

Synopsis
Plasma cell dyscrasia (PCD) is a heterogeneous disease which has seen a tremendous change in outcomes due to improved therapies. Over the last few decades, multiparametric flow cytometry has played an important role in the detection and monitoring of PCDs. Flow cytometry is a high sensitivity assay for early detection of minimal residual disease (MRD) that correlates well with progression-free survival and overall survival. Before flow cytometry can be effectively implemented in the clinical setting sample preparation, panel configuration, analysis, and gating strategies must be optimized to ensure accurate results. Current consensus methods and reporting guidelines for MRD testing are discussed.
Keywords: Plasma cell dyscrasia, plasma cell neoplasm, multiparametric flow cytometry, multiple myeloma, minimal residual disease, MRD, panel optimization, high sensitivity assay
INTRODUCTION
Plasma cells (PC) are terminally differentiated and non-dividing immune cells arising from B cells whose primary function is to secrete antibodies to fight infection.1–3 PCs can live for a long period, even a life time, in the bone marrow if survival signals such as interleukin-6 (IL-6), A proliferation-inducing ligand (APRIL), and B cell activating factor (BAFF) are provided by stromal cells and various hematopoietic cells such as eosinophil.4–9 PCs can only produce a single kind of antibody in a single class of immunoglobulin but each cell can produce several thousand antibodies per second, making it an integral part of the humoral immune response.10 PCs like all other leukocytes are susceptible to transformation. Most plasma cell dyscrasias (PCDs) develop after affinity maturation has occurred in the germinal center, as the gene sequence of most myeloma cells are hypermutated, exhibit phenotypic features similar to those of long-lived PCs, and are usually distributed in multiple compartments of the bone marrow.1,11–13 Once a clonal PC population is established, it has the potential to behave in a number of ways not all of which will require treatment. Currently, there are several subtypes of PCDs including (i) monoclonal gammopathy of undetermined significance (MGUS), (ii) asymptomatic myeloma, (iii) multiple myeloma, (iv) PC leukemia, (v) plasmacytoma, and (vi) amyloidosis.14,15
Plasma Cell Disorders
Normal PCs generate a spectrum of antibodies with different heavy chain (IgM, IgG, IgA, IgD, and IgE) and light chain (kappa and lambda) characteristics. With the exception of a very few non-secreting cases, a common early finding in early PCDs is the presence of a monoclonal heavy and light chain restricted antibody, referred to as an a paraprotein or M protein. These often appear before any relevant clinical symptoms are discerned. A monoclonal gammopathy is defined as any situation in which a clonal M protein is present in the blood or urine and is reflective of a clonal plasma or lymphoid cell proliferation. Purely reactive proliferations will often generate a polyclonal immunoglobulin response but these are by definition not monoclonal M proteins. Since both PC and lymphoproliferative disorders can produce M proteins, making the distinction between which type of disorder is responsible for the M protein is critically important and flow cytometry can assist in this process. Diseases of PCs include MGUS, multiple myeloma, and PC leukemia.
Monoclonal Gammopathy of Undetermined Significance (MGUS)
MGUS is the most common PC disorder. Its incidence rises rapidly with age and affects 3% of individuals 50 years of age or older and 10% of individuals over the age of 70.16–19 Increasingly more sensitive assays and more frequent testing have resulted in increased diagnosis. MGUS is characterized by a serum M protein of less than 30 g/L with fewer than 10% PCs in the bone marrow and no evidence of bone or organ damage.14 Although it reflects the presence of an expanded clone of immunoglobulin secreting cells, it is not considered a malignancy and no immediate therapeutic intervention is required. Approximately only 1% of MGUS patients will progress to myeloma each year.20 The immunophenotype of MGUS frequently shows two populations of CD38br, CD138br PCs, one with a normal CD19+, CD56– phenotype that is polyclonal and a second that is either CD19– and CD56+ or CD56– and monoclonal.21
Asymptomatic Plasma Cell Myeloma
Asymptomatic myeloma is characterized by an M protein concentration of greater than 30 g/L, with more than 10% PCs in the bone marrow, but with no related tissue or end organ damage or clinical sequelae such as hypercalcemia, renal failure, anemia, and bone lesions.14,22 The presence of radiographically detected bone lesions, even if not symptomatic, would exclude a patient from this category because these are an indication for treatment. About 10% of asymptomatic myeloma patients will progress to multiple myeloma each year.23,24 Because smoldering and indolent PC myeloma are a continuum, neither can be reliably diagnosed prospectively, and both are asymptomatic, thus the term asymptomatic PC myeloma is preferred to describe both.
Multiple Myeloma
Multiple myeloma is characterized by an M protein concentration of >30 g/L, greater than 10% PCs in the bone marrow, and evidence of organ and tissue damage.14 It is also characterized by calcium elevation, renal insufficiency, anemia, and bone lesions due to the accumulation of clonal PCs in the bone marrow. Unlike PC leukemia, the PCs in multiple myeloma circulate in the blood at such a low frequency they cannot be readily detected morphologically. Each year in the United States, nearly 27,000 people are diagnosed with multiple myeloma which accounts for 1% of all malignancies and 10% of all hematological neoplasms.25 Normally, PCs make up less than 5% of leukocytes in the bone marrow but if they transform, PCs can fill up the bone marrow, and also cause damage to the bone through the release of osteoclast-activating factors26. Over time, they collect and form tumors in multiple areas of the bones giving rise to the term “multiple” myeloma.
Plasma Cell Leukemia
PC leukemia is similar to multiple myeloma but differs in presentation due to the presence of PCs in the peripheral blood circulation. It is defined as a population of at least 2 × 103 PCs/μL or representing greater than 20% of circulating leukocytes.16 PC leukemia may be present at the time of initial diagnosis but more commonly evolves as a late feature of multiple myeloma (secondary PC leukemia). Primary PC leukemia is found in 2 - 5% of myeloma cases and has a poor prognosis.27–29 PC leukemias are usually associated with hypercalcemia, renal failure, and anemia, but not bone lesions. Phenotypically, it is more often CD56− than multiple myelomas.
Plasmacytoma
Plasmacytoma is a discrete solid PC tumor found either in the bone (solitary bone plasmacytoma) or soft tissues (extramedullary plasmacytoma). Approximately 80% of extramedullary plasmacytomas occur in the upper respiratory tract including the oropharynx, nasopharynx, sinuses, or larynx though they can occur in any soft tissue. Approximately 20% of cases have a small M protein, most typically IgA. Symptoms are generally related to the tumor mass and compression of adjacent tissues. Plasmacytomas are characterized by a similar range of immunophenotypic and genotypic abnormalities to those encountered in multiple myeloma.30
Primary Amyloidosis
Primary amyloidosis is a rare disorder caused by a PC which produces intact immunoglobulins or most commonly immunoglobulin light chain fragments and rarely heavy chain fragments. These build up in tissues and organs forming β-amyloid sheets. Renal and cardiac amyloid deposition can cause serious organ impairment and death. The immunophenotype of PCs causing primary amyloidosis, like myeloma, are typically light chain restricted, CD38 and CD138 positive, CD45 negative or dim, with the majority being CD56+, CD117+, and CD19−.31,32
DIAGNOSIS
The diagnosis of PCD is challenging. For instance, studies have reported similar genetic alteration between pre-malignant MGUS and multiple myeloma11. Accordingly, diagnosis is usually based on a number of factors. A standard workup includes total serum protein, serum and urine protein electrophoresis, immunofixation in serum and urine, detection of serum free light chains (sFLC), and the following additional parameters: complete blood count, serum creatinine, electrolytes (including calcium), lactate dehydrogenase, and β2 microglobulin. In a patient with suspected multiple myeloma, a bone marrow biopsy is normally obtained.
Multiparametric flow cytometry (MFC) is a powerful technique that has been routinely employed in clinical settings to characterize, diagnose, and monitor hematological malignancies.33–39 Though the assessment of myeloma cells in the bone marrow compartment by flow cytometry has been conducted since the 1990s, the technology has only recently been widely accepted as a routine clinical test for multiple myeloma.40–44 This delayed acceptance was due primarily to the unavailability of specific markers that could be used for reliable detection of PCs and selective loss of PCs while making a single cell suspension from bone marrow.45,46 Technical variables including PC baseline autofluorescence levels, inconsistent staining profiles, and the use of different monoclonal antibody (mAb) clones and fluorochromes all contributed to the slow adoption of MFC for the detection of PCDs.42,45–50
With the increased understanding of human cell biology and the expansion of mAb choices, recent reports published by independent investigators have shown that the expression of several markers can be used to distinguish normal from abnormal PCs.37,43–45,48,51–54 As such, greater than 95% of the multiple myeloma patients are found to have an aberrant phenotype associated with their neoplastic PCs. The remainder of this chapter will focus on the utilization of MFC immunophenotyping and how it can be helpful in the diagnosis, prognosis and disease monitoring of PCDs. Specifically, the methodological considerations involved during panel design, including the selection and validation of informative mAbs, and data analysis will be elaborated.
Methodological Considerations for Evaluating Abnormal Plasma Cell
MFC is a reliable technique that enables concurrent and correlated detection and analysis of multiple parameters at the single cell level. It permits the study of large number of cells within a relatively short period of time, storage of that information for further evaluation, quantitative evaluation of antigen expression levels, and combined detection of surface and intracellular antigens.36,45,55,56 Before flow cytometry can be applied to clinical diagnosis of PCDs, optimizing sample preparation, panel configuration, and analysis and gating strategies is critical.57–63 This topic has been previously addressed in detail and the reader is referred to the Cytometry B Special Issue dedicated to the validation of cell-based fluorescence assays from the International Council for Standardization of Haematology and the International Clinical Cytometry Society.57,59,62,64 Major efforts over the last couple of years have also been invested into reaching a consensus over which mAb panels should be run on bone marrow samples from newly diagnosed multiple myeloma patients and when testing them for minimal residual disease (MRD).58,65,66 Presently, the lack of External Quality Assessment (EQA)/Proficiency Testing (PT) programs for PCD testing constrains the evaluation of inter-laboratory reproducibility and concordance.55,67 At the time of this writing, the United Kingdom National External Quality Assessment Service (UK NEQAS) group had begun a prototype EQA/PT program which evaluated the reproducibility of multiple myeloma MRD detection among 8 participating laboratories. Before such programs become widely available, the optimization and validation of a flow cytometry-based PCD assay should be taken with care, using current published consensus protocols as a guideline and validated by comparing results with a laboratory that has a well-established assay. This section describes in detail sample handling, mAb and fluorochrome selection, panel design, sample staining, data analysis, evaluation of sample quality, and reporting.
Panel selection
In clinical flow cytometry, cell populations are considered as abnormal if they have an atypical differentiation patterns, increased or decreased expression of normal antigens, asynchronous maturational patterns, or the expression of aberrant antigens. Currently, the most commonly used markers for the discrimination of normal plasma and myeloma cells from other leukocytes include CD38, CD138, CD45, CD19, CD56, and cytoplasmic immunoglobulin light chains in combination with light scatter.37,41 Practically, an assay for detecting PCD should be able to identify disease in all tested patients; which is a strength of flow cytometry when compared to other technologies. It does not necessarily have to fully characterize the disease but should retain the capability of detecting the presence of diseased cells with appropriate specificity and sensitivity. The minimum mAbs recommended for initial assessment of PCDs are CD38, CD138, CD45, CD19, and CD56, with the expression of secondary antigens considered to be informative added as necessary.58 The expression patterns of CD38, CD138, and CD45 antigens are recommended by the European Myeloma Net to be used as the backbone markers for the identification and enumeration of PCs in all tubes.66,68 The unique expression patterns of CD19, CD20, CD27, CD28, CD56, CD81, CD117, and cytoplasmic immunoglobulin light chains have all been employed for the discrimination between normal or reactive versus clonal PCs (Table 1).45,48,68,69
Table 1.
Comparison of antigen expression patterns between normal and abnormal plasma cells.
| Antigen(s) | Normal Plasma Cells | Abnormal Plasma Cells | ||
|---|---|---|---|---|
| Expression Level | Percentage | Expression Level | Percentage | |
| (a) Identification of normal and abnormal plasma cells in bone marrow | ||||
| * CD45 | +++ | 94% | − | 73 – 80% |
| * CD38 | +++ | 100% | + | 80% |
| CD138 | ++ | 98% | ++ | 98% |
| (b) Markers useful for the distinction of normal and abnormal plasma cells in bone marrow | ||||
| CD19 | ++ | >70% | − | 95% |
| CD56 | − | >85% | +++ | 60 – 75% |
| CD117 | − | 100% | ++ | 30 – 32% |
| CD27 | +++ | 100% | − to + | 40 – 68% |
| CD81 | ++ | 100% | − to + | 55% |
| CD28 | − to + | <15% dim cells | +++ | 15 – 45% |
| CD20 | − | >95% | ++ | 17 – 30% |
| CD200 | − | N/A | +++ | 70% |
+Dimly Positive
++Moderately Positive
+++Strongly Positive
-Negative
Can be used to identify and distinguish normal and abnormal plasma cells
An effective panel that has been proposed and validated by the ICCS multiple myeloma minimal residual disease consensus group and the EuroFlow consortium consists of two 8-color tubes that measure a total of 10 specific antigens.53,70 The final panel utilized by these groups uses 8 surface markers in the first tube (Tube 1), while the second tube (Tube 2) is comprised of 6 surface and 2 intracellular markers (Table 2). In our configuration, Tube 1 and Tube 2 have a backbone of CD38, CD138, and CD45 which in combination with light scatter provides the best approach for identification of PCs and their discrimination from all other cells in the sample. Both tubes also contain CD19, CD56, and CD27. In combination with CD38 and CD45, that can be aberrantly dim or absent, the lack of CD19 expression and/or strong CD56 expression and/or dim CD27 expression differentiates in most cases neoplastic PCs from their normal counterpart. Tube 1 also has CD81 and CD117 while in Tube 2 these mAbs are substituted with cytoplasmic anti-kappa and anti-lambda polyclonal antibodies. The merit of using two 8-color tubes with redundancy in the design allows for the confirmation of the presence and number of abnormal PCs in acquired samples from a statistical and internal quality control standpoint. As the costs associated with the two-tube approach are higher and the technical aspect of clinical flow cytometer continues to improve to accommodate more fluorescent parameters, a few labs have explored using a single 10-color tube.71,72 Regardless, both approaches are in harmony. When directly compared, the single 10-color tube had a slight reduction in total cell number, although there was no apparent loss of either normal or abnormal PCs. To perform a viability assessment and to establish background autofluorescent levels, the authors’ laboratory utilizes a third tube, which contains a Fixable Live Dead reagent with anti-CD38, anti-CD138, and anti-CD45.
