

Abstract
The relative (to BEt3) hydride ion affinity (HIA) of a series of acridine borenium salts has been calculated, with some HIAs found to be similar to that for [Ph3C]+. The HIA at the acridine C9 position is controlled by both acridine and the boron substituents, the latter presumably affecting the strength of the B=N bond in the acridane-BY2 products from hydride transfer. Through a range of hydride abstraction benchmarking reactions against organic hydride donors the experimental HIA of [F5acr-BCat]+ (cat = catechol, F5acr = 1,2,3,4,7-pentafluoroacridine) has been confirmed to be extremely high and closely comparable to that of [Ph3C]+. The high HIA of [F5acr-BCat]+ enables H2 and alkene activation in a FLP with 2,6-di-tert-butylpyridine. Finally, the HIA of pyridine and quinoline borenium cations has been determined, with the HIA at boron in [PinB(amine)]+ (pin = pinacol, amine = pyridine or quinoline) found to be relatively low. This enabled the hydroboration of pyridine and quinoline by HBPin to be achieved through the addition of 5–10 mol % of bench-stable cationic carbon Lewis acids such as 2-phenyl-N,N-dimethylimidazolium salts.
Introduction
Borenium cations are three-coordinate boron compounds possessing a unit positive charge which often results in significant Lewis acidity at the boron center.1,2 Recently, these cations have been applied in numerous stoichiometric (including hydroboration,3 C–H borylation,4 and carboboration5) and catalytic transformations (e.g., as chiral Lewis acid catalysts6 and in frustrated Lewis pair (FLP) mediated reductions).7 These applications exploit the boron center as the locus of electrophilic reactivity. However, the electron-deficient nature of the {BY2}+ fragment in an [L→BY2]+ borenium cation also results in considerable modulation of the electronic structure of the neutral donor ligand L. This activates the donor L by enhancing the electrophilicity and/or the Brønsted acidity at various positions (Scheme 1, right). The use of {BY2}+ fragments to activate a donor substrate for a chemical transformation is underexplored, particularly in contrast to the well-documented use of [L→BY2]+ to bind and activate substrates (Scheme 1, left; when not monocationic, e.g. R = BH3, these structures can be represented containing borocation subunits).6,8 A limited number of notable exceptions have used {BY2}+ fragments as Lewis acid activators for the functionalization of L: for example, in hydroboration chemistry (via [(imine)BPin]+ and [(pyridine)BPin]+) and the deprotonation of [(2,6-lutidine)BY2]+.9 However, no studies probing the factors affecting Lewis acidity at both the boron and carbon centers (in the donor substrate, L) in [(N-heterocycle)→BY2]+ cations has been performed. While we and others have separately investigated the Lewis acidity of a range of borenium cations and found considerable variance in calculated hydride ion affinity/hydride donor ability of up to 37 kcal mol–1 on modifying both L and Y, this only considered the boron center as the locus of reactivity.9c,10
Scheme 1. Donor Substrate (Shown in Blue) Activation by [LBY2]+ (Left) and [BY2]+ (Right).

Due to the highly electron deficient nature of the formally four-valence-electron boron center in {BY2}+ the degree of activation of a substrate bound to {BY2}+ should be greater than the activation of a substrate bound to formally six-valence-electron Lewis acids (e.g., {H3C}+, {R3Si}+, {L→BY2}+). One notable example highlighting the considerable electrophilicity of {BY2}+ moieties is provided by [B(mesityl)2]+ binding and deoxygenating CO2.11 Furthermore, a modified Gutmann–Beckett method test12 revealed greater substrate activation by the formally four-valence-electron fragment {CatB}+ (Cat = catechol), with [CatB(OPEt3)]+ having δ(31P) 106.9 ppm, a value considerably downfield relative to that for [CatB(NEt3)(OPEt3)]+ (δ(31P) 88.8 ppm) and to that for [Et3Si(OPEt3)]+ (δ(31P) 91.2 ppm) (Figure 1).9c,12b
Figure 1.

Modified Gutmann–Beckett method test δ(31P) values.
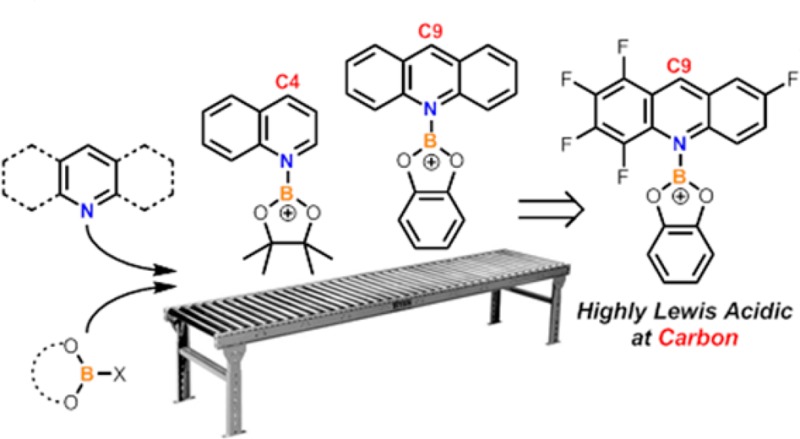
In other studies [acridine-BCl2]+ ([1]+) was demonstrated to be highly Lewis acidic at the C9 carbon (Scheme 2) and using hydride ion affinity (HIA) calculations it was found to have a much higher Lewis acidity at C9 in comparison to that of the N-Me-acridinium cation [2]+.13 The particularly high calculated HIA value for the acridine C9 position in [1]+ was attributed to the formation of B=N multiple-bond character in the acridane-BX2 product formed after hydride transfer. Recently, [1][AlCl4] has been exploited as a hydride abstractor and demonstrated to be a stronger hydridophile than B(C6F5)3.14
Scheme 2. Hydride Transfer Reactivity of [1]+/[HB(C6F5)3]−.

Due to the significant current interest in the transition-metal-free reduction of N-heterocycles, including examples mediated by borocations, we were interested in assessing the Lewis acidity at boron and carbon of a range of borocation salts containing acridine, quinoline, and pyridine groups. Herein we report our findings and demonstrate that the Lewis acidity at the boron and carbon centers can be readily tuned across a large range by variation of the borane and N-heterocyclic moieties, even leading to one example which has Lewis acidity toward hydride comparable to that of [Ph3C]+.
