

Introduction
The life cycle of most marine species is characterized by high fecundity, dispersal during a planktonic larval phase and a sessile or sedentary adult phase with large census population size (Figure 1). Many articles on marine population genetics start by recalling these main characteristics and ensuing expectations, namely: high genetic diversity within population, low genetic differentiation between populations, high influx of effectively selected advantageous and deleterious mutations, and a high migration load opposing local adaptation. Most of the time, this reminder serves to better emphasize the paradox that observations deviate from these expectations, the most popular being genetic differentiation despite larval dispersal, challenging the idea that marine populations are open and well connected, just followed by pervasive local adaptation, challenging the idea that high migration rates prevent the establishment of locally adapted alleles in the sea, and finally low effective population size (Ne) despite large census size (N). The list of such counterexamples became so long that what was initially thought paradoxical finally became a shift to a new paradigm (Hauser and Carvalho 2008): (1) larval dispersal would be less effective than expected to homogenize allele frequencies between populations with high rates of self-recruitment, as, for example, in coral reef fishes (Palumbi and Warner 2003; Jones et al. 2005), (2) selection would be rampant in genomes and easy to identify with genome scans, as, for example, adaptation to brackish waters of the Baltic Sea that would target nothing less than ∼500 loci homogeneously distributed in the herring genome (Lamichhaney et al. 2012; Martinez Barrio et al. 2016), and (3) genetic drift would be stronger than thought owing to a very low proportion of individuals effectively contributing to the following generation, as, for example, in exploited fish and shellfish stocks (Hauser et al. 2002; Ovenden et al. 2007; Pinsky and Palumbi 2014). However, we should not elude too hastily that the list is also long of examples that confirmed the predictions. Genetic panmixia is often reported at large spatial scales (Addison et al. 2008; Faure et al. 2008), sometimes despite sampling across emblematic barriers to dispersal or in remote islands (Launey et al. 2002; Aurelle et al. 2003; Gérard et al. 2008) or in the deep sea (Teixeira et al. 2012). Selection is sometimes very difficult to identify with genome scans outside recognized hotspot of differentiation that hide a complex history (Bierne et al. 2011; Gagnaire et al. 2015), as, for example, in recently introduced species (Riquet et al. 2013; Rohfritsch et al. 2013). Despite the seemingly overwhelming evidence that the Ne/N ratio could locally be very low in marine populations, one may suspect a publication bias toward low Ne/N ratios, and estimates are subject to a variety of downward biases (Waples 2016). In addition, marine animals with a bentho-pelagic life cycle have among the highest genetic diversity (Figure 2) together with terrestrial species sharing similar life-history traits (Romiguier et al. 2014), suggesting the long-term species-wide Ne is rather high, and has never been in these species as low as in species with smaller fecundity and strong parental investment. Although the simple predictions initially put forward a few decades ago were certainly too naïve, and the study of the genetics of marine populations has unveiled many surprises, we are still not confident enough to decide whether marine population genetics has some peculiarities that makes it really different, or not, from what is found in terrestrial species.
Figure 1.
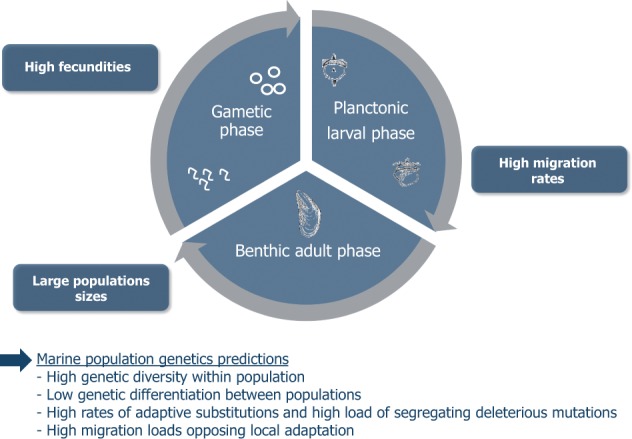
A typical bentho-pelagic life cycle, its main characteristics, and the 4 basic predictions of marine population genetics.
Figure 2.

Nonsynonymous diversity as a function of synonymous diversity in animals (data from Galtier 2016). Marine species are depicted by blue squares and continental species (terrestrial and freshwater) by green circles. According to Romiguier et al. (2016) species have been classified into 2 categories: “offspring quantity” species (dark symbols) with high fecundity and small propagule size, and “offspring quality” species (light symbols) with low fecundity and large propagule size. The 4 pictures of offsprings (mussel and mosquito larvae, seahorse and tortoise offsprings) illustrate the 2 categories in the 2 environments.