Table 2.
Composition of panel used for the characterization of plasma cells
| Antigen | Fluorochrome | Clone | Source |
|---|---|---|---|
| (a) Tube 1 | |||
| CD138 | BV421 | MI15 | Becton Dickinson (San Jose, CA) |
| CD27 | BV510 | O323 | BioLegend (San Diego, CA) |
| CD38 | FITC | T16 | Beckman Coulter (Brea, CA) |
| CD56 | PE | C5.9 | Cytognos (Salamanca, Spain) |
| CD45 | PcPCy5.5 | HI30 | BioLegend (San Diego, CA) |
| CD19 | PECy7 | J3-119 | Beckman Coulter (Brea, CA) |
| CD117 | APC | 104D2 | Becton Dickinson (San Jose, CA) |
| CD81 | APC-C750 | M38 | Cytognos (Salamanca, CA) |
| (B) Tube 2 | |||
| CD138 | BV421 | MI15 | Becton Dickinson (San Jose, CA) |
| CD27 | BV510 | O323 | BioLegend (San Diego, CA) |
| CD38 | FITC | T16 | Beckman Coulter (Brea, CA) |
| CD56 | PE | C5.9 | Cytognos (Salamanca, Spain) |
| CD45 | PcPCy5.5 | HI30 | BioLegend (San Diego, CA) |
| CD19 | PECy7 | J3-119 | Beckman Coulter (Brea, CA) |
| cKappa | APC | poly | Dako (Carpinteria, CA) |
| cLambda | APC-C750 | poly | Cytognos (Salamanca, Spain) |
| (C) Tube 3 | |||
| CD138 | BV421 | MI15 | Becton Dickinson (San Jose, CA) |
| Live Dead Aqua | – | – | Thermo Fisher Scientific (Grand Island, NY) |
| CD38 | FITC | T16 | Beckman Coulter (Brea, CA) |
| CD45 | PcPCy5.5 | H130 | BioLegend (San Diego, CA) |
Abbreviations: APC, Allophycocyanin; APC-C750, Allophycyanin Tandem 750; BV421, Brilliant Violet 421; BV510, Brilliant Violet 510; FITC, Fluorescein Isothiocyanate; PerCPCy5.5, Peridinin Chlorophyll Cyanine 5.5; PE, Phycoerythrin; PECy7, Phycoerythrin Cyanine 7.
The significance of CD19, CD20, CD27, CD28, CD38, CD45, CD56, CD81, CD117, and CD138 as they relate to PCDs are described below:
CD19 is normally expressed at all stages of B cell development ranging from pro-B to PCs. In patients with MGUS, the expression of CD19 on PCs is normally high, while PCs in multiple myeloma are most often either negative or dim for CD19 expression.24 The decreased expression of CD19 is usually caused by the altered expression of PAX-5.63 The lack of expression of CD19 has been a facilitating feature when identifying malignant PCs as greater than 95% of abnormal PCs are negative for CD19 whereas it is noteworthy that CD19 is positively expressed only by approximately 70% of normal PCs.62
CD20 expression occurs later in early B cell maturation while CD34 is concurrently downregulated. As B cells differentiate into normal PC, they lose CD20 but it is expressed on roughly one-third of abnormal PCs. The CD20 antigen has been associated with shorter patient survival however, the prognostic significance of CD20 expression is unclear.73 CD20 is associated with a mature morphology and often with a small PC, lymphoplasmacytic morphology with the t(11:14) translocation.69
CD27 plays a role in helping B cells differentiate into PCs. In the B cell lineage, CD27 is considered as a memory marker because its expression is limited to germinal center cells, memory B cells and PCs.69 In MGUS, the expression of CD27 on PCs is usually high. In patients with multiple myeloma, the expression of CD27 on abnormal PCs is often dimmer than normal PCs. When CD27 is expressed at normal intensities on abnormal PCs, it is usually correlated with a better prognosis and loss of CD27 expression has been associated with shorter PFS and OS.74,75
CD28 is normally not expressed on PCs and rarely in patients with MGUS, but in one third of multiple myeloma patients it is found on their abnormal PCs. The expression of CD28 is associated with an aggressive myeloma phenotype. When used concurrently with CD117, it has been found that CD28 can be used to stratify multiple myeloma cases into 3 risk categories49: (i) good prognosis CD28−/CD117+, (ii) intermediate prognosis CD28−/CD117−, and (iii) poor prognosis CD28+/CD117−. As the disease progresses, CD28 expression increases and it is often associated with relapse.76 Interestingly, while the expression of CD28 can induce antigen-independent T cell activation, it does not induce proliferation in neoplastic PCs; instead, it is speculated to act as a pro-survival signal to myeloma cells by ligating CD80/CD86 on myeloid DC facilitating the secretion of supportive cytokines such as IL-6 and IL-8,69,77,78 as well as the immunosuppressive enzyme IDO.79
CD38 is present on many cells including B cell progenitors and germinal center B cells.80,81 Both abnormal and normal PCs brightly express CD38, with neoplastic cells often expressing it at a slightly lower intensity than normal PC.82 Antibodies to CD38 are currently being used therapeutically for the treatment of multiple myeloma which can block the binding of many mAb clones used in flow cytometry, causing the myeloma in samples from these patients to appear CD38 negative.
CD45 is a well-known marker that is expressed at variable levels on leukocyte subsets. Normal PCs while generally positive can also be negative for CD45.82 In patients with MGUS, heterogeneous distribution of CD45+ normal and CD45− abnormal PCs in bone marrow have been observed.83 In multiple myeloma patients, however, the expression of CD45 on neoplastic PCs is negative to dim in the majority of cases. The expression of CD45 with other markers such as CD19 or CD27 allows further refinement of the PC identification process.84
CD56, also known as the neural cell adhesion molecule (NCAM), is a common marker used for the identification of natural killer (NK) and NKT cells as well as a valuable marker that can be utilized to define an abnormal phenotype of neoplastic PCs. It is an adhesion molecule involved in anchorage of myelomatous PC to the bone marrow stroma and its absence of expression is associated in some studies with extramedullary spreading and more aggressive disease.49 Positive CD56 expression can be found on PCs in the bone marrow in the majority of multiple myeloma patients. Myeloma cells circulating into the peripheral blood usually lack CD56, whereas myeloma cells located in pleural or ascetic effusions are usually CD56+. The down-modulation of CD56 by a formerly positive myeloma may indicate extramedullary diffusion of the disease,85 whereas its initial absence has been associated with the presence of extra-marrow involvement, a tendency to leukemization, a lower frequency of osteolytic lesions, and plasmablastic morphology.86–88 When combined with CD19, the expression of CD56 can provide substantial diagnostic value.69 Lack of CD19 expression and/or strong CD56 expression differentiates neoplastic PCs from their normal counterpart in most cases. However, it should be noted that with improving outcomes and increased sensitivity of flow-MRD methods, it is now well-established that a subset of normal PCs can be CD19− and/or CD56+.69
CD81 is a tetraspanin family member broadly expressed on hematopoietic cells with the exception of erythrocytes, platelets, and neutrophils.89 It is expressed on all B cells where it forms a multi-molecular complex with CD19 and CD21, which together are involved in signaling of B cell maturation and antibody production. The expression of CD81 on immature B cells is brighter than on mature B cells. In approximately half of the patients with multiple myeloma, the expression of CD81 is dim to negative in comparison with normal PCs.37 It has been found that patients with CD81 expression on myeloma cells have shorter progression-free survival (PFS) and overall survival (OS) when compared to those who do not express it.90
CD117, also known as the proto-oncogene c-Kit, is commonly found on myeloid, erythroid and megakaryocytic progenitors, and mast cells, whereas normal PCs are CD117−.91–93 In MGUS, 50% of the cases express CD117; in multiple myeloma, about one third of patients are CD117+.93 The expression of CD117 on PCs predicted better outcome and it can be used in combination with CD28 to allow for risk stratification (see CD28 above).
CD138, also known as syndecan-1, is a transmembrane heparin sulfate proteoglycan that is expressed during PC stage of B cell maturation. CD138 functions as the alpha receptor for collagen, fibronectin and thrombospondin.94 In addition, CD138 can also be shed into the extracellular matrix to trap growth-promoting and proangiogenic cytokines.95,96 Both abnormal and normal PC in MGUS and multiple myeloma express high levels of CD138.82 Currently, CD138 serves as a universal marker for PCs detection and it can provide a basis to quantify or assess disease burden. The staining intensity of CD138 mAb, may be attenuated when samples are exposed to sodium heparin, stored for long periods, refrigerated, or frozen.95,97,98
Performance Evaluation of Fluorochrome-conjugated mAb
When selecting the mAbs to be incorporated into a multiple myeloma panel, the cytometrist is presented with a variety of choices in clones, fluorochromes, and sources. Table 2 presents a list of markers found to give the best resolution of PCs from other leukocytes with excellent discrimination between normal and abnormal PCs. This table should be used as a general guide for laboratories designing their own panels to diagnose and monitor PCDs. The strategies used to assess the performance of individual mAbs such as fluorescence intensity, percentage of negative versus positive cells, signal-to-noise ratio, and stain index will vary from lab-to-lab, however we have found signal-to-noise and Stain Index to be the most informative. Building on the experience of the EuroFlow group,70 we evaluated several additional clones and fluorochromes to derive the panel described in Table 2. The most significant adjustment was to replace CD138 Pac Blue with BV421 and CD27 Pac Orange with BV510, changes that were also adopted by the EuroFlow group. We also found CD38 clone T16 from Beckman Coulter gave a slightly higher signal-to-noise ratio. The detection of CD38 is becoming problematic with the introduction and clinical use of daratumumab, a humanized IgG1 kappa mAb targeting CD38 and shown to significantly improve outcomes in refractory multiple myeloma. Daratumumab blocks the binding of most anti-CD38 mAbs used in flow cytometry interfering with the assessment of PCDs. To date the only partial solution found has been to use a multi-epitope anti-CD38 containing a cocktail of mAbs. Other approaches under investigation have been to look at substituting or combining mAbs to CD38 with mAb to CD54, CD229, and CD319.54
In panel development, to confirm that the fluorochromes selected for each mAb do not have significant signal broadening or cross-laser excitation artifact that would compromise the resolution of important populations we use a fluorescence minus one (FMO) approach.99 This consists of an mAb cocktail in which the mAbs included are identical to those in the test article, except for the exclusion of the one fluorochrome that is being controlled. To completely validate the PCD tubes, we control for all fluorochromes. In the 8-color cocktails shown in Table 2, we set up 9 tubes for each cocktail; the first tube containing all the mAbs except for the FITC conjugated mAb, the second minus the PE conjugated mAb, continuing this process for all fluorochromes and finishing with a tube containing all the mAbs. Each FMO is compared to the fully-stained panel ensuring that all populations of interest are well separated.
The panel in Table 2 represent a consensus approach which can be readily adopted.66,70 Labs desiring to modify the consensus approach should demonstrate that their panel is comparable or better than the consensus panel. A strategy to determine the performance of an alternative mAb is to evaluate its staining resolution in conjunction with all the other mAbs used in the panel, rather than using the mAb alone This is best accomplished by defining a negative and positively stained population and calculating their signal-to-noise ratio as first described by Rawston and colleagues100. As an example (Fig. 1), when evaluating the performance of CD81, the signal-to-noise ratio may be calculated by dividing the fluorescence intensity of an internal positive population (e.g. T lymphocytes defined as CD19−/CD56−/CD45br/SSClo) by the fluorescence intensity of an internal negative population (e.g. granulocytes defined as CD45dim/SSChi). This approach can be performed on both peripheral blood and bone marrow samples, but it should be noted that the specimen employed should mimic the condition and type of the specimens to be tested. In Table 3 and Table 4, the data for all mAbs from our analysis of 10 bone marrow samples with no hematological disease is provided. Laboratories initiating a validation of their PCD panel may use this table as a guideline to measure the performance of each mAb in their tube(s). CD138 was an exception. Due to the paucity of CD138 positive cells in normal bone marrow we used a CD-CHEX CD103™ PLUS control reagent (Streck, Omaha, NE) which contains a CD138 positive control population and compared its fluorescence intensity to B cells defines as CD19+/CD45br/SSClo.
Figure 1. Signal-to-noise ratio performance assessment strategy for mAbs.