Results and Discussion
Hydride Ion Affinity Calculations on Acridine Borocations
Calculations were performed to assess the HIA of a range of acridine borocations to expand on our earlier work.13 Calculations were carried out at the M06-2X/6-311G(d,p) level with a DCM solvation model (PCM, DCM), and the HIA at boron and the C9 position was determined relative to BEt3 (ΔH HIA values were determined from the reactions between [HBEt3]— and the Lewis acids as previously reported).15 In addition to acridine (acr), F5acr (F5acr = 1,2,3,4,7-pentafluoroacridine) was used, as it is a readily synthesized acridine that has electronics dramatically different from those of the parent perprotio acridine.16 It should be noted that attempts to calculate the HIA at boron for acridine-BPin borenium cations led instead to dissociation of acr and F5acr from HBPin; thus, these values cannot be included in Figure 2. The six calculated borocation structures shown in Figure 2 all contain trigonal-planar boron centers and are unremarkable, being closely comparable to that previously reported for [1]+.13 Replacement of acr for F5acr has minimal effects (e.g., B–N elongation of <0.03 Å for the fluorinated congener), the only notable effect being an increase in the C–N–B–Y dihedral angle for the bulkier F5acr congeners (which have one F orientated toward the {BY2}+ moiety). The calculated HIAs at boron for the six borenium cations were in the range −30 kcal mol–1 ([Acr-BBN]+) to −61 kcal mol–1 ([F5acr-BCl2]+), with the difference being primarily due to changes in boron substitution, rather than the acridine. For example, when the acridine borenium analogues [Acr-BCl2]+ ([1]+) and [Acr-BCat]+ ([3]+) are considered, upon exchanging −BCat for −BCl2 the HIA at boron increases from −33 to −60 kcal mol–1, as expected on the basis of relative π donor ability and consistent with previous calculations. In contrast, pentafluorination of acridine led only to a small increase in HIA at boron (<5 kcal mol–1). This is in part due to the approximately orthogonal arrangement of the {BY2}+ and acridine moieties (for all borocations the calculated average C–N–B–Y dihedral angles are 64–88°). This presumably reduces the effect of the acridine substituents on the energy of orbital(s) containing significant B pz character. Furthermore, the lower σ donor ability of F5acr relative to acridine (expected to enhance the HIA at boron) will be offset by the increase in steric environment around boron (particularly the F substituent at the acridine C1 position), which disfavors pyramidalization at B and thus will reduce the Lewis acidity at boron.
Figure 2.

Hydride ion affinity at boron (relative to BEt3), Legend: (a) from ref (13).
The calculated HIA values at the acridine C9 position were found to be higher than those at boron by 14–50 kcal mol–1 for all of the borenium cations investigated, with HIA values at C9 ranging from −61 kcal mol–1 ([Acr-BPin]+) to −85 kcal mol–1 ([F5acr-BCl2]+, [4]+) (Figure 3). These are all significantly higher than the HIA values calculated for the N-Me-acridinium cation [2]+, previously reported (HIA = −53 kcal mol–1).17 This is attributed to B=N multiple-bond formation on hydride transfer, as indicated by a significant reduction in the B–N bond distance in each case on acridine to acridane conversion (B–N distances in the borenium cations are 1.50–1.53 Å, which contract to 1.40–1.43 Å in the acridane-BY2 compounds, with the latter having effectively coplanar Y2BNR2 arrangements). It is notable that the HIA at C9 of the boronium cation [(Acr)2BCl2]+ (−59 kcal mol–1) is considerably lower than that at C9 of its borenium analogue [1]+ (−75 kcal mol–1), demonstrating the disparate degrees of activation of N-heterocycles in borenium and boronium salts. The lower HIA for [2]+ also emphasizes the dramatic difference in N-heterocycle activation by formally four-valence-electron (e.g., {BY2}+) versus six-valence-electron species ({CH3}+).
Figure 3.

Calculated relative (to BEt3) HIA values (kcal mol–1) of a range of carbon Lewis acids. The value for [1]+ is taken from ref (13) and that for [2]+ from ref (17).
The magnitude of HIA at C9 was affected by both the boron and acridine substituents. The boron substituents will modify the strength of the B=N bond formed after hydride transfer. This phenomenon is exemplified by F5acridane-BCl2, where there is less effective Cl→B π donation, having a shorter calculated B=N distance (1.407 Å) than F5acridane-BCat (B=N = 1.420 Å), where there is more effective π donation from O to B. Acridine pentafluorination also leads to a ca. 10 kcal mol–1 increase in Lewis acidity toward hydride, with fluorination of acridine lowering the LUMO energy, which significantly enhances the HIA at C9. Indeed, there is a good correlation between the calculated HIA values at C9 and the LUMO energies (Figure S48 in the Supporting Information; in all of these borenium cations the LUMO has significant character at the C9 position). The HIAs calculated for a number of borenium salts are comparable to that calculated for [Ph3C]+,17 suggesting that these borocations should exhibit a broad scope of hydride abstraction reactivity. Finally, the HIA of F5acr-AlCl3 adduct 6 was calculated, due to the potential for this compound to form alongside borenium cations when combinations of weakly basic amines/ClBY2/AlCl3 are used. Notably the HIA of 6 is considerably lower than that of [4]+, with the origin of this disparity attributed to (i) the ability to form B=N bonds on addition of hydride for the latter and (ii) the relative energy of the LUMO, which is calculated to be considerably higher for 6 relative to [4]+ (by 16 kcal mol–1), further indicating the ability of the {BY2}+ fragment to strongly activate N-heterocycles.
Synthesis of Acridine Borocations
To further assess the HIA values relative to those of established carbocations, a number of acridine borenium cations were synthesized from the acridine chloroborane Lewis adducts by B–Cl heterolysis with either AlCl3 (Scheme 3) or Na[B(C6F5)4] (the synthesis of Acr-BCl3 and [1][AlCl4] has been previously reported).13 When acridine and chlorocatecholborane (CatBCl) were mixed in DCM, a yellow solid rapidly precipitated and proved extremely poorly soluble in organic solvents. However, upon addition of AlCl3 the resulting [Acr-BCat][AlCl4] salt [3][AlCl4] was soluble in DCM, allowing for characterization by NMR spectroscopy, which revealed a δ(11B) value of 28.2 ppm consistent with a borenium salt formulation.
Scheme 3. Synthesis of Acridine Borocations [3]+ and [5]+.

The solid-state structure of [3][AlCl4] (Figure 4) revealed the geometry at boron to be trigonal planar (angles at boron sum to 360°) with no close Cl3Al–Cl···B contacts (all >3.881(7) Å). The B–N bond distance in [3]+ (B–N 1.489(10) Å) is comparable to that calculated (1.490 Å) and to that in [1]+ (1.503(3) Å).13 The BCat and acridine rings in [3]+ are mutually twisted by 63.5(2)°, precluding any significant B–N π character (for comparison, in the BCl2 analogue [1]+ the angle is 77° due to the larger Cl–B–Cl angle relative to the smaller O–B–O angle).
Figure 4.

Solid-state structures of [3][AlCl4] (left), [5][AlCl4] (center), and 6 (right) with thermal ellipsoids at the 50% probability level and H atoms omitted for clarity. Selected bond distances (Å) and angles (deg): for [3][AlCl4], B1–N1 1.489(6), B1–O1 1.363(10), B1–O2 1.346(10), N1–C 1.385(10) and 1.379(10), O1–B1–O2 114.2(6), angle between the acridine and BCat planes 63.5(2)°; for [5][AlCl4] B1–N1 1.462(19), B1–O1 1.353(19), B1–O2 1.34(2), N1–C 1.402(15) and 1.360(19), O1–B1–O2 116.2(13), angle between the acridine and BCat planes 87.8(5); for 6, N1–Al1 1.999(5), N1–C 1.366(7) and 1.376(7), Al–N–C9 151.1(3).