To illustrate this, we asked during 20 years the gimmick question—are the predictions of marine population genetics verified or falsified?—to the successive cohorts of invited speakers and students of a PhD course devoted to this issue, bringing fresh look to old questions. The answer is never clear-cut. Although important progresses have been made (Hellberg 2009; Hedgecock and Pudovkin 2011) and much hope is put in the forthcoming genomic data obtained by massively parallel sequencing (Nielsen et al. 2009; Kelley et al. 2016), nothing is yet settled and the reality is certainly not as simple as the impression one could obtain from a hasty overview of the literature. For this special issue, we wanted to gather at the same place a set of papers that would provide an up-to-date and balanced appraisal of the best knowledge available on marine population genetics. Our main wish was that concepts and theoretical aspects be frontally addressed and carefully explained. The readers will likely appreciate that contributors have followed this recommendation. Because population genetics is quickly moving toward NGS-based analyses, the objective was also to address how genome sequencing could allow making progress in the near future. When preparing this special issue we had in mind young undergraduate students beginning a PhD in marine population genomics. We wanted to provide them with the required material for a safe start, freed from too much equivocal statement. Probably their first anxiety will be to find their way in the bioinformatics pipelines to analyze their data. This is understandable, but shall not keep them away from essential conceptual issues. This column is aimed at guiding them through the literature and helping them shaping their own opinion.
Contributions to This Issue
The plan of the special issue roughly follows topics with an increasing spatiotemporal scale for the first 5 contributions, starting with small scale genetic patchiness (Eldon et al. 2016) and finishing with speciation genomics (Pogson 2016), passing through genetic load (Plough 2016), seascape genomics (Riginos et al. 2016) and local adaptation (Gagnaire and Gaggiotti 2016). The issue ends with 2 contributions addressing the genetic consequences of anthropogenic activities, aquaculture escapees and restocking with hatchery-propagated individuals (Waples et al. 2016) and marine invasions (Viard et al. 2016). Not only shall we understand how humans are modifying the marine environment, but these human-induced introductions also represent life-size natural experiments from which we can gain a better understanding of marine population evolution.
Eldon et al. (2016) begins the special issue with chaotic genetic patchiness (CGP). CGP has been introduced by Johnson and Black (1982, 1984) to describe the unexpected observation of temporally instable, slight but significant, genetic differentiation at a fine-grained spatial scale (i.e., below the dispersal distance permitted by planktonic larval dispersal). The most puzzling in CGP is that the level of differentiation observed at a small scale can be of the same order of magnitude as the level of differentiation observed at a very large spatial scale over thousands of kilometers (David et al. 1997). Therefore, the process that generates CGP does not only need to induce genetic differentiation at short distance but should not lead to stronger genetic differentiation at long distance. Although direct selection on some of the loci analyzed was proposed during allozyme time (Mitton 1997), the observation of CGP with noncoding DNA markers allowed rejecting the hypothesis of direct selection. CGP, therefore, needs genetic drift to be enhanced despite large census population sizes. We can think of 2 related processes, both being known as Hedgecock’s hypothesis of “sweepstake reproduction success” (Hedgecock and Pudovkin 2011). The first one is the stochastic sampling of parental pools in species with high fecundity, when lucky winners of the reproduction sweepstake reproduce at the right time and at the right place (Hedgecock 1994). The other is of genetic origin, and is sometimes termed genetic draft following Gillespie (2000), when lucky winners have a selective advantage (Plough et al. 2016). Both processes have received supports, and the second paper of this issue addressed the hypothesis that rampant genome-wide selective events could explain the temporary selective success of a few families in marine invertebrates (Plough 2016). This kind of selection is different from local adaptation at specific loci, although it may contribute to it. It is better understood as a stochastic process, which can be approximated on the long term as reduced effective population size (Gillespie 2000; Charlesworth 2009), although this verbal explanation remains approximate as detailed by Eldon et al. (2016). In the sixth paper, Waples et al. (2016) identify the conditions of a human-induced sweepstakes effect when a stock enhancement program is successful and a fraction of individuals of the population eventually traces back to the relatively few parents used for captive rearing. Eldon et al. (2016) provides a thoughtful review of the theory of the consequence of skewed