressing GAL4‐BD fusions and Y187 strain expressing GAL4‐AD fusions were mated. Diploids were plated on selective medium missing Leu and Trp (‐LT) to control homogeneous growth of the yeast and on medium missing Leu, Trp, His and adenine (‐LTHAd) to detect interactions. MuRF1‐MuRF3, MuRF3‐MuRF3 and p53‐LargeT interactions were used as positive controls. GAL4‐AD, fusions with the Activation Domain of GAL4; GAL4‐BD, fusions with the DNA Binding Domain of GAL4; D1, day 1; D6, day 6. (b) Confirmation of GST‐MuRF1‐telethonin interaction using GST pulldown assay. His‐telethonine was co‐expressed with GST‐MuRF1 or GST in BL21 (DE3) E. coli. Bacteria were lyzed and extracts were submitted to GST pulldown. Immunoblotting against MuRF1 (upper panel) and telethonin (lower panel) showed that telethonin was pulled down with GST‐MuRF1 but not with GST alone (arrows). Lys, lysat; FT, Flowthrough; W1 and W3, first and third washes; Elu, eluate; IB, immunoblot
Supplemental figure 4 Representative wide field images of split‐GFP assays performed in Fig. 5
Supplemental table S1 Compilation of skeletal muscle E2s data