

Abstract
Rationale: The mechanisms underlying formation of lung lymphoid follicles (LF) in chronic obstructive pulmonary disease (COPD) are unknown. The chemokine receptor CXCR3 regulates immune responses in secondary lymphoid structures elsewhere in the body and is highly expressed by Th1 lymphocytes in the airway in COPD. Because chemokine receptors control inflammatory cell homing to inflamed tissue, we reasoned that CXCR3 may contribute to LF formation in COPD.
Objectives: We assessed the expression of CXCR3 and its ligands (IP-10/CXCL10, Mig/CXCL9, and ITAC/CXCL11) by LF cells in never-smokers, smokers without COPD, and subjects with COPD.
Methods: CXCR3, IP-10, Mig, and ITAC expression were assessed in lung sections from 46 subjects (never-smokers, smokers without COPD [S], and subjects with COPD in GOLD stages 1–4) by immunohistochemistry.
Measurements and Main Results: CXCR3-expressing T cells (CD8+ or CD4+) and B cells (CD20+) were topographically distributed at the follicle periphery and center, respectively. The percentage of immunohistochemically identified CXCR3+ cells increased progressively while proceeding from S through GOLD 3–4 (P < 0.01 for GOLD 3–4 vs. S). Moreover, the number of CXCR3+ follicular cells correlated inversely with FEV1 (r = 0.60). The CXCR3 ligands IP-10 and Mig were expressed by several cell types in and around the follicle, including CD68+ dendritic cells/ macrophages, airway epithelial cells, endothelial cells, and T and B cells.
Conclusions: These results suggest that LF form in the COPD lung by recruitment and/or retention of CXCR3-expressing T and B lymphocytes, which are attracted to the region through production of CXCR3 ligands IP-10 and Mig by lung structural and follicular cells.
Keywords: lung inflammation, chemokines, cell recruitment
AT A GLANCE COMMENTARY
Scientific Knowledge on the Subject
The chemokine receptor CXCR3 regulates immune responses in secondary lymphoid structures and is highly expressed by Th1 lymphocytes in the airway in chronic obstructive pulmonary disease (COPD). In contrast, the mechanisms underlying formation of lung lymphoid follicles in COPD are unknown.
What This Study Adds to the Field
We report that the chemokine receptor CXCR3 and its ligands Mig/CXCL9 and IP-10/CXCL10 are highly expressed by cells in and around lymphoid follicles in COPD. These findings suggest that CXCR3 contributes to follicle formation in COPD.
Chronic obstructive pulmonary disease (COPD) is an inflammatory disease in which the cellular infiltrate is comprised primarily of CD8+/Tc1 and CD4+/Th1 lymphocytes and macrophages (1–6). This infiltrate, which persists long after cigarette smoking is ended (7, 8), is diffusely distributed throughout the lung, including the small airways, submucosal glands, lung parenchyma, and pulmonary arteries (1, 3, 5, 9–11). In addition, T cells, B cells, macrophages, and dendritic cells aggregate into organized lymphoid follicles in close proximity to the airways and within the lung parenchyma (12–14). It has been reported that the number of airways containing lymphoid follicles increases in severe COPD (i.e., in GOLD stages 3 and 4) when compared with subjects in GOLD stages 0 to 2 (12).
Lung lymphoid follicles (LF) are known to participate in adaptive humoral and T-cell–mediated responses to antigen (15). However, the mechanisms that contribute to their formation in COPD are poorly understood. Nonetheless, the basic processes governing the targeting and recruitment of inflammatory cells into tissues are now well understood (16, 17). Specifically, inflammatory cell recruitment requires a multistep process in which receptors expressed by inflammatory cells interact with complementary receptors expressed on endothelial cells of venules in the target tissue. The first step—loose rolling and attachment of inflammatory cells to the vessel wall—requires activation of addressins on lymphocytes and selectins on endothelial cells. In contrast, the second and third steps, firm cell attachment to the vessel wall followed by diapedesis into inflamed tissue, are mediated by G-protein—coupled chemokine receptors on the surface of inflammatory cells, which are activated by their cognate ligands produced in the target tissue. Receptor activation in turn up-regulates and enhances the affinity of integrins expressed by the inflammatory cell. Selectivity of recruitment of T- and B-cell subsets to different secondary lymphoid tissues is achieved by cell-type specific expression of discrete chemokine receptors and expression of their ligands in the target tissue (16, 17).
Considerable knowledge exists about the mechanisms that mediate CD4+ and CD8+ T-cell recruitment into the airways in subjects with COPD (2, 4, 18, 19). Specifically, the majority of CD4+ and CD8+ T cells in the airways of subjects with COPD are of the Th1/Tc1 phenotype and primarily express the chemokine receptor CXCR3 and, to a lesser extent, CCR5. In contrast, the chemokine/chemokine receptors contributing to lung lymphoid follicle formation in COPD are unstudied. CXCR3 is expressed by T cells and B cells in secondary lymphoid structures in other parts of the body, where it plays an important role in type I cellular and humoral immune responses (20–22). We hypothesized that recruitment and possibly retention of T cells and B cells to LF is mediated by CXCR3 expression in response to production of its cognate ligands by cells in close proximity to the follicle.
In the present study, we examined the expression of CXCR3 and its ligands by lymphoid follicle cells in the COPD lung and examined the effects of cigarette smoking on lymphoid follicle CXCR3 expression by comparing results obtained in subjects with COPD with smokers without COPD and never-smokers. Finally, because specific chemokine receptors mediate T-cell recruitment into lymphoid follicles in other parts of the body, we examined LF expression of CCR9 and CCR10, the chemokine receptors that mediate T-cell recruitment into lymphoid follicles in the skin (i.e., CCR9) and gut (i.e., CCR10) (23–26).