offspring distribution on the genetic diversity, expanding the Kingman’s coalescent—based on the Wright–Fisher population model—through the introduction of multiple merger coalescence events, delivering new expectations that can explain CGP. Interestingly the effect of skewed offspring distribution is now also addressed beyond the marine world, for instance in partially clonal organisms where a lucky clone can disproportionally contribute to the next sexual reproduction event (Tellier and Lemaire 2014) and even in viruses (Irwin et al. 2016). While we now better understand how genetic drift/draft can locally and transiently be enhanced by sweepstakes reproduction, Eldon et al. (2016) underline that this is probably not sufficient alone to completely explain CGP. We need to understand how the effect of drift can be uncoupled between small and large spatial scales. The authors propose 2 explanations: (1) collective dispersal (Broquet et al. 2013) and (2) asynchronous local population dynamics. Both processes are able to generate kin aggregations required for a pattern of CGP, and due to their extremely similar signatures, are together fitting the “variable source hypothesis”. These are difficult hypotheses to test and tease apart in the wild, yet Eldon et al. (2016) provide an exhaustive review of the evidence accumulated to date. In the fourth paper, Gagnaire and Gaggiotti (2016) provide an interesting additional example: among a sample of 250 wild caught sea breams analyzed at 34,000 SNPs, they identified 2 pairs of individuals with a half-sib relatedness and 6 pairs with a first cousins relatedness (see Figure 4 in Gagnaire and Gaggiotti 2016). This result suggests that a few families have a disproportionate contribution to the current generation, although most of the sampled fishes are unrelated, in accordance with the “Eldon–Wakeley” model (Eldon and Wakeley 2006). As a last word about CGP, it is important to keep in mind that enhanced drift and viscous population structures affect the genetic diversity only locally and sporadically, while genetic drift is probably very slow at larger scale. For instance, 2 populations of mussels have been shown to be demographically independent for thousands of years while not departing from apparent genetic panmixia (Faure et al. 2008; Fraïsse et al. 2016), despite they might still exhibit CGP at a microscale within populations.
Plough (2016) addresses the question of the possibly high genetic load in marine animals and its possible origins. Besides revealing that we are still lacking data in many groups, this timely review shows that the evidence to date suggests marine bivalves in particular rather than marine animals in general suffer a high genetic load. Even in bivalves, the data are limited to 2 species of oysters, Ostrea edulis and Crassostrea gigas, and more data would be needed in other bivalve species and more generally in marine invertebrate taxa. Nonetheless the review also reveals bivalves share a high genetic load with long-lived plants, awakening a prediction initially made by Williams (1975) in his “elm-oyster” model while drawing parallels between the population biology of oysters and highly fecund plants. Williams (1975) hypothesized that a high segregation load could be the consequence of the high and variable early mortalities observed in these species. Skewed offspring distribution proposed to interpret CGP may thus also explain a high genetic load, though as explained above and in Eldon et al. (2016) a high genetic load would also, in turn, contribute to elevate the variance of the reproductive success (Plough et al. 2016), generating a positive feedback loop between the 2 processes. More in depth theoretical developments are clearly lacking as the few ones proposed to date have mainly addressed neutral processes. However, sweepstakes reproduction has been found to increase the probability of fixation of advantageous alleles (Der et al. 2012), and is likely to increase the polymorphic sojourn time of deleterious mutations (Harrang et al. 2013). In addition, high dispersal is likely to favor a dynamic of perpetual replacement of swamping-prone locally adapted alleles (Yeaman 2015). As for theory, we are also lacking experimental investigations, the genome scan literature having to date mainly concentrated its effort on large effect size mutations (Rockman 2012; Gagnaire and Gaggiotti 2016), often between differentiated genetic backgrounds already entered into the speciation process (Bierne et al. 2011). Molecular evolution provides interesting promises to better characterize the load in marine invertebrates (e.g., Harrang et al. 2013). In addition, more experiments as the one done by Plough et al. (2016) should be conducted in the future. The analysis of transmission ratio distortions should be conducted in other species, but also should be replicated as much as possible in the same species to better understand if the same mutations are involved in different families or if we witness the tip of the iceberg of a rampant flux of transient-selected polymorphisms. The next 2 papers of the issue also propose improvements to better understand selection and adaptation in wild populations of marine organisms.