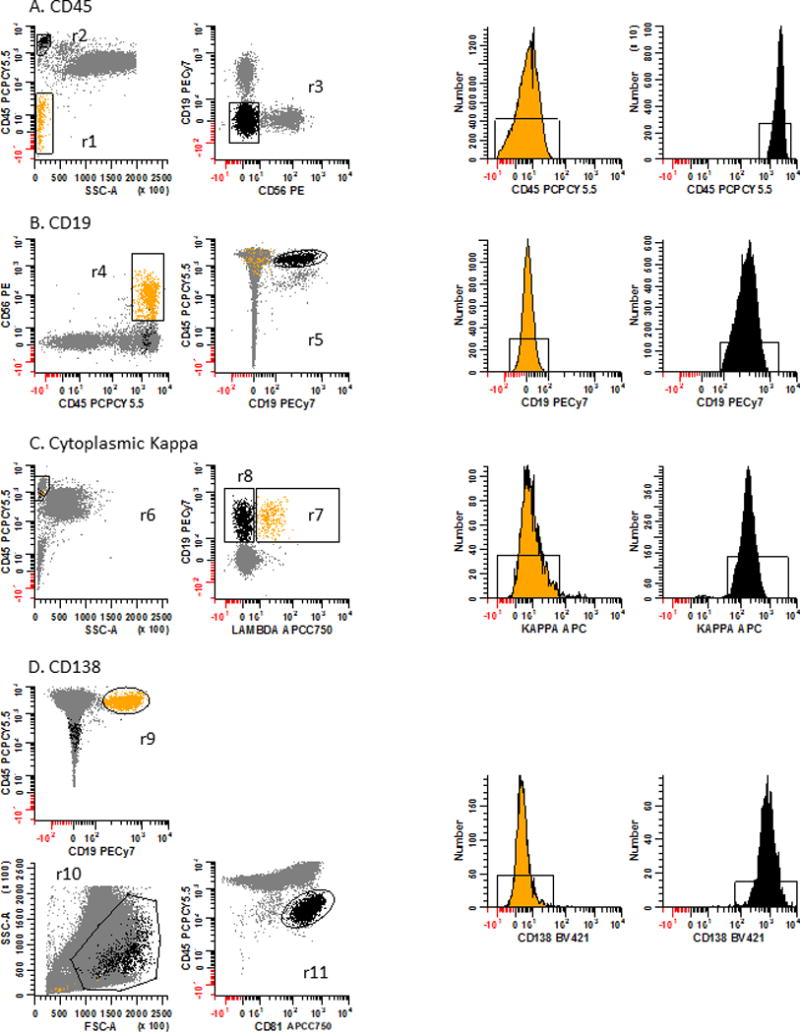
The strategy to assess the performance of each mAb in the PCD panel relies on defining a negatively and positively staining population for each mAb. These populations are defined in Table 3. Next, the median fluorescence intensity (MFI) is determined for each population and the signal-to-noise ratio calculated by dividing the positive population’s MFI by the negative population’s MFI. To qualify as an acceptable mAb, the calculated signal-to-noise value should be greater than the recommend value in Table 4. Panel 1A: For CD45, erythroid precursors are used as the negative population defined as SSClo/CD45− events (r1: yellow dots and corresponding histogram) and T cells as the positive population defined as CD45br/SSClo (r2) and CD19−/CD56− events (r3: black dots and corresponding histogram). Panel 1B: For CD19, NK cells are used as the negative population defined as CD45br/CD56+ events (r4: yellow dots and corresponding histogram) and B cells as the positive population defined as CD19+/CD45br events (r5: black dots and corresponding histogram). Panel 1C: For cKappa light chain, cLambda light chain+ B cells are used as the negative population defined as CD45br/SSC-Alo (r6) and CD19+/cLambda light chain+ events (r7: yellow dots and corresponding histogram) and cLambda light chain− cells as the positive population defined as CD45br/SSC-Alo (r6) and CD19+/cLambda light chain− events (r8: black dots and corresponding histogram). Panel 1D: For CD138 a procedural control for immunophenotyping, CD-Chex CD103™ Plus cells which contains a CD138 positive population are spiked into a bone marrow sample. B cells are used as the negative population defined as CD45br/CD19+ events (r9: yellow dots and corresponding histogram) and spiked control cells as the positive population defined by FSC-A and SSC-A (r10) and as CD45dim/CD81+ events (r11: black dots and corresponding histogram). Note these data are all gated on R1 & R2 & R3, ‘Total Leukocytes’ as defined in Fig. 2. In data not shown, for the CD19 negative and positive populations, and CD138 negative population a FSC-A vs SSC-A plot was used to define lymphocytes.
Table 3.
Determination of internal negative and positive populations using bone marrow sample for the selection of optimal monoclonal antibody combination
| Tested Markers | Negative Population | Positive Population | ||
|---|---|---|---|---|
| Phenotype | Generic Name | Phenotype | Generic Name | |
| CD45 | CD45−/SSClo | Erythroid Cells | CD19−/CD56−/CD45br/SSClo | T Cells |
| CD19 | CD45br/CD56+/SSClo | NK Cells | CD19+/CD45br/SSClo | B Cells |
| CD27 | CD117+/CD45dim | Mast Cells | CD19−/CD56−/CD45br/SSClo | T Cells |
| CD81 | CD45dim/SSChi | Granulocytes | CD19−/CD56−/CD45br/SSClo | T Cells |
| CD56 | CD19+/CD56−/CD45br/SSClo | B Cells | CD19−/CD56+/CD45br/SSClo | NK Cells |
| CD117 | CD19−/CD56−/CD45br/SSClo | T Cells | CD45dim/CD117+ | Mast Cells |
| CD138 | CD19+/CD45br/SSClo | B Cells | CD45dim/CD81+ | Spiked Controla |
| CD38 | CD19+/CD81− | Mature B | CD19+/CD81+ | B-progenitors |
| cKappa | CD19+/CD45br/cLambda+/SSClo | cLambda+ B Cells | CD19+/CD45br/cLambda−/SSClo | Lambda− B Cells |
| cLambda | CD19+/CD45br/cKappa+/SSClo | cKappa+ B cells | CD19+/CD45br/cKappa−/SSClo | Kappa− B cells |
A procedural control for immunophenotyping, CD-Chex CD103™ Plus (Streck, Omaha, NE) which contains CD138, was spiked into the bone marrow sample and used as the positive population
Table 4.
Evaluation of staining performance of monoclonal antibody in the context of multicolor analysis
| Tested Markers | Evaluated Sample, na | Signal-to-Noise | ||
|---|---|---|---|---|
| Mean ± SD | Range | Recommended | ||
| CD45 | 10 | 323.7 ± 101 | 226 – 573.2 | >100 |
| CD19 | 10 | 134.5 ± 56.6 | 37.6 – 215.3 | >10 |
| CD27 | 10 | 20.7 ± 6.9 | 10.3 – 32.1 | >5 |
| CD81 | 10 | 25.3 ± 12.2 | 11.8 – 49.4 | >5 |
| CD56 | 10 | 50.1 ± 25.1 | 32 – 114.9 | >10 |
| CD117 | 10 | 46.1 ± 31 | 15.2 – 99.9 | >10 |
| CD138 | 10 | 83.9 ± 46.3 | 30.9 – 144.3 | >10 |
| CD38 | 10 | 19.5 ± 9.8 | 4.8 – 36.0 | >3 |
| cKappa | 10 | 18 ± 13.2 | 4.9 – 45.2 | >3 |
| cLambda | 10 | 13.5 ± 5.4 | 5.8 – 25.4 | >3 |
Ten random patient bone marrow samples received for flow cytometric evaluation which had no evidence of hematological disease by flow cytometry or histopathological assessment
Sample Storage and Quality
Bone marrow aspirate, blood, and fine needle aspirate (FNA) are appropriate specimens for the routine detection of PCs. Multiple myeloma MRD testing has been established on bone marrow aspirates. The detection and assessment of abnormal PCs in peripheral blood at initial diagnosis and 2 weeks prior to an autologous stem cell collection may become a routine test as studies have demonstrated these can be clinically relevant.101,102 Regardless of the types of specimen received for testing, the importance of having good quality samples prior to processing cannot be overemphasized.
As the age of a specimen is a critical factor for any test carried out using flow cytometry, the sample tube should be labeled with the collection date and time. It is recommended that blood and bone marrow specimen to be stored at room temperature.103 Standard practice is to process specimens as soon as possible and preferably within 48 hours of collection. Samples may be collected in either an EDTA or sodium heparin vacutainer tube, though EDTA is preferable due to attenuation of CD138 signal when stored in heparin. Tissue samples may be placed in a small volume of RPMI as a holding medium and should be processed immediately.
A compromised specimen such as one that is excessively hemodilute, partially clotted, too warm or cold to the touch, or displays sign of hemolysis will not yield the same results as when optimal specimens are processed. For this reason, the quality and integrity of a bone marrow specimen should be qualitatively assessed upon receipt via physical observations. Hemodilution can be determined by confirming the presence of normal PCs, B cell progenitors, mast cells, and nucleated RBC in the bone marrow specimen.65 The viability and the presence of PCs in a specimen can be checked using the staining Tube 3. Samples with less than 85% viability are deemed suboptimal for flow cytometry testing. In such cases, it is recommended that each report include a statement about sample quality including a notation of hemodilution and viability. In the event an irreplaceable and rare specimen such as bone marrow is compromised due to unforeseen circumstances, it should not be rejected for flow cytometry testing. Instead a notation must be included clearly stating that the result may have been compromised and why.
Staining Procedure to Label Leukocyte Subsets
Depending on the level of sensitivity required, there are two staining procedures that can be used for the assessment of PCDs, these are (i) the routine PCD test (e.g. low sensitivity assay) and (ii) the MRD high sensitivity test. The reason for two different staining procedures is due to the difference in frequency of abnormal cells that are present between overt disease (e.g. at the time of diagnosis) and rare cell detection (e.g. after the patients have undergone treatment). The routine test is less labor intensive but lacks the sensitivity of the MRD assay. This section will discuss the procedure for routine PCD testing. High sensitivity MRD testing will be described in the section below entitled “Minimal Residual Disease Testing”. In both procedures, the panel described in Table 2 is applicable. For routine testing, a less comprehensive panel consisting of at least CD38, CD138, CD45, CD19, and CD56 can be employed with secondary mAbs to CD27, CD117, cytoplasmic light chain reagents, and other mAbs added as required.
Details of our routine testing procedures can be found elsewhere.104 Briefly, bone marrow samples received for PCD testing are filtered through a 70 μm cell strainer (Corning, Manassas, VA) to exclude spicules from the samples. An absolute cell count on blood, bone marrow and FNA is performed using an automated cell counter (e.g. Sysmex XS-1000i, Lincolnshire, IL). The specimen is then transferred to a 15 mL conical tube and washed twice using flowcytometry (FCM) buffer (containing 0.5% bovine serum albumin, 0.1% sodium azide, 0.04 g/L and sodium EDTA in PBS) to remove plasma immunoglobulins. A second cell count is performed and approximately 1 × 106 cells are transferred to three labeled 12 × 75 mm polystyrene tubes. Cocktails of fluorochrome-conjugated mAbs are added to each tube according to Table 2 and the samples are incubated for 30 minutes at room temperate in the dark. After surface labeling, 2 mL of FACS™ Lysing Solution (BD Biosciences, San Jose, CA) is added to each tube and allowed to stand at room temperature for 10 minutes. All the tubes are then centrifuged at 540 × g for 5 minutes and washed once using FCM buffer.
Tubes to be labeled with surface mAbs only (e.g. Tube 1 and Tube 3) are resuspended in 500 μL of 0.5% methanol free formaldehyde (Polysciences, Warrington, PA) in PBS. For intracellular staining with anti-kappa and anti-lambda reagents (e.g. Tube 2), following the first wash in FCM buffer, the cells are resuspended in 300 μL of 2% formaldehyde and incubated in the dark for 20 minutes at room temperature. Fixed cells are then washed with 3 mL of FCM buffer and resuspended in 50 μL of Permeabilization Medium B (Thermo Fisher Scientific, Carlsbad, CA). Saturating amounts of kappa and lambda antibodies are added and the sample is incubated in the dark for 30 minutes at room temperature. Finally, this tube is washed once using FCM buffer and resuspended in 500 μL of FCM buffer or 0.5% formaldehyde for data acquisition.
Data Analysis and Gating Strategy
The analysis approach recommended by the ICCS Consensus Group utilizes the expression of CD38, CD138, CD45, and light scatter characteristics to define PCs, while excluding contaminant lymphocytes, doublets, and debris. Such ‘fit-for-purpose’ gating strategy can be easily adapted to any multiple myeloma analysis, regardless of the instrument, panel, or type of flow cytometric analysis software package used. The recommended gating steps for an inclusive identification, enumeration, and characterization of PCs are as follows:
I. Identification of total plasma cells
Place a rectangular region (R1) on a bivariate plot (A) of Time vs. SSC-A to circumscribe all events collected in continuity (Fig. 2). This dot plot can be used to assess the chronologic heterogeneity of the acquisition by eliminating any invalid events such as air bubbles which may occur during the run.
On a bivariate plot (B) of FSC-A vs. FSC-H, create a rectangular region (R2) to include the singlet cell population. Doublets will have more FSC-A than FSC-H. Gate this bivariate histogram on (R1).
On a bivariate plot (C) of FSC-A vs. SSC-A, create an irregular region (R3) to circumscribe the cell population of interest and exclude aggregates, debris, and dead or apoptotic events. Gate this bivariate histogram on (R1 & R2). Cells gated on (R1 & R2 & R3) define ‘Total Leukocytes’.
Create 3 separate bivariate plots (D: CD138 vs. CD38), (E: CD45 vs. CD38), and (F: CD45 vs. CD138). Gate each of these bivariate plots on (R1 & R2 & R3). An irregular region (R4) is drawn on plot D circumscribing the CD138+/CD38+ events; another irregular region (R5) is drawn on plot E circumscribing the CD45+/-/CD38+ events; a third irregular region (R6) is drawn on plot F circumscribing the CD45+/-/CD138+ events. CD45 is helpful for gating but should not be used as a marker for cell exclusion since both normal and clonal PCs can exhibit variable CD45 expression. When setting regions consider that CD138 expression may be attenuated and/or that PCs from patient on anti-CD38 immunotherapy may appear to be CD38 negative by flow cytometry. Since CD45 on PCs is generally dim to negative, evaluating it in combination with CD38 and CD138 can help in these situations. The combined Boolean gate definition (R1 & R2 & R3 & R4 & R5 & R6) defines both normal and neoplastic PCs (‘Total PCs’) that are present in the sample for subsequent analysis.
Figure 2. Gating strategy used for the identification of normal and abnormal plasma cells.
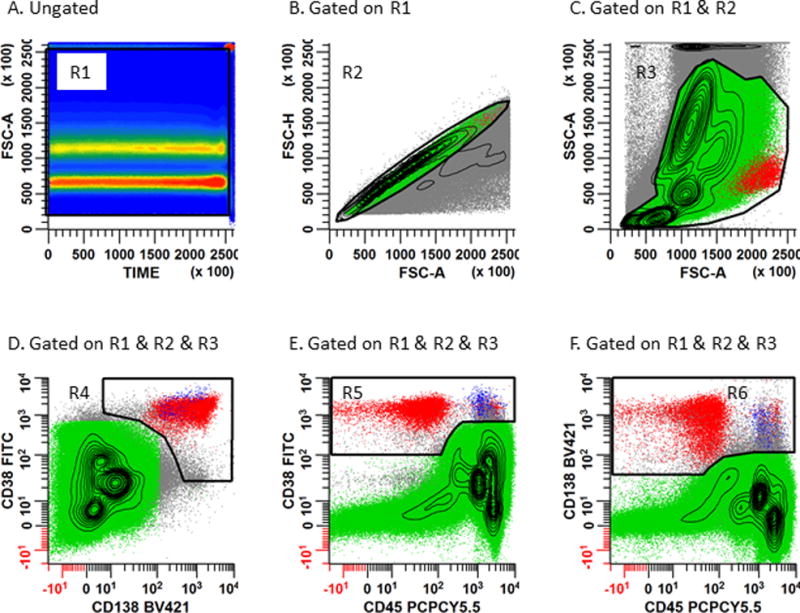
Panel 2A: A rectangular region (R1) is placed on the bivariate plot of Time vs. SSC-A to circumscribe all events collected in continuity. This dot plot can be used to assess the chronologic heterogeneity of the acquisition by eliminating any invalid events such as air bubbles which occur during the run. Panel 2B: Serial gating is performed by applying the region R1 to a bivariate plot of FSC-A vs. FSC-H. A rhomboid region (R2) is then created to include the singlet cell population. Caution should be exercised not to exclude hyperdiploid or tetraploid plasma cells which may exhibit aberrantly high light scatter characteristics. Panel 2C: Gate a bivariate plot of FSC-A vs. SSC-A on (R1 and R2). An irregular region (R3) is created to circumscribe the cell population of interest and exclude aggregated events, debris, and dead and apoptotic events. Create 3 separate bivariate plots (Panel 2D: CD138 vs. CD38), (Panel 2E: CD45 vs. CD38), and (Panel 2F: CD45 vs. CD138). Gate each of these bivariate plots on ‘Total Leukocytes’ (R1 & R2 & R3). An irregular region (R4) is drawn on Panel 2D circumscribing the CD138+/CD38+ events; another irregular region (R5) is drawn on Panel 2E circumscribing the CD45+/-/CD38+ events; a third irregular region (R6) is drawn on Panel 2F circumscribing the CD45+/-/CD138+ events. CD45 is helpful for defining PCs and identifying any CD38− or CD138− PC populations. The Boolean gate (R1 & R2 & R3 & R4 & R5 & R6) defines both normal (blue) and abnormal (red) PCs for subsequent immunophenotyping.
