

Correction
After publication of our article [1] it was brought to our attention that a line of code was missing from our program to combine the within-replicate variance and between-replicate variance. This led to an overestimation of the standard errors calculated using the Enrich2 random-effects model.
This programming error has been corrected in v1.2.0 of the software, available from [2].
We have reanalyzed the data in the paper using v1.2.0. The results are very similar and do not significantly affect any of the manuscript’s conclusions. Notably, none of the numeric values quoted in the main text change.
Updated versions of Figs. 4, 5, 6 and 7 are provided below. Updated versions of Additional file 2 Figures S1, S2 and S4 and Additional files 3, 4 and 5 are available from the links below.
Fig. 4.

A random-effects model for scoring replicate selections. Variant scores for 20 randomly selected variants from the BRCA1 E3 ubiqutin ligase dataset are shown. The replicate scores (green) were determined for each variant using Enrich2 weighted regression. Combined variant score estimates were determined using a fixed-effect model (orange) or the Enrich2 random-effects model (blue). In all cases, error bars show +2 or −2 standard errors
Fig. 5.

Standard error-based filtering improves replicate correlation. Variant scores from two replicates of the C2 domain dataset are shown. Each panel plots the top quartile of variants selected by standard error from the random-effects model (leftmost column, blue points), standard error from the fixed-effect model (middle-left column, green points), input library count (middle-right column, orange points), or total count in all libraries (rightmost column, purple points). Scores and standard errors are calculated using only the input and final round of selection (top row) or using all three rounds (bottom row). The dashed line is the best linear fit and the Pearson correlation coefficient is shown
Fig. 6.
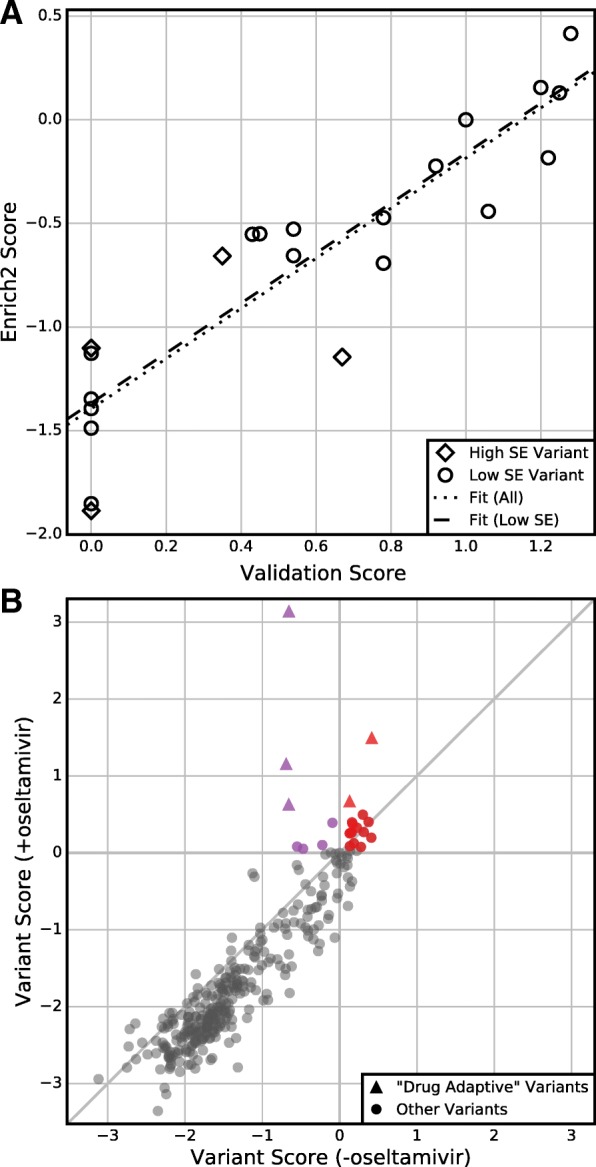
Standard errors enable hypothesis testing. a Enrich2 variant scores are plotted against single-variant growth assay scores for the 22 individually validated variants of the neuraminidase dataset. Four (18%) of these variants have Enrich2 standard errors larger than the median standard error. The dotted line shows the best linear fit for all variants and the dashed line shows the best linear fit for variants with standard errors less than the median. b Enrich2 variant scores are plotted for selections performed in the presence or absence of the small molecule inhibitor oseltamivir. Colored points indicate variants that significantly outperformed wild-type in the drug’s presence. Red points also scored significantly higher than wild-type in the drug’s absence. Triangles indicate the five “drug-adaptive” mutations identified originally [3]
Fig. 7.

Variant scoring for growth and binding experiments using simulated data. a Enrich2 variant effect scores derived from simulated data are plotted against expected Enrich2 scores based on true variant effects in the simulation. Enrich2 accurately scores variants in both simulated binding assays (left) and growth assays (right). Shading indicates point density from low (blue) to high (white). b Noisy variants were generated by randomizing their true effect in one replicate selection (green line). Noisy variants have higher overall standard errors than other variants (dashed gray line) in both binding and growth assay simulations. c The percentage of variants removed at each standard error percentile cutoff (5% intervals) is plotted. Standard error filtering preferentially removes noisy variants (green points)
We would like to thank Jörn Schmiedel for alerting us to this issue, helping us reproduce it and testing the fix.
Additional files
Supplementary figures. (PDF 423 kb)
Replicate correlation tables. (XLSX 14 kb)
Individually validated variants of the neuraminidase gene. (XLSX 12 kb)
Variants with higher scores than wild-type in the presence of oseltamivir. (XLSX 15 kb)
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13059-018-1391-7) contains supplementary material, which is available to authorized users.
The original article can be found online at https://doi.org/10.1186/s13059-017-1272-5.
References
- 1.Rubin AF, Gelman H, Lucas N, Bajjalieh SM, Papenfuss AT, Speed TP, et al. A statistical framework for analyzing deep mutational scanning data. Genome Biol. 2017;18(1):150. doi: 10.1186/s13059-017-1272-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.https://doi.org/10.5281/zenodo.890269
- 3.Jiang L, Liu P, Bank C, Renzette N, Prachanronarong K, Yilmaz LS, et al. A balance between inhibitor binding and substrate processing confers influenza drug resistance. J Mol Biol. 2016;428:538–53. doi: 10.1016/j.jmb.2015.11.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures. (PDF 423 kb)
Replicate correlation tables. (XLSX 14 kb)
Individually validated variants of the neuraminidase gene. (XLSX 12 kb)
Variants with higher scores than wild-type in the presence of oseltamivir. (XLSX 15 kb)