Riginos et al. (2016) review the seascape genomics program and propose improvements. At first sight, the approach might seem straightforward: a vector with spatial positions of samples, a matrix with as many ecological variables as available, a matrix of population connectivity predicted from a hydrodynamic model, a matrix of genotypes with lots of markers, a method to correlate factors while partialling out others, and ready-to-publish outputs are there, with a list of candidate loci for adaptation to temperature, another for salinity and some again for other ecological factors. Riginos et al. (2016) nicely tackle all the pitfalls of such a seemingly simple approach while reviewing the last developments that should improve inferences at every stage of the analysis. They particularly emphasize how important is the quantification of seascape dynamics and patterns, but also how prone to high false discovery rates and misinterpretation outlier tests can be (Bierne et al. 2013; Lotterhos and Whitlock 2014). Although the task of achieving meaningful seascape genomics analysis might seem challenging, Riginos et al. (2016) highlight promising new strategies to overcome tricky problems, including a strategy to spatially optimize sampling. To date, the interpretations of the seascape genomics program have probably been hampered by complex histories at recognized hotspots of genetic differentiation such as the North Sea–Baltic Sea or the Atlantic Ocean–Mediterranean Sea transitions (Gagnaire et al. 2015). Yet, efficient methods to reconstruct the demographic history of populations can now reveal unsuspected histories of secondary contact even when the spatial distribution is patchy (Martin et al. 2015; Rougemont et al. 2016; Rougeux et al. 2016). Seascape genomics is now able to uncover genetic–environment associations outside recognized biogeographic boundaries (Benestan et al. 2016). Riginos et al. (2016), as well as Gagnaire and Gaggiotti (2016) in the following article, provide pertinent and complementary advices to tackle such a complex and challenging task.
Gagnaire and Gaggiotti (2016) address some methodological challenges associated with the detection of polygenic selection using molecular approaches. The performance of population genomic methods strongly depends on the genetic architecture of underlying fitness traits, the migration–selection–drift balance, the length of the chromosomal region affected by selection and the density of markers used. If one agrees with these basic expectations, one should recognize that the genome scan literature has to date been mainly biased toward the detection of large effect QTLs (Rockman 2012) and the detection of large genomic islands of differentiation resulting from coupling among many selected genes of various sorts (Bierne et al. 2011), and might have discussed too many false positives (Lotterhos and Whitlock 2014) while missing many if not most of the loci really involved in local adaptation, either because their differentiation was not strong enough (Le Corre and Kremer 2012) or because the density of markers was insufficient (Hoban et al. 2016). Much remains to be done in the study of local adaptation in the sea. Gagnaire and Gaggiotti (2016) propose an innovative agenda to do so. They first provide an overview of cutting edge genome scan methods to detect selection using genetic differentiation between samples or between phenotypes, including when demography and spatial population structure are complex. Second, they highlight the potential of quantitative genetics theory and approaches to supplement the genome scan analysis. More precisely they propose to circumvent the lack of power of single locus tests by the use of polygenic scores. The combined approach allows assessing the proportion of the genetic variance explained by candidate loci in the study, and use genomic predictions to gain useful information about the strength and directionality of local adaptation (Berg and Coop 2014). Although experimental crossing in the lab would facilitate the analysis, the possible existence of groups of individuals with recent relatedness in populations enduring sweepstake reproduction success described in the first article (Eldon et al. 2016) could potentially make possible quantitative genetics in wild marine populations.
Pogson (2016) also proposed a mixed integrative approach to the study of speciation in marine species. This time the idea is to use molecular evolution approaches that allow estimating the rate of adaptive substitution and the deleterious load (Eyre-Walker and Keightley 2007; Galtier 2016) and specific codons under adaptive evolution in proteins (Yang 2004). Molecular evolution has rarely been used in speciation genomics of either marine or terrestrial species. Thanks to the democratization of high-throughput sequencing in nonmodel species, molecular evolution is likely to be increasingly combined with the study of the genomic landscape of differentiation (Christe et al. 2016). The issue addressed in Pogson’s article is of consequential importance in speciation research: is adaptive protein evolution a common cause of reproductive incompatibilities? So-called “speciation genes” identified to date often exhibit signals of positive Darwinian selection, although most probably driven by genomic conflicts rather than by adaptation to the external environment (Presgraves 2010). It would be premature, however, to conclude that this is a general property of speciation genes. Pogson offers to systematically test that regions of high differentiation between incipient species connected by contemporary gene flow are enriched for fast adaptively evolving genes. The article takes the form of a research roadmap with detailed explanations of every step. Molecular evolution of coding and noncoding sequences at known position of a sequenced genome, together with the analysis of admixture and clines in contact zones, should allow testing the hypothesis that genomic regions resisting introgression are enriched for fast-evolving proteins involved in Reproductive Isolation (RI). Conversely, this approach also allows testing the hypothesis that introgressed chromosomal tracts are enriched with segregating deleterious amino acid mutations (Christe et al. 2016). Another merit of Pogson’s approach is to stimulate the comparison of long- and short-term evolution, by comparing the divergence between well-diverged species that did not exchange genes for very long time, and the differentiation, cline shapes and admixture patterns between taxa at earlier stages of the speciation process. For instance, many population genomics studies of the Northern Atlantic would likely benefit from the comparative analysis with related species from the Pacific Ocean (Väinölä 2003; Laakkonen et al. 2015).