II. Phenotypic characterization of normal and abnormal plasma cells
As PCD is a heterogeneous disease where neoplastic PCs can express variable antigen levels, defining abnormal PCs can be challenging (Fig. 3). Currently, there is not a single marker that can be used alone to reliably distinguish abnormal PCs from normal PCs. However, provided that a sufficient number of events are acquired, the combination of markers used in both Tube 1 and Tube 2 are capable of resolving abnormal PCs in a high percentage of cases. For this reason, all permutation of bivariate plots should be created and gated on ‘Total PCs’, which is identified above as cells circumscribed using the aforementioned Boolean gate definition (R1 & R2 & R3 & R4 & R5 & R6).
Figure 3. Immunophenotypic profiles of normal and abnormal PCs.
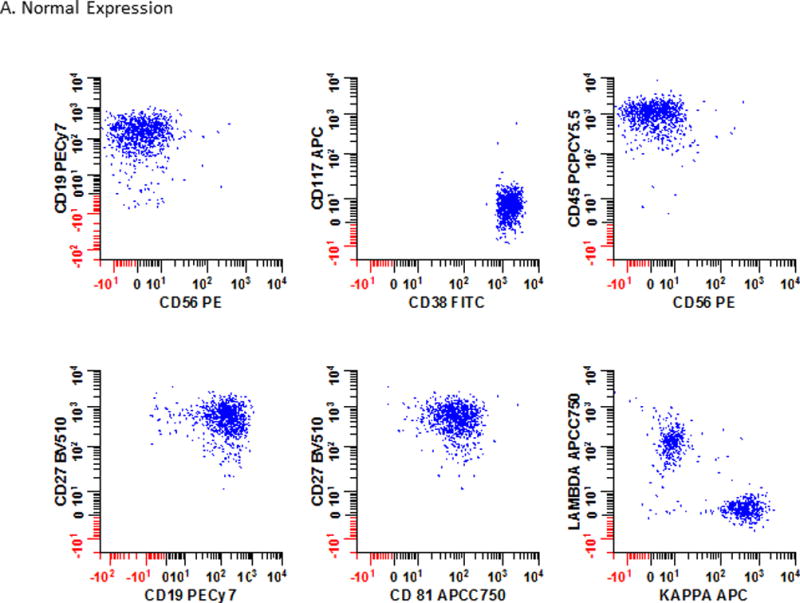
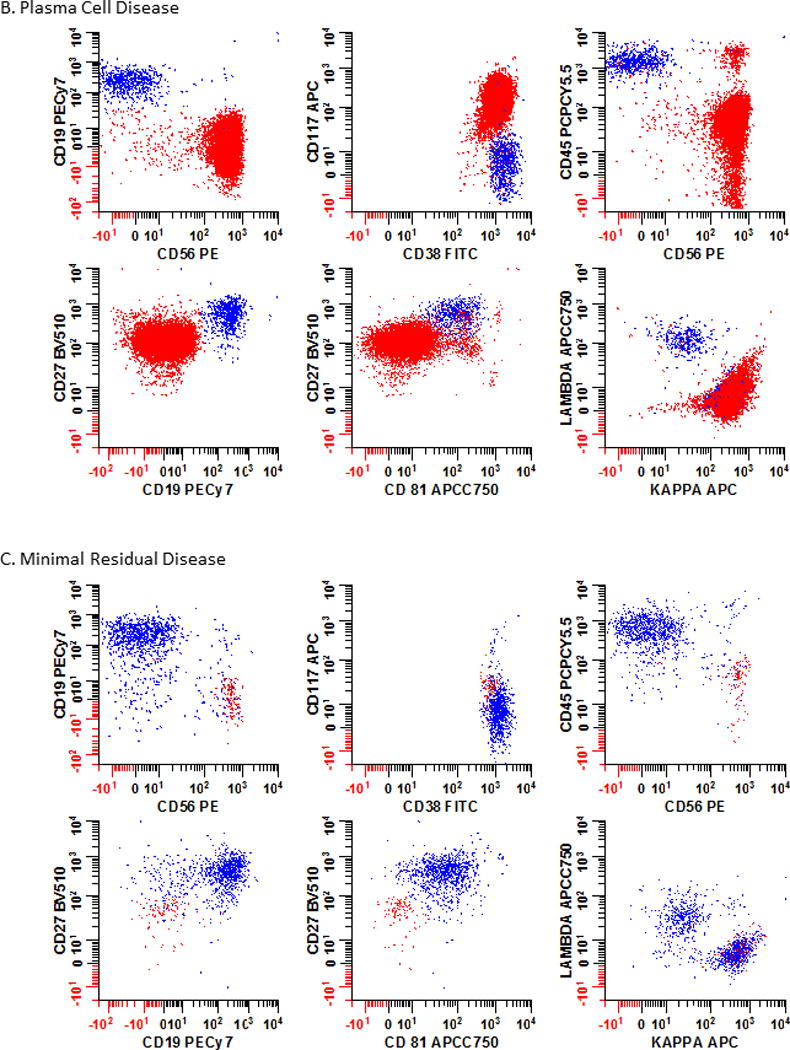
The immunophenotypic profiles of bone marrow from a patient (Panel 3A) with no obvious hematological disease at testing; (Panel 3B) a patient with PCD; and (Panel 3C) a patient with multiple myeloma MRD are shown. As PCD is a heterogeneous disease, no single marker can be reliably used to identify all abnormal cell populations. Instead the interpretation is based on all the markers included in the analysis. In this example, 6 different bivariate plots that were each separately gated on plasma cells using the strategy defined in Fig. 2 are shown. Blue: normal plasma cells; Red: neoplastic plasma cells.
An individual is considered to have a PCD if the PCs express two or more aberrant cell markers, which are often some combination of CD38dim, CD45- to dim, CD19−, CD56+, CD27dim, CD81dim, CD117+ and clonally restricted. Non-neoplastic PCs exhibit heterogeneous expression of CD45 and CD19, are mostly negative for CD20 and CD117, and invariably show homogeneously bright expression of CD81. Moreover, dim or absent CD81 expression is only observed in abnormal PCs, with 95% sensitivity and 100% specificity. The expression of CD56 and CD27 is observed in a subset of non-neoplastic PCs (between 5 - 20% of all PCs) with the latter more frequently expressed in post-treatment bone marrow samples. Given the nature of clonal PCs, antigen expression is often more uniform in myeloma cells rather than present across a heterogeneous spectrum as is seen in normal PCs. Neoplastic PCs will often show additional aberrancies, such as CD117+ or CD27dim expression; and these aberrancies will be co-expressed within the same cell population, whereas immunophenotypic variations in normal PCs tend to be heterogeneous and/or distributed among different subsets. Finally, provided a sufficient number of PCs are acquired, analysis of cytoplasmic light chain expression within subsets showing a myeloma-like aberrant phenotypes (e.g. CD19− or CD56+ PCs), while not routinely required, can be a valuable additional step in confirming the diagnosis.
Representative cases to compare and contrast the immunophenotypic profile of PCs in bone marrow samples are illustrated in Fig. 3. Data from a patient with no hematological disease is presented in Fig. 3A. Note the positive expression of CD19, CD38, CD27, and CD81. The PCs are also polytypic for cytoplasmic light chains. In Fig. 3B, an obvious population of abnormal PCs (red) expresses CD56 and CD117; are dim for CD45 and CD27; negative for CD19 and CD81; and are cytoplasmic kappa light chain restricted. In Fig. 3C, an MRD population (red) is intermixed with normal PCs (blue) with a phenotype similar to that seem in Fig. 3B but note the slightly dimmer expression of CD38 on the abnormal cells.
III. Quality assessment of bone marrow aspirates
Tube 1 in the panel described here is designed to assess if the patient bone marrow aspirate is hemodilute by evaluating for the presence of erythroid precursors, mast cells, and hematogones (Fig. 4). If abnormal PCs are found or these populations are present in the aspirate, then the sample should be reported as adequately representative of bone marrow based. Conversely, if no PCs are found and these populations are absent or reduced then the sample should be reported out as hemodilute. The following gating parameters are used in the identification of mast cells, hematogones, and erythroblasts.
Figure 4. Presence of mast cells, hematogones and erythroid precursors can be used for quality assessment of bone marrow aspirates.
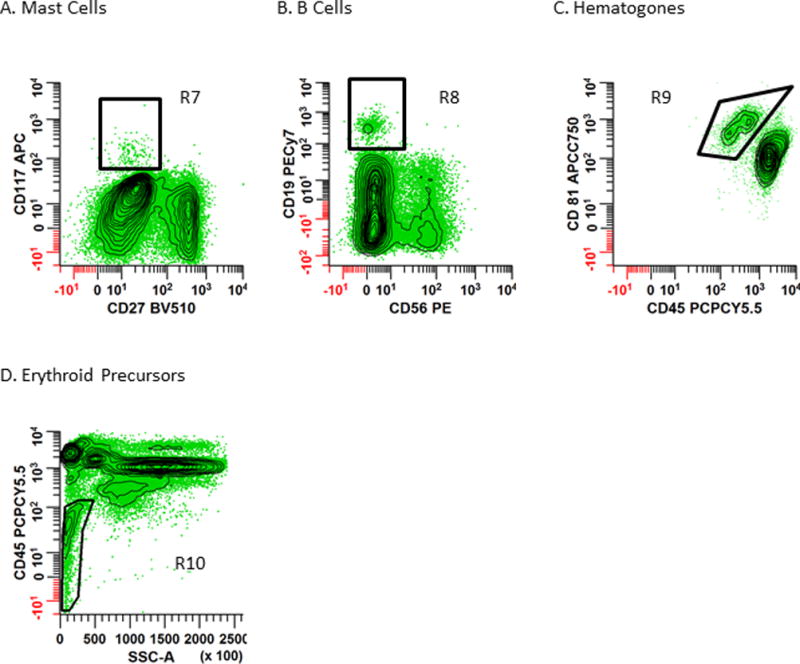
Panel 4A: Mast cells are identified by drawing a rectangular region (R7) to circumscribe CD27−/CD117+ events. Panel 4B – C: B cells are first identified by creating a region (R8) on a bivariate plot of CD56 vs. CD19 to circumscribe CD56−/CD19+ cells. Then a bivariate plot of CD45 vs. CD81, gated on R8 and Total Leukocytes is used to define mature and immature B cells. Hematogones (immature B cells) are defined by R9 as the CD45dim/CD81br population. Panel D: Erythroid precursors are defined by R10 on a bivariate plot of SSC-A vs. CD45 which circumscribes the CD45−/dim/SSClo population. Note: these histograms are all gated on ‘Total Leukocytes’ identified using the Boolean strategy (R1 & R2 & R3) defined in Fig. 2.
Using the bivariate plots shown in Fig. 2, total leukocytes are identified using the Boolean strategy R1 & R2 & R3.
As shown in Fig. 4A, draw a rectangular region (R7) to circumscribe the CD27−/CD117+ events to identify the mast cells.
To define hematogones, B cells are first identified by creating a region (R8) on a bivariate plot of CD56 vs. CD19 to circumscribe CD56−/CD19+ cells. This region (R8) is then applied to a bivariate plot of CD45 vs. CD81, and the CD45dim/CD81br cells (R9) identify the hematogones.
As shown in Fig. 4D, erythroid precursors (R10) are identified as CD45dim/SSC-Alo events.
MINIMAL RESIDUAL DISEASE TESTING
In the routine diagnoses of PCDs the presence of monoclonal proteins are important for the evaluation of disease. They are measured in the urine and serum and can be identified by immunofixation electrophoresis (IFE) and quantified by protein electrophoresis (urine protein electrophoresis or serum protein electrophoresis). Changes in monoclonal protein concentrations are used to determine response to therapy or disease progression. Other assessments include the morphological evaluation of PC concentration in the bone marrow and whether there is evidence of new lytic bone lesions or plasmacytomas.105 A complete response (CR) prior to the use of novel agents was uncommon and responses were divided into several disease states, including stable, minimal, partial, very good partial, and complete responses. Improvements in therapy and methodologies to detect monoclonal proteins allows better stratification of good responses. Patients fulfilling the definition of CR may still have disease detected by IFE, which is defined as a near CR (nCR).105,106 Similarly, utilization of sensitive sFLC assays may detect remaining imbalances of kappa and lambda concentrations suggestive of persistent disease in patients who were otherwise deemed to be in CR.107 The rationale for adding the sFLC ratio to the multiple myeloma response criteria was to provide a more sensitive and precise indication of CR for use in comparative clinical trials by enabling the detection of small quantities of abnormal proteins in patients with little or no detectable monoclonal protein on serum protein electrophoresis and IFE. The International Myeloma Working Group defined a stringent CR as a patient who satisfied the criteria for a CR and was negative for sFLC.108
Achievement of stringent complete response (sCR) is associated with a significantly longer OS and PFS than CR or nCR are regardless of the therapy utilized.109–114 With novel therapeutics such as immunomodulatory drugs, proteasome inhibitors, and immunotherapy evolving at an unprecedented rate there is a need for even more sensitive methods to evaluation response to therapy. The development and testing of new therapeutics have been limited because almost all patients achieve an sCR and consequently randomized Phase 3 clinical trials take years to show benefits since PFS and OS are used as study endpoints.55,115–118 Due to the long latency between drug development and approval to be considered as a therapeutic option, the measurement of MRD by MFC as an independent method to predict PFS and OS for patients diagnosed with PCD has been championed.119,120 All flow cytometric studies to date have strongly correlated with PFS and OS at the Day 100 time point, reducing the time required to reach meaningful clinical outcomes from years to months.121–124
There are currently 3 methods for detecting MRD that focus on the malignant multiple myeloma clone: immunophenotyping by flow cytometry, allele-specific oligonucleotide real time quantitative polymerase chain reaction (ASO-qPCR), and next-generation sequencing (NGS). Flow cytometry and ASO-qPCR have about the same sensitivity, while NGS is about 10 times more sensitive. Whereas flow cytometry is applicable for virtually all multiple myeloma patients (≥95% of cases), ASO-qPCR and NGS have a more restricted applicability (50 – 90% of cases). This is mainly due to the high number of somatic hypermutations in the complementarity-determining regions (CDR) of the B cell immunoglobulin gene which cause variable levels of primer annealing with unpredictable amplification and quantitation of results.