The results of the present study indicate that the number of CXCR3+ T and B cells in lymphoid follicles increases in COPD but not in smokers without COPD. Moreover, the CXCR3 ligands IP-10/CXCL10 and Mig/CXCL9 are expressed by lung follicular cells and the overlying airway epithelium. Finally, LF do not express the chemokine receptors expressed in lymphoid follicles in the skin and gut (i.e., CCR9 and CCR10).
METHODS
Study Population
Lung tissue was obtained from 27 subjects with COPD and 19 non-COPD subjects. In 33 subjects, tissue samples were obtained during thoracotomy for the purpose of lung cancer resection, lung volume reduction surgery, or lung transplantation. In 13 subjects, samples were obtained by the region's lung transplant organization, Gift of Life, Inc.
Pulmonary Function
Severity of COPD was assessed according to the Global Initiative for Obstructive Lung Disease (GOLD) score based on their preoperative postbronchodilatory spirometry results (27). Measurements of FEV1, FVC, and FEV1/FVC were obtained using a spirometer that met American Thoracic Society standards. Lung function tests were not available for lungs provided by the Gift of Life (n = 13). In these subjects, obstructive lung disease was excluded based on a life-long nonsmoking history or, in the case of smokers, age < 45 years.
Immunohistochemistry
Lungs were fixed in 4% formalin, embedded in paraffin, and cut into serial sections 5 μm thick. Sections were heated at 60°C for 40 minutes and deparaffinized by passage through xylene (3 × 10 minutes) followed by serial ethanol for hydration. Antigen retrieval was performed by heating in citrate or EDTA buffer, depending on the antigen/antibody combination being studied (see Table E1 in the online supplement for details). Sections were treated with 3% hydrogen peroxide in methanol for 20 minutes to quench endogenous peroxidase and blocked with 2.5% normal horse serum for 1 hour. Serial sections were stained for 1 hour at room temperature for CXCR3, CD4, CD8, CD20, CD68, Mig, IP-10, ITAC, CCR9, or CCR10. For each antigen, the intensity and specificity of staining was optimized relative to an appropriate IgG isotype control by varying antibody concentration and incubation period (Table E1). After PBS wash, slides were incubated with horseradish peroxidase polymer–conjugated antimouse IgG (ImPress kit; Vector Labs, Burlingame, CA) for 30 minutes and with DAB substrate for 3 to 5 minutes (Dako, Glostrup, Denmark) or with NovaRED substrate for 5 to 7 minutes (Vector Labs). Slides were then stained with hematoxylin, dehydrated, and mounted.
Several antibodies have been used to assess CXCR3 immunoreactivity in human tissues (2, 28, 29). In preliminary studies in lung tissue from five subjects with COPD, we compared two widely used anti-CXCR3 antibodies (clone 49,801.111 [R&D Systems, Minneapolis, MN] and clone 1C6 [BD Biosciences, San Diego, CA]). Both antibodies showed a high degree of specific staining compared with isotype control antibodies. In serial sections, the percentage of follicular cells staining positively with the 49,801.111 and 1C6 antibodies was similar (P > 0.25). Accordingly, only the 49,801.111 clone was used in the remaining subjects, and the results reported are with this antibody.
Lymphoid follicles were identified in hematoxylin-stained sections at 100 to 200× magnification as an aggregate of contiguous mononuclear cells. Follicles were photographed for analysis using a digital camera at maximum resolution (3,840 × 3,072 pixels) (Nikon E-600 microscope, Digital Eclipse DXM-1200 camera, Nikon ACT-1 software; Nikon, Tokyo, Japan). Image postprocessing was performed using Adobe Photoshop CS2 (Adobe Software, San Jose, CA) to optimize contrast, color saturation, and sharpness. In sections containing multiple follicles, a minimum of two follicles were photographed.
Follicle boundaries were delineated by tracing the perimeter and calculating the area using Image J software (NIH, Bethesda, MD) (Figure 1). Follicle cell number was determined from measurements of follicle area using the following equation: cell number = 18,563.4 × follicle area in mm2 (r = 0.99). A data set of eight lymphoid follicles having a 10-fold range of area was used to derive this equation.
Figure 1.
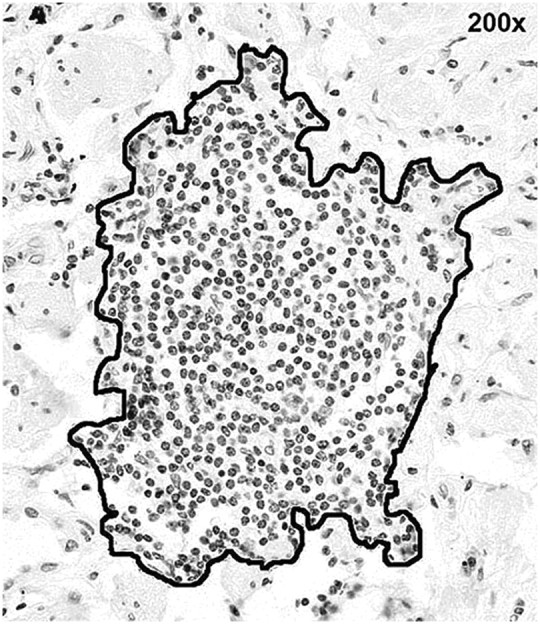
Lung lymphoid follicle. Follicle boundary (i.e., perimeter) was traced freehand to delineate follicular cells. Original magnification: 200×.
Cells staining for CXCR3 on their membrane were counted using the crosshair tool in Image J, which keeps a running count of positive cells identified by mouse click. For CXCR3+ cells, counting was performed by two independent observers. Counts of the two observers for the same follicles were highly correlated and yielded similar results (r > 0.95).