Waples et al. (2016) addressed the issue of the release in the wild of individuals reared in captivity either intentionally for marine stock enhancement or involuntarily as a consequence of aquaculture escapees. One likely effect, known as the Ryman–Laikre (R–L) effect, is an increase in inbreeding and a reduction of the effective population size. Ryman and Laikre (1991) provided a simple expression of the effective population size of the captive–wild system as a function of 3 variables: x, the contribution of captive-reared individuals to the next generation; Ne(C), the effective size of the captive population; and Ne(W), the effective size of the wild population. To limit the R–L effect one basically needs to maintain the effective size of the captive population as high as possible, which is often hardly possible, and to limit the contribution of the captive stock to the wild population. To account for the characteristics of marine populations, Waples et al. (2016) have decomposed the basic R–L equation into key components of its 3 variables, and among them Ne/N ratios of the captive (αC) and wild (αW) populations. They identified a key parameter, β, which is the ratio of Ne/N in captive and wild components (β = αC/αW). Unless the captive contribution is almost nil captive propagation will sharply reduce effective size unless β is very large (>103). Let us imagine αW is truly tiny in standard marine populations with very large census size, despite this is disputable (Waples 2016), and in addition that the breeding program is efficient enough to maintain a high αC, which also is disputable, in this case one does not expect a strong R–L effect. However, not only restocking is little an issue in this case, but the captive contribution x can hardly be anything than small; the release of hatchery-propagated stocks is but a drop in the bucket. Stock enhancement programs typically seek to enhance harvest opportunities of declining fisheries, or to increase abundance of endangered species. In this situation, αW cannot be tiny or the survival of the wild stock would be desperate, and so would be a stock enhancement program, on the long term. In addition, stock enhancement programs often require sustained mass release of hatchery-propagated seeds to be successful in the sea (Morvezen et al. 2016). Therefore, we might, possibly a bit abusively, contend that a strong R–L effect is always expected for any successful stock enhancement program in the sea. The parallel with Hedgecock’s sweepstake effects is enlightening here: the few parents used for captive rearing can be seen as lucky winners that, thanks to human activities, escaped the high mortalities of early developmental stages. Even if the process also happens in the wild, stock enhancement programs can only exacerbate the sweepstake effect.
On a similarly reduced temporal scale, but potentially large spatial one, Viard et al. (2016) address the issue of biological invasions in the sea. Even more than on lands genetic tools have been essential to identify invasive populations in the sea and discriminate them from native congeners, retrace the invasion pathways, monitor the invasion, and study the demography and adaptation as well as the eco-evolutionary processes leading to a successful invasion. The more progresses made with molecular techniques, the more we discover hidden invasions and witness the big melting pot of exotic species that humans have spread worldwide. If it is any consolation, the study of marine invasions has deepened our understanding of marine population genetics. Viard et al. (2016) confirm in their review that invasion is rarely accompanied by a strong signal of genetic bottleneck. Population differentiation with the native range and also within the colonization range is often very low, even while the invading species spread through recognized barriers to dispersal, biogeographic boundaries and ecoregions (Riquet et al. 2013). Finally, genome scans have failed to identify candidate loci for adaptation during invasion. All these results suggest that bentho-pelagic marine species with invasive potential are finally in compliance with the initial predictions of marine population genetics. Marine invasion genetics can, therefore, be taken as argument to oppose the claims that marine species disperse less, have lower effective size, and adapt more easily than initially thought.
Acknowledgments
We thank Zhi-Yun Jia for the invitation to guest edit a special column on marine population genomics in Current Zoology. We are of course very much indebted to all the contributors of the column for the beautiful work they did. We are also grateful to the students and invited speakers of the Doctoral Training Course “Génétique et Evolution des Organismes Marins” (Doctoral Programme GAIA, University of Montpellier) for all the discussions we had during these 20 years of courses. We are also thankful to colleagues with whom we had stimulating discussions at meetings and workshops supported by the research group on Marine Connectivity GDR CNRS – Ifremer 3445 MarCo.
Funding
This work was supported by a Languedoc-Roussillon Region “Chercheur(se)s d'avenir” grant to N.B. (Connect7 project) and by the GDR CNRS – Ifremer 3445 MarCo.