Two large studies have reported the correlation of immunophenotyping by flow cytometry with PFS and OS in uniformly treated patients. Paiva and colleagues124 assessed MRD status at Day 100 by MFC in 295 newly diagnosed multiple myeloma patients treated with induction therapy followed by autologous hematopoietic stem cell transplant (aHSCT) in the GEM2000 protocol. Compared to MRD positive patients at Day 100, those with MRD negative status had longer PFS (median 71 vs. 37 months; p< 0.001) and OS (median not reached vs. 89 months; p=0.002).124 In multivariate analyses, MRD status by MFC at Day 100 after transplant was the most important independent prognostic factor for both PFS and OS. Rawstron and colleague122 evaluated the role of MFC in assessing MRD after induction therapy (n= 378) and at Day 100 after transplant (n=397) in the MRC Myeloma IX Study. MRD status at Day 100 after transplant was highly predictive of PFS (28.6 vs. 15.5 months; p< 0.001) and OS (80.6 vs. 59 months; p= 0.0183) for MRD-negative and MRD-positive patients, respectively. In a smaller Phase 2 trial of 31 patients, the Intergroupe Francophone du Myélome reported 21 (68%) of patients became MRD negative by flow cytometry after treatment. After 39 months of follow-up, none of the flow MRD negative patients had relapsed. Thus, from each of these studies it can be concluded that MRD testing by flow cytometry represents a sensitive, easily and quickly performed, surrogate method of predicting PFS and OS within months of therapy.
Measuring Minimal Residual Disease by Flow Cytometry
Previously, we described our routine or low sensitivity flow cytometry method for detecting PCDs. The high sensitivity assay discussed in this section is basically a modification of the lower sensitivity assay designed to acquire more cells. Since abnormal PCs are often present in very low numbers in post-treatment bone marrow aspirates it is critical to acquire many events. The current recommendation of the ICCS Multiple Myeloma Consensus Group is acquire a minimum of 2 × 106 cells and preferably 5 × 106 cells. Obviously, using 100 μL of bone marrow would be insufficient to achieve these numbers. Therefore, it is necessary to use high volumes of sample. Two methodologies have been utilized for this, namely the ‘Pre-lysis’ and ‘Pooled-tube’ approaches.
In the Pre-lysis approach, approximately 30 × 106 cells are treated with Ammonium-Chloride-Potassium (ACK) lysing buffer (155 mM ammonium chloride, 10 mM potassium bicarbonate, and 0.2 mM EDTA) for 10 minutes at room temperature (at a 1 to 9 sample:ACK lysing buffer ratio).125 The cells are then washed with FCM buffer and adjusted to approximately 5 × 107 cells/mL. To tubes containing the mAbs described in Table 2, 100 - 200 μL of sample is added and the cells are incubated in the dark for 30 minutes at room temperature. After incubation, the residual erythrocytes are lysed a second time with saponin based BD FACS™ Lysing Solution for 10 minutes and then washed with FCM Buffer and processed as above for flow cytometric acquisition. Intracellular staining for Tube 2 is performed as previously described.
In the Pooled-tube approach, the sample is washed, counted and adjusted to 1 × 107 cells/mL. Six replicates for each of the mAb cocktails described in Table 2 are set up. To each tube 200 μL of washed sample are added and the cells are incubated, lysed, and washed as described in the routine assay above. During the last wash, the cells from replicate samples are combined for flow cytometric acquisition. Intracellular staining is separately performed on the pooled sample.
Either method is satisfactory and capable of staining a sufficient number of cells to easily acquire 5 × 106 events. The major advantage of the Pre-lysis approach is that while it requires more mAb than conventional staining to achieve saturation of the cells, it does not require as much mAb as the Pooled-tube approach. The advantage of the Pooled-tube method is that it more readily fits into the standard workflow of a laboratory. The Pre-lysis method can adversely impact surface staining of some antigens particularly CD138 and may facilitate the breakdown of some tandem dyes. Ficoll Hypaque enrichment must never be used as it may significantly reduce PC numbers and it accelerates antigen loss from PCs, especially CD138126; in addition, it will compromise PC quantitation due to differential cell enrichment during gradient density centrifugation.
Assay Sensitivity Metrics and Reporting
We (submitted] and others120 have clearly established that the more events that are acquired, the more sensitive the assay becomes, and the higher the predictive value for PFS. In most clinical laboratories, the detection sensitivity of MFC to identify neoplastic PCs in the bone marrow ranges from 10−4 (1 in 10,000 cells) to 10−5 (1 in 100,000 cells).127 Even though multicenter data has demonstrated that a minimum detection sensitivity of 0.01% is of clear prognostic value for MRD monitoring by MFC, it is evident that as assay sensitivity has increased, so has the correlation between MRD status and clinical outcomes. Today a minimum sensitivity of 0.001% is the accepted norm and the threshold for abnormal PCs used in the determination of MRD ranges from 20 – 100 cells.121 There are numerous studies demonstrating that 20 events are a conservative value for the smallest number of a homogeneous and clustered population of cells that can be reliably detected by an experienced cytometrist.128,129 Using this value, the limit of detection (LOD) is calculated as:
For example, if the minimum threshold for is 20 events and 5 × 106 ‘Total Leukocytes’ are acquired, then the LOD is 0.0004%. It is generally accepted that between 50 - 100 events represent the minimum number of events required to accurately quantify a cell population130,131. Thus, the lower limit of quantification (LLOQ) is calculated as:
If 50 events represent the LLOQ threshold and 5×106 ‘Total Leukocytes’ are collected, then the LLOQ is 0.001%. Regardless of the theoretical values for LOD and LLOQ, the College of American Pathologists requires that each laboratory experimentally confirm these values in their assay.
Finally, it is important that a PCD MRD report contain the following information:
The number and percentage of normal and abnormal PCs
The LOD, LLOQ, or both
Optionally the total number of leukocytes evaluated after excluding doublets and debris
The phenotype of the abnormal cells
A statement about the quality of the bone marrow aspirate (ie hemodilution and viability as necessary)
DISCUSSION
PCD is a heterogeneous disease with a spectrum of severities and clinical outcomes. While overall it is not a prevalent form of cancer, PCDs represent the second most-common hematological disorder and the focal, anatomically-isolated nature of the neoplasm constitutes a considerable challenge for medical practice. A relatively large percentage of patients with PCDs exhibit indolent forms of the disease (e.g. MGUS), however many suffer with aggressive malignancy (e.g. multiple myeloma or plasma cell leukemia) which manifests with symptoms of organ failure, bone destruction, and perturbed hematopoiesis. Most types of PCD are developed after affinity maturation has already taken place, as the genetic sequences of myeloma cells have been found to be consistently hypermutated, and exhibit phenotypic features similar to those of long-lived PCs.132–134 The oversecretion of M protein, the elevated percentage of plasma cells in the bone marrow, and the presence of organ damage are typically considered when determining the specific subtype of PCD.
The various manifestations of PCD require different treatment regiments. Historically, many patients respond to first-line agents, however most become refractory to therapy and eventually relapse. The treatment of multiple myeloma has evolved significantly over time. Initial therapies developed in the 1960s involved the use of hematopoietic stem cell transplants (HSCT) along with chemotherapeutic agents such as melphalan, vincristine, doxorubicin, or cyclophosphamide in combination with prednisone. While these treatments modestly improved patient survival, their efficacy was limited due to the low mitotic index of PCs.135–137
Based upon these findings, alternative systemic approaches to the treatment of PCDs were explored; primarily involving the use of thalidomide, which functions as an inhibitor of angiogenesis and also acts as an immune modulator by opposing the activity of IL-6. This cytokine otherwise promotes the survival of malignant plasma cells by upregulating cell adhesion molecules, tumor promoting cytokines, and down-regulating tumor suppressor proteins. Modest success with this approach eventually led to the development and implementation of the next generation of therapies. Such drugs include bortezomib (a selective proteosome inhibitor), dexamethasone (an anti-inflammatory agent), and the thalidomide analogue lenalidomide (an immune modulator and anti-angiogenic) which further extended OS in treated patients. When these medicines were employed in concert with HSCT, OS increased to an unprecedented, but still grim 5 years for 45% of multiple myeloma cases.138
The conventional medical age has added targeted therapies to the armamentarium of drugs used to treat PCDs. Immunotherapeutic agents such as anti-PD-1 (CD279), anti-PD-L1 (CD274), and anti-CD38 (daratumumab) have been employed; with their successes driving the development of additional immunotherapies.139 These novel therapeutics are quickly advancing but their mainstream transition has been limited because randomized Phase 3 clinical trials take years to show benefits when PFS and OS are used as study endpoints.55,115–118 Due to the long latency between drug development and the approval to be considered as therapeutic options, the measurement of MRD by MFC as an independent method to predict PFS and OS for patients diagnosed with PCD has been employed.119,120 All flow cytometric studies to date have strongly correlated the measurement of MRD with PFS and OS at the Day 100 time point, reducing from years to months the time required to reach meaningful clinical outcomes.121–124
While the use of mAbs to target cell surface molecules on PCs has expanded as an innovative therapeutic choice, their use can obstruct the detection of PCs by MFC. Daratumumab and isatuximab can impair the measurement of CD38,70,139 likewise indatuximab ravtansine may interfere with CD138 detection.139 Even though the discovery of these mAb therapies offer a new avenue by which we can approach the detection of multiple myeloma, they have made the search for alternative markers to identify both normal and neoplastic PCs by MFC a priority.140 Independent investigators have reported the expression of CD54, CD229, CD269, and CD319 to be valuable for this purpose.54,141,142 In particular, CD269 and CD319 were found to be more versatile markers and withstood storage longer than CD138.141 It will be interesting to see how these markers can be integrated into the clinical test setting of MRD by MFC.
Besides MFC, alternatives for the detection of multiple myeloma MRD are ASO-qPCR and NGS whose assay performance characteristics are contrasted in Table 5. Before NGS, ASO-qPCR was regarded as one of the most sensitive assays to detect PCDs with a sensitivity limit of 10−5 to 10−6.127 ASO-qPCR involves the design of specific primers complementary to the clonal rearrangement in the CDR genes of mature B cells. Puig and colleagues143 analyzed data from three consecutive myeloma trials that utilized both ASO-qPCR and 4-color MFC to evaluate the MRD negativity and clinical outcome in 170 patients. The results showed both technologies correlated well with MRD-negativity to predict PFS and OS. However, 58% of the patients could not be evaluated by ASO-qPCR due to the failure of clonal detection, unsuccessful sequencing, or suboptimal performance of primer or probe sets. These technical shortcomings are due to the highly mutated CDR region, as well as the heterogeneous infiltration of disease into the marrow.143,144 As a potential solution, personalized primer and probe sets are required to detect the somatic-mutated sequence for each patient. Therefore, this finding suggests that even though ASO-qPCR is sensitive and specific, it is only applicable to a subset of patients. Furthermore, it may be more laborious and time-consuming that MFC, indicating that MFC is probably a more practical and feasible tool for routine MRD assessment than ASO-qPCR.34
Table 5.
Comparison of flow cytometry and molecular techniques for MRD analysis in multiple myeloma
| Parameters | Flow Cytometry | Molecular Techniques | ||
|---|---|---|---|---|
| 2008 EMN Consensus (4 – 6 colors) | 2016 ICCS Consensus (≥ 8 colors) | ASO-qPCR | Next-generation Sequencing | |
| Applicability, % | > 95 | > 99 | 50 – 90 | 80 – 90 |
| LLOQ, % | 0.01 | > 0.001 | > 0.001 | > 0.001 |
| Number of cells/Amount of DNA required for LLOQ | 0.5 × 106 cells/tube | 2 – 5 × 106 cells/tube | 500 ng (1,000,000 cells for triplicate analysis) | 14 ug (2,000,000 cells for triplicate analysis) |
| LOD, % | 0.0040 | 0.0004 | 0.0001 | 0.0001 |
| Reproducibility | High | High | Low | High |
| Pretreatment Evaluation | Required | Not Required | Required | Required |
| Fresh Sample | Required, < 48 h old storage | Recommended, < 48 h before DNA extraction | ||
| Diagnostic Sample | Useful, but not required | Required | ||
| Quantitative | Yes | No | ||
| Sample Quality Assurance | Not Required | Additional tests are required | ||
| Cost | Low | High (at diagosis) Medium (follow-up) | High | |
| Turnaround | Can take up to 1 d | Can take several days | ||
| Availability | Widely-available | Intermediate | Limited | |
| Harmonization | Yes (EMN) | Yes (ICCS/ESCCA) | Yes | Ongoing (EuroMRD) |
Abbreviations: ASO-qPCR, allele-specific oligonucleotide real time quantitative polymerase chain reaction; EMN, European Myeloma Network; ESCCA, European Society for Clinical Cell Analysis; ICCS, International Clinical Cytometry Society; LLOQ, lower limit of quantification;
LOD, limit of detection.