To access acridine borocation salts with more negative HIA values, borenium salts were synthesized containing F5acr. The pentafluorination of acridine reduces the Lewis basicity; therefore, upon mixing of BCl3 and F5acr 11B{1H} NMR spectroscopy revealed no observable coordination. However, upon addition of AlCl3 a broad signal consistent with [F5acr-BCl2]+ ([4]+, (δ(11B) 46.5 ppm) was observed. Despite the formation of [4][AlCl4], the in situ 1H and 11B{1H} NMR spectra indicated incomplete consumption of starting materials; thus, [4]+ exists in equilibrium in solution with F5acr, BCl3, and the Lewis adduct 6. This is comparable to the reactivity observed with other weak pyridyl nucleophiles: for example, 2,6-dichloropyridine/BCl3/AlCl3 mixtures previously reported.18 Interestingly, upon reaction of F5acr with CatBCl and AlCl3, [F5acr-BCat][AlCl4] ([5][AlCl4]) was formed as the major product with the in situ 11B NMR spectrum showing predominantly [5][AlCl4] (δ(11B) 27.9 ppm, broad) and a small amount of CatBCl (δ(11B) 28.7 ppm, sharp). Crystals of [5][AlCl4] suitable for X-ray diffraction were grown from a concentrated DCM solution layered with pentanes. The solid-state structure of [5][AlCl4] (Figure 4) revealed a B–N bond distance of 1.50(2) Å, again comparable to the calculated structure of [5]+ and to those observed in [1]+ and [3]+. However, in [5]+ the BCat and acridine planes are almost orthogonal (87.8(5)°) due to the greater steric bulk of the pentafluorinated acridine. Again the geometry at boron in [5]+ is trigonal planar with no close contacts between the boron center and the anion and no evidence for any interaction between the boron center and the proximal fluoride on acridine. Finally, F5acr-AlCl3 (6) was produced by mixing equimolar amounts of F5acr and AlCl3, and the identity of the compound was confirmed by NMR spectroscopy and X-ray diffraction. The molecular structure obtained for 6 revealed an N–Al bond length of 1.999(5) Å, comparable to the 1.959(6) Å N–Al bond distance reported for quinoline-AlCl3,19 with the Al–Cl bond distances ranging from 2.1166(8) to 2.1361(19) Å. The solid-state structure (Figure 4) revealed the AlCl3 group to lie out of the acridine plane, with an Al–N–C9 angle of 151.1(3)°, due to steric pressures between the fluorinated acridine and the AlCl3.
Hydride Abstraction Tests
To experimentally benchmark acridine borocation salts against more established carbon Lewis acids, stoichiometric hydride transfer reactions were performed. Initially the greater HIA of [3]+ in comparison to N-Me-acridinium, [2]+, was confirmed through the reaction of [3]+ with N-Me-acridane, which led to rapid hydride transfer to form acridane-BCat and [2]+. Additional carbon Lewis acids were selected, guided by Mayr electrophilicity (E) parameters (although as noted by Mayr and co-workers the kinetic analysis derived E values can be affected by steric hindrance).20 The organic hydrides/carbon Lewis acids chosen were cycloheptatriene/tropylium (E parameter −3.72), dibenzosuberene/dibenzotropylium (E parameter −0.63), and triphenylmethane/Ph3C+ (E parameter 0). The HIA values for these three carbon Lewis acids were also calculated and are shown in Figure 3.
Cycloheptatriene was added to a solution of [3][AlCl4], with NMR spectroscopy revealing the partial formation of [tropylium][AlCl4] (δ(1H) 9.26 ppm) and acridane-BCat (δ(1H) 3.83 ppm) after 5 min. After 16 h at room temperature complete consumption of [3][AlCl4] had occurred with formation of further tropylium-[AlCl4] and acridane-BCat confirming that the HIA of [3]+ is greater than that of [tropylium]+. As [3]+ and dibenzotropylium have HIA values within 2 kcal mol–1, it was unclear which would experimentally be more Lewis acidic toward hydride. When [3][AlCl4] was mixed with dibenzosuberene, no reaction was observed, even upon prolonged heating at 60 °C. To assess if the absence of hydride transfer is due to unfavorable thermodynamics or a significant kinetic barrier to hydride transfer, the hydride abstraction of acridane-BCat by dibenzotropylium was carried out. This resulted in rapid hydride transfer from acridane-BCat to dibenzotropylium, generating [3]+ and dibenzosuberene, indicating that dibenzotropylium is a stronger Lewis acid toward hydride in comparison to [3]+. Finally, as expected [Ph3C][B(C6F5)4] rapidly abstracted hydride from acridane-BCat to form triphenylmethane and [3]+. The relative HIAs from these experiments are summarized in Figure 5.
Figure 5.

Relative hydride ion affinity from transfer reactions.
For [1]+, 5 min of mixing with cycloheptatriene was sufficient for the complete consumption of [1]+ with tropylium cation formation observed by 1H NMR spectroscopy, indicating rapid hydride transfer. However, no 1H NMR resonance corresponding to the acridane-BCl2 CH2 group was observed; instead, a mixture of acridane products derived from the substituent scrambling reaction of acridane-BCl2 with [1]+ was apparent by multinuclear NMR spectroscopy as previously reported.13 Regardless, the rapid formation of tropylium-[AlCl4] confirms the relative Lewis acidity toward hydride. Hydride abstraction from dibenzosuberene using [1][AlCl4] was attempted next, but after 17 h at room temperature no reaction was observed. The reaction mixture was subsequently heated at 60 °C for 5 h, in which time acridane CH2 resonances were observed in the 1H NMR spectrum, indicating some hydride transfer, with redistribution reactions of acridane-BCl2 again occurring. Notably, on prolonged heating significant quantities of [1]+ transform to multiple byproducts not derived from hydride transfer (including acr-AlCl3, acr-BCl3, and BCl3 as previously reported using other amine/BCl3/AlCl3 derived borenium cations).13 While the side reactivity using [1]+ complicated the analysis, the observation of some acridane species indicated that [1]+ at least has Lewis acidity toward hydride comparable to that of dibenzotropylium. The hydride abstraction from triphenylmethane using [1][AlCl4] also was attempted, but after heating at 60 °C for 20 h no [Ph3C]+ was observed, with heating again leading to redistribution reactivity of [1][AlCl4], resulting in a mixture of [1][AlCl4], acr-BCl3, acr-AlCl3, and BCl3, as detected by multinuclear NMR spectroscopy. While the rapid hydride abstraction from cycloheptatriene by [1][AlCl4] along with partial hydride abstraction from dibenzosuberene is indicative of a significant HIA for [1]+, it is less Lewis acidic than Ph3C+. Furthermore, the propensity of [1]+ and acridane-BCl2 to react further (e.g., by substituent scrambling) significantly complicates the use of this reagent.