Immunofluorescence Microscopy
In subsequent experiments, the identity of CXCR3+ cells was confirmed by double labeling with immunofluorescent antibodies against CD20 (a B-cell antigen), CD8, or CD4 using a commercially available Mouse on Mouse labeling kit (Vector Labs). After antigen retrieval, formalin-fixed tissue sections were incubated with avidin-biotin blocking reagent and reacted with anti-CXCR3 antibody (clone 49,801.111). Biotinylated antimouse IgG antibody was added, followed by Cy5-conjugated streptavidin. Tissues were incubated again with avidin-biotin blocking reagent and reacted with CD8, CD4, or CD20 antibody. Biotinylated antimouse IgG antibody was then added, followed by fluorescein-conjugated avidin. Tissue sections were cover-slipped with mounting material containing 4′,6′-diamidine-2-phenylindole dilactate (DAPI) to label cell nuclei (Vectashield, Vector Labs) and stored in the dark at 4°C. Follicles were photographed using a Nikon E-800 fluorescent microscope with attached Retiga EXi digital camera (Qimaging Inc, Surrey, BC, Canada) and processed in Photoshop CS2.
Statistics
Statistical analysis was performed using SigmaStat (Systat Software, Chicago, IL). Group data are expressed as mean ± SEM. Statistical significance of differences in group mean values was determined on normally distributed data by one-way ANOVA and on nonnormally distributed data by Kruskal-Wallis one-way ANOVA on ranks. Post-hoc, pair-wise comparisons were performed using Dunn's Method and unpaired t test. A P value < 0.05 was considered significant.
RESULTS
Study Population
The demographic characteristics, smoking history, and pulmonary function data of the study subjects are shown in Table 1. Gender ratio was similar across groups. FEV1 % predicted, FEV1/FVC, and DlCO/VA % predicted were significantly different across the several GOLD groups (P < 0.001 for each parameter) as expected. Cigarette pack-years and age were greater in GOLD 1–2 and 3–4 than in smokers without COPD (S) (P ≤ 0.01 for each comparison).
TABLE 1.
SUBJECT CHARACTERISTICS
|
Subject Groups |
NS |
S |
G1-2 |
G3-4 |
P Value |
|---|---|---|---|---|---|
| Gender, male/female | 5/4 | 5/5 | 7/4 | 8/8 | 0.90 |
| Age, year | 51 ± 6 | 47 ± 5 | 61 ± 2 | 59 ± 2 | 0.02 |
| Smoking history, pack-year | — | 28 ± 5 | 45 ± 4 | 54 ± 6 | 0.01 |
| FEV1, % predicted | — | 91 ± 5 | 72 ± 4 | 28 ± 3 | <0.001 |
| FEV1/FVC, % | — | 78 ± 4 | 62 ± 2 | 36 ± 3 | <0.001 |
| DlCO/VA, % predicted |
— |
85 ± 9 |
83 ± 5 |
51 ± 6 |
<0.001 |
Definition of abbreviations: G=Global lnitiative for Obstructive Lung Disease score (subjects with COPD); NS=never-smokers; S=smokers without COPD.
Most of the subjects in all GOLD stages had stopped smoking at the time of lung resection. For example, 70% of smokers without COPD (S), 50% of subjects in GOLD 1–2, and 100% of subjects in GOLD 3–4 were exsmokers.
Lymphoid Follicle Number Increases in Smokers and Subjects with COPD
Lymphoid follicles were more frequent in smokers or exsmokers compared with never-smokers. For example, follicle number per unit area of tissue (follicles/cm2) was 0.4 ± 0.1 in never-smokers, 1.2 ± 0.2 in smokers without COPD, 2.0 ± 0.6 in GOLD 1–2, and 4.9 ± 1.1 in GOLD 3–4 (P < 0.015 for the comparison of each GOLD group to never-smokers).
Expression of CXCR3 and its Ligands by Follicular Cells
Lymphoid follicles demonstrated a characteristic topographical arrangement of cells (Figure 2). CD20+ B cells, which comprised the majority of cells, were localized to the center of the follicle. The smaller numbers of CD4+ and CD8+ T cells were located primarily at the periphery of the follicle (Figure 2).
Figure 2.
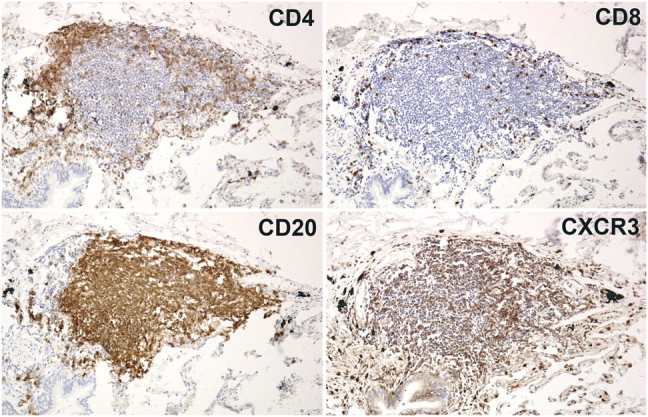
Lymphoid follicle topography. A representative lymphoid follicle (GOLD 1) was immunohistochemically stained for CD4, CD8, CD20, or CXCR3 (brown stain). Serial sections show CD20+ B cells concentrated in the follicle center, CD4+ and CD8+ T cells localized at the periphery, and CXCR3+ cells diffusely distributed throughout the follicle. Original magnification: 200×.