The other approach to MRD detection is NGS. When compared to ASO-qPCR, both technologies have a similar detection sensitivity of 10−5, but under ideal circumstances NGS may detect as few as 1 neoplastic cell in a million. NGS also has the advantage of being applicable to more patients, as it does not require patient-specific primers.145,146 In a study conducted by Martinez-Lopez and colleagues,147 patients who were MRD-negative by NGS had a longer PFS and increased OS. Summarily, the concordance rate between NGS and MFC, and also NGS and ASO-qPCR were 83% and 85%, respectively. As myeloma cells become rarer, NGS is better than MFC for detecting MRD but it has the disadvantage of being susceptible to the evolving heterogeneity of myeloma commensurate with therapy. A recent study published by Munshi and colleagues148 suggested that several evolved clonotypes were measured in 37.6% of the post-treatment multiple myeloma samples which could confound the ability of NGS but not MFC to detect MRD. NGS remains the least studied MRD testing modality and continued clinical correlations will be required before widespread adoption is possible.
Multiple MFC consensus panels have been proposed in the last two decades, but they typically included largely overlapping lists of CD markers for each disease category. Virtually all consensus proposals lack information about reference antibody clones for the proposed CD markers and they only provide limited information on the most appropriate combinations of relevant markers for multicolor mAb panels. In 2013, the ICCS Multiple Myeloma MRD Consensus Group was formed and tasked with the development of consensus documents for the detection of MRD by MFC. These documents were reviewed with the FDA in March 2014 and published in January 2015. It was proposed that MRD testing by flow cytometry needs to be integrated now into the response criteria for multiple myeloma and determined that for regulatory approval, a surrogate must be shown to be reasonably likely to predict clinical benefit. This has already been accomplished by Paiva and colleagues124 and Rawstron and colleagues68. Therefore, the FDA concluded that “MRD assessment in multiple myeloma has the potential to be a surrogate clinical endpoint that could be used to support regulatory purposes for drug review” with a standardized approach.149 Therefore, there is an urgent need for adoption of consensus protocols within the multiple myeloma community for inclusion of MRD negativity by MFC as a surrogate endpoint in clinical trials.
Key Point.
Plasma cell dyscrasia is a hematological disorder in which normal plasma cells become transformed in the bone marrow and soft tissues
Multiparametric flow cytometry is a reliable tool to evaluate plasma cell dyscrasias
Flow cytometry is a high sensitivity assay that can be used to detect minimal residual disease which has been shown to correlate with progression-free survival and overall survival
Acknowledgments
Flow cytometry services were provided by the Flow and Image Cytometry Core Facility at the Roswell Park Cancer Institute which is supported in part by the NCI Cancer Center Support Grant 5P30 CA016056.
Footnotes
Disclosure Statement:
The authors have no commercial or financial conflicts of interest to disclose.
References
- 1.Shapiro-Shelef M, Calame K. Plasma cell differentiation and multiple myeloma. Current Opinion in Immunology. 2004;16(2):226–234. doi: 10.1016/j.coi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5(3):230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- 3.Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. 2015;15(3):160–171. doi: 10.1038/nri3795. [DOI] [PubMed] [Google Scholar]
- 4.Sze DM, Toellner KM, Garcia de Vinuesa C, Taylor DR, MacLennan IC. Intrinsic constraint on plasmablast growth and extrinsic limits of plasma cell survival. J Exp Med. 2000;192(6):813–821. doi: 10.1084/jem.192.6.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu VT, Beller A, Nguyen TT, Steinhauser G, Berek C. The long-term survival of plasma cells. Scandinavian journal of immunology. 2011;73(6):508–511. doi: 10.1111/j.1365-3083.2011.02544.x. [DOI] [PubMed] [Google Scholar]
- 6.Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 7.Moreaux J, Legouffe E, Jourdan E, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004;103(8):3148–3157. doi: 10.1182/blood-2003-06-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belnoue E, Pihlgren M, McGaha TL, et al. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood. 2008;111(5):2755–2764. doi: 10.1182/blood-2007-09-110858. [DOI] [PubMed] [Google Scholar]
- 9.Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. 2002;169(8):4213–4221. doi: 10.4049/jimmunol.169.8.4213. [DOI] [PubMed] [Google Scholar]
- 10.Kierszenbaum A. Histology and cell biology: an introduction to pathology. St. Louis: Mosby; 2002. p. 275. [Google Scholar]
- 11.Kuehl WM, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer. 2002;2(3):175–187. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]
- 12.Anderson KC, Carrasco RD. Pathogenesis of myeloma. Annu Rev Pathol. 2011;6:249–274. doi: 10.1146/annurev-pathol-011110-130249. [DOI] [PubMed] [Google Scholar]
- 13.Bakkus MH, Heirman C, Van Riet I, Van Camp B, Thielemans K. Evidence that multiple myeloma Ig heavy chain VDJ genes contain somatic mutations but show no intraclonal variation. Blood. 1992;80(9):2326. [PubMed] [Google Scholar]
- 14.Kyle RA, Rajkumar SV. Leukemia: official journal of the Leukemia Society of America. 1. Vol. 23. Leukemia Research Fund, U.K; 2009. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma; pp. 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slovak ML. Multiple myeloma: current perspectives. Clin Lab Med. 2011;31(4):699–724, x. doi: 10.1016/j.cll.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Group TIMW. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121(5):749–757. [PubMed] [Google Scholar]
- 17.Kyle RA, Finkelstein S, Elveback LR, Kurland LT. Incidence of monoclonal proteins in a Minnesota community with a cluster of multiple myeloma. Blood. 1972;40(5):719–724. [PubMed] [Google Scholar]
- 18.Saleun JP, Vicariot M, Deroff P, Morin JF. Monoclonal gammopathies in the adult population of Finistère, France. Journal of Clinical Pathology. 1982;35(1):63–68. doi: 10.1136/jcp.35.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Axelsson U, Bachmann R, Hallen J. Frequency of pathological proteins (M-components) on 6,995 sera from an adult population. Acta Med Scand. 1966;179(2):235–247. doi: 10.1111/j.0954-6820.1966.tb05453.x. [DOI] [PubMed] [Google Scholar]
- 20.Bianchi G, Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood. 2015;125(20):3049–3058. doi: 10.1182/blood-2014-11-568881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pittaluga S, Wlodarska I, Pulford K, et al. The monoclonal antibody ALK1 identifies a distinct morphological subtype of anaplastic large cell lymphoma associated with 2p23/ALK rearrangements. Am J Pathol. 1997;151(2):343–351. [PMC free article] [PubMed] [Google Scholar]
- 22.Rajkumar SV, Landgren O, Mateos M-V. Smoldering multiple myeloma. Blood. 2015;125(20):3069. doi: 10.1182/blood-2014-09-568899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez-Persona E, Vidriales MB, Mateo G, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110(7):2586–2592. doi: 10.1182/blood-2007-05-088443. [DOI] [PubMed] [Google Scholar]
- 24.Campana D, Coustan-Smith E. Minimal residual disease studies by flow cytometry in acute leukemia. Acta Haematol. 2004;112(1-2):8–15. doi: 10.1159/000077554. [DOI] [PubMed] [Google Scholar]
- 25.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 26.Noll JE, Williams SA, Tong CM, et al. Myeloma plasma cells alter the bone marrow microenvironment by stimulating the proliferation of mesenchymal stromal cells. Haematologica. 2014;99(1):163–171. doi: 10.3324/haematol.2013.090977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Avet-Loiseau H, Daviet A, Brigaudeau C, et al. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: a study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myelome and the Groupe Francais de Cytogenetique Hematologique. Blood. 2001;97(3):822–825. doi: 10.1182/blood.v97.3.822. [DOI] [PubMed] [Google Scholar]
- 28.Dimopoulos MA, Palumbo A, Delasalle KB, Alexanian R. Primary plasma cell leukaemia. Br J Haematol. 1994;88(4):754–759. doi: 10.1111/j.1365-2141.1994.tb05114.x. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Sanz R, Orfao A, Gonzalez M, et al. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy, and cytogenetic characteristics. Blood. 1999;93(3):1032–1037. [PubMed] [Google Scholar]
- 30.Boll M, Parkins E, O’Connor SJ, Rawstron AC, Owen RG. Extramedullary plasmacytoma are characterized by a ‘myeloma-like’ immunophenotype and genotype and occult bone marrow involvement. Br J Haematol. 2010;151(5):525–527. doi: 10.1111/j.1365-2141.2010.08386.x. [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Wang M, Chen Y, et al. Immunophenotypic analysis of abnormal plasma cell clones in bone marrow of primary systemic light chain amyloidosis patients. Chinese medical journal. 2014;127(15):2765–2770. [PubMed] [Google Scholar]
- 32.Paiva B, Vidriales MB, Perez JJ, et al. The clinical utility and prognostic value of multiparameter flow cytometry immunophenotyping in light-chain amyloidosis. Blood. 2011;117(13):3613–3616. doi: 10.1182/blood-2010-12-324665. [DOI] [PubMed] [Google Scholar]
- 33.Ocqueteau M, Orfao A, Almeida J, et al. Immunophenotypic characterization of plasma cells from monoclonal gammopathy of undetermined significance patients. Implications for the differential diagnosis between MGUS and multiple myeloma. Am J Pathol. 1998;152(6):1655–1665. [PMC free article] [PubMed] [Google Scholar]
- 34.Sarasquete ME, Garcia-Sanz R, Gonzalez D, et al. Minimal residual disease monitoring in multiple myeloma: a comparison between allelic-specific oligonucleotide real-time quantitative polymerase chain reaction and flow cytometry. Haematologica. 2005;90(10):1365–1372. [PubMed] [Google Scholar]
- 35.Orfao A, Ortuno F, de Santiago M, Lopez A, San Miguel J. Immunophenotyping of acute leukemias and myelodysplastic syndromes. Cytometry A. 2004;58(1):62–71. doi: 10.1002/cyto.a.10104. [DOI] [PubMed] [Google Scholar]
- 36.Vidriales MB, San-Miguel JF, Orfao A, Coustan-Smith E, Campana D. Minimal residual disease monitoring by flow cytometry. Best Pract Res Clin Haematol. 2003;16(4):599–612. doi: 10.1016/s1521-6926(03)00067-7. [DOI] [PubMed] [Google Scholar]
- 37.van Dongen JJ, Lhermitte L, Bottcher S, et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia. 2012;26(9):1908–1975. doi: 10.1038/leu.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foon KA, Todd RF., 3rd Immunologic classification of leukemia and lymphoma. Blood. 1986;68(1):1–31. [PubMed] [Google Scholar]
- 39.van Dongen JJ, Adriaansen HJ, Hooijkaas H. Immunophenotyping of leukaemias and non-Hodgkin’s lymphomas. Immunological markers and their CD codes. Neth J Med. 1988;33(5-6):298–314. [PubMed] [Google Scholar]
- 40.Terstappen LW, Johnsen S, Segers-Nolten IM, Loken MR. Identification and characterization of plasma cells in normal human bone marrow by high-resolution flow cytometry. Blood. 1990;76(9):1739–1747. [PubMed] [Google Scholar]
- 41.Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328–346. doi: 10.1016/S1470-2045(16)30206-6. [DOI] [PubMed] [Google Scholar]
- 42.San Miguel JF, Gonzalez M, Gascon A, et al. Immunophenotypic heterogeneity of multiple myeloma: influence on the biology and clinical course of the disease. Castellano-Leones (Spain) Cooperative Group for the Study of Monoclonal Gammopathies. Br J Haematol. 1991;77(2):185–190. doi: 10.1111/j.1365-2141.1991.tb07975.x. [DOI] [PubMed] [Google Scholar]
- 43.Rawstron AC, Davies FE, DasGupta R, et al. Flow cytometric disease monitoring in multiple myeloma: the relationship between normal and neoplastic plasma cells predicts outcome after transplantation. Blood. 2002;100(9):3095–3100. doi: 10.1182/blood-2001-12-0297. [DOI] [PubMed] [Google Scholar]
- 44.San Miguel JF, Almeida J, Mateo G, et al. Immunophenotypic evaluation of the plasma cell compartment in multiple myeloma: a tool for comparing the efficacy of different treatment strategies and predicting outcome. Blood. 2002;99(5):1853–1856. doi: 10.1182/blood.v99.5.1853. [DOI] [PubMed] [Google Scholar]
- 45.Paiva B, Almeida J, Perez-Andres M, et al. Utility of flow cytometry immunophenotyping in multiple myeloma and other clonal plasma cell-related disorders. Cytometry B Clin Cytom. 2010;78(4):239–252. doi: 10.1002/cyto.b.20512. [DOI] [PubMed] [Google Scholar]
- 46.Nadav L, Katz BZ, Baron S, et al. Diverse niches within multiple myeloma bone marrow aspirates affect plasma cell enumeration. Br J Haematol. 2006;133(5):530–532. doi: 10.1111/j.1365-2141.2006.06068.x. [DOI] [PubMed] [Google Scholar]
- 47.Ng AP, Wei A, Bhurani D, Chapple P, Feleppa F, Juneja S. The sensitivity of CD138 immunostaining of bone marrow trephine specimens for quantifying marrow involvement in MGUS and myeloma, including samples with a low percentage of plasma cells. Haematologica. 2006;91(7):972–975. [PubMed] [Google Scholar]
- 48.Harada H, Kawano MM, Huang N, et al. Phenotypic difference of normal plasma cells from mature myeloma cells. Blood. 1993;81(10):2658–2663. [PubMed] [Google Scholar]