Due to the limited Lewis acidity found for [3][AlCl4] and the issues with side reactivity when [1]+ was used, [F5acr-BCat][AlCl4] ([5][AlCl4]) was explored, with the chelation of catechol preventing any substituent scrambling of the acridaneBY2 species. The reaction between [5][AlCl4] and cycloheptatriene resulted in complete conversion of the borenium [5]+ to F5-acridane-BCat within 5 min of mixing. [5][AlCl4] then was reacted with dibenzosuberene, and after 10 min of mixing in DCM the 1H NMR spectrum exhibited the formation of F5-acridane-BCat (CH2 δ(1H) 3.87 ppm), and dibenzotropylium (CH δ(1H) 10.41 ppm), with longer reaction times at room temperature leading to complete consumption of dibenzosuberene and further formation of F5-acridane-BCat. The hydride abstraction from dibenzosuberene confirms that the fluorinated acridine in [5]+ provides a marked increase in Lewis acidity at the C9 position in comparison to [3]+.
The borenium [5][AlCl4] and [Ph3C]+ have similar calculated HIA values (−77 and −75 kcal mol–1, respectively), and [5][AlCl4] and triphenylmethane initially showed no reaction on mixing. However, after the reaction mixture was heated at 100 °C for 16 h, a small amount of hydride transfer was observed, on the basis of the formation of F5-acridane-BCat by 1H NMR spectroscopy. Integration of the Ph3CH and F5-acridane-CH2 signals revealed a conversion of ca. 10% (from [5][AlCl4]). The reaction mixture was heated for a further 72 h, during which time no further hydride abstraction occurred, suggesting that the reaction had reached equilibrium. To confirm this, a hydride transfer from F5-acridane-BCat to [Ph3C][B(C6F5)4] was utilized. After this mixture was heated at 100 °C for 16 h, integration of the Ph3CH and F5-acridane-CH2 signals in the 1H NMR spectrum showed 86% hydride transfer to form Ph3CH, with retention of 14% F5-acridane-BCat. Upon further heating no changes were observed, again indicating that the reaction had reached equilibrium. This confirms that [5]+ and [Ph3C]+ have similar Lewis acidities toward hydride (and a ΔG° value for hydride transfer of only 7 kJ mol–1), with the relative HIA reactivity summarized in Figure 6. The significant hydride Lewis acidity of [5]+ means that it should be effective for H2 activation in a frustrated Lewis pair (FLP) with suitably hindered bases. However, using [5]+/2,6-di-tert-butylpyridine (DBP), H2 activation was extremely slow even at 100 °C (33% conversion of [5]+ to F5-acridane-BCat after 72 h under 4 bar of H2 in o-dichlorobenzene (o-DCB); Scheme 4); thus, acridine fluorination actually increases the kinetic barrier to H2 activation in this case (relative to that previously observed for [1]+ in an FLP with DBP).13
Figure 6.

Relative hydride ion affinity from stoichiometric transfer reactions.
Scheme 4. [5][AlCl4]/2,6-DBP FLP Activation of H2.

To assess the relative reactivity of these acridine borenium salts toward other soft nucleophiles, 1,1-diphenylethylene was utilized, due to the precedence for reaction of acridine/[2]+ mixtures with vinyl ethers at the C9 position (vinyl ethers themselves are not compatible with highly electrophilic borocations).21 1,1-Diphenylethene was added to a mixture of [5][AlCl4] and DBP in DCM, which caused a rapid color change from orange to dark green. Analysis of the green solution by 1H NMR spectroscopy revealed the formation of a new product displaying a coupled AB doublet system (δ(1H) 6.01 and 5.00 ppm (d, 3JHH = 10 Hz)), suggesting the formation of a new alkene-based product. After hydrolysis and workup the acridine-derived product was characterized as compound 7, formed via the alkene attacking the highly Lewis acidic C9 site of [5][AlCl4] followed by deprotonation of the alkene-derived CH2 (Scheme 5) to form 7 on B–N hydrolysis. An identical reaction was carried out with the less electrophilic borenium [3][AlCl4], but no acridane alkene product was generated. This lack of reactivity displayed by [3][AlCl4] is markedly different from that of [5]+, due to [3]+ being a weaker electrophile.
Scheme 5. Reaction of 1,1-Diphenylethylene and [5][AlCl4].

Finally, despite the propensity for substituent scrambling, we attempted to assess the HIA of the most Lewis acidic of the borenium salts, [F5acr-BCl2]+, by hydride transfer reactions. A sample of the equilibrium mixture containing [4][AlCl4] was generated (which is present in solution along with BCl3 and F5acr-AlCl3 (6)) and then Ph3CH was added. Mixing for 40 h (with periodic analysis by NMR spectroscopy) at room temperature led to a small amount of an F5-acridane species (suggesting that hydride abstraction from Ph3CH does occur); however, [4]+ had predominantly converted to the protonated acridine and an unidentified species at δ(11B) 33.7 ppm. The formation of F5-acridane was confirmed by GC-MS analysis of the hydrolyzed reaction mixture. The complex mixture present in this reaction raises the question as to whether other F5-acridine species, e.g. 6, are capable of behaving as potent hydride acceptors. To confirm that 6 is not the hydride abstracting species, a F5acr-AlCl3/ Ph3CH reaction mixture was subjected to conditions identical with those for the partially successful [4][AlCl4]/Ph3CH hydride transfer reaction, but in this case using 6 no hydride transfer was observed. This is consistent with calculated HIA values and thus indicates that the borenium [4]+ is required in order to abstract hydride from triphenylmethane but that its considerable electrophilicity at boron and carbon results in currently unidentified side reactions.
HIA Studies on Pyridine and Quinoline Borocations
With the HIA values confirmed experimentally for acridine borocations the Lewis acidities at B and C in quinoline (quin) and pyridine (py) borocations were studied. In contrast to acridines these N-heterocycles provide less steric bulk around the boron center and can be reduced at two carbon positions (C2 and C4). Consistent with the reduced steric bulk of quinoline, the calculated structures of [quinBCat]+ and [quinBPin]+ have coplanar quinoline and {BO2} moieties. It is particularly notable that the HIA at boron for [quinBCat]+ is −40 kcal mol–1, more negative than for [3]+ (−33 kcal mol–1) despite [quinBCat]+ being planar (thus enabling π donation from quinoline to the B pz orbital). The relative HIA values are attributed to the greater steric effect of acridine, which results in a higher pyramidalization energy and thus a lower HIA at boron in [3]+; in addition, quinoline is less basic than acridine, which may contribute to a higher HIA (based on the pKa values of the conjugate acids of quinoline (4.9) and acridine (5.6)).22 As expected, the HIA at boron for the pinacol congener [quinBPin]+ ([8]+) is less negative (−27 kcal mol–1) than that of the catechol analogue due to more O→B π donation in the former. The HIA at boron in [8]+ is closely comparable to that calculated for [pyBPin]+ ([9]+; −26.5 kcal mol–1).