CXCR3+ cells were highly prevalent within the follicle and widely distributed in the B- and T-cell areas, suggesting that all three major inflammatory cell types expressed the receptor (Figure 2). This was confirmed by fluorescent double-labeling, which demonstrated that CXCR3+ cells also stained positively for CD20 and CD8 (Figures 3 and 4). CXCR3+/CD20+ double-positive cells were detected in B-cell areas (Figure 3), whereas CXCR3+/CD8+ double-positive cells were detected in the periphery of the follicle (Figure 4). Similar results were obtained for CXCR3/CD4 (data not shown).
Figure 3.

CXCR3 is expressed by CD20+ B cells. Sections were double-labeled for CXCR3 (Cy5-streptavidin, red) and CD20 (fluorescein-avidin, green). Cell nuclei were stained with DAPI (blue). The top row shows lymphoid follicle cells viewed in the Cy5 channel (red) (left), the FITC channel (green) (middle), or as an overlay of images 1 and 2 (right). The middle row shows insets at higher power. The bottom row shows isotype controls. Original magnification: 600–1,000×.
Figure 4.

CXCR3 is expressed by CD8+ T cells. Sections were double-labeled for CXCR3 (Cy5-streptavidin, red) and CD8 (fluorescein-avidin, green). Cell nuclei were stained with DAPI (blue). The top row shows lymphoid follicle cells viewed in the Cy5 channel (red) (left), the FITC channel (green) (middle), or as an overlay of images 1 to 2 (right). The middle row shows insets at higher power. The bottom row shows isotype controls. Original magnification: 400–1,000×.
IP-10 and Mig immunoreactivity were present in a high percentage of large, irregularly shaped CD68+ dendritic/macrophage cells and smaller mononuclear cells that resembled B and T lymphocytes (Figure 5). Mig immunoreactivity, and to a lesser extent IP-10 and ITAC immunoreactivity, were also seen in the overlying airway epithelium and high endothelial venules (see Figure E3).
Figure 5.
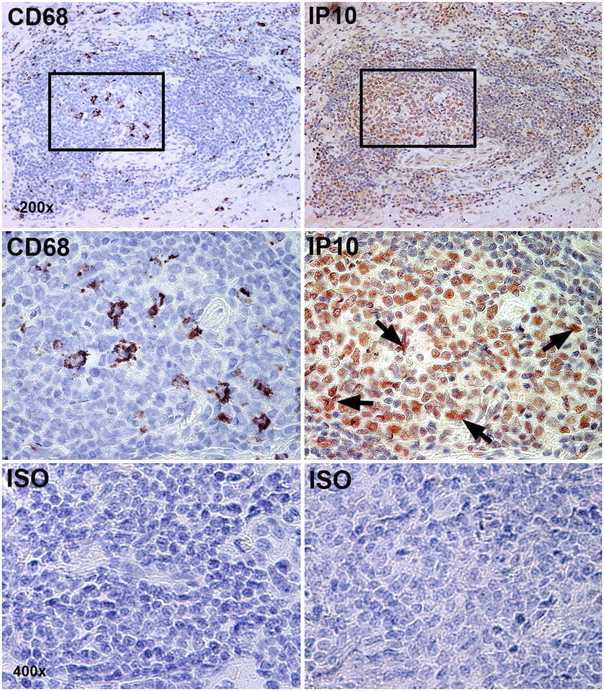
IP-10 and CD68 are expressed in lymphoid follicles. The top row of images shows adjacent serial sections of a single lymphoid follicle stained for CD68 and IP-10. The middle row shows insets at higher power. The bottom row shows isotype controls. Black arrows show IP-10 staining of stellate dendritic cells amid smaller mononuclear cells in lymphoid follicle germinal center. Original magnification: 200–400×.
In contrast to CXCR3, no cells positive for CCR9 or CCR10 were seen in lung follicles, although isolated stained cells were present in Peyer's patches and tonsillar tissue used as positive controls (Figures E1 and E2).
CXCR3 Expression in LF is Increased in Subjects with COPD
The percentage of CXCR3+ cells within lung follicles varied across the four groups (P < 0.001 by ANOVA) (Figures 6 and 7). Specifically, the percentage of CXCR3+ cells was 33 ± 6% in never-smokers (NS), 45 ± 8% in smokers without COPD (S), 54 ± 5% in GOLD 1–2, and 73 ± 6% in GOLD 3–4 (Figure 7). The percentage of CXCR3+ cells tended to be greater in smokers without COPD (S) than in never-smokers (NS) (i.e., 44 vs. 32%), but differences did not reach statistical significance (P > 0.2). In contrast, the percentage of CXCR3+ cells increased significantly with increasingly severe COPD. For example, CXCR3+ cells were significantly greater in GOLD 3–4 than in subjects in GOLD 1–2 and smokers without COPD (S) (P < 0.02 for both comparisons). The smoking histories were similar in subjects in GOLD 3–4 and GOLD 1–2 (P > 0.2).
Figure 6.

Representative lymphoid follicles stained for CXCR3. Follicle cells stained for CXCR3 (brown stain) in representative subjects from never-smokers (NS), smokers without COPD (S), and GOLD 4 groups. Isotype controls shown for comparison.
Figure 7.

CXCR3+ cells in lymphoid follicles—individual data. Scatterplot shows the percentage of CXCR3+ follicular cells in individual never-smokers (NS), smokers without chronic obstructive pulmonary disease (COPD) (S), and subjects with COPD in GOLD stages 1 to 4. Horizontal bars show group mean values. The percentage of CXCR3+ cells increased significantly across all groups (P < 0.001) and with increasing COPD severity (P = 0.016 for G3–4 vs. S by post-hoc t test).
In agreement with the above results of CXCR3+ cells with increasing GOLD score, the percentage of CXCR3+ cells correlated inversely with FEV1 (r = 0.60; Figure 8).