- 49.Mateo G, Montalban MA, Vidriales MB, et al. Prognostic value of immunophenotyping in multiple myeloma: a study by the PETHEMA/GEM cooperative study groups on patients uniformly treated with high-dose therapy. J Clin Oncol. 2008;26(16):2737–2744. doi: 10.1200/JCO.2007.15.4120. [DOI] [PubMed] [Google Scholar]
- 50.Mateo Manzanera G, San Miguel Izquierdo JF, Orfao de Matos A. Immunophenotyping of plasma cells in multiple myeloma. Methods Mol Med. 2005;113:5–24. doi: 10.1385/1-59259-916-8:5. [DOI] [PubMed] [Google Scholar]
- 51.Pellat-Deceunynck C, Bataille R. Normal and malignant human plasma cells: proliferation, differentiation, and expansions in relation to CD45 expression. Blood Cells Mol Dis. 2004;32(2):293–301. doi: 10.1016/j.bcmd.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 52.Cannizzo E, Bellio E, Sohani AR, et al. Multiparameter immunophenotyping by flow cytometry in multiple myeloma: The diagnostic utility of defining ranges of normal antigenic expression in comparison to histology. Cytometry B Clin Cytom. 2010;78(4):231–238. doi: 10.1002/cyto.b.20517. [DOI] [PubMed] [Google Scholar]
- 53.Flores-Montero J, de Tute R, Paiva B, et al. Immunophenotype of normal vs. myeloma plasma cells: Toward antibody panel specifications for MRD detection in multiple myeloma. Cytometry B Clin Cytom. 2016;90(1):61–72. doi: 10.1002/cyto.b.21265. [DOI] [PubMed] [Google Scholar]
- 54.Pojero F, Flores-Montero J, Sanoja L, et al. Utility of CD54, CD229, and CD319 for the identification of plasma cells in patients with clonal plasma cell diseases. Cytometry B Clin Cytom. 2016;90(1):91–100. doi: 10.1002/cyto.b.21269. [DOI] [PubMed] [Google Scholar]
- 55.Oldaker TA, Wallace PK, Barnett D. Flow cytometry quality requirements for monitoring of minimal disease in plasma cell myeloma. Cytometry B Clin Cytom. 2016;90(1):40–46. doi: 10.1002/cyto.b.21276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.San-Miguel JF, Vidriales MB, Orfao A. Immunological evaluation of minimal residual disease (MRD) in acute myeloid leukaemia (AML) Best Pract Res Clin Haematol. 2002;15(1):105–118. doi: 10.1053/beha.2001.0193. [DOI] [PubMed] [Google Scholar]
- 57.Wood B, Jevremovic D, Bene MC, Yan M, Jacobs P, Litwin V. Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part V - assay performance criteria. Cytometry B Clin Cytom. 2013;84(5):315–323. doi: 10.1002/cyto.b.21108. [DOI] [PubMed] [Google Scholar]
- 58.Wood BL, Arroz M, Barnett D, et al. 2006 Bethesda International Consensus recommendations on the immunophenotypic analysis of hematolymphoid neoplasia by flow cytometry: optimal reagents and reporting for the flow cytometric diagnosis of hematopoietic neoplasia. Cytometry B Clin Cytom. 2007;72(Suppl 1):S14–22. doi: 10.1002/cyto.b.20363. [DOI] [PubMed] [Google Scholar]
- 59.Barnett D, Louzao R, Gambell P, De J, Oldaker T, Hanson CA. Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part IV - postanalytic considerations. Cytometry B Clin Cytom. 2013;84(5):309–314. doi: 10.1002/cyto.b.21107. [DOI] [PubMed] [Google Scholar]
- 60.Davis BH, Wood B, Oldaker T, Barnett D. Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part I - rationale and aims. Cytometry B Clin Cytom. 2013;84(5):282–285. doi: 10.1002/cyto.b.21104. [DOI] [PubMed] [Google Scholar]
- 61.Stetler-Stevenson M, Davis B, Wood B, Braylan R. 2006 Bethesda International Consensus Conference on flow cytometric immunophenotyping of hematolymphoid neoplasia. Cytometry B Clin Cytom. 2007;72(Suppl 1):S3. doi: 10.1002/cyto.b.20362. [DOI] [PubMed] [Google Scholar]
- 62.Davis BH, Dasgupta A, Kussick S, Han JY, Estrellado A. Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part II - preanalytical issues. Cytometry B Clin Cytom. 2013;84(5):286–290. doi: 10.1002/cyto.b.21105. [DOI] [PubMed] [Google Scholar]
- 63.Purvis NB, Oldaker T. Validation and quality control in clinical flow cytometry. In: Kottke-Marchant K, Davis BH, editors. Laboratory Hematology Practice. Oxford, UK: Wiley-Blackwell; 2012. pp. 115–130. [Google Scholar]
- 64.Tanqri S, Vall H, Kaplan D, et al. Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part III - analytical issues. Cytometry B Clin Cytom. 2013;84(5):291–308. doi: 10.1002/cyto.b.21106. [DOI] [PubMed] [Google Scholar]
- 65.Arroz M, Came N, Lin P, et al. Consensus guidelines on plasma cell myeloma minimal residual disease analysis and reporting. Cytometry B Clin Cytom. 2016;90(1):31–39. doi: 10.1002/cyto.b.21228. [DOI] [PubMed] [Google Scholar]
- 66.Stetler-Stevenson M, Paiva B, Stoolman L, et al. Consensus guidelines for myeloma minimal residual disease sample staining and data acquisition. Cytometry B Clin Cytom. 2016;90(1):26–30. doi: 10.1002/cyto.b.21249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kalina T, Flores-Montero J, Lecrevisse Q, et al. Quality assessment program for EuroFlow protocols: summary results of four-year (2010-2013) quality assurance rounds. Cytometry A. 2015;87(2):145–156. doi: 10.1002/cyto.a.22581. [DOI] [PubMed] [Google Scholar]
- 68.Rawstron AC, Orfao A, Beksac M, et al. Report of the European Myeloma Network on multiparametric flow cytometry in multiple myeloma and related disorders. Haematologica. 2008;93(3):431–438. doi: 10.3324/haematol.11080. [DOI] [PubMed] [Google Scholar]
- 69.Bataille R, Jego G, Robillard N, et al. The phenotype of normal, reactive and malignant plasma cells. Identification of “many and multiple myelomas” and of new targets for myeloma therapy. Haematologica. 2006;91(9):1234–1240. [PubMed] [Google Scholar]
- 70.Flores-Montero J, Sanoja-Flores L, Paiva B, et al. Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia. 2017 doi: 10.1038/leu.2017.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Royston DJ, Gao Q, Nguyen N, Maslak P, Dogan A, Roshal M. Single-tube 10-fluorochrome analysis for efficient flow cytometric evaluation of minimal residual disease in plasma cell myeloma. Am J Clin Pathol. 2016;146(1):41–49. doi: 10.1093/ajcp/aqw052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roshal M, Flores-Montero JA, Gao Q, et al. MRD detection in multiple myeloma: comparison between MSKCC 10-color single-tube and EuroFlow 8-color 2-tube methods. Blood Advances. 2017;1(12):728. doi: 10.1182/bloodadvances.2016003715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ruiz-Arguelles GJ, San Miguel JF. Cell surface markers in multiple myeloma. Mayo Clin Proc. 1994;69(7):684–690. doi: 10.1016/s0025-6196(12)61350-0. [DOI] [PubMed] [Google Scholar]
- 74.Guikema JE, Hovenga S, Vellenga E, et al. CD27 is heterogeneously expressed in multiple myeloma: low CD27 expression in patients with high-risk disease. Br J Haematol. 2003;121(1):36–43. doi: 10.1046/j.1365-2141.2003.04260.x. [DOI] [PubMed] [Google Scholar]
- 75.Moreau P, Robillard N, Jego G, et al. Lack of CD27 in myeloma delineates different presentation and outcome. Br J Haematol. 2006;132:168–170. doi: 10.1111/j.1365-2141.2005.05849.x. [DOI] [PubMed] [Google Scholar]
- 76.Robillard N, Jego G, Pellat-Deceunynck C, et al. CD28, a marker associated with tumoral expansion in multiple myeloma. Clin Cancer Res. 1998;4(6):1521–1526. [PubMed] [Google Scholar]
- 77.Jackson N, Ling NR, Ball J, Bromidge E, Nathan PD, Franklin IM. An analysis of myeloma plasma cell phenotype using antibodies defined at the IIIrd International Workshop on Human Leucocyte Differentiation Antigens. Clinical and Experimental Immunology. 1988;72(3):351–356. [PMC free article] [PubMed] [Google Scholar]
- 78.Shapiro VS, Mollenauer MN, Weiss A. Endogenous CD28 expressed on myeloma cells up-regulates interleukin-8 production: implications for multiple myeloma progression. Blood. 2001;98(1):187–193. doi: 10.1182/blood.v98.1.187. [DOI] [PubMed] [Google Scholar]
- 79.Nair JR, Carlson LM, Koorella C, et al. Journal of immunology. 3. Vol. 187. Baltimore, Md: 2011. CD28 expressed on malignant plasma cells induces a pro-survival and immunosuppressive microenvironment; pp. 1243–1253. 1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reinherz EL, Kung PC, Goldstein G, Levey RH, Schlossman SF. Discrete stages of human intrathymic differentiation: analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc Natl Acad Sci U S A. 1980;77(3):1588–1592. doi: 10.1073/pnas.77.3.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reinherz EL, Schlossman SF. The characterization and function of human immunoregulatory T lymphocyte subsets. Immunol Today. 1981;2(4):69–75. doi: 10.1016/0167-5699(81)90019-0. [DOI] [PubMed] [Google Scholar]
- 82.Bataille R, Robillard N, Pellat-Deceunynck C, Amiot M. A cellular model for myeloma cell growth and maturation based on an intraclonal CD45 hierarchy. Immunol Rev. 2003;194:105–111. doi: 10.1034/j.1600-065x.2003.00039.x. [DOI] [PubMed] [Google Scholar]
- 83.Kumar S, Rajkumar SV, Kyle RA, et al. Prognostic value of circulating plasma cells in monoclonal gammopathy of undetermined significance. J Clin Oncol. 2005;23(24):5668–5674. doi: 10.1200/JCO.2005.03.159. [DOI] [PubMed] [Google Scholar]
- 84.Moreau P, Robillard N, Avet-Loiseau H, et al. Patients with CD45 negative multiple myeloma receiving high-dose therapy have a shorter survival than those with CD45 positive multiple myeloma. Haematologica. 2004;89(5):547–551. [PubMed] [Google Scholar]
- 85.Pellatdeceunynck C, Barille S, Puthier D, et al. Adhesion molecules on human myeloma cells - significant changes in expression related to malignancy, tumor spreading, and immortalization. Cancer Research. 1995;55(16):3647–3653. [PubMed] [Google Scholar]
- 86.Pellat-Deceunynck C, Barille S, Jego G, et al. The absence of CD56 (NCAM) on malignant plasma cells is a hallmark of plasma cell leukemia and of a special subset of multiple myeloma. Leukemia. 1998;12(12):1977–1982. doi: 10.1038/sj.leu.2401211. [DOI] [PubMed] [Google Scholar]
- 87.Ely SA, Knowles DM. Expression of CD56/neural cell adhesion molecule correlates with the presence of lytic bone lesions in multiple myeloma and distinguishes myeloma from monoclonal gammopathy of undetermined significance and lymphomas with plasmacytoid differentiation. Am J Pathol. 2002;160(4):1293–1299. doi: 10.1016/S0002-9440(10)62556-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sahara N, Takeshita A. Prognostic significance of surface markers expressed in multiple myeloma: CD56 and other antigens. Leuk Lymphoma. 2004;45(1):61–65. doi: 10.1080/1042819031000149377. [DOI] [PubMed] [Google Scholar]
- 89.Langebrake C, Brinkmann I, Teigler-Schlegel A, et al. Immunophenotypic differences between diagnosis and relapse in childhood AML: Implications for MRD monitoring. Cytometry B Clin Cytom. 2005;63(1):1–9. doi: 10.1002/cyto.b.20037. [DOI] [PubMed] [Google Scholar]
- 90.Paiva B, Gutierrez NC, Chen X, et al. Clinical significance of CD81 expression by clonal plasma cells in high-risk smoldering and symptomatic multiple myeloma patients. Leukemia. 2012;26(8):1862–1869. doi: 10.1038/leu.2012.42. [DOI] [PubMed] [Google Scholar]
- 91.Bataille R, Pellat-Deceunynck C, Robillard N, Avet-Loiseau H, Harousseau JL, Moreau P. CD117 (c-kit) is aberrantly expressed in a subset of MGUS and multiple myeloma with unexpectedly good prognosis. Leuk Res. 2008;32(3):379–382. doi: 10.1016/j.leukres.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 92.Schmidt-Hieber M, Pérez-Andrés M, Paiva B, et al. CD117 expression in gammopathies is associated with an altered maturation of the myeloid and lymphoid hematopoietic cell compartments and favorable disease features. Haematologica. 2011;96(2):328–332. doi: 10.3324/haematol.2010.031872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kraj M, Poglod R, Kopec-Szlezak J, Sokolowska U, Wozniak J, Kruk B. C-kit receptor (CD117) expression on plasma cells in monoclonal gammopathies. Leuk Lymphoma. 2004;45(11):2281–2289. doi: 10.1080/10428190412331283279. [DOI] [PubMed] [Google Scholar]
- 94.Ridley RC, Xiao H, Hata H, Woodliff J, Epstein J, Sanderson RD. Expression of syndecan regulates human myeloma plasma cell adhesion to type I collagen. Blood. 1993;81(3):767–774. [PubMed] [Google Scholar]
- 95.Yang Y, Borset M, Langford JK, Sanderson RD. Heparan sulfate regulates targeting of syndecan-1 to a functional domain on the cell surface. Journal of Biological Chemistry. 2003;278(15):12888–12893. doi: 10.1074/jbc.M209440200. [DOI] [PubMed] [Google Scholar]
- 96.Dhodapkar MV, Kelly T, Theus A, Athota AB, Barlogie B, Sanderson RD. Elevated levels of shed syndecan-1 correlate with tumour mass and decreased matrix metalloproteinase-9 activity in the serum of patients with multiple myeloma. Br J Haematol. 1997;99(2):368–371. doi: 10.1046/j.1365-2141.1997.3893203.x. [DOI] [PubMed] [Google Scholar]
- 97.Jourdan M, Ferlin M, Legouffe E, et al. The myeloma cell antigen syndecan-1 is lost by apoptotic myeloma cells. Br J Haematol. 1998;100(4):637–646. doi: 10.1046/j.1365-2141.1998.00623.x. [DOI] [PubMed] [Google Scholar]
- 98.San Antonio JD, Karnovsky MJ, Gay S, Sanderson RD, Lander AD. Interactions of syndecan-1 and heparin with human collagens. Glycobiology. 1994;4(3):327–332. doi: 10.1093/glycob/4.3.327. [DOI] [PubMed] [Google Scholar]
- 99.Mahnke YD, Roederer M. Optimizing a Multi-colour Immunophenotyping Assay. Clinics in laboratory medicine. 2007;27(3):469-v. doi: 10.1016/j.cll.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rawstron AC, Fazi C, Agathangelidis A, et al. A complementary role of multiparameter flow cytometry and high-throughput sequencing for minimal residual disease detection in chronic lymphocytic leukemia: an European Research Initiative on CLL study. Leukemia. 2016;30(4):929–936. doi: 10.1038/leu.2015.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nowakowski GS, Witzig TE, Dingli D, et al. Circulating plasma cells detected by flow cytometry as a predictor of survival in 302 patients with newly diagnosed multiple myeloma. Blood. 2005;106(7):2276–2279. doi: 10.1182/blood-2005-05-1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dingli D, Nowakowski GS, Dispenzieri A, et al. Flow cytometric detection of circulating myeloma cells before transplantation in patients with multiple myeloma: a simple risk stratification system. Blood. 2006;107(8):3384–3388. doi: 10.1182/blood-2005-08-3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin P, Owens R, Tricot G, Wilson CS. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am J Clin Pathol. 2004;121(4):482–488. doi: 10.1309/74R4-TB90-BUWH-27JX. [DOI] [PubMed] [Google Scholar]
- 104.Tario JD, Wallace PK. Reagents and cell staining for immunophenotyping by flow cytometry. In: McManus LM, Mitchell RN, editors. Pathobiology of Human Disease. San Diego: Elsevier; 2014. pp. 3678–3701. [Google Scholar]
- 105.Lahuerta JJ, Martinez-Lopez J, Serna JD, et al. Remission status defined by immunofixation vs. electrophoresis after autologous transplantation has a major impact on the outcome of multiple myeloma patients. Br J Haematol. 2000;109(2):438–446. doi: 10.1046/j.1365-2141.2000.02012.x. [DOI] [PubMed] [Google Scholar]
- 106.Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 107.Bradwell AR, Carr-Smith HD, Mead GP, et al. Highly sensitive, automated immunoassay for immunoglobulin free light chains in serum and urine. Clin Chem. 2001;47(4):673–680. [PubMed] [Google Scholar]
- 108.Blade J, Rosinol L, Cibeira MT, Rovira M, Carreras E. Hematopoietic stem cell transplantation for multiple myeloma beyond 2010. Blood. 2010;115(18):3655–3663. doi: 10.1182/blood-2009-08-238196. [DOI] [PubMed] [Google Scholar]
- 109.Attal M, Harousseau J-L, Stoppa A-M, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. New England Journal of Medicine. 1996;335(2):91–97. doi: 10.1056/NEJM199607113350204. [DOI] [PubMed] [Google Scholar]
- 110.Barlogie B, Jagannath S, Vesole DH, et al. Superiority of tandem autologous transplantation over standard therapy for previously untreated multiple myeloma. Blood. 1997;89(3):789–793. [PubMed] [Google Scholar]
- 111.Kyle RA, Leong T, Li S, et al. Complete response in multiple myeloma: clinical trial E9486, an Eastern Cooperative Oncology Group study not involving stem cell transplantation. Cancer. 2006;106(9):1958–1966. doi: 10.1002/cncr.21804. [DOI] [PubMed] [Google Scholar]
- 112.Lahuerta JJ, Mateos MV, Martinez-Lopez J, et al. Influence of pre- and post-transplantation responses on outcome of patients with multiple myeloma: sequential improvement of response and achievement of complete response are associated with longer survival. J Clin Oncol. 2008;26(35):5775–5782. doi: 10.1200/JCO.2008.17.9721. [DOI] [PubMed] [Google Scholar]
- 113.Niesvizky R, Richardson PG, Rajkumar SV, et al. The relationship between quality of response and clinical benefit for patients treated on the bortezomib arm of the international, randomized, phase 3 APEX trial in relapsed multiple myeloma. Br J Haematol. 2008;143(1):46–53. doi: 10.1111/j.1365-2141.2008.07303.x. [DOI] [PubMed] [Google Scholar]
- 114.Hoering A, Crowley J, Shaughnessy JD, Jr, et al. Complete remission in multiple myeloma examined as time-dependent variable in terms of both onset and duration in Total Therapy protocols. Blood. 2009;114(7):1299–1305. doi: 10.1182/blood-2009-03-211953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chou T. Multiple myeloma : recent progress in diagnosis and treatment. J Clin Exp Hematop. 2012;52(3):149–159. doi: 10.3960/jslrt.52.149. [DOI] [PubMed] [Google Scholar]
- 116.Laubach J, Hideshima T, Richardson P, Anderson K. Clinical translation in multiple myeloma: from bench to bedside. Semin Oncol. 2013;40(5):549–553. doi: 10.1053/j.seminoncol.2013.07.009. [DOI] [PubMed] [Google Scholar]
- 117.Laubach JP, Voorhees PM, Hassoun H, Jakubowiak A, Lonial S, Richardson PG. Current strategies for treatment of relapsed/refractory multiple myeloma. Expert Rev Hematol. 2014;7(1):97–111. doi: 10.1586/17474086.2014.882764. [DOI] [PubMed] [Google Scholar]
- 118.Kocoglu M, Badros A. The role of immunotherapy in multiple myeloma. Pharmaceuticals. 2016;9(1):3. doi: 10.3390/ph9010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Landgren O, Gormley N, Turley D, et al. Flow cytometry detection of minimal residual disease in multiple myeloma: Lessons learned at FDA-NCI roundtable symposium. Am J Hematol. 2014;89(12):1159–1160. doi: 10.1002/ajh.23831. [DOI] [PubMed] [Google Scholar]
- 120.Paiva B, Cedena M-T, Puig N, et al. Minimal residual disease monitoring and immune profiling in multiple myeloma in elderly patients. Blood. 2016;127(25):3165. doi: 10.1182/blood-2016-03-705319. [DOI] [PubMed] [Google Scholar]
- 121.Roschewski M, Stetler-Stevenson M, Yuan C, Mailankody S, Korde N, Landgren O. Minimal residual disease: what are the minimum requirements? Journal of Clinical Oncology. 2014;32(5):475–476. doi: 10.1200/JCO.2013.52.1955. [DOI] [PubMed] [Google Scholar]
- 122.Rawstron AC, Child JA, de Tute RM, et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J Clin Oncol. 2013;31(20):2540–2547. doi: 10.1200/JCO.2012.46.2119. [DOI] [PubMed] [Google Scholar]
- 123.Landgren O, Devlin S, Boulad M, Mailankody S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: a meta-analysis. Bone Marrow Transplant. 2016;51(12):1565–1568. doi: 10.1038/bmt.2016.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Paiva B, Vidriales MB, Cervero J, et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood. 2008;112(10):4017–4023. doi: 10.1182/blood-2008-05-159624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jasper GA, Arun I, Venzon D, et al. Variables affecting the quantitation of CD22 in neoplastic B cells. Cytometry B Clin Cytom. 2011;80(2):83–90. doi: 10.1002/cyto.b.20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Morice WG, Hanson CA, Kumar S, Frederick LA, Lesnick CE, Greipp PR. Novel multi-parameter flow cytometry sensitively detects phenotypically distinct plasma cell subsets in plasma cell proliferative disorders. Leukemia. 2007;21(9):2043–2046. doi: 10.1038/sj.leu.2404712. [DOI] [PubMed] [Google Scholar]
- 127.Nishihori T, Song J, Shain KH. Minimal Residual Disease Assessment in the Context of Multiple Myeloma Treatment. Current Hematologic Malignancy Reports. 2016;11:118–126. doi: 10.1007/s11899-016-0308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hedley BD, Keeney M. Technical issues: flow cytometry and rare event analysis. Int J Lab Hematol. 2013;35(3):344–350. doi: 10.1111/ijlh.12068. [DOI] [PubMed] [Google Scholar]
- 129.Subira D, Castanon S, Aceituno E, et al. Flow cytometric analysis of cerebrospinal fluid samples and its usefulness in routine clinical practice. Am J Clin Pathol. 2002;117(6):952–958. doi: 10.1309/123P-CE6V-WYAK-BB1F. [DOI] [PubMed] [Google Scholar]
- 130.Rawstron AC, Bottcher S, Letestu R, et al. Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC) update on the international harmonised approach for flow cytometric residual disease monitoring in CLL. Leukemia. 2013;27(1):142–149. doi: 10.1038/leu.2012.216. [DOI] [PubMed] [Google Scholar]
- 131.Nieto WG, Almeida J, Romero A, et al. Increased frequency (12%) of circulating chronic lymphocytic leukemia-like B-cell clones in healthy subjects using a highly sensitive multicolor flow cytometry approach. Blood. 2009;114(1):33–37. doi: 10.1182/blood-2009-01-197368. [DOI] [PubMed] [Google Scholar]
- 132.Hallett WH, Jing W, Drobyski WR, Johnson BD. Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biol Blood Marrow Transplant. 2011;17(8):1133–1145. doi: 10.1016/j.bbmt.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 133.Walker BA, Wardell CP, Johnson DC, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. 2013;121(17):3413–3419. doi: 10.1182/blood-2012-12-471888. [DOI] [PubMed] [Google Scholar]
- 134.Genadieva-Stavric S, Cavallo F, Palumbo A. New approaches to management of multiple myeloma. Curr Treat Options Oncol. 2014;15(2):157–170. doi: 10.1007/s11864-014-0276-6. [DOI] [PubMed] [Google Scholar]
- 135.Liebisch P, Dohner H. Cytogenetics and molecular cytogenetics in multiple myeloma. Eur J Cancer. 2006;42(11):1520–1529. doi: 10.1016/j.ejca.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 136.Blade J, Samson D, Reece D, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102(5):1115–1123. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- 137.Harousseau J-L, Moreau P. Autologous hematopoietic stem-cell transplantation for multiple myeloma. New England Journal of Medicine. 2009;360(25):2645–2654. doi: 10.1056/NEJMct0805626. [DOI] [PubMed] [Google Scholar]
- 138.Rajkumar SV. Treatment of multiple myeloma. Nat Rev Clin Oncol. 2011;8(8):479–491. doi: 10.1038/nrclinonc.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.O’Donnell EK, Raje NS. New monoclonal antibodies on the horizon in multiple myeloma. Therapeutic Advances in Hematology. 2017;8(2):41–53. doi: 10.1177/2040620716682490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Khagi Y, Mark TM. Potential role of daratumumab in the treatment of multiple myeloma. Onco Targets Ther. 2014;7:1095–1100. doi: 10.2147/OTT.S49480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Frigyesi I, Adolfsson J, Ali M, et al. Robust isolation of malignant plasma cells in multiple myeloma. Blood. 2014;123(9):1336–1340. doi: 10.1182/blood-2013-09-529800. [DOI] [PubMed] [Google Scholar]
- 142.Veillette A, Guo H. CS1, a SLAM family receptor involved in immune regulation, is a therapeutic target in multiple myeloma. Crit Rev Oncol Hematol. 2013;88(1):168–177. doi: 10.1016/j.critrevonc.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 143.Puig N, Sarasquete ME, Balanzategui A, et al. Critical evaluation of ASO RQ-PCR for minimal residual disease evaluation in multiple myeloma. A comparative analysis with flow cytometry. Leukemia. 2014;28(2):391–397. doi: 10.1038/leu.2013.217. [DOI] [PubMed] [Google Scholar]
- 144.Rasmussen T, Poulsen TS, Honore L, Johnsen HE. Quantitation of minimal residual disease in multiple myeloma using an allele-specific real-time PCR assay. Exp Hematol. 2000;28(9):1039–1045. doi: 10.1016/s0301-472x(00)00514-2. [DOI] [PubMed] [Google Scholar]
- 145.Ladetto M, Bruggemann M, Monitillo L, et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia. 2014;28(6):1299–1307. doi: 10.1038/leu.2013.375. [DOI] [PubMed] [Google Scholar]
- 146.Avet-Loiseau H, Corre J, Lauwers-Cances V, et al. Evaluation of minimal residual disease (MRD) by next Generation Sequencing (NGS) Is highly predictive of progression free survival in the IFM/DFCI 2009 trial. Blood. 2015;126(23):191. [Google Scholar]
- 147.Martinez-Lopez J, Lahuerta JJ, Pepin F, et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood. 2014;123(20):3073–3079. doi: 10.1182/blood-2014-01-550020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Munshi NC, Minvielle S, Tai Y-T, et al. Deep Igh sequencing identifies an ongoing somatic hypermutation process with complex and evolving clonal architecture in myeloma. Blood. 2015;126(23):21. [Google Scholar]
- 149.Gormley NJ, Turley DM, Dickey JS, et al. Regulatory perspective on minimal residual disease flow cytometry testing in multiple myeloma. Cytometry B Clin Cytom. 2016;90(1):73–80. doi: 10.1002/cyto.b.21268. [DOI] [PubMed] [Google Scholar]
- 150.Salem D, Stetler-Stevenson M, Yuan C, Landgren O. Myeloma minimal residual disease testing in the United States: Evidence of improved standardization. American Journal of Hematology. 2016;91(12):E502–E503. doi: 10.1002/ajh.24540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Logan AC, Zhang B, Narasimhan B, et al. Minimal residual disease quantification using consensus primers and high-throughput IGH sequencing predicts post-transplant relapse in chronic lymphocytic leukemia. Leukemia. 2013;27(8):1659–1665. doi: 10.1038/leu.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wu D, Sherwood A, Fromm JR, et al. High-throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med. 2012;4(134):134ra163. doi: 10.1126/scitranslmed.3003656. [DOI] [PubMed] [Google Scholar]
- 153.van der Velden VH, Hochhaus A, Cazzaniga G, Szczepanski T, Gabert J, van Dongen JJ. Detection of minimal residual disease in hematologic malignancies by real-time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia. 2003;17(6):1013–1034. doi: 10.1038/sj.leu.2402922. [DOI] [PubMed] [Google Scholar]
- 154.Paiva B, Gutierrez NC, Rosinol L, et al. High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood. 2012;119(3):687–691. doi: 10.1182/blood-2011-07-370460. [DOI] [PubMed] [Google Scholar]
- 155.Paiva B, Martinez-Lopez J, Vidriales MB, et al. Comparison of immunofixation, serum free light chain, and immunophenotyping for response evaluation and prognostication in multiple myeloma. J Clin Oncol. 2011;29(12):1627–1633. doi: 10.1200/JCO.2010.33.1967. [DOI] [PubMed] [Google Scholar]
- 156.Rawstron AC, Paiva B, Stetler-Stevenson M. Assessment of minimal residual disease in myeloma and the need for a consensus approach. Cytometry B Clin Cytom. 2016;90(1):21–25. doi: 10.1002/cyto.b.21272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Paiva B, van Dongen JJM, Orfao A. New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood. 2015;125(20):3059. doi: 10.1182/blood-2014-11-568907. [DOI] [PMC free article] [PubMed] [Google Scholar]