From the perspective of N-heterocycle reduction the HIA values at C2 and C4 are more significant (Figure 7), particularly for the BPin analogues, as HBpin is widely used as a reductant in this field.9a,23 For [8]+ and [9]+ the HIA at C4 is marginally more Lewis acidic than that at C2 by 1 and 3 kcal mol–1, respectively (Figure 7), with each being higher than the respective HIA at boron. Furthermore, as observed with the acridine analogues, the HIA at carbon of [8]+ and [9]+ is higher than that for methylated quinoline and pyridine as well as for the respective boronium cations and a silylated pyridine (with pyridine reduction via N-silylated pyridines also reported).24 This further confirms the enhanced N-heterocycle activation achieved by a formally four-valence-electron fragment. For comparison the activation of quinoline by a neutral borane, BF3, has dramatically less effect on the Lewis acidity at C2/C4 even in comparison to the boronium cation [(quin)2BPin]+, consistent with the greater Lewis acidity of borocations versus simple neutral boranes.
Figure 7.
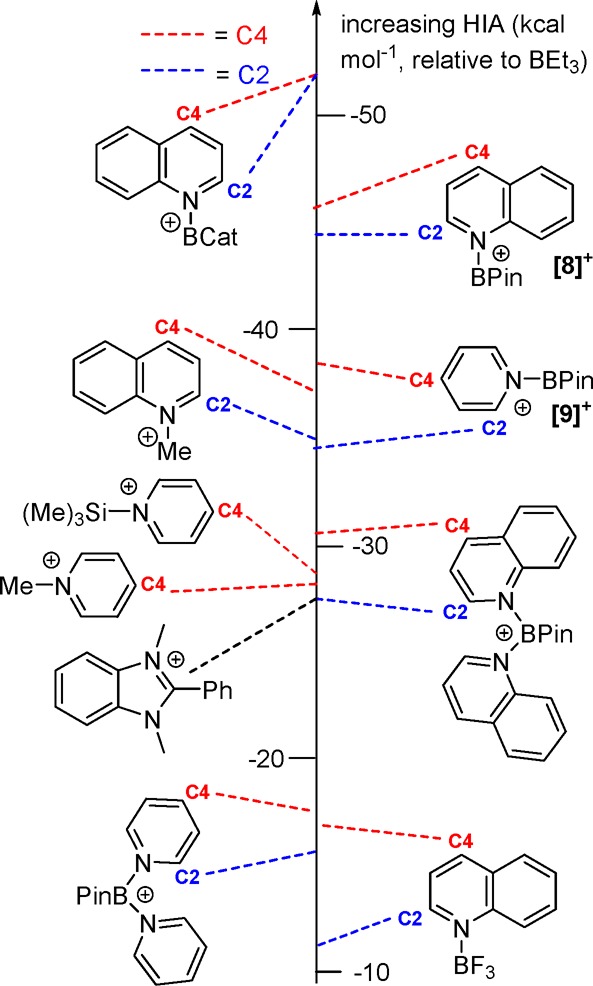
HIA values of Lewis acid activated pyridine and quinoline compounds.
The relatively low HIA values at boron for [8]+ and [9]+ indicated that a range of bench-stable carbon Lewis acids would be able to abstract hydride from the PinBH(amine) adducts. Due to the relative HIAs at boron and carbon in [8]+ and [9]+ these borocations then should be reduced by additional amounts of PinBH(amine) to form borylated dihydroquinolines/dihydropyridines. Indeed, addition of 10 mol % of [2]+ (HIA = 53 kcal mol–1) or 5 mol % of 2-phenyl-N,N-dimethylimidazolium (HIA = 27 kcal mol–1)25 salts to mixtures of quinoline or pyridine and HBPin resulted in successful reduction (Scheme 6).26 Hydroboration proved selective for pyridine (generating the 1,4-hydroborated product exclusively); however, with quinoline a mixture of the 1,2- and 1,4-hydroborated products were formed (Figures S41–S44 in the Supporting Information). While hydride transfer from HBPin(amine) to the borenium cations is favorable on the basis of relative HIA values, the role of boronium cations in the reduction process cannot be precluded, as these cations also have appreciable HIA values. For example, the HIA at C4 for [(quin)2BPin]+ is more negative than the HIA at B for [quinBPin]+, indicating that hydride transfer to the boronium cation from (quin)(H)BPin should be favored.
Scheme 6. Cationic Carbon Lewis Acid Catalyzed Hydroboration of Pyridine and Quinoline.

In conclusion, through the judicious choice of N-heterocycle and borane substituents it is possible to generate N-heterocycle borenium salts that exhibit an extremely wide range of Lewis acidities toward hydride at boron and carbon. The Lewis acidity at carbon can be tuned over a 50 kcal mol–1 range from relatively low values, e.g. [pyridine-BPin]+, through to the highly Lewis acidic [F5acridine-BCl2]+. A number of acridine borenium cations with extremely high HIAs can be readily synthesized and have potential for use as an alternative to trityl salts in hydride abstraction reactions. Furthermore, the formation of N-heterocycle borenium salts is a simple method of strongly activating the N-heterocycle group toward reduction (and other) processes, leading for example to the facile synthesis of acridane and hydroborated quinoline and pyridine compounds.
Experimental Section
Unless otherwise indicated, all manipulations were conducted under inert conditions either using standard Schlenk techniques or in a MBraun UniLab glovebox (<0.1 ppm of H2O/O2). Dichloromethane-d2 and o-dichlorobenzene were dried over CaH2 and distilled prior to storage over 3 Å molecular sieves. Protio dichloromethane was dried using an Innovative Technology SPS system and stored over activated molecular sieves. Unless otherwise stated, all compounds were purchased from commercial sources and used as received. NMR spectra were recorded on Bruker AvanceIII-400 and Bruker Ascend-400 spectrometers. Chemical shifts are reported as dimensionless δ values and are frequency referenced relative to residual protio impurities in the NMR solvents for 1H and 13C{1H} respectively, while 11B{1H} and 19F{1H} shifts are referenced relative to external BF3·Et2O and hexafluorobenzene, respectively. Coupling constants J are given in hertz (Hz) as positive values regardless of their real individual signs. The multiplicities of the signals are indicated as “s”, “d”, “t” “q” “pent”, “sept”, and “m” for singlet, doublet, triplet, quartet, pentet, septet, and multiplet, respectively. All calculations were conducted at the M06-2X/6-311G(d,p) level with a solvation model (PCM, CH2Cl2) using the Gaussian software package.27 For all calculations the optimized energies were confirmed as true minima by frequency analysis and the absence of any imaginary frequencies. Purity was indicated by multinuclear NMR spectroscopy in organic solvents (in which the sample fully dissolved) and supported by MS and/or elemental analysis.