Figure 8.

CXCR3+ cells in lymphoid follicles correlate with FEV1. Scatterplot showing individual results of percentage of CXCR3+ follicular cells versus FEV1% predicted. Solid circles: smokers without COPD; inverted open triangles: GOLD 1–2; solid triangles: GOLD 3–4. FEV1 was inversely related (2nd order polynomial) to percentage of CXCR3+ cells (r = 0.60).
DISCUSSION
In humans, LF are induced by chronic inflammation or infection (30–35), and the number of LF is increased in advanced COPD (12–14). However, the cellular and molecular mechanisms that contribute to the formation of LF in COPD are not well understood.
The present study addressed this issue by examining expression of the chemokine receptor CXCR3, which mediates type I (Th1/Tc1) cell responses in secondary lymphoid structures in other parts of the body (20, 22, 36, 37). Lung lymphoid follicle cells were studied in subjects with COPD (GOLD stages 1–4), chronic smokers without COPD and never-smokers. Standard immunohistochemical techniques were used to examine expression of CXCR3 by follicular T cells (CD4+ and CD8+ cells), B cells (CD20+), and dendritic cells/macrophages (CD68+ cells).
Our results are in agreement with those of other researchers who have demonstrated that the number of LF is increased in chronic cigarette smokers (32) and even more so in subjects with advanced COPD (12, 13, 32). Our results also confirm prior studies that have demonstrated that LF are comprised primarily of B cells, with a smaller number of CD4+ and CD8+ T cells topographically arranged at the periphery (14, 15, 31).
In this study, we demonstrate for the first time that a majority of T and B cells in lung follicles express CXCR3 and that the percentage of CXCR3-expressing cells is greater in subjects with COPD than in chronic smokers without COPD. Moreover, within the COPD population, the percentage of CXCR3+ cells increases with increasing GOLD score and decreasing FEV1.
Cigarette smoking contributes to the formation of LF and the development of COPD (14, 32, 39). In our study, CXCR3+ cells tended to increase in smokers compared with never-smokers. However, we believe that the increased percentage of CXCR3+ cells in LF in COPD reflects factors in addition to cigarette smoking per se for the following two reasons: (1) CXCR3+ cells were greater in GOLD 3–4 than in GOLD 1–2 despite a similar smoking history; and (2) most subjects in the several GOLD stages were exsmokers including 100% of subjects in GOLD 3–4, the group with the highest percentage of CXCR3+ cells. Rather, we think it likely that CXCR3+ T and B cells in LF reflect the contribution of additional pathogenetic mechanisms to the development of COPD. For example, viral infection and/or immune responses to autoantigen(s) have been implicated in the pathogenesis of the inflammatory response characteristic of COPD (18, 40–42). Immune responses to both types of antigens involve CXCR3+ Th1 lymphocytes (31, 43, 44).
In this study, we also demonstrate for the first time that several cell types within and close to the lymphoid follicles express the cognate CXCR3 ligands IP-10/CXCL10 and Mig/CXCL9. Specifically, T and B lymphocytes, CD68+ dendritic cells/macrophages, endothelial cells, and the airway epithelial cells overlying follicles expressed IP-10 or Mig. Production of these CXCR3 ligands by cells in and around the follicle is the likely mechanism for recruitment and possibly retention of CXCR3-expressing T and B cells, as has been reported in other secondary lymphoid structures (22, 45).
The present study suggests that CCR9 and CCR10, the respective chemokine receptors that recruit T and B cells to secondary lymphoid structures in the gut and skin, are not expressed in LF (23–25).
Previous studies indicate that in COPD most CD4+ and CD8+ cells in the airway wall and lung parenchyma express CXCR3 and are of the Th1 phenotype (i.e., they express IFN-γ rather than IL-4) (2, 4, 18, 43). Moreover, lung T cells from subjects with COPD produce greater amounts of IFN-γ, IP-10, and Mig than T cells from smokers without COPD, suggesting that CXCR3 or its signaling pathway are up-regulated (43).
Although the present study indicates that CXCR3 expression is enhanced in lymphoid follicle cells in COPD, it is unlikely that the CXCR3 chemoreceptor is solely responsible for inflammatory cell recruitment into these structures. For example, Th1 cells generally coexpress CXCR3 and CCR5 (2, 20, 24, 38, 43, 46). In fact, in lung homogenates, the numbers of CCR5+, CXCR6+, and CXCR3+ T cells are increased in COPD compared with smokers (47). However, in that study, the population of CXCR3+ cells greatly exceeded (∼10-fold excess) the CCR5+ or CXCR6+ populations, suggesting a predominant role for CXCR3 (47).
Overexpression of CXCR3 ligands by airway and alveolar epithelial cells is believed to explain the increase in Th1/Tc1 cells in COPD (4, 18, 19, 43). In fact, IP-10, Mig, and ITAC are up-regulated in COPD in small airway epithelial cells and whole lung lysates (4, 47–49). IP-10, Mig, and ITAC concentrations in sputum are increased in COPD compared with nonsmokers (50). Moreover, airway and alveolar epithelial cells release IP-10, Mig, and I-TAC in response to cytokines present in COPD (i.e., IFN-γ, TNF-α, and IL-1) and, in the case of IP-10, in response to viral infection (51). Our data extend these findings to LF and support the hypothesis that in COPD, CXCR3-expressing T and B cells are recruited to and possibly retained in LF in response to IP-10 and/or Mig produced by inflammatory cells and structural cells inside and outside the follicle.