References
- Addison JA, Ort BS, Mesa KA, Pogson GH, 2008. Range-wide genetic homogeneity in the California sea mussel Mytilus californianus: a comparison of allozymes, nuclear DNA markers, and mitochondrial DNA sequences. Mol Ecol 17:4222–4232. [DOI] [PubMed] [Google Scholar]
- Aurelle D, Guillemaud T, Afonso P, Morato T, Wirtz P. et al. , 2003. Genetic study of Coris julis (Osteichtyes, Perciformes, Labridae) evolutionary history and dispersal abilities. CR Biol 326:771–785. [DOI] [PubMed] [Google Scholar]
- Benestan L, Quinn BK, Maaroufi H, Laporte M, Clark FK. et al. , 2016. Seascape genomics provides evidence for thermal adaptation and current-mediated population structure in American lobster Homarus americanus. Mol Ecol 25:5073–5092. [DOI] [PubMed] [Google Scholar]
- Berg JJ, Coop G, 2014. A population genetic signal of polygenic adaptation. PLoS Genet 10:e1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne N, Roze D, Welch JJ, 2013. Pervasive selection or is it … ? Why are FST outliers sometimes so frequent? Mol Ecol 22:2061–2064. [DOI] [PubMed] [Google Scholar]
- Bierne N, Welch J, Loire E, Bonhomme F, David P, 2011. The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20:2044–2072. [DOI] [PubMed] [Google Scholar]
- Broquet T, Viard F, Yearsley JM, 2013. Genetic drift and collective dispersal can result in chaotic genetic patchiness. Evolution 67:1660–1675. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, 2009. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet 10:195–205. [DOI] [PubMed] [Google Scholar]
- Christe C, Stölting KN, Paris M, Fraïsse C, Bierne N. et al. , 2016. Adaptive evolution and segregating load contribute to the genomic landscape of divergence in two tree species connected by episodic gene flow. Mol Ecol. doi: 10.1111/mec.13765. [DOI] [PubMed] [Google Scholar]
- David P, Perdieu MA, Pernot AF, Jarne P, 1997. Fine-grained spatial and temporal population genetic structure in the marine bivalve Spisula ovalis. Evolution 51:1318–1322. [DOI] [PubMed] [Google Scholar]
- Der R, Epstein C, Plotkin JB, 2012. Dynamics of neutral and selected alleles when the offspring distribution is skewed. Genetics 191:1331–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldon B, Riquet F, Yearsley J, Jollivet D, Broquet T, 2016. Current hypotheses to explain genetic chaos under the sea. Curr Zool 62:551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldon B, Wakeley J, 2006. Coalescent processes when the distribution of offspring number among individuals is highly skewed. Genetics 172:2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD, 2007. The distribution of fitness effects of new mutations. Nat Rev Genet 8:610–618. [DOI] [PubMed] [Google Scholar]
- Faure MF, David P, Bonhomme F, Bierne N, 2008. Genetic hitchhiking in a subdivided population of Mytilus edulis. BMC Evol Biol 8:164.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraïsse C, Belkhir K, Welch J, Bierne N, 2016. Local inter-species introgression is the main cause of outlying levels of intra-specific differentiation in mussels. Mol Ecol 25:269–286. [DOI] [PubMed] [Google Scholar]
- Gagnaire PA, Broquet T, Aurelle D, Viard F, Souissi A. et al. , 2015. Using neutral, selected and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evol Appl 8:769–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnaire P-A, Gaggiotti OE, 2016. Detecting polygenic selection in marine populations by combining population genomics and quantitative genetics approaches. Curr Zool 62:603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, 2016. Adaptive protein evolution in animals and the effective population size hypothesis. PLoS Genet 12:e1005774.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard K, Bierne N, Borsa P, Chenuil A, Féral J-P, 2008. Pleistocene separation of mitochondrial lineages of Mytilus spp. mussels from Northern and Southern Hemispheres and strong genetic differentiation among southern populations. Mol Phyl Evol 49:84–91. [DOI] [PubMed] [Google Scholar]