[Acr-BCat][AlCl4] ([3][AlCl4])
An oven-dried Schlenk tube was loaded with acridine (500 mg, 2.79 mmol) which was dried under vacuum for 1 h prior to the addition of chlorocatechol–borane (430 mg, 2.79 mmol). Dichloromethane (10 mL) was added to the combined solids, which led to an immediate bright orange precipitate, and stirring was continued for 1 h at ambient temperature. After this time, all volatiles were removed to afford a bright orange free-flowing solid tentatively assigned as [Acr-BCatCl]. Due to the poor solubility of [Acr-BCatCl] in common solvents it was not possible to obtain NMR spectroscopic data. A Schlenk flask was charged with “[Acr-BCatCl]” (150 mg, 0.45 mmol) and AlCl3 (60 mg, 0.45 mmol) before DCM (5 mL) was added. The resulting mixture was stirred overnight, after which time an orange solution formed. The solution was concentrated under vacuum and subsequently layered with pentane. After standing for 24 h a crop of brown crystalline needles had grown, which were isolated, washed with pentane, and dried under vacuum (168 mg, 80% yield). Anal. Calcd for C19H13AlBCl4NO2: C, 48.88; H, 2.81; N, 3.00. Found: C, 48.69; H, 2.77; N, 3.15. 1H NMR (500 MHz, methylene chloride-d2): δ 10.01 (s, 1H, Acr-C9H), 8.63 (d, J = 8 Hz, 2H, Acr-CH), 8.40 (t, J = 8 Hz, 2H, Acr-CH), 8.30 (d, J = 9 Hz, 2H, Acr-CH), 8.07 (t, J = 8 Hz, 2H, Acr-CH), 7.66 (dd, J = 8, 4 Hz, 2H, Cat-CH), 7.49 (dd, J = 6, 4 Hz, 2H, Cat-CH). 13C{1H} NMR (126 MHz, methylene chloride-d2): δ 154.8 (Acr-C9), 147.8 (Cat-C-O), 142.7 (Acr-C), 141.2 (Acr-CH), 132.6 (Acr-CH), 129.7 (Acr-CH), 126.9 (Acr-C), 126.1 (Cat-CH), 120.2 (Acr-CH), 114.8 (Cat-CH). 11B{1H} NMR (160 MHz, methylene chloride-d2): δ 28.2. 27Al{1H} NMR (104 MHz, methylene chloride-d2): δ 104.0.
[F5acr-BCat][AlCl4] ([5][AlCl4])
An oven-dried J. Young tube was loaded with F5-acridine (25.9 mg, 0.1 mmol), CatBCl (14.9 mg, 0.1 mmol), and dichloromethane (1 mL). AlCl3 (12.8 mg, 0.1 mmol) was added to the reaction mixture followed by agitation at ambient temperature for 5 min to afford a bright yellow homogeneous solution, which was confirmed being as the desired borocation by 1H, 11B{1H}, 27Al, and 19F{1H} NMR spectroscopy (the conversion was quantitative by NMR spectroscopy). The product was isolated after drying, washing with pentane, and drying under vacuum, to yield [F5acr-BCat][AlCl4] ([5][AlCl4]) (35 mg, 0.63 mmol, 65% yield). Anal. Calcd for C19H8AlBCl4F5NO2: C, 40.98; H, 1.45; N, 2.52. Found: C, 40.84; H, 1.50; N, 2.60. 1H NMR (500 MHz, CD2Cl2, 298 K): δ 10.20 (s, 1H, Acr-C9H), 8.37–8.30 (m, 1H, Acr-CH), 8.29–8.23 (m, 1H, Acr-CH), 8.06 (dd, J = 10, 4 Hz, 1H, Acr-CH), 7.62 (dd, J = 6, 3 Hz, 2H, Cat–CH), 7.47 (dd, J = 6, 3 Hz, 2H, Cat–CH). 13C{1H} NMR (126 MHz, CD2Cl2, 298 K): δ 162.9 (Acr-C), 160.9 (Acr-C), 148.9 (Acr-C9H), 147.7 (Cat-C-O), 141.52 (Acr-C), 134.3 (d, J = 28 Hz, Acr-CH), 129.2 (Acr-C), 128.2 (d, J = 11 Hz, Acr-C), 126.2 (Cat-CH), 122.4 (d, J = 9 Hz, Acr-CH), 116.2 (Acr-C), 116.1 (d, J = 23 Hz, Acr-CH), 114.9 (Cat-CH). 19F{1H} NMR (376 MHz, CD2Cl2, 298 K): δ −102.84 (s), −129.52 (tdd, J = 19, 9, 3 Hz), −136.84 (td, J = 16, 10 Hz), −147.14 (dd, J = 18, 15 Hz), −148.81 (t, J = 18 Hz). 11B{1H} NMR (128 MHz, CD2Cl2, 298 K): δ 27.9. 27Al NMR (104 MHz, methylene chloride-d2): δ 103.4.
F5acr-AlCl3 (6)
An oven-dried J. Young NMR tube was loaded with F5-acridine (20.2 mg, 0.075 mmol) before d2-dichloromethane (0.5 mL) was added. The sample was agitated before AlCl3 (10.0 mg, 0.075 mmol) was added, causing an instantaneous color change to yellow. The sample was sonicated for 15 min. Crystals of F5-acridine-AlCl3 suitable for X-ray crystallography were grown from a concentrated DCM solution of the product layered with hexanes. 1H NMR (500 MHz, CD2Cl2): δ 9.63 (s, 1H, Acr-C9-H), 8.87 (dd, J = 10, 4 Hz, 1H, Acr-CH), 8.06–7.99 (m, 1H, Acr-CH), 7.94 (dd, J = 7, 3 Hz, 1H, Acr-CH). 13C{1H} NMR (126 MHz, CD2Cl2): δ 162.6, 160.5, 146.9, 142.9, 142.0 (Acr-C9H), 140.8, 134.9, 129.9 (d, J = 9 Hz, Acr-CH), 128.8 (d, J = 11 Hz), 128.4 (d, J = 28 Hz, Acr-CH), 112.3 (d, J = 23 Hz, Acr-CH). 19F{1H} NMR (376 MHz, CD2Cl2): δ −106.0, −129.6 (t, J = 16 Hz), −139.3 (t, J = 18 Hz), −143.8 – −145.3 (m), −152.5 (t, J = 18 Hz). 27Al NMR (104 MHz, CD2Cl2): δ 104.1 (AlCl3), 102.3 (br, F5acr-AlCl3).
Activation of H2 with [F5acr-BCat][AlCl4] and 2,6-Di-tert-butylpyridine
A J. Young NMR tube was equipped with a benzene-d6 filled capillary and charged with F5-acridine (20.2 mg, 0.075 mmol) before drying under vacuum. The F5-acridine was dissolved in o-DCB (0.5 mL), before CatBCl (11.6 mg, 0.075 mmol) and AlCl3 (10.0 mg, 0.075 mmol) were added. The sample was agitated for 5 min before 2,6-di-tert-butylpyridine (16.8 μL, 0.075 mmol) was added to the reaction mixture. The reaction mixture was subsequently degassed and placed under 4 atm of H2. The reaction was set to heat at 100 °C with periodic monitoring by NMR spectroscopy (see Figures S30–S32 in the Supporting Information).