Previous studies on secondary lymphoid structures outside the lung have elucidated several important immunological functions of CXCR3 in addition to chemotaxis. These studies suggest that CXCR3+ cells likely contribute to humoral and cell-mediated type I immune reactions to antigen in LF (21, 22, 39). For example, in the presence of antigen, CXCR3+ memory Th1/Tc1 cells (but not naive T cells) and CXCR3+ memory B cells are rapidly activated and produce cytokines and chemokines, which amplify the immune response (20–22, 37). Exposure to antigen also up-regulates CXCR3 expression in these cells (37, 38). In addition, antibody production by memory B cells is markedly impaired in Mig knockout mice, indicating that CXCR3+ CD4+ T cells assist B cells in producing antibody (22). Moreover, antigen presentation as a result of physical contact between dendritic cells and CD4+ T cells is augmented by dendritic cell release of IP-10 (22, 45).
Activation of CXCR3 in T cells induces pleiotropic responses in addition to chemotaxis, which likely foster lymphoid follicle growth and promote lung inflammation. For example, IP-10, Mig, or I-TAC enhance proliferation of antigen-stimulated Th1 cells and augment their production of IFN-γ, thereby activating dendritic cells and macrophages (28, 38, 52). Finally, IP-10 and Mig released by lung CD4+ and CD8+ T cells stimulate alveolar macrophage production of matrix metalloproteinase-12, which digests lung elastin and has been linked to emphysema (40, 43).
Although the precise role of LF in COPD is unclear and is not elucidated in the present study, follicles are known to mediate humoral and cell-mediated adaptive immune responses (15, 31). LF protect against infectious agents (e.g., influenza virus) but may also contribute to lung damage in autoimmune diseases (e.g., rheumatoid lung disease) (15, 31). Recently, it has been suggested that COPD is an autoimmune disease and that LF participate in this process (14, 43, 53, 54). Lung T cells from emphysematous subjects proliferate and release cytokine in the presence of elastin fragments, and circulating antibodies against elastin fragments are increased in proportion to the severity of emphysema (53). Also, LF in COPD are comprised of an oligoclonal population of immunoglobulin-secreting B cells (14). It has been suggested that these oligoclonal B cells secrete autoantibodies directed against epitopes on matrix proteins or tobacco smoke products (14). Our results indicate that CXCR3 expression is increased in proportion to the severity of COPD. Because previous studies indicate that CXCR3 expression in immune cells is up-regulated by exposure to antigen (37, 38), the findings of our study are compatible with ongoing stimulation of LF cells by an autoantigen and /or alloantigen in subjects with COPD.
The results of the present study and previous studies in animal models in which CXCR3 is inhibited or knocked out suggest a potential therapeutic role for CXCR3 antagonists alone or in combination with CCR5 antagonists in COPD (44, 55–59). For example, CXCR3 knockout mice demonstrate attenuated inflammatory responses to short-term cigarette smoke exposure (56). Further, CXCR3/CCR5 double knockout mice demonstrate impaired entry of antigen-specific CD8 cells into the lung during influenza virus infection (44). Finally, small-molecule CXCR3/CCR5 antagonists inhibit allograft inflammation and rejection in animal models (55).
In conclusion, the present study indicates that the chemokine receptor CXCR3 is highly expressed by CD4+ and CD8+ T cells and CD20+ B cells in lymphoid follicles of subjects with COPD and that CXCR3 expression in lymphoid follicles increases with worsening COPD.
Supported by Temple University.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.200807-1089OC on February 12, 2009
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Di Stefano A, Turato G, Maestrelli P, Mapp CE, Ruggieri MP, Roggeri A, Boschetto P, Fabbri LM, Saetta M. Airflow limitation in chronic bronchitis is associated with T-lymphocyte and macrophage infiltration of the bronchial mucosa. Am J Respir Crit Care Med 1996;153:629–632. [DOI] [PubMed] [Google Scholar]
- 2.Panina-Bordignon P, Papi A, Mariani M, Di Lucia P, Casoni G, Bellettato C, Buonsanti C, Miotto D, Mapp C, Villa A, et al. The C–C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J Clin Invest 2001;107:1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE, Maestrelli P, Ciaccia A, Fabbri LM. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;157:822–826. [DOI] [PubMed] [Google Scholar]
- 4.Saetta M, Mariani M, Panina-Bordignon P, Turato G, Buonsanti C, Baraldo S, Bellettato CM, Papi A, Corbetta L, Zuin R, et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002;165:1404–1409. [DOI] [PubMed] [Google Scholar]
- 5.Saetta M, Turato G, Facchini FM, Corbino L, Lucchini RE, Casoni G, Maestrelli P, Mapp CE, Ciaccia A, Fabbri LM. Inflammatory cells in the bronchial glands of smokers with chronic bronchitis. Am J Respir Crit Care Med 1997;156:1633–1639. [DOI] [PubMed] [Google Scholar]
- 6.Turato G, Zuin R, Miniati M, Baraldo S, Rea F, Beghe B, Monti S, Formichi B, Boschetto P, Harari S, et al. Airway inflammation in severe chronic obstructive pulmonary disease: relationship with lung function and radiologic emphysema. Am J Respir Crit Care Med 2002;166:105–110. [DOI] [PubMed] [Google Scholar]
- 7.Willemse BW, ten Hacken NH, Rutgers B, Lesman-Leegte IG, Timens W, Postma DS. Smoking cessation improves both direct and indirect airway hyperresponsiveness in COPD. Eur Respir J 2004;24:391–396. [DOI] [PubMed] [Google Scholar]
- 8.Gamble E, Grootendorst DC, Hattotuwa K, O'Shaughnessy T, Ram FS, Qiu Y, Zhu J, Vignola AM, Kroegel C, Morell F, et al. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur Respir J 2007;30:467–471. [DOI] [PubMed] [Google Scholar]
- 9.Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, Mapp CE, Fabbri LM, Donner CF, Saetta M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 1998;158:1277–1285. [DOI] [PubMed] [Google Scholar]
- 10.Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med 1995;152:1666–1672. [DOI] [PubMed] [Google Scholar]
- 11.Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, Cavallesco G, Tropeano G, Mapp CE, Maestrelli P, et al. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999;160:711–717. [DOI] [PubMed] [Google Scholar]
- 12.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004;350:2645–2653. [DOI] [PubMed] [Google Scholar]
- 13.Hogg JC, Chu FS, Tan WC, Sin DD, Patel SA, Pare PD, Martinez FJ, Rogers RM, Make BJ, Criner GJ, et al. Survival after lung volume reduction in chronic obstructive pulmonary disease: insights from small airway pathology. Am J Respir Crit Care Med 2007;176:454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Strate BW, Postma DS, Brandsma CA, Melgert BN, Luinge MA, Geerlings M, Hylkema MN, van den Berg A, Timens W, Kerstjens HA. Cigarette smoke-induced emphysema: a role for the B cell? Am J Respir Crit Care Med 2006;173:751–758. [DOI] [PubMed] [Google Scholar]
- 15.Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, Woodland DL, Lund FE, Randall TD. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med 2004;10:927–934. [DOI] [PubMed] [Google Scholar]
- 16.Ebert LM, Schaerli P, Moser B. Chemokine-mediated control of T cell traffic in lymphoid and peripheral tissues. Mol Immunol 2005;42:799–809. [DOI] [PubMed] [Google Scholar]
- 17.Kehrl JH. Chemoattractant receptor signaling and the control of lymphocyte migration. Immunol Res 2006;34:211–227. [DOI] [PubMed] [Google Scholar]
- 18.Curtis JL, Freeman CM, Hogg JC. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc 2007;4:512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panina P, Mariani M, D'Ambrosio D. Chemokine receptors in chronic obstructive pulmonary disease (COPD). Curr Drug Targets 2006;7:669–674. [DOI] [PubMed] [Google Scholar]
- 20.Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med 1998;187:875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park MK, Amichay D, Love P, Wick E, Liao F, Grinberg A, Rabin RL, Zhang HH, Gebeyehu S, Wright TM, et al. The CXC chemokine murine monokine induced by IFN-gamma (CXC chemokine ligand 9) is made by APCS, targets lymphocytes including activated B cells, and supports antibody responses to a bacterial pathogen in vivo. J Immunol 2002;169:1433–1443. [DOI] [PubMed] [Google Scholar]
- 22.Rabin RL, Alston MA, Sircus JC, Knollmann-Ritschel B, Moratz C, Ngo D, Farber JM. CXCR3 is induced early on the pathway of CD4+ T cell differentiation and bridges central and peripheral functions. J Immunol 2003;171:2812–2824. [DOI] [PubMed] [Google Scholar]
- 23.David R, Marelli-Berg FM. Regulation of T-cell migration by co-stimulatory molecules. Biochem Soc Trans 2007;35:1114–1118. [DOI] [PubMed] [Google Scholar]
- 24.Kunkel EJ, Boisvert J, Murphy K, Vierra MA, Genovese MC, Wardlaw AJ, Greenberg HB, Hodge MR, Wu L, Butcher EC, et al. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am J Pathol 2002;160:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiss Y, Proudfoot AE, Power CA, Campbell JJ, Butcher EC. CC chemokine receptor CCR4 and the CCR10 ligand cutaneous T cell-attracting chemokine (CTACK) in lymphocyte trafficking to inflamed skin. J Exp Med 2001;194:1541–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zabel BA, Agace WW, Campbell JJ, Heath HM, Parent D, Roberts AI, Ebert EC, Kassam N, Qin S, Zovko M, et al. Human G protein-coupled receptor GRP-9–6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. J Exp Med 1999;190:1241–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R, van Weel C, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: gold executive summary. Am J Respir Crit Care Med 2007;176:532–555. [DOI] [PubMed] [Google Scholar]
- 28.Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest 1998;101:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romagnani P, Annunziato F, Lasagni L, Lazzeri E, Beltrame C, Francalanci M, Uguccioni M, Galli G, Cosmi L, Maurenzig L, et al. Cell cycle-dependent expression of cxc chemokine receptor 3 by endothelial cells mediates angiostatic activity. J Clin Invest 2001;107:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gould SJ, Isaacson PG. Bronchus-associated lymphoid tissue (BALT) in human fetal and infant lung. J Pathol 1993;169:229–234. [DOI] [PubMed] [Google Scholar]
- 31.Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest 2006;116:3183–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richmond I, Pritchard GE, Ashcroft T, Avery A, Corris PA, Walters EH. Bronchus associated lymphoid tissue (BALT) in human lung: its distribution in smokers and non-smokers. Thorax 1993;48:1130–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suda T, Chida K, Hayakawa H, Imokawa S, Iwata M, Nakamura H, Sato A. Development of bronchus-associated lymphoid tissue in chronic hypersensitivity pneumonitis. Chest 1999;115:357–363. [DOI] [PubMed] [Google Scholar]
- 34.Tschernig T, Pabst R. Bronchus-associated lymphoid tissue (BALT) is not present in the normal adult lung but in different diseases. Pathobiology 2000;68:1–8. [DOI] [PubMed] [Google Scholar]
- 35.Bienenstock J, McDermott MR. Bronchus- and nasal-associated lymphoid tissues. Immunol Rev 2005;206:22–31. [DOI] [PubMed] [Google Scholar]
- 36.Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol 2002;168:3195–3204. [DOI] [PubMed] [Google Scholar]
- 37.Bonecchi R, Bianchi G, Bordignon PP, D'Ambrosio D, Lang R, Borsatti A, Sozzani S, Allavena P, Gray PA, Mantovani A, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 1998;187:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol 1998;28:3696–3705. [DOI] [PubMed] [Google Scholar]
- 39.Bosken CH, Hards J, Gatter K, Hogg JC. Characterization of the inflammatory reaction in the peripheral airways of cigarette smokers using immunocytochemistry. Am Rev Respir Dis 1992;145:911–917. [DOI] [PubMed] [Google Scholar]
- 40.Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003;22:672–688. [DOI] [PubMed] [Google Scholar]
- 41.Hogg JC. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am J Respir Crit Care Med 2001;164:S71–S75. [DOI] [PubMed] [Google Scholar]
- 42.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 2007;87:1047–1082. [DOI] [PubMed] [Google Scholar]
- 43.Grumelli S, Corry DB, Song LZ, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis DE, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med 2004;1:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fadel SA, Bromley SK, Medoff BD, Luster AD. CXCR3-deficiency protects influenza-infected CCR5-deficient mice from mortality. Eur J Immunol 2008;38:3376–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoneyama H, Narumi S, Zhang Y, Murai M, Baggiolini M, Lanzavecchia A, Ichida T, Asakura H, Matsushima K. Pivotal role of dendritic cell-derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J Exp Med 2002;195:1257–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sallusto F, Kremmer E, Palermo B, Hoy A, Ponath P, Qin S, Forster R, Lipp M, Lanzavecchia A. Switch in chemokine receptor expression upon TCR stimulation reveals novel homing potential for recently activated T cells. Eur J Immunol 1999;29:2037–2045. [DOI] [PubMed] [Google Scholar]
- 47.Freeman CM, Curtis JL, Chensue SW. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8+ cells correlates with chronic obstructive pulmonary disease severity. Am J Pathol 2007;171:767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hardaker EL, Bacon AM, Carlson K, Roshak AK, Foley JJ, Schmidt DB, Buckley PT, Comegys M, Panettieri RA Jr, Sarau HM, et al. Regulation of TNF-alpha- and IFN-gamma-induced CXCL10 expression: participation of the airway smooth muscle in the pulmonary inflammatory response in chronic obstructive pulmonary disease. FASEB J 2004;18:191–193. [DOI] [PubMed] [Google Scholar]
- 49.Sauty A, Dziejman M, Taha RA, Iarossi AS, Neote K, Garcia-Zepeda EA, Hamid Q, Luster AD. The T cell-specific CXC chemokines IP-10, MIG, and I-TAC are expressed by activated human bronchial epithelial cells. J Immunol 1999;162:3549–3558. [PubMed] [Google Scholar]
- 50.Costa C, Rufino R, Traves SL, Lapa ESJR, Barnes PJ, Donnelly LE. CXCR3 and CCR5 chemokines in induced sputum from patients with COPD. Chest 2008;133:26–33. [DOI] [PubMed] [Google Scholar]
- 51.Spurrell JC, Wiehler S, Zaheer RS, Sanders SP, Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol 2005;289:L85–L95. [DOI] [PubMed] [Google Scholar]
- 52.Whiting D, Hsieh G, Yun JJ, Banerji A, Yao W, Fishbein MC, Belperio J, Strieter RM, Bonavida B, Ardehali A. Chemokine monokine induced by IFN-gamma/CXC chemokine ligand 9 stimulates T lymphocyte proliferation and effector cytokine production. J Immunol 2004;172:7417–7424. [DOI] [PubMed] [Google Scholar]
- 53.Lee SH, Goswami S, Grudo A, Song LZ, Bandi V, Goodnight-White S, Green L, Hacken-Bitar J, Huh J, Bakaeen F, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med 2007;13:567–569. [DOI] [PubMed] [Google Scholar]
- 54.Taraseviciene-Stewart L, Burns N, Kraskauskas D, Nicolls MR, Tuder RM, Voelkel NF. Mechanisms of autoimmune emphysema. Proc Am Thorac Soc 2006;3:486–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akashi S, Sho M, Kashizuka H, Hamada K, Ikeda N, Kuzumoto Y, Tsurui Y, Nomi T, Mizuno T, Kanehiro H, et al. A novel small-molecule compound targeting CCR5 and CXCR3 prevents acute and chronic allograft rejection. Transplantation 2005;80:378–384. [DOI] [PubMed] [Google Scholar]
- 56.Nie L, Xiang R, Zhou W, Lu B, Cheng D, Gao J. Attenuation of acute lung inflammation induced by cigarette smoke in CXCR3 knockout mice. Respir Res 2008;9:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hasegawa H, Inoue A, Kohno M, Lei J, Miyazaki T, Yoshie O, Nose M, Yasukawa M. Therapeutic effect of CXCR3-expressing regulatory T cells on liver, lung and intestinal damages in a murine acute GVHD model. Gene Ther 2008;15:171–182. [DOI] [PubMed] [Google Scholar]
- 58.Turner JE, Steinmetz OM, Stahl RA, Panzer U. Targeting of Th1-associated chemokine receptors CXCR3 and CCR5 as therapeutic strategy for inflammatory diseases. Mini Rev Med Chem 2007;7:1089–1096. [DOI] [PubMed] [Google Scholar]
- 59.Hildebrandt GC, Corrion LA, Olkiewicz KM, Lu B, Lowler K, Duffner UA, Moore BB, Kuziel WA, Liu C, Cooke KR. Blockade of CXCR3 receptor:ligand interactions reduces leukocyte recruitment to the lung and the severity of experimental idiopathic pneumonia syndrome. J Immunol 2004;173:2050–2059. [DOI] [PubMed] [Google Scholar]