- Gillespie JH, 2000. Genetic drift in an infinite population. The pseudohitchhiking model. Genetics 155:909–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrang E, Lapègue S, Morga B, Bierne N, 2013. A high genetic load contributes to high protein diversity in a marine bivalve. G3 Genes Genomes Genet 3:333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser L, Adcock GJ, Smith PJ, Bernal Ramírez JH, Carvalho GR, 2002. Loss of microsatellite diversity and low effective population size in an overexploited population of New Zealand snapper Pagrus auratus. Proc Natl Acad Sci 99:11742–11747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser L, Carvalho GR, 2008. Paradigm shifts in marine fisheries genetics: ugly hypotheses slain by beautiful facts. Fish Fisher 9:333–362. [Google Scholar]
- Hedgecock D, 1994. Does variance in reproductive success limit effective population sizes of marine organisms? In: Beaumont AR, editor. Genetics and Evolution of Aquatic Organisms. London: Chapman & Hall, 122–134. [Google Scholar]
- Hedgecock D, Pudovkin AI, 2011. Sweepstakes reproductive success in highly fecund marine fish and shellfish: a review and commentary. Bull Mar Sci 87:971–1002. [Google Scholar]
- Hellberg ME, 2009. Gene flow and isolation among populations of marine animals. Annu Rev Ecol Evol Syst 40:291–310. [Google Scholar]
- Hoban S, Kelley JL, Lotterhos KE, Antolin MF, Bradburd G. et al. , 2016. Finding the genomic basis of local adaptation: pitfalls, practical solutions, and future directions. Am Nat 188:379–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin KK, Laurent S, Matuszewski S, Vuilleumier S, Ormond L. et al. , 2016. On the importance of skewed offspring distributions and background selection in virus population genetics. Heredity. doi:10.1038/hdy.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MS, Black R, 1982. Chaotic genetic patchiness in an intertidal limpet, Siphonria sp. Mar Biol 70:157–164. [Google Scholar]
- Johnson MS, Black R, 1984. Pattern beneath the chaos: the effect of recruitment on genetic patchiness in an intertidal limpet. Evolution 38:1371–1383. [DOI] [PubMed] [Google Scholar]
- Jones GP, Planes S, Thorrold SR, 2005. Coral reef fish larvae settle close to home. Curr Biol 15:1314–1318. [DOI] [PubMed] [Google Scholar]
- Kelley JL, Brown AP, Therkildsen NO, Foote AD, 2016. The life aquatic: advances in marine vertebrate genomics. Nat Rev Genet 17:523–534. [DOI] [PubMed] [Google Scholar]
- Laakkonen HM, Strelkov P, Lajus DL, Väinölä R, 2015. Introgressive hybridization between the Atlantic and Pacific herrings (Clupea harengus and C. pallasii) in the north of Europe. Mar Biol 162:39–54. [Google Scholar]
- Lamichhaney S, Barrio AM, Rafati N, Sundström G, Rubin C-J. et al. , 2012. Population-scale sequencing reveals genetic differentiation due to local adaptation in Atlantic herring. Proc Natl Acad Sci 109:19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launey S, Ledu C, Boudry P, Bonhomme F, Naciri-Graven Y, 2002. Geographic structure in the European flat oyster, Ostrea edulis L, as revealed by microsatellite polymorphism. J Hered 93:331–351. [DOI] [PubMed] [Google Scholar]
- Le Corre V, Kremer A, 2012. The genetic differentiation at quantitative trait loci under local adaptation. Mol Ecol 21:1548–1566. [DOI] [PubMed] [Google Scholar]
- Lotterhos KE, Whitlock MC, 2014. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol Ecol 23:2178–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CH, Cutler JS, Friel JP, Dening Touokong C, Coop G. et al. , 2015. Complex histories of repeated gene flow in Cameroon crater lake cichlids cast doubt on one of the clearest examples of sympatric speciation. Evolution 69:1406–1422. [DOI] [PubMed] [Google Scholar]
- Martinez Barrio A, Lamichhaney S, Fan G, Rafati N, Pettersson M. et al. , 2016. The genetic basis for ecological adaptation of the Atlantic herring revealed by genome sequencing. eLife 5:e12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitton JB, 1997. Selection in natural populations. New York: Oxford University Press. [Google Scholar]
- Morvezen R, Boudry P, Laroche J, Charrier G, 2016. Stock enhancement or sea ranching[quest] Insights from monitoring the genetic diversity, relatedness and effective population size in a seeded great scallop population (Pecten maximus). Heredity 117:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen EE, Hemmer-Hansen J, Larsen PF, Bekkevold D, 2009. Population genomics of marine fishes: identifying adaptive variation in space and time. Mol Ecol 18:3128–3150. [DOI] [PubMed] [Google Scholar]
- Ovenden JR, Peel D, Street R, Courtney AJ, Hoyle SD. et al. , 2007. The genetic effective and adult census size of an Australian population of tiger prawns (Penaeus esculentus). Mol Ecol 16:127–138. [DOI] [PubMed] [Google Scholar]
- Palumbi SR, Warner RR, 2003. Why gobies are like hobbits. Science 299:51–52. [DOI] [PubMed] [Google Scholar]
- Pinsky ML, Palumbi SR, 2014. Meta-analysis reveals lower genetic diversity in overfished populations. Mol Ecol 23:29–39. [DOI] [PubMed] [Google Scholar]
- Plough LV, 2016. Genetic load in marine animals: a review. Curr Zool 62:567–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plough LV, Shin G, Hedgecock D, 2016. Genetic inviability is a major driver of type III survivorship in experimental families of a highly fecund marine bivalve. Mol Ecol 25:895–910. [DOI] [PubMed] [Google Scholar]
- Pogson GH, 2016. Studying the genetic basis of speciation in high gene flow marine invertebrates. Curr Zool 62:643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC, 2010. The molecular evolutionary basis of species formation. Nat Rev Genet 11:175–180. [DOI] [PubMed] [Google Scholar]
- Riginos C, Crandall ED, Liggins L, Bongaerts P, Treml EA, 2016. Navigating the currents of seascape genomics: how spatial analyses can augment population genomic studies. Curr Zool 62:581–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquet F, Daguin-Thiébaut C, Ballenghien M, Bierne N, Viard F, 2013. Contrasting patterns of genome-wide polymorphism in the native and invasive range of the marine mollusc Crepidula fornicata. Mol Ecol 22:1003–1018. [DOI] [PubMed] [Google Scholar]
- Rockman MV, 2012. The QTN program and the alleles that matter for evolution: all that’s gold does not glitter. Evolution 6:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohfritsch A, Bierne N, Boudry P, Heurtebise S, Cornette F. et al. , 2013. Population genomics shed light on the demographic and adaptive histories of European invasion in the Pacific oyster, Crassostrea gigas. Evol Appl 6:1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romiguier J, Gayral P, Ballenghien M, Bernard A, Cahais V. et al. , 2014. Comparative population genomics in animals uncovers the determinants of genetic diversity. Nature 515:261–263. [DOI] [PubMed] [Google Scholar]
- Rougemont Q, Gagnaire P-A, Perrier C, Genthon C, Besnard A-L. et al. , 2016. Inferring the demographic history underlying parallel genomic divergence among pairs of parasitic and nonparasitic lamprey ecotypes. Mol Ecol. doi:10.1111/mec.13664. [DOI] [PubMed] [Google Scholar]
- Rougeux C, Bernatchez L, Gagnaire P-A, 2016. Modeling the multiple facets of speciation-with-gene-flow towards improving divergence history inference of a recent fish adaptive radiation. bioRxiv. doi:10.1101/068932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman N, Laikre L, 1991. Effects of supportive breeding on the genetically effective population size. Conserv Biol 5:325–329. [Google Scholar]
- Teixeira S, Serrao EA, Arnaud-Haond S, 2012. Panmixia in a fragmented and unstable environment: the hydrothermal shrimp Rimicaris exoculata disperses extensively along the Mid-Atlantic Ridge. PLoS ONE 7:e38521.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier A, Lemaire C, 2014. Coalescence 2.0: a multiple branching of recent theoretical developments and their applications. Mol Ecol 23:2637–2652. [DOI] [PubMed] [Google Scholar]
- Väinölä R, 2003. Repeated trans-Arctic invasions in littoral bivalves: molecular zoogeography of the Macoma balthica complex. Mar Biol 143:935–946. [Google Scholar]
- Viard F, David P, Darling JA, 2016. Marine invasions enter the genomic era: three lessons from the past, and the way forward. Curr Zool 62:629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waples RS, 2016. Tiny estimates of the Ne/N ratio in marine fishes: are they real? J Fish Biol. doi:10.1111/jfb.13143. [DOI] [PubMed] [Google Scholar]
- Waples RS, Hindar K, Karlsson S, Hard JJ, 2016. Evaluating the Ryman–Laikre effect for marine stock enhancement and aquaculture. Curr Zool 62:617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC, 1975. Sex and Evolution. Princeton (NJ: ): Princeton University Press. [Google Scholar]
- Yang Z, 2004. Adaptive molecular evolution In: Balding DJ, Bishop M, Cannings C, editors. Handbook of statistical genetics. New York: John Wiley & Sons, Ltd, 237–350. [Google Scholar]
- Yeaman S, 2015. Local adaptation by alleles of small effect. Am Nat 186(Suppl 1):S74–S89. [DOI] [PubMed] [Google Scholar]