Reaction of [F5acr-BCat][AlCl4] and 2,6-Di-tert-butylpyridine with 1,1-Diphenylethylene: Formation of Compound 7
A J. Young NMR tube was equipped with a benzene-d6 filled capillary and charged with F5-acridine (20.2 mg, 0.15 mmol) before drying under vacuum. The F5-acridine was dissolved in DCM-d2 (0.5 mL), before CatBCl (11.6 mg, 0.075 mmol) and AlCl3 (10.0 mg, 0.075 mmol) were added. The sample was agitated for 5 min, 2,6-di-tert-butylpyridine (16.8 μL, 0.075 mmol) was added, and the sample was mixed, before 1,1-diphenylethylene (13.2 μL, 0.075 mmol) was also placed in the tube. When the sample was mixed, it instantly became dark green; the reaction mixture was mixed for 16 h, in which time the sample turned turquoise and colorless crystals formed (assumed to be [2,6-di-tert-butylpyridinium][AlCl4]). The reaction mixture was then hydrolyzed by washing with water (3 × 2 mL), and the organic fraction was dried over MgSO4 and filtered to give a pale yellow solution. The solvent was removed under vacuum, leaving a yellow oily residue. The nonpolar reaction products were extracted by washing with pentane, before the pentane was removed in vacuo to leave a yellow oil containing a mixture of compound 7, 2,6-di-tert-butylpyridine, and traces of F5-acridine. The identity of compound 7 was confirmed by 1H, 13C{1H}, and 19F{1H} NMR spectroscopy as well as accurate mass spectrometry. 1H NMR (500 MHz, methylene chloride-d2): δ 7.58–7.52 (m, 2H), 7.49–7.44 (m, 1H), 7.43–7.39 (m, 2H), 7.33 (s, 2H), 7.21–7.18 (m, 2H), 7.16–7.14 (m, 1H), 6.92–6.86 (m, 1H), 6.84–6.80 (m, 1H), 6.76 (d, J = 9 Hz, 1H), 6.37 (s, 1H), 6.13 (d, J = 10 Hz, 1H), 5.03 (d, J = 10 Hz, 1H). 13C NMR (126 MHz, methylene chloride-d2): δ 160.0, 158.1, 141.9, 140.5, 139.6, 134.1, 130.5 (d, J = 2 Hz), 129.2, 128.7, 128.3 (d, J = 5 Hz), 128.1 (d, J = 11 Hz), 127.6, 125.7, 123.0 (d, J = 7 Hz), 116.2 (d, J = 8 Hz), 115.6 (d, J = 23 Hz), 115.3 (d, J = 23 Hz), 106.9 (d, J = 20 Hz), 36.47. 19F NMR (376 MHz, methylene chloride-d2): δ −122.0, −143.7 (dd, J = 22, 10 Hz), −160.5 (t, J = 21 Hz), −164.9 (ddd, J = 21, 10, 5 Hz), −170.7 (td, J = 22, 5 Hz). MS (accurate mass, ESI+): m/z calcd [C27H16F5N]+, 449.1197; found, 449.1195
Reduction of Pyridine with HBPin and Catalytic Benzimidazolium Salt
A J. Young NMR tube equipped with a benzene-d6 capillary was charged with pyridine (8.1 μL, 0.1 mmol), DCM (0.5 mL), and HBPin (12.7 μL, 0.12 mmol). Solid [N,N-dimethyl-2-phenylbenzimidazolium][B(3,5-C6H4Cl2)4] (4.1 mg, 0.005 mmol) was added to the mixture. The reaction mixture was heated at 60 °C for 24 h, after which time it was found that the reaction had proceeded to 75% hydroboration of pyridine (see Figures S43 and S44 in the Supporting Information).
Acknowledgments
This work was made possible by financial support from the EPSRC (EP/K03099X/1 and EP/M023346/1), the ERC under framework 7 (Grant number 305868), and the Royal Society (for the award of a University Research Fellowship). Additional research data supporting this publication are available as Supporting Information accompanying this publication. Dr. G. Whitehead is thanked for assistance with X-ray crystallography.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.organomet.7b00779.
Accession Codes
CCDC 1580954–1580956 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Koelle P.; Nöth H. Chem. Rev. 1985, 85, 399. 10.1021/cr00069a004. [DOI] [Google Scholar]
- a Piers W. E.; Bourke S. C.; Conroy K. D. Angew. Chem., Int. Ed. 2005, 44, 5016. 10.1002/anie.200500402. [DOI] [PubMed] [Google Scholar]; b Ingleson M. J. Top. Organomet. Chem. 2015, 49, 39–71. 10.1007/978-3-319-13054-5_2. [DOI] [Google Scholar]; c Lawson J. R.; Melen R. L. Inorg. Chem. 2017, 56, 8627. 10.1021/acs.inorgchem.6b02911. [DOI] [PubMed] [Google Scholar]; d Eisenberger P.; Crudden C. M. Dalton Trans. 2017, 46, 4874. 10.1039/C6DT04232E. [DOI] [PubMed] [Google Scholar]
- For select examples of hydroboration, see:; a Prokofjevs A.; Boussonniere A.; Li L.; Bonin H.; Lacôte E.; Curran D. P.; Vedejs E. J. Am. Chem. Soc. 2012, 134, 12281. 10.1021/ja305061c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pan X.; Boussonnière A.; Curran D. P. J. Am. Chem. Soc. 2013, 135, 14433. 10.1021/ja407678e. [DOI] [PubMed] [Google Scholar]; c Boussonnière A.; Pan X.; Geib S. J.; Curran D. P. Organometallics 2013, 32, 7445. 10.1021/om400932g. [DOI] [Google Scholar]
- For select examples of C–H borylation, see:; a De Vries T. S.; Prokofjevs A.; Harvey J. N.; Vedejs E. J. Am. Chem. Soc. 2009, 131, 14679. 10.1021/ja905369n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bagutski V.; Del Grosso A.; Ayuso Carrillo J.; Cade I. A.; Helm M. D.; Lawson J. R.; Singleton P. J.; Solomon S. A.; Marcelli T.; Ingleson M. J. J. Am. Chem. Soc. 2013, 135, 474. 10.1021/ja3100963. [DOI] [PubMed] [Google Scholar]; c Stahl T.; Müther K.; Ohki Y.; Tatsumi K.; Oestreich M. J. Am. Chem. Soc. 2013, 135, 10978. 10.1021/ja405925w. [DOI] [PubMed] [Google Scholar]; d Chernichenko K.; Lindqvist M.; Kotai B.; Nieger M.; Sorochkina K.; Papai I.; Repo T. J. Am. Chem. Soc. 2016, 138, 4860. 10.1021/jacs.6b00819. [DOI] [PubMed] [Google Scholar]; e Yin Q.; Klare H. G.; Oestreich M. Angew. Chem., Int. Ed. 2017, 56, 3712. 10.1002/anie.201611536. [DOI] [PubMed] [Google Scholar]
- For select examples of carboboration, see:; a Devillard M.; Brousses R.; Miqueu K.; Bouhadir G.; Bourissou D. Angew. Chem., Int. Ed. 2015, 54, 5722. 10.1002/anie.201500959. [DOI] [PubMed] [Google Scholar]; b Cade I. A.; Ingleson M. J. Chem. - Eur. J. 2014, 20, 12874. 10.1002/chem.201403614. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lawson J. R.; Fasano V.; Cid J.; Vitorica-Yrezabal I.; Ingleson M. J. Dalton Trans. 2016, 45, 6060. 10.1039/C5DT03003J. [DOI] [PubMed] [Google Scholar]; d Wilkins L. C.; Lawson J. R.; Wieneke P.; Rominger F.; Hashmi A. S. K.; Hansmann M. M.; Melen R. L. Chem. - Eur. J. 2016, 22, 14618. 10.1002/chem.201602719. [DOI] [PubMed] [Google Scholar]
- For a review, see:Corey E. Angew. Chem., Int. Ed. 2009, 48, 2100. 10.1002/anie.200805374. [DOI] [PubMed] [Google Scholar]
- For select examples of the activation of H2 with borocations, see:; a Farrell J. M.; Hatnean J. A.; Stephan D. W. J. Am. Chem. Soc. 2012, 134, 15728. 10.1021/ja307995f. [DOI] [PubMed] [Google Scholar]; b Eisenberger P.; Bestvater B. P.; Keske E. C.; Crudden C. M. Angew. Chem., Int. Ed. 2015, 54, 2467. 10.1002/anie.201409250. [DOI] [PubMed] [Google Scholar]; c Lam J.; Günther B. A. R.; Farrell J. M.; Eisenberger P.; Bestvater B. P.; Newman P. D.; Melen R. L.; Crudden C. M.; Stephan D. W. Dalton Trans. 2016, 45, 15303. 10.1039/C6DT02202B. [DOI] [PubMed] [Google Scholar]; d Devillard M.; Mallet-Ladeira S.; Bouhadir G.; Bourissou D. Chem. Commun. 2016, 52, 8877. 10.1039/C6CC03183H. [DOI] [PubMed] [Google Scholar]
- De Vries T. S.; Prokofjevs A.; Vedejs E. Chem. Rev. 2012, 112, 4246. 10.1021/cr200133c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Eisenberger P.; Bailey A. M.; Crudden C. M. J. J. Am. Chem. Soc. 2012, 134, 17384. 10.1021/ja307374j. [DOI] [PubMed] [Google Scholar]; b Fan X.; Zheng J.; Hua Li Z.; Wang H. J. Am. Chem. Soc. 2015, 137, 4916. 10.1021/jacs.5b03147. [DOI] [PubMed] [Google Scholar]; c Clark E. R.; Del Grosso A.; Ingleson M. J. Chem. - Eur. J. 2013, 19, 2462. 10.1002/chem.201203318. [DOI] [PubMed] [Google Scholar]; d Zheng J.; Fan X.; Zhou B.; Hua Li Z.; Wang H. Chem. Commun. 2016, 52, 4655. 10.1039/C6CC00347H. [DOI] [PubMed] [Google Scholar]
- a Heiden Z. M.; Lathem A. P. Organometallics 2015, 34, 1818. 10.1021/om5011512. [DOI] [Google Scholar]; b Lathem A. P.; Heiden Z. M. Dalton Trans 2017, 46, 5976. 10.1039/C7DT00777A. [DOI] [PubMed] [Google Scholar]
- Shoji Y.; Tanaka N.; Mikami K.; Uchiyama M.; Fukushima T. Nat. Chem. 2014, 6, 498. 10.1038/nchem.1948. [DOI] [PubMed] [Google Scholar]
- a Beckett M. A.; Strickland G. C.; Holland J. R.; Varma K. S. Polymer 1996, 37, 4629. 10.1016/0032-3861(96)00323-0. [DOI] [Google Scholar]; b Del Grosso A.; Pritchard R. G.; Muryn C. A.; Ingleson M. J. Organometallics 2010, 29, 241. 10.1021/om900893g. [DOI] [Google Scholar]
- Clark E. R.; Ingleson M. J. Organometallics 2013, 32, 6712. 10.1021/om400463r. [DOI] [Google Scholar]
- a Litters S.; Kaifer E.; Himmel H. J. Angew. Chem., Int. Ed. 2016, 55, 4345. 10.1002/anie.201600296. [DOI] [PubMed] [Google Scholar]; Litters S.; Kaifer E.; Himmel H. J. Eur. J. Inorg. Chem. 2016, 2016, 4090. 10.1002/ejic.201600629. [DOI] [Google Scholar]
- Mock M. T.; Potter R. G.; Camaioni D. M.; Li J.; Dougherty W. G.; Kassel W. S.; Twamley B.; DuBois D. L. J. Am. Chem. Soc. 2009, 131, 14454. 10.1021/ja905287q. [DOI] [PubMed] [Google Scholar]
- Adamson A. J.; Banks R. E.; Tipping A. E. J. J. Fluorine Chem. 1993, 64, 5. 10.1016/S0022-1139(00)80058-5. [DOI] [Google Scholar]
- Clark E. R.; Ingleson M. J. Angew. Chem., Int. Ed. 2014, 53, 11306. 10.1002/anie.201406122. [DOI] [PubMed] [Google Scholar]
- Del Grosso A.; Ayuso Carrillo J.; Ingleson M. Chem. Commun. 2015, 51, 2878. 10.1039/C4CC10153G. [DOI] [PubMed] [Google Scholar]
- Engelhardt L. M.; Junk P. C.; Raston C. L.; Skelton B. W.; White A. H. J. Chem. Soc., Dalton Trans. 1996, 15, 3297. 10.1039/dt9960003297. [DOI] [Google Scholar]
- a Mayr H.; Patz M. Angew. Chem., Int. Ed. Engl. 1994, 33, 938. 10.1002/anie.199409381. [DOI] [Google Scholar]; b Mayr’s Database Of Reactivity Parameters: http://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/.
- Takano R.; Ishihara Y.; Saito H.; Kitihara T.; Takano J. J. Heterocycl. Chem. 1996, 33, 1403. 10.1002/jhet.5570330465. [DOI] [Google Scholar]
- Brown H. C.; McDaniel D. H.; Häfliger O. In Determination of Organic Structures by Physical Methods; Braude E. A., Nachod F. C., Eds.; Academic Press: New York, 1955. [Google Scholar]
- For a review of this area, see:Park S.; Chang S. Angew. Chem., Int. Ed. 2017, 56, 7720. 10.1002/anie.201612140. [DOI] [PubMed] [Google Scholar]
- Gutsulyak D. V.; van der Est A.; Nikonov G. I. Angew. Chem., Int. Ed. 2011, 50, 1384. 10.1002/anie.201006135. [DOI] [PubMed] [Google Scholar]
- Fasano V.; Radcliffe J. E.; Curless L. D.; Ingleson M. J. Chem. - Eur. J. 2017, 23, 187. 10.1002/chem.201604613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During the course of this work the catalytic reduction of pyridine through the use of HBPin and ammonium tetraphenylborate as an initiator was reported, which will form borenium/boronium cations identical with those discussed in this work. On the basis of this recent publication no substrate scope investigations were carried out upon our pyridine and quinoline reduction systems.Keyzer E. N.; Kang S. S.; Hanf S.; Wright D. S. Chem. Commun. 2017, 53, 9434. 10.1039/C7CC04988A. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision D.01; Gaussian, Inc., Wallingford